
Intermolecular oxidative radical fluoroalkylfluorosulfonylation of unactivated alkenes with (fluoroalkyl)trimethylsilane, silver fluoride, sulfur dioxide and N-fluorobenzenesulfonimide†
Qiongzhen
Lin‡
ab,
Yongan
Liu‡
b,
Zhiwei
Xiao
b,
Liping
Zheng
c,
Xiumiao
Zhou
c,
Yong
Guo
 b,
Qing-Yun
Chen
b,
Changge
Zheng
*a and
Chao
Liu
*b
b,
Qing-Yun
Chen
b,
Changge
Zheng
*a and
Chao
Liu
*b
aSchool of Chemical Engineering, Xinjiang Agricultural University, Urumqi, Xinjiang Uygur Autonomous Region 830052, China. E-mail: cgzheng@jiangnan.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: chaoliu@sioc.ac.cn
cSchool of Chemical Engineering and Food Science, Zhengzhou Institute of Technology, 18 Yingcai Street, Zhengzhou 450044, China
First published on 19th December 2018
Abstract
An intermolecular oxidative radical fluoroalkylfluorosulfonylation reaction of unactivated alkenes with convenient and commercially available (fluoroalkyl)trimethylsilane, silver fluoride, sulfur dioxide and N-fluorobenzenesulfonimide (NFSI) is described. This transformation efficiently affords various fluoroalkyl-containing alkyl sulfonyl fluorides with good functional group tolerance under mild conditions. Silver fluoroalkyl complexes as the fluoroalkyl radical source generated from (fluoroalkyl)trimethylsilane and silver fluoride may be the key intermediate.
The fluoroalkyl group is one of the most important fluorine-containing groups with unique physical and chemical properties. Its selective introduction into diverse classes of organic molecules commonly has beneficial and profound effects on the properties of parent organic molecules.1 Although impressive progress has been made in direct fluoroalkylation reactions in recent years,2 there is still a high demand for rapid and efficient installation of a range of fluoroalkyl groups to target molecules using convenient and commercial available fluoroalkylation reagents. Additionally, due to the unusual stability of the SVI–F bond, fluorosulfonyl (FSO2) groups have unique reactivity–stability patterns and have broad applications in the field of chemical biology, organic synthesis and materials.3 Notably, although most sulfonyl fluorides studied or utilized in the literature are aromatic sulfonyl fluorides,4 the corresponding aliphatic derivatives have also shown promising results as tool compounds in chemical biology3a,5 and as better alternatives to sulfonyl chlorides in sulfonylation reactions, especially for parallel synthesis.6 The underestimation of aliphatic sulfonyl fluorides in the literature is probably due to the lack of direct and efficient synthetic approaches to them. To address these issues, an efficient and promising method is to concomitantly introduce a fluoroalkyl group and a fluorosulfonyl group into valuable synthetic targets in one step, and the fluoroalkylfluorosulfonylation of alkenes via a radical pathway should be a desirable entry to fulfil the task since the intermolecular radical 1,2-difunctionalization type fluoroalkylation of alkenes has emerged as a powerful tool for the introduction of versatile fluoroalkyl groups into target molecules.7
Recently, we developed a novel intermolecular oxidative radical trifluoromethylfluorosulfonylation reaction of unactivated alkenes with readily available Ag(O2CCF2SO2F) and N-fluorobenzenesulfonimide (NFSI) (Scheme 1a).8 Although the reaction efficiently resulted in CF3-containing alkyl sulfonyl fluorides, only the CF3 group can be incorporated. Preliminary mechanistic experiments showed that AgCF3 species may be involved in the reaction mechanism as the key intermediate. Also inspired by the recent advance in radical insertion reactions of sulfur dioxide,9 we then envision that if the key silver fluoroalkyl complexes (AgRF)10 generated from convenient and commercially available reagents TMSRF and AgF can be utilized in similar reactions in combination with sulfur dioxide, introduction of various fluoroalkyl groups and fluorosulfonyl groups into unactivated alkenes may be expected (Scheme 1b). As a continuation of our research interest in radical fluoroalkylation and sulfur dioxide utilization,8,11 we herein present the results.
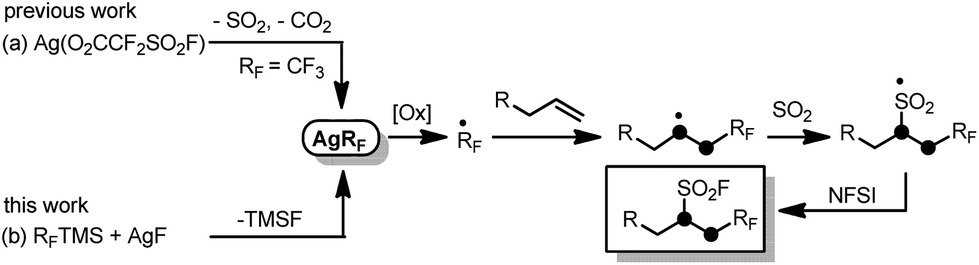 | ||
| Scheme 1 Intermolecular oxidative radical fluoroalkylfluorosulfonylation of unactivated alkenes. | ||
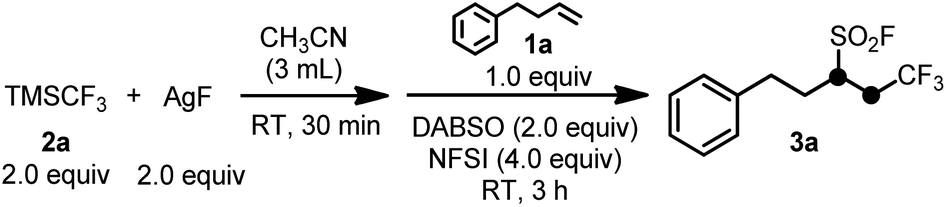
Initial studies were carried out using (trifluoromethyl)trimethylsilane (Me3SiCF3 or TMSCF3, commonly known as the Ruppert–Prakash reagent, widely-used and a commercially available nucleophilic trifluoromethylating agent)12 and silver fluoride to generate the key AgCF3 species,10 4-phenyl-1-butene (1a) as the model alkene substrate, 1,4-diazabicyclo[2.2.2]octane-bis(sulfur dioxide) adduct (DABSO)13 as a convenient and commercially available solid source of SO2, and NFSI as an electrophilic fluorination reagent. In our optimization studies of the reaction conditions, it was found that 2.0 equiv. of TMSCF3, 2.0 equiv. of AgF, 1.0 equiv. of 1a, 2.0 equiv. of DABSO, and 4.0 equiv. of NFSI in 3 mL of CH3CN at room temperature were the suitable conditions to afford the desired product 3a in excellent yield (86%, Table 1, entry 1). Replacement of CH3CN with other common reaction solvents, such as DMF, DMSO NMP or THF resulted in lower yields of 3a (Table 1, entries 2–4). Notably, water was found to be harmful for the desired reaction since using small amounts of water as the co-solvent resulted in no formation of 3a (Table 1, entry 5). An attempt at increasing the yield of the desired product by utilizing some additives met with failure (Table 1, entries 6–13). While increased reaction temperatures had a deleterious effect on the desired reaction, room temperature or 0 °C led to an excellent yield of the target product 3a (Table 1, entries 1, 14 and 15). Finally, the increased concentration of TMSCF3 and AgF did not have a significant effect on the reaction (Table 1, entry 16).
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yield of 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a General reaction conditions: TMSCF3 (0.4 mmol) and AgF (0.4 mmol) in CH3CN (3 mL) were stirred at room temperature under an Ar atmosphere for 30 min, and then 4-phenyl-1-butene (1a, 0.2 mmol), DABSO (0.4 mmol) and NFSI (0.8 mmol) were added in turn and stirred for 3 h. b Yields were determined by 19F NMR spectroscopy using 1-methoxy-4-(trifluoromethoxy)benzene as an internal standard. | ||
| 1 | None | 86 |
| 2 | DMF instead of CH3CN | 42 |
| 3 | DMSO instead of CH3CN | 49 |
| 4 | NMP or THF instead of CH3CN | 0 |
| 5 | 0.1 mL of H2O as a co-solvent | 0 |
| 6 | 1.0 equiv. of pyridine as an additive | 19 |
| 7 | 1.0 equiv. of 2,6-dimethylpyridine as an additive | 63 |
| 8 | 1.0 equiv. of 2,2′-bipyridine as an additive | 11 |
| 9 | 1.0 equiv. of o-phenanthroline as an additive | 10 |
| 10 | 1.0 equiv. of 2,6-di-tert-butylpyridine as an additive | 72 |
| 11 | 1.0 equiv. of PPh3 as an additive | 65 |
| 12 | 1.0 equiv. of Et3N as an additive | 73 |
| 13 | 1.0 equiv. of 2,4,6-collidine as an additive | 75 |
| 14 | 0 °C | 86 |
| 15 | 50 °C | 76 |
| 16 | 4.0 equiv. of TMSCF3 and 4.0 equiv. of AgF | 87 |
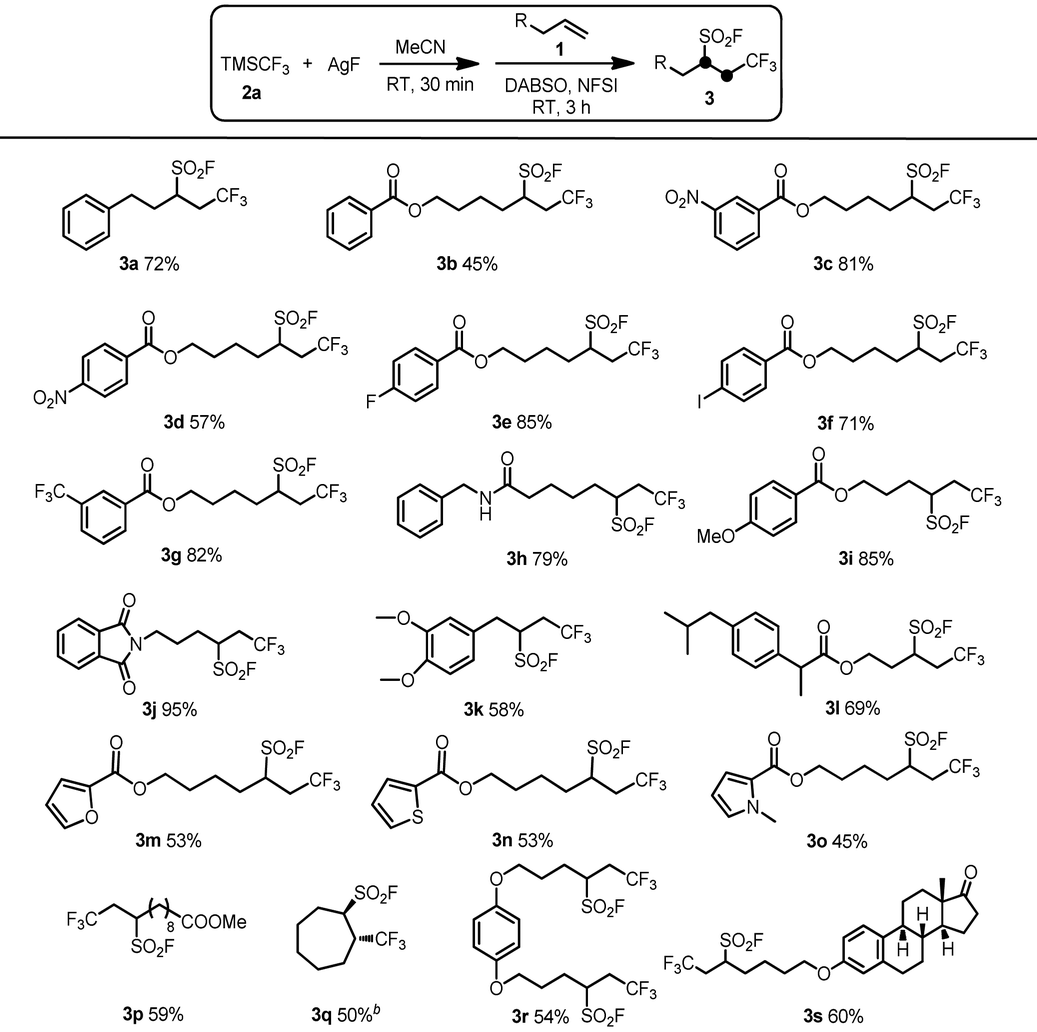
With the optimal reaction conditions established, the substrate scope of the intermolecular oxidative radical fluoroalkylfluorosulfonylation reactions with respect to unactivated alkenes was explored. As shown in Table 2, a range of unactivated alkenes participated in our protocol, providing good yields of the desired products 3. Various functional groups including nitro (3c, 3d), halogen (3e, 3f), amide (3h), phthalimide (3j), ether (3i, 3k, 3r, 3s), ester (3b–i, 3l–p), and heterocyclic (3m–o) were well tolerated under the reaction conditions providing the corresponding target products in good yields. In particular, the iodo group in substrate 1f can survive the standard reaction conditions, affording the desired product 3f in good yield. Substrate 1r with two terminal alkenyl groups was smoothly applied to the fluoroalkylfluorosulfonylation reaction to result in the desired product 3r in an acceptable yield. Moreover, an estrone derivative with an alkenyl group was also a suitable partner for this transformation to successfully produce the desired product 3s.
| a Reaction conditions: TMSCF3 (0.6 mmol) and AgF (0.6 mmol) in CH3CN (4.5 mL) were stirred at room temperature under an Ar atmosphere for 30 min, and then the alkene 1 (0.3 mmol), DABSO (0.6 mmol) and NFSI (1.2 mmol) were added in turn and stirred for 3 h. Yields refer to chromatographically pure material unless otherwise noted. b Yields were determined by 19F NMR spectroscopy with 1-methoxy-4-(trifluoromethoxy)benzene as an internal standard. |
|---|
|
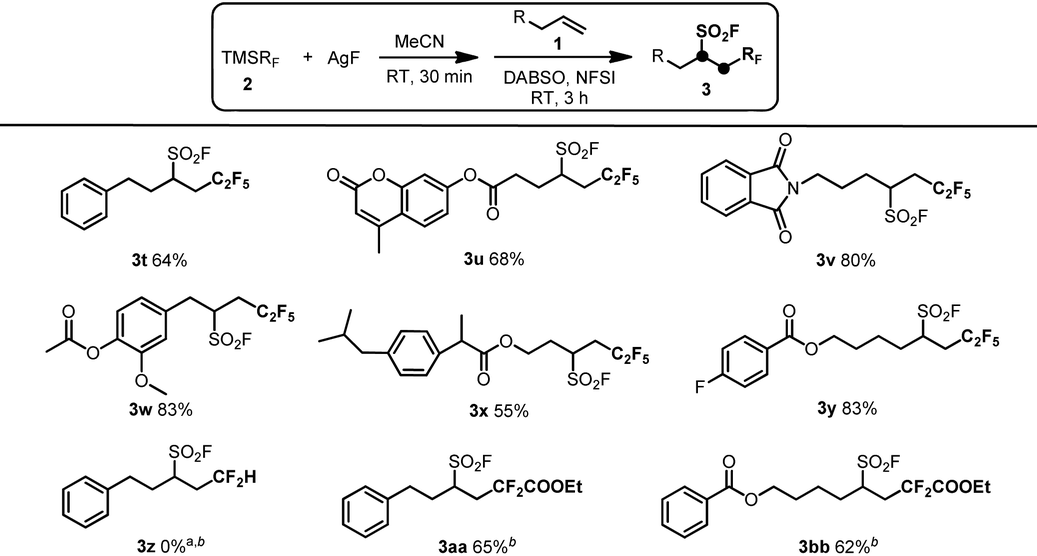
To further explore the application of this protocol, various (fluoroalkyl)trimethylsilanes (TMSRF) were employed under the optimized reaction conditions (Table 3). As expected, replacement of CF3 in TMSCF3 with other perfluoroalkyl groups like the C2F5 group resulted in the desired pentafluoroethylfluorosulfonylation products in good yields under similar reaction conditions (3t–y). However, the use of TMSCF2H instead of TMSCF3 under the standard or modified reaction conditions resulted in no formation of the desired product 3z, probably due to the relative unstability of the key intermediate AgCF2H and the decreased electrophilic ability of the CF2H radical compared with the perfluoroalkyl radical such as the CF3 or C2F5 radical. To our surprise, the use of TMSCF2COOEt as the reactant does not appear to be effective under the standard reaction conditions and led to lower yields of the target products 3aa and 3bb. Notably, good yields of the desired products 3aa and 3bb were achieved when the formation of the key intermediate AgCF2COOEt was performed at 0 °C in 15 min, which might be ascribed to the increased stability of AgCF2COOEt at lower reaction temperatures. All these experimental results showed that the intermolecular oxidative radical fluoroalkylfluorosulfonylation reaction of unactivated alkenes is sensitive to the stability of the key intermediate AgRF and the electronic properties of the corresponding fluoroalkyl radical generated from TMSRF and AgF.
| a Reaction conditions: TMSRF (0.6 mmol) and AgF (0.6 mmol) in CH3CN (4.5 mL) were stirred at room temperature under an Ar atmosphere for 30 min, and then the alkene 1 (0.3 mmol), DABSO (0.6 mmol) and NFSI (1.2 mmol) were added in turn and stirred for 3 h. Yields of isolated products were reported. b Reaction conditions: TMSRF (0.6 mmol) and AgF (0.6 mmol) in CH3CN (4.5 mL) were stirred at 0 °C under an Ar atmosphere for 15 min, and then the alkene 1 (0.3 mmol), DABSO (0.6 mmol) and NFSI (1.2 mmol) were added in turn and stirred for 3 h. Yields of isolated products were reported. |
|---|
|
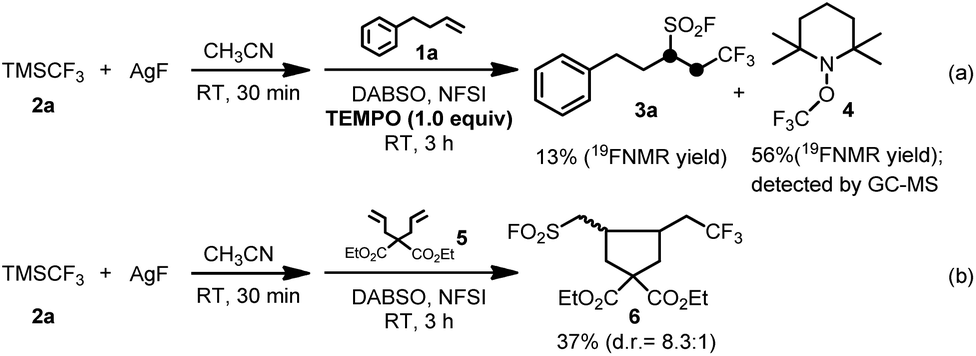
Two sets of control experiments were carried out to shed light on the reaction mechanism. In the first experiment, 2,2,6,6-tetramethyl-1-piperidyloxy (TEMPO) as a radical scavenger was used to trap the possible radical intermediates generated in the reaction system. The corresponding TEMPO-trapped complex 4 was obtained in 56% yield on the basis of 19F NMR spectroscopic analysis along with only 13% yield of the desired product 3a (Scheme 2a). In the second experiment, alkene 5 was subjected to the standard reaction conditions to generate the ring-closed product 6 in 37% isolated yield (Scheme 2b). We reasoned that the alkyl radical generated in situ from the addition of the CF3 radical to alkene 5 undergoes an irreversible intramolecular cyclization at a much faster rate than that of the consequent radical insertion of sulfur dioxide and a rapid fluorination process. The above experiments strongly suggested that a radical reaction pathway may be involved in the fluoroalkylfluorosulfonylation reaction of unactivated alkenes, and the corresponding silver fluoroalkyl species as the key intermediate can produce a fluoroalkyl radical to initiate the desired reaction, and the resulting alkylsulfonyl radical derived from the radical insertion of sulfur dioxide is rapidly fluorinated by NFSI to give the final desired product (Scheme 1).
 | ||
| Scheme 2 Control experiments. | ||
In conclusion, we have reported an intermolecular oxidative radical fluoroalkylfluorosulfonylation reaction of unactivated alkenes with convenient and commercially available (fluoroalkyl)trimethylsilane, silver fluoride, sulfur dioxide and N-fluorobenzenesulfonimide. This transformation efficiently affords various fluoroalkyl-containing alkyl sulfonyl fluorides with good functional group tolerance under mild conditions. Silver fluoroalkyl complexes generated from (fluoroalkyl)trimethylsilane and silver fluoride may be the key intermediate as the fluoroalkyl radical source, and the alkyl radical produced from the addition of a fluoroalkyl radical to an alkene undergoes radical insertion with sulfur dioxide and the consequent rapid fluorination with NFSI to afford the final product.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21421002, 21302207, 21871283), the Science and Technology Commission of Shanghai Municipality (No. 17ZR1437000), the Foundation of Science and Technology on Sanming Institute of Fluorochemical Industry (FCIT201701BR), and the Henan Province Science and Technology Open Cooperation Program (18210600017).Notes and references
- For selected reviews, see: (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31 RSC.
- For selected reviews on fluoroalkylation reactions, see: (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS; (b) Y. Lu, C. Liu and Q.-Y. Chen, Curr. Org. Chem., 2015, 19, 1638 CrossRef CAS; (c) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed; (d) G. Landelle, A. Panossian, S. Pazenok, J.-P. Vors and F. R. Leroux, Beilstein J. Org. Chem., 2013, 9, 2476 CrossRef PubMed; (e) B. Lantaño, M. R. Torviso, S. M. Bonesi, S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2015, 285, 76 CrossRef; (f) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed; (g) Q. Liu, C. Ni and J. Hu, Natl. Sci. Rev., 2017, 4, 303 CrossRef; (h) H.-X. Song, Q.-Y. Han, C.-L. Zhao and C.-P. Zhang, Green Chem., 2018, 20, 1662 RSC; (i) S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287 CrossRef CAS.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430 CrossRef CAS; (b) B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083 CrossRef CAS PubMed; (c) Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919 CrossRef CAS PubMed; (d) T. J. Guo, G. Y. Meng, X. J. Zhan, Q. Yang, T. C. Ma, L. Xu, K. B. Sharpless and J. Dong, Angew. Chem., Int. Ed., 2018, 57, 2605 CrossRef CAS PubMed.
- For selected recent examples, see: (a) L. Tang, Y. Yang, L. Wen, X. Yang and Z. Wang, Green Chem., 2016, 18, 1224 RSC; (b) A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233 RSC; (c) Y. Chen and M. C. Willis, Chem. Sci., 2017, 8, 3249 RSC; (d) A. L. Tribby, I. Rodríguez, S. Shariffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294 CrossRef CAS PubMed.
- (a) S. O. Alapafuja, S. P. Nikas, I. T. Bharathan, V. G. Shukla, M. L. Nasr, A. L. Bowman, N. Zvonok, J. Li, X. Shi, J. R. Engen and A. Makriyannis, J. Med. Chem., 2012, 55, 10074 CrossRef CAS PubMed; (b) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650 RSC.
- A. V. Bogolubsky, Y. S. Moroz, P. K. Mykhailiuk, S. E. Pipko, A. I. Konovets, I. V. Sadkova and A. Tolmachev, ACS Comb. Sci., 2014, 16, 192 CrossRef CAS PubMed.
- For selective reviews on radical 1,2-difunctionalization type fluoroalkylation, see: (a) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (c) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS PubMed; (d) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS PubMed; (e) X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821 CrossRef CAS; (f) X. Wu, S. Wu and C. Zhu, Tetrahedron Lett., 2018, 59, 1328 CrossRef CAS. For selected examples on radical 1,2-difunctionalization type fluoroalkylation, see: (g) X. Wu, L. Chu and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 2198 CrossRef CAS PubMed; (h) Y.-F. Wang, G. H. Lonca and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1067 CrossRef CAS PubMed; (i) G. Ma, W. Wan, J. Li, Q. Hu, H. Jiang, S. Zhu, J. Wang and J. Hao, Chem. Commun., 2014, 50, 9749 RSC; (j) W. Yu, X.-H. Xu and F.-L. Qing, Adv. Synth. Catal., 2015, 357, 2039 CrossRef CAS; (k) Y.-B. Wu, G.-P. Lu, T. Yuan, Z.-B. Xu, L. Wan and C. Cai, Chem. Commun., 2016, 52, 13668 RSC; (l) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388 CrossRef CAS; (m) Y.-F. Cheng, X.-Y. Dong, Q.-S. Gu, Z.-L. Yu and X.-Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 8883 CrossRef CAS PubMed; (n) Q. Zhao, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2017, 56, 11575 CrossRef CAS PubMed; (o) J. Yu, Z. Wu and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 17156 CrossRef CAS PubMed; (p) X.-T. Li, Q.-S. Gu, X.-Y. Dong, X. Meng and X.-Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 7668 CrossRef CAS PubMed.
- Y. Liu, H. Wu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Angew. Chem., Int. Ed., 2017, 56, 15432 CrossRef CAS PubMed.
- For recent reviews on sulfur dioxide radical insertion reactions: (a) D. Zheng and J. Wu, Sulfur Dioxide Insertion Reactions for Organic Chemisty, Springer, Singapore, 2017 CrossRef; (b) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691 RSC; (c) K. Hofman, N.-W. Liu and G. Manolikakes, Chem. – Eur. J., 2018, 24, 11852 CrossRef CAS PubMed.
- (a) W. Tyrra and D. Naumann, J. Fluorine Chem., 2004, 125, 823 CrossRef CAS; (b) Y. Zeng, L. Zhang, Y. Zhao, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955 CrossRef CAS PubMed; (c) Y. Ye, S. H. Lee and M. S. Sanford, Org. Lett., 2011, 13, 5464 CrossRef CAS PubMed.
- (a) H. Wu, Z. Xiao, J. Wu, Y. Guo, J.-C. Xiao, C. Liu and Q.-Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 4070 CrossRef CAS PubMed; (b) B. He, Z. Xiao, H. Wu, Y. Guo, Q.-Y. Chen and C. Liu, RSC Adv., 2017, 7, 880 RSC; (c) Z. Xiao, Y. Liu, L. Zheng, C. Liu, Y. Guo and Q.-Y. Chen, J. Org. Chem., 2018, 83, 5836 CrossRef CAS PubMed; (d) Z. Xiao, Y. Liu, L. Zheng, X. Zhou, Y. Xie, C. Liu, Y. Guo and Q.-Y. Chen, Tetrahedron, 2018, 74, 6213 CrossRef CAS; (e) Y. Liu, Q. Lin, Z. Xiao, C.-G. Zheng, Y. Guo, Q.-Y. Chen and C. Liu, Chem. – Eur. J., 2019, 25 DOI:10.1002/chem.201805526.
- (a) G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757 CrossRef CAS; (b) G. K. S. Prakash, R. Krishnamurti and G. A. Olah, J. Am. Chem. Soc., 1989, 111, 393 CrossRef CAS; (c) I. Ruppert, K. Schlich and W. Volbach, Tetrahedron Lett., 1984, 25, 2195 CrossRef CAS; (d) S. Krishnamoorthy and G. K. S. Prakash, Synthesis, 2017, 49, 3394 CrossRef CAS.
- (a) P. S. Santos and M. T. S. Mello, J. Mol. Struct., 1988, 178, 121 CrossRef CAS; (b) B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372 CrossRef CAS PubMed; (c) H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876 CrossRef CAS PubMed; (d) E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo01192c |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2019 |