
Multimicelle aggregate mechanism for spherical multimolecular micelles: from theories, characteristics and properties to applications
Meiwei
Qi
and
Yongfeng
Zhou
 *
*
School of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China. E-mail: yfzhou@sjtu.edu.cn; Fax: +86 21 54741297
First published on 20th August 2019
Abstract
Spherical micelles are the most commonly and widely used supramolecular structures in the self-assembly studies of amphiphilic polymers and for applications in areas such as sensing, drug delivery, diagnostic and functional materials. According to their structure and size, spherical micelles can be classified as small simple micelles (with size less than 30 nm) and large complex micelles (with size more than 30–50 nm); the formation of small simple micelles can generally be explained by a “star micelle” mechanism; however, this mechanism cannot be used to explain the formation of large complex micelles. Currently, the mechanisms to explain the formation of large complex micelles include “large compound micelle”, “multicompartment micelle”, “supermicelle” and “multimicelle aggregate” (MMA) mechanisms. The MMA mechanism can be divided into unimolecular micelle aggregate (UMA) and small micelle aggregate (SMA) mechanisms and is quite different from other mechanisms in terms of the structure and property of micelles; in addition, although MMA was proposed to explain the formation of large multimolecular micelles from hyperbranched polymers at the beginning, to date, it has been proved to be applicable for explaining the formation of large micelles from other amphiphilic polymers such as dendrimers, comb polymers, graft polymers, star polymers, linear polymers and even small molecules. Furthermore, due to their special structure, MMAs have shown unique potential applications in light-emitting materials, light-harvesting systems, nanoporous materials and biomedical areas; in this review, we have summarized the recent progress in the mechanism, precursors, structural characteristics and properties of MMAs as well as their applications.
 Meiwei Qi | Meiwei Qi completed her BSc and MSc degrees at the Harbin Institute of Technology. In 2014, she joined Prof. Yongfeng Zhou's group as a PhD student in Chemistry at Shanghai Jiao Tong University. Currently, she is working on the synthesis, self-assembly and cytomimetic applications of responsive polymers. |
 Yongfeng Zhou | Yongfeng Zhou completed his BSc and MSc degrees in Chemistry at Harbin Institute of Technology. In 2005, he obtained his PhD degree in Polymer Chemistry and Physics from Shanghai Jiao Tong University. Then, he worked in the same university as a lecturer and as an associate professor. In2010, he was promoted as a full professor there. His current research interests include the controllable synthesis, self-assembly, computer simulation and cytomimetic application of hyperbranched polymers or alternating copolymers. |
1. Introduction
Micelle, a term from biology, is a core–corona supramolecular aggregate of amphiphilic molecules dispersed in a liquid. A typical micelle possesses a solvophobic inner core and a solvophilic outer corona. The inner core is composed of the solvophobic segments of amphiphiles, creating an encapsulation space for the solubilisation of hydrophobic guest molecules. The outer corona is formed from the solvophilic segments, contacting the surrounding solvent closely to stabilize the micelle.1 Moreover, micelles are approximately spherical in shape. Surfactant micelles and polymer micelles are two basic categories of spherical micelles. In an aqueous solution, when the surfactant concentration is above the critical micelle concentration (CMC), surfactant molecules tend to aggregate together with the hydrophilic heads on the surface of micelles in contact with the surrounding water molecules and the hydrophobic tails accumulating in the micelle centre, sequestered from the external water molecules.2 Due to low molecular weight of surfactant molecules (a few hundreds of grams per mole), the size of a surfactant micelle is generally small i.e. about the length of two molecules. Polymers, with high molecular weight, have distinct advantages, such as much lower CMC, high disassembly resistance upon dilution in some cases and a large adjustable range of size, over surfactants for the formation of micelles. Thus, polymer micelles have received significant attention from both academia and industry. They can be divided into unimolecular micelles and multimolecular micelles. The former are quite simple, whereas the latter are really complex and cannot be easily discerned; to date, several mechanisms have been proposed to explain the formation of polymeric multimolecular micelles, which have been discussed in the present review article. Herein, the multimolecular micelles are subdivided into two types according to the micelle size: micelles smaller than 30–50 nm, called small simple micelles, and micelles larger than 50 nm, called large complex micelles (>50 nm). The small simple micelles generally involve a simple one-step aggregation process; however, the formation process of large complex micelles is usually accompanied by multi-step aggregation.The formation of small simple micelles is very common during the self-assembly of amphiphilic linear diblock copolymers. Fig. 1a shows the typical structure of these micelles. Generally, the micelle cores are formed by the aggregation of hydrophobic blocks, and the shells are formed by the hydrophilic blocks of the amphiphilic block copolymers.3,4 These micelles are similar to the surfactant micelles, and their diameter is about the length of two molecules and often less than 30–50 nm. Generally, these small simple micelles are called “star micelles”.
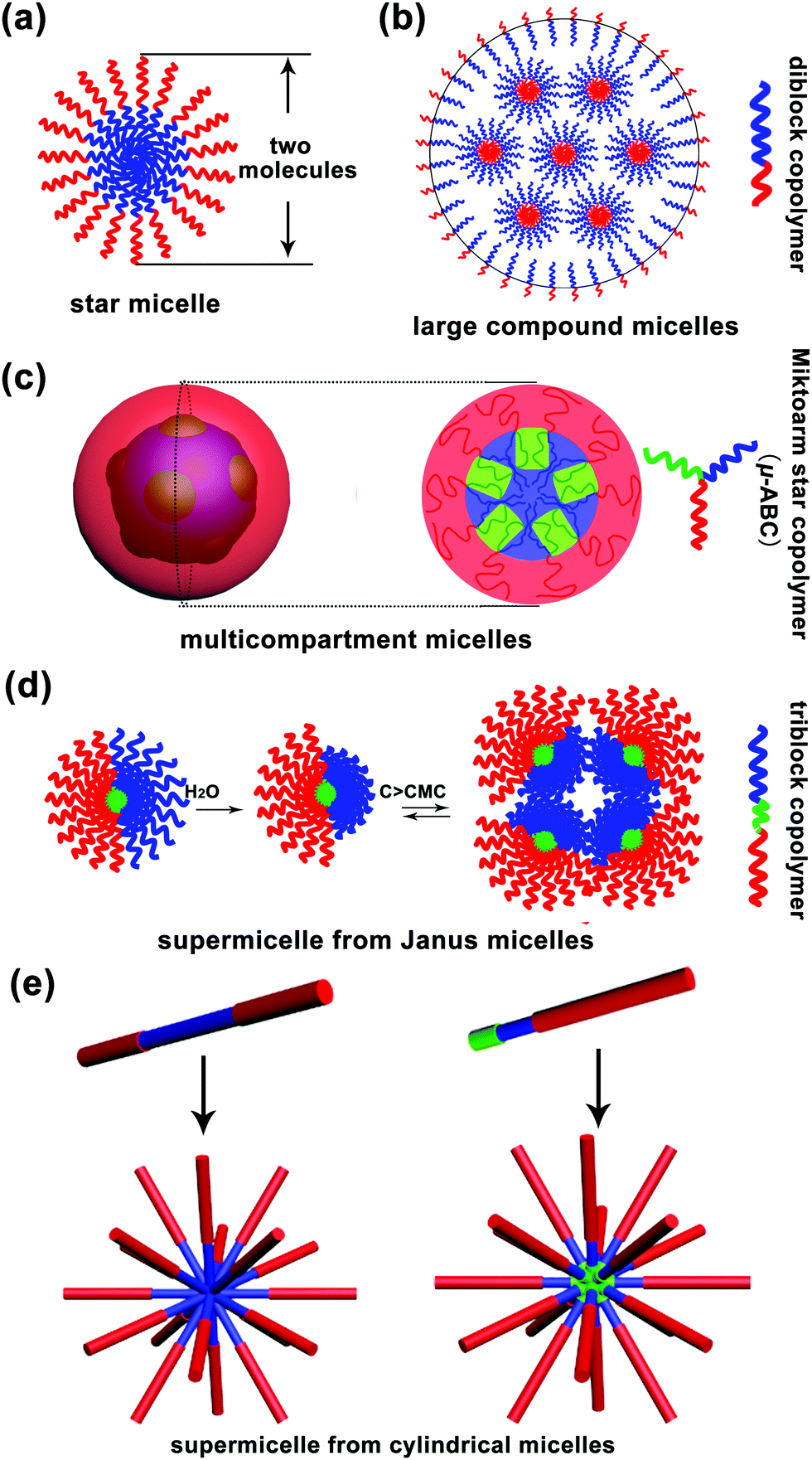 | ||
| Fig. 1 Schematic of the various self-assembly mechanisms of spherical micelles: (a) star micelle, (b) large compound micelle, (c) multicompartment micelle, and (d) and (e) supermicelle mechanisms. Red indicates the hydrophilic segments, blue indicates the hydrophobic segments and green indicates incompatible segments with the abovementioned two segments. | ||
In addition to the above mentioned small simple micelles, multimolecular micelles above 50 nm are also widely observed in a polymer self-assembly, which have been termed large complex micelles herein. For example, Eisenberg and coworkers found that polystyrene-b-poly acrylic acid (PS-b-PAA) diblock copolymers could self-assemble into large micelles of up to 1200 nm in a solvent; however, the formation of these large micelles could not be explained by the “star micelle” mechanism;5 they put forward the famous “large compound micelle (LCM)” mechanism to explain their formation.5,6 The key feature of LCMs is that they have hydrophilic surfaces and are filled with microphase-separated reverse micelle aggregates (Fig. 1b). Note that LCMs are generally formed from “crew-cut” amphiphilic diblock copolymers with short hydrophilic blocks and long hydrophobic blocks.
Furthermore, amphiphilic tri- or multi-block copolymers can self-assemble into large complex micelles with size above 50 nm. In 2004, Lodge, Hillmyer and coworkers reported the formation of multicompartment micelles through the self-assembly of μ-typed ABC star copolymers according to cryo-transmission electron microscopy (cryo-TEM) measurements.7 After this, a significant number of multicompartment micelles have been designed and reported.7 Multicompartment micelles are generally defined as a supramolecular assembly composed of a hydrophilic shell and a hydrophobic core with self-segregated domains.8,9 However, an observable phase separation inside the micelle core is not sufficient to define a micelle as a multicompartment system, and the key is that various domains formed by phase separation must enable the selective confinement of different hydrophobic molecules.8 According to the abovementioned definitions, the copolymers that have the ability to form multicompartment micelles are mainly triblock copolymers of the form ABC, where A is a hydrophilic block, and B and C are hydrophobic blocks, such as hydrocarbon-dominated or fluorocarbon-dominated hydrophobic blocks, and highly incompatible with each other. To date, these triblock copolymers include miktoarm star μ-ABC, star-block ABC, linear triblock copolymer ABC, graft copolymer A-graft-B/C and graft block A-graft-B-b-C copolymers. A typical schematic for the formation of a multicompartment micelle from the miktoarm star μ-ABC copolymers is shown in Fig. 1c.10
In addition to the multicompartment micelle mechanism, there is another special self-assembly mechanism, named as “supermicelle mechanism”, that explains the formation of large complex micelles; “supermicelles” are based on the specially designed “Janus micelles”, which have two chemically different half spheres in the corona.9 Due to their special geometry, these Janus micelles exhibit a strong tendency to aggregate into larger micelles that are regarded as “supermicelles”.11 These “supermicelles” generally have a large size of more than 100 nm and even around 500 nm. In 2003, Müller and coworkers12 first prepared amphiphilic Janus micelles by cross-linking the middle block of a microphase-separated polystyrene-b-polybutadiene-b-poly(methyl methacrylate) (PS-b-PB-b-PMMA) triblock copolymer in the bulk state, followed by alkaline hydrolysis of the PMMA ester groups. Above the critical aggregation concentration (CAC), these amphiphilic Janus micelles aggregated into “supermicelles” in an aqueous solution (Fig. 1d). Subsequently, many “supermicelles” obtained from different “Janus micelles” have been reported.13–17 The formation of “supermicelles” requires many qualified conditions. Most importantly, the block copolymers should self-assemble into “Janus micelles” before finally forming “supermicelles”.18 Typically, for the ABC-triblock copolymers, the middle block B is physically or chemically cross-linked to form the core of the micelle, and the incompatible blocks A and C separate into two hemispheres.19–21
In addition to the abovementioned supermicelles obtained from spherical Janus micelles, there is another kind of “supermicelles” assembled from long cylindrical micelles, as first reported by Manners and coworkers in 2012.22 These supermicelles are a hierarchical supramolecular assembly, and the primary cylindrical micelles are block co-micelles prepared by a living crystallization-driven self-assembly (CDSA) in solution. Through the introduction of hydrogen bonds, electrostatic or solvophobic interactions, the primary cylindrical micelles self-assembled into supermicelles with various patterns such as star-, cross-, brush-, train track-, windmill-like and even more complex patterns (Fig. 1e);22–27 however, due to the limitations of CDSA, to date, these supermicelles have been mainly obtained from copolymers containing a polyferrocenydimethylsilane (PFS) block. Note that in this review, we have only considered the “supermicelles” obtained from spherical Janus micelles, and those obtained from cylindrical micelles have not been included.
In addition to linear block copolymers, dendritic polymers, including dendrimers and hyperbranched polymers, are very promising polymer precursors in self-assembly. Generally, amphiphilic dendritic copolymers only assemble into unimolecular micelles in dilute solutions. However, in 2005, our group synthesized a series of hyperbranched multiarm copolyethers with a hydrophobic hyperbranched poly[3-ethyl-3-(hydroxymethyl)oxetane] core (HBPO) and many hydrophilic poly(propylene oxide) (PPO) arms (HBPO-star-PPO) and found that these copolyethers could form size-controlled large complex micelles with size in the range from 100 to 300 nm depending on the molar ratios of the PPO arms to the HBPO cores. Due to the special spherical and highly branched topological structure of hyperbranched copolymers, the abovementioned self-assembly mechanisms of “star micelles”, “LCMs”, “multicompartment micelles” or “supermicelles” are not suitable for HBPO-star-PPO large micelles. To explain the formation of these large spherical micelles, we utilized nuclear magnetic resonance (NMR) and variable temperature Fourier transform infrared spectroscopy (FTIR) to analyse the large micelles and first suggested that these large spherical micelles might be a kind of multimicelle aggregates (MMAs) formed by the aggregation of small micelles associated by intermicellar interactions such as hydrogen bonds (Fig. 2a).28 Moreover, the equilibrium between the inter-micellar interactions and micellar solvation determines the size of the MMAs. Subsequently, our group synthesized a hyperbranched multiarm copolymer with an HBPO core and many poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) arms (HBPO-star-PDMAEMA) (Fig. 2b) and found that these copolymers could assemble into large multimolecular micelles. The magnified TEM image (Fig. 2d) evidently indicates that the large micelles have been built from the small spherical building units with the size of around 4.7 nm.29 Due to the small size of the building units, the large micelles should be a kind of MMAs obtained from the aggregation of unimolecular micelles without phase separation. Haag's group independently proved the MMA mechanism for the self-assembly of large micelles from dendritic copolymers.30 In subsequent experiments, our group found that adamantyl-terminated hyperbranched polyglycerol (HPG-AD) (Fig. 2c) could also form large MMAs from the aggregation of small micelles that were around 20–30 nm in size and larger than unimolecular micelles (Fig. 2e).31 Furthermore, the MMA mechanism was verified by dissipative particle dynamics (DPD) simulations.32
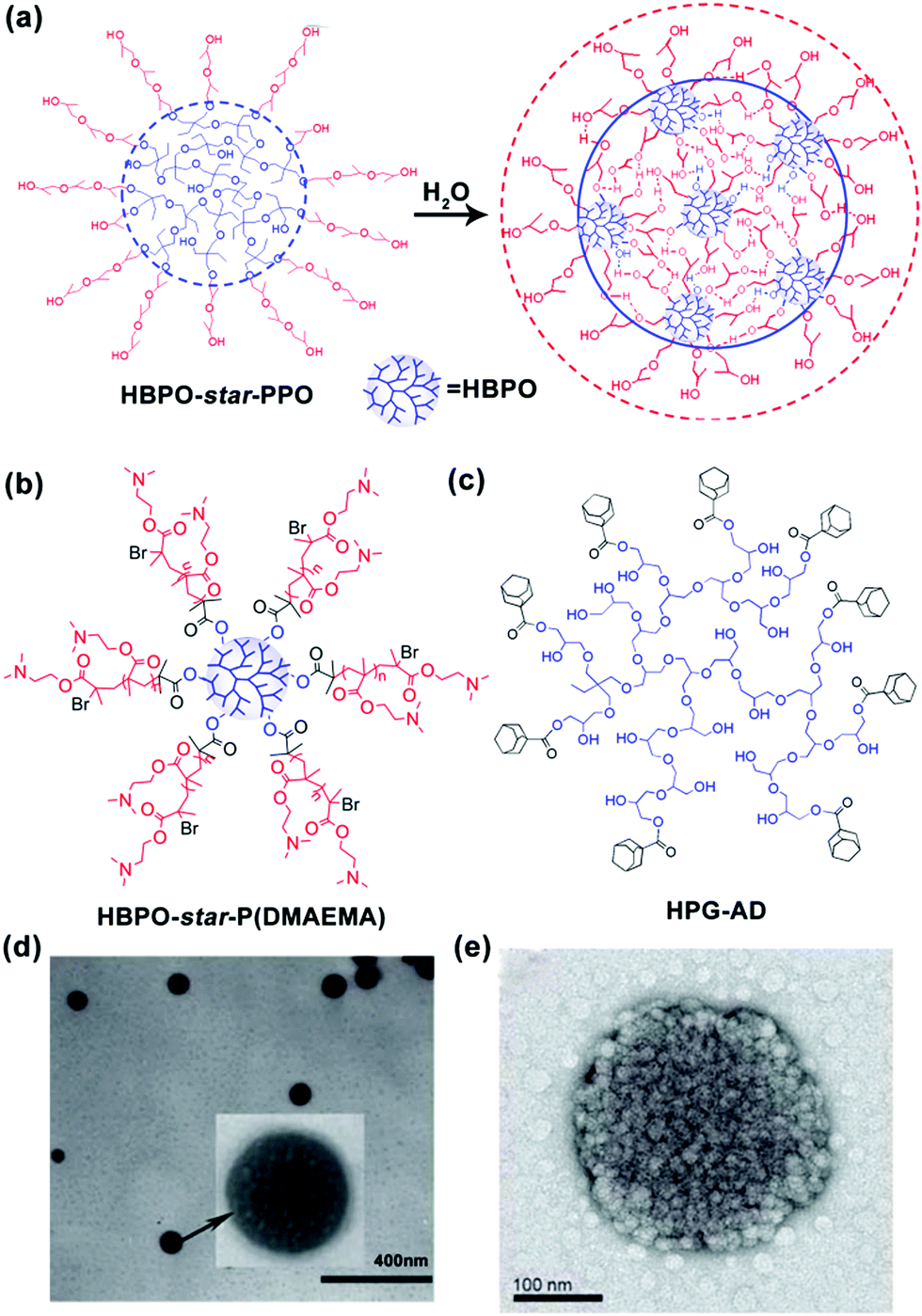 | ||
| Fig. 2 (a) Schematic for the formation of MMAs from HBPO-star-PPO. (b) Chemical structure of HBPO-star-P(DMAEMA), (c) Chemical structure of HPG-AD, (d) TEM image of MMAs obtained from HBPO-star-P(DMAEMA), and (e) TME image of MMA obtained from HPG-AD. Reproduced with permission from ref. 28. Copyright 2005, American Chemical Society. Reproduced with permission from ref. 29 and 31. Copyright 2007 and 2010, Wiley-VCH. | ||
As abovementioned, the MMAs originate from hyperbranched polymers; however, subsequently, the MMA mechanism has also been proved to be universal and can be used to explain the self-assembly of large spherical micelles from dendritic polymers, comb polymers, star polymers, linear polymers and even small molecules.33–36 A variety of MMAs assembled from different precursors have been prepared and applied in a broad range of fields such as biomedical fields, optical nanotechnology and functional materials.37,38 The MMA mechanism has become a general mechanism in the self-assembly of large micelles, and MMAs have demonstrated significant potential in various applications.
This review provides an overview of MMAs, including their self-assembly mechanism, unique characteristics, applicable precursors and potential applications. Especially, the unique advantages of MMAs in their applications have also been elaborated in detail. We aimed to elucidate the structural characteristics of MMAs and their hierarchical designability and properties and deepen the understanding of the relationship between their structural design and applications. We hope that this review will stimulate new ideas and inspire continuous endeavours in this emerging research area.
2. Mechanisms for the formation of multimicelle aggregates (MMAs)
The MMA formation mechanism was first proposed and proved by experiments. Subsequently, dissipative particle dynamics (DPD) simulations provided more information, revealing that amphiphilic dendritic multiarm copolymers assemble into three kinds of micelles: UMs (generally around 10 nm), microphase-separated small micelles (ms-SMs, around 20–50 nm) and large multimolecular micelles (>100 nm).32 The large multimolecular micelles are a kind of MMAs with two formation mechanisms: one is called the unimolecular micelle aggregate (UMA) mechanism (Fig. 3a) and another called the small micelle aggregate (SMA) mechanism (Fig. 3b). | ||
| Fig. 3 Schematic for the multimicelle aggregate (MMA) mechanism. (a) Unimolecular aggregation (UMA) and (b) small micelle aggregation (SMA). Red indicates the hydrophilic segments, and blue indicates the hydrophobic segments. | ||
2.1 Unimolecular micelle aggregates (UMAs)
Generally, dendritic multiarm copolymers tend to form UMs in dilute solutions without any intermolecular aggregation. Especially, when the dendritic multiarm copolymers have long hydrophilic arms, UMs can be easily obtained. However, when the hydrophobic/hydrophilic ratio of the dendritic copolymers is moderate and there are interaction forces between hydrophilic arms, the UMs tend to aggregate into large multimolecular micelles, named as UMAs (Fig. 3a). Note that UMAs are formed by the direct aggregation of UMs without any micro-phase separation during the assembly process. Each UM is an independent building unit of UMAs, which are smaller than 20 nm (generally less than 10 nm) due to their limited molecular weight (<100 kDa).2.2 Small micelle aggregates (SMAs)
When the hydrophobic core and hydrophilic arms of the dendritic multiarm copolymer are strongly incompatible, the spherical dendritic copolymers first microphase-separate into a cone-shaped structure and then further aggregate into small micelles, which are coined as ms-SMs (Fig. 3b). This micelle formation mechanism is totally different from that of UMAs, whereas it is somewhat similar to that of the “star micelles” formed by linear block copolymers, which also involves the spontaneous segregation of hydrophobic and hydrophilic blocks. Moreover, the ms-SMs have a limited size of two extended molecules in diameter (20–50 nm), which is similar to that of the “star micelle”. If the hydrophilicity of the arms of the dendritic multiarm copolymers is further decreased, secondary aggregation of the ms-SMs occurs to form large micelles, named as SMAs (Fig. 3b).2.3 Assembly and disassembly process of the MMAs
Corresponding to the abovementioned simulation results, the assembly and disassembly processes of MMAs have also been observed in experiments; this further verifies and elucidates the mechanism of MMA formation; to prove the UMA mechanism, we report the case of a pH-responsive micelle obtained from carboxyl-terminated hyperbranched polymers (HBPO-COOHs). At a high pH (12.21), almost all the –COOH groups of the HBPO-COOH polymers were ionized, and the Coulombic repulsion between –COO− ions stabilized the polymers into UMs (10 nm). However, with a decrease in pH, the ionization of –COOH decreased, which increased the multiple hydrogen bonding interactions between the coronas of UMs. Finally, the small UMs aggregated to form large UMAs (500 nm, pH 2.34).39 This process was reversible. When the pH was increased to 12.21, the UMAs disaggregated into small UMs. All the pH-responsive assembly and disassembly processes of UMAs were proved by dynamic light scattering (DLS), TEM and atomic force microscopy (AFM) measurements.In addition to the UMA mechanism, the assembly and disassembly processes of SMAs were verified by experiment. Jiang and coworkers40 synthesized amphiphilic anthracene-functionalized β-cyclodextrins (CD-ANs) through Cu(I)-catalyzed azide–alkyne click reactions. Their results showed that the obtained amphiphilic CD-ANs first self-assembled into primary ms-SMs of several tens of nm in diameter, which then segregated hydrophilic cyclodextrin shells from hydrophobic anthracene cores because of microphase separation (Fig. 4a). With an increase in water content, the primary core–shell ms-SMs further aggregated into SMAs with the diameter of around 600–700 nm (Fig. 4b). Ingeniously, through the photo dimerization of anthracene, the anthracene cores of ms-SMs could be photo-cross-linked under 365 UV illumination. As a result, the cross-linked ms-SMs could be dissociated from SMAs in good solvents (DMF or THF) (Fig. 4c); this confirmed the SMA mechanism very well.
 | ||
| Fig. 4 (a) Self-assembly and disassembly process of the CD-ANs. (b) The SEM image of the SMAs assembled from CD-ANs. (c) The SEM image of the disassembled ms-SMs after UV cross-linking in the good solvents DMF or THF. Reproduced with permission from ref. 40. Copyright 2016, Wiley-VCH. | ||
2.4 Characteristics of the MMA mechanism
According to the abovementioned descriptions, the MMA mechanism can be divided into UMA and SMA. The difference between the UMA and SMA lies in the building blocks: the UMA comprises UMs, whereas the SMA consists of ms-SMs. The MMA mechanism can well explain the formation of large multimolecular micelles with diameter larger than two molecules, for example, micelles larger than 50 nm. However, there is a self-limiting growth mechanism for MMAs.41 When the building blocks of UMs or ms-SMs want to join the growing MMA, two driving forces dominate the process: one is the attractive force, such as H-bonds, electrostatic force and hydrophobic interaction, that facilitates the aggregation of building blocks and the other is the repulsive force, for example, the hydration force due to the solvation process, that keeps the building blocks away. When the attractive and repulsive forces reach a balance, the growth of MMAs is stabilized, and thus, the size of the MMAs remains stable. Attributed to this self-limiting growth process, the finally obtained MMAs generally have a narrow size distribution and even the building blocks are polydisperse in nature.28The UMA mechanism is quite unique when compared with the other multimolecular micelle mechanisms. The “star micelles”, “LCMs”, “multicompartment micelles” and “supermicelles” involve a microphase separation process; however, this is not necessary for UMAs. The UMA mechanism is generally suitable for amphiphilic polymers that are stable in solution as UMs. Then, when the concentration is above the critical aggregation concentration, the UMs spontaneously further aggregate into UMAs. From this aspect, UMAs are more suitable for highly branched polymers such as dendritic polymers, star polymers, brush polymers or graft polymers.
Similar to that of the LCMs, “multicompartment micelles” and “supermicelles”, the mechanism of SMAs also involves the microphase separation process and the secondary aggregation of small micelles. However, there are still some basic differences between SMAs and other multimolecular micelles. First, the building blocks of LCMs are reverse micelles, whereas they are normal micelles in SMAs, “multicompartment micelles” and “supermicelles”. Second, the microphase separation level in the building blocks of SMAs is much lower than that in the cases of “multicompartment micelles” and “supermicelles”. In SMAs, the building blocks are normal core–shell ms-SMs like the “star micelles”, and there is no further phase separation in the cores and shells of ms-SMs; however the building blocks are “Janus micelles” with segregated shells in the case of “supermicelles” and complex core–shell micelles with segregated cores in the case of “multicompartment micelles”. Thus, compared to “multicompartment micelles” and “supermicelles”, SMAs are suitable for amphiphiles, such as dendritic multiarm copolymers, comb polymers, star polymers, diblock copolymers and even small molecules, with one hydrophobic part and one hydrophilic part. In the following sections, we have focused on the structure of MMAs obtained from various precursors, and the applications of MMAs as functional materials.
3. Precursor for MMAs
As abovementioned, the assembly process of MMAs is composed of two stages: the formation of UMs or SMs and their further secondary aggregation. In general, it is very difficult to investigate these dynamic two stages for most of the large multimolecular micelles due to their fast assembly process. Thus, the formation of MMAs is verified from the TEM images of the large micelles consisting of many small building blocks inside them. According to the size of the building blocks, UMAs and SMAs can be discriminated: the building blocks with a size of around 10 nm are for the UMAs and those with a size of 20–50 nm are for the SMAs. Fortunately, the responsive micelles show advantages in terms of the observation of these two stages under different external stimuli through a series of measurements such as DLS, UV spectroscopy and TEM; directly, the change in Dh during the assembly of UMs or SMs to MMAs can be detected by DLS under various stimulus conditions. In addition, the disassembly process from MMAs to UMs or ms-SMs can be used to prove the MMA structure. In this section, we mainly focused on the MMA structure obtained from various assembly precursors including small molecules, linear polymers, H-shaped polymers, star polymers, comb polymers and dendritic polymers (Fig. 5). Furthermore, the dynamic assembly process of MMAs has been discussed based on the responsive micelles.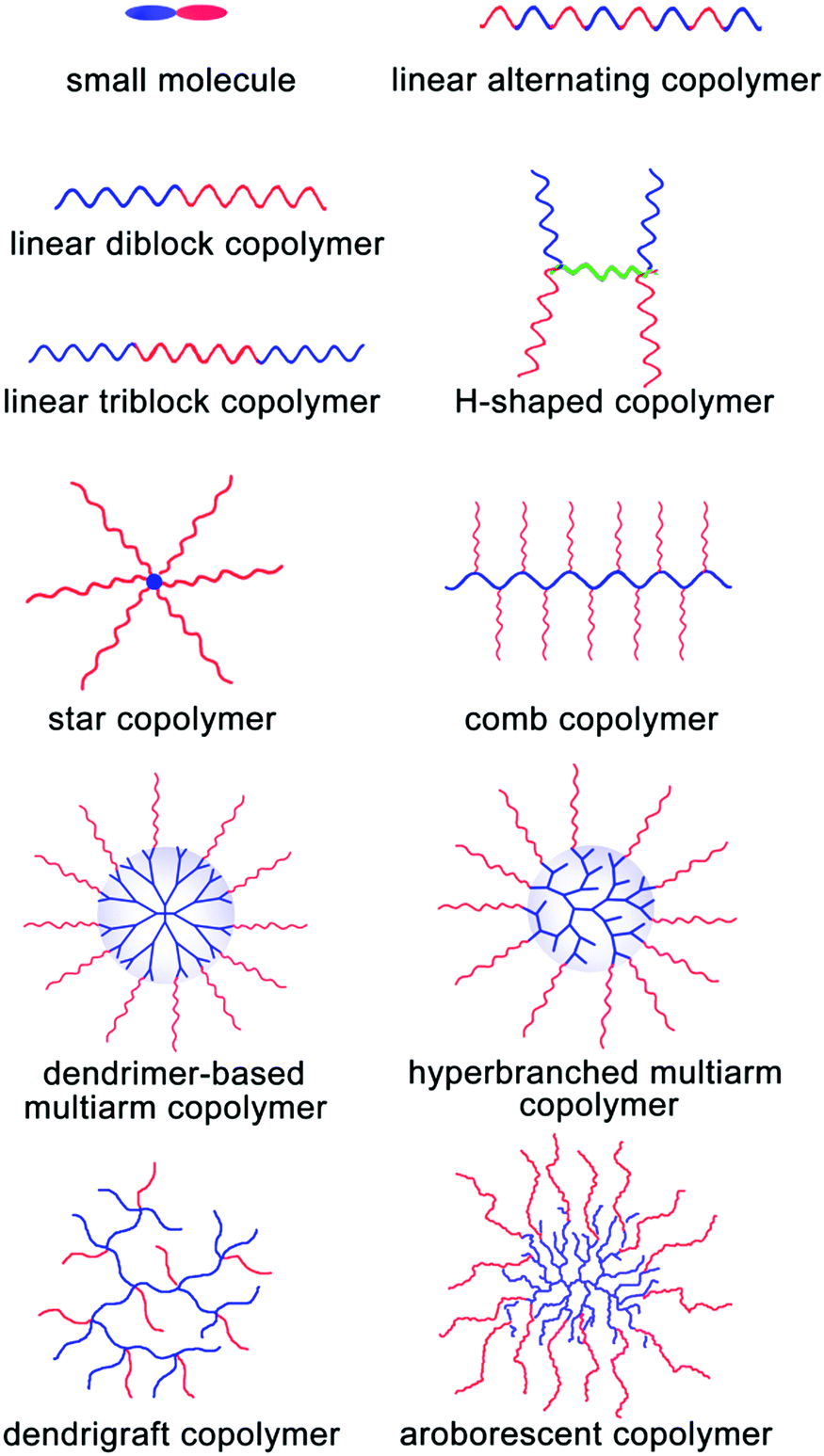 | ||
| Fig. 5 Schematic of the incorporation of various precursors for MMAs. Red blocks are hydrophilic and blue blocks are hydrophobic. | ||
3.1 MMAs obtained from small molecules
Amphiphilic small molecules assemble into large multimolecular micelles, in accordance with the SMA mechanism. Due to the inherent amphiphilicity of small molecules, they self-assemble into small “star micelles”, and then, these small micelles aggregate into large SMAs. We reported a drug self-delivery system based on the self-assembly of an amphiphilic drug–drug conjugate (ADDC) from the hydrophilic anticancer drug irinotecan (Ir) and the hydrophobic anticancer drug chlorambucil (Cb) via a hydrolyzable ester linkage. The results show that the Ir–Cb ADDCs first self-assemble into small micelles (around 3.4 nm) with segregated Cb cores and Ir shells, and then, these small micelles aggregate into large micelles (around 75.7 nm) (Fig. 6). The large nanoparticles exhibit longer blood retention half-life and overcome the multidrug resistance of tumour cells efficiently, which result in an excellent anticancer activity in vitro and in vivo.42 Similarly, other ADDCs, such as irinotecan-bendamustine,43 camptothecin-floxuridine,44 fluxuridine-bendamustine,45 and methotrexate-gemcitabine,46 and even mixed ADDCs, such as irinotecan-chlorambucil and gemcitabine-chlorambucil,35 also obey the SMA mechanism and assemble into large nanoparticles. In addition, bolaamphiphilic molecules composed of a heptyl alkyl chain with two tyrosine end groups assembled into unusual spherical interior-filled structures through an aggregation-assembly process, which was also consistent with the SMA mechanism.47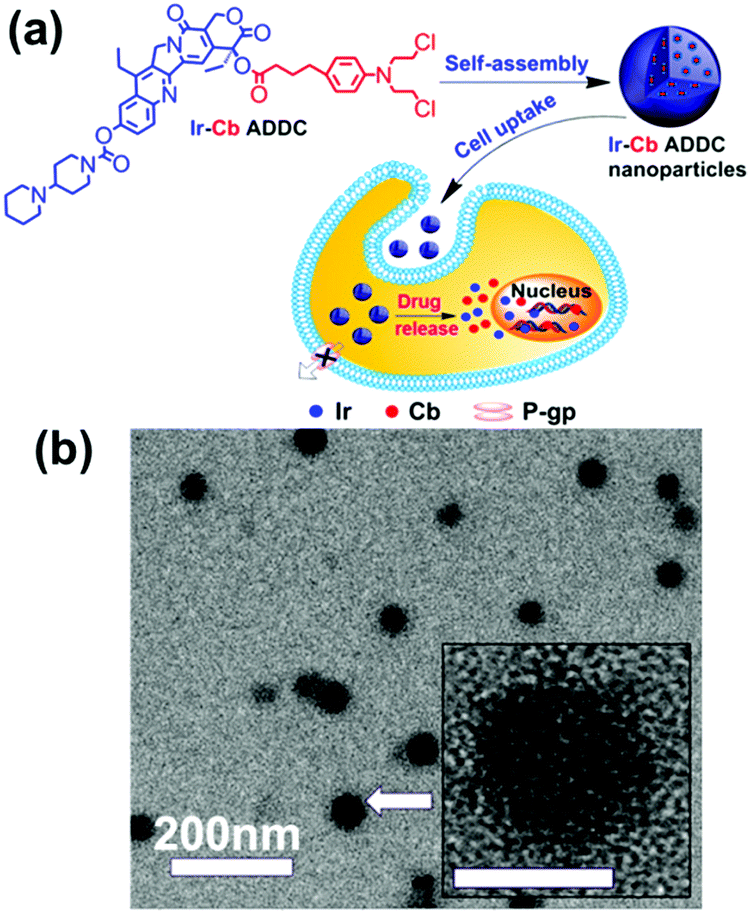 | ||
| Fig. 6 (a) Schematic of the fabrication, self-assembly, and self-delivery of an amphiphilic ADDC. (b) TEM image of Ir–Cb ADDC MMAs. Inset: The amplified image of a MMA. Reproduced with permission from ref. 42. Copyright 2014, American Chemical Society. | ||
3.2 MMAs obtained from linear block copolymers
The concentration of the linear block copolymer and external stimulus can exert a strong influence on the size and structure of the resultant micelles. Under certain conditions, amphiphilic linear diblock copolymers can also form MMAs. An and coworkers studied the thermoresponsive micellization of poly(ethylene glycol)-b-poly(N-isopropylacrylamide) (PEG-b-PNIPAM) with a low and narrowly distributed molecular weight of the PNIPAM block in water. The results showed that above the critical aggregation temperature, PEG110-b-PNIPAM44 favoured the formation of large MMAs at lower concentrations.48 Ma and coworkers studied the thermoresponsive behaviors of “schizophrenic” micelles obtained from poly(acrylic acid)-b-poly(2-dimethylamino) ethyl methacrylate (PAA156-b-PDMAEMA175). The results showed that at a high pH value (10.6), as the temperature was elevated, the Dh of the PDMAEMA-core micelles increased greatly from 220 nm to more than 1000 nm, indicating the formation of SMAs.49 Mandal and coworkers synthesized peptide–polymer hybrid bioconjugates containing PMMA chains and oligopeptide molecules through atom transfer radical polymerization (ATRP) using functionalized peptide-initiators.50 The results indicated that the obtained peptide–PMMA polymer assembled into MMAs in different organic solvents, and the average size of the MMAs decreased with an increase in the solvent polarity. Moreover, Wei and coworkers prepared an electroactive ABA triblock copolymer of poly (ethylene glycol) (PEG) and aniline pentamer (AP) (PEG-b-AP-b-PEG). This amphiphilic PEG-b-AP-b-PEG could self-assemble spontaneously into uniform MMAs when added directly to an aqueous solution. Moreover, the size of the MMAs could be adjusted from around 70 nm to 150 nm by changing the solution pH or the oxidation state of the AP block.513.3 MMAs obtained from alternating copolymers
Alternating copolymers are regular linear polymers with two species of alternating monomeric units, and the alternating units are small molecular monomers. Featuring a special structure, the alternating copolymers exhibit many unique characteristics including a large number of alternating segments, strict uniform alternating segments and small-molecular-weight alternating segments. Due to these features, alternating copolymers show unique characteristics and advantages in the assembly process. Recently, we synthesized a new kind of amphiphilic alternating copolymers through an epoxy–thiol click chemistry reaction and found that well-defined ultrathin nanotubes and vesicles could be obtained from these alternating copolymers.52–55 Moreover, through DPD simulations, we proved that the alternating copolymers could assemble into not only tubes and vesicles, but also spherical micelles, micelle networks, worm-like micelles, disk-like micelles, bicontinuous micelles, channelized micelles and MMAs. As shown in Fig. 7, MMAs were obtained based on spherical micelles and micelle networks. When the volume fraction of solvophilic A (fA) ≥ 3/4 and the effect of solvent-selectivity of solvophilic A aAS = 26–28, spherical micelles and micelle networks tend to aggregate together and no fusions happen; this leads to the formation of MMAs.56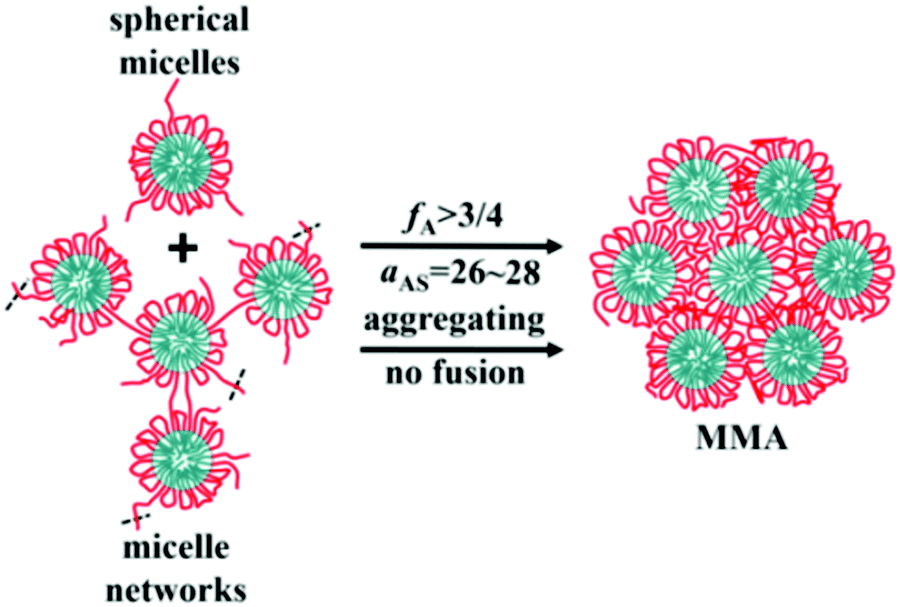 | ||
| Fig. 7 Schematic for the formation of solid MMAs assembled from alternating copolymers. Red lines are for the solvophilic units of the copolymer and cyan are for the solvophobic units. The black dash lines mean that there would be more linked chains or micelles. Reproduced with permission from ref. 56. Copyright 2019, Springer. | ||
The MMA model of alternating copolymers was also proved by experiment. We synthesized an alternating copolymer, poly(9,9′-bis(4-glycidyloxy phenyl)fluorene-alt-2,3-dihydroxy-butylene dithioether) (P(BGF-a-DHBDT)), via an epoxy–thiol click reaction between 1,4-dithiothreitol and 9,9′-bis(4-glycidyloxyphenyl)fluorine (Fig. 8a). The results showed that this alternating copolymer self-assembled into hollow polymeric spheres (HPSs) in an N,N-dimethylformamide (DMF)/H2O solvent mixture. The self-assembly mechanism of the wall of HPSs followed the MMA model. At first, P(BGF-a-DHBDT) self-assembled into small micelles with a hydrophobic BGF core (around 2 nm) (Fig. 8b and c) and hydrophilic DHBDT corona. Then, the micelles further aggregated through intermicellar H-bonding.57 These HPSs can further be prepared into hollow carbon spheres with uniform micropores; this has been discussed in Section 4.2.
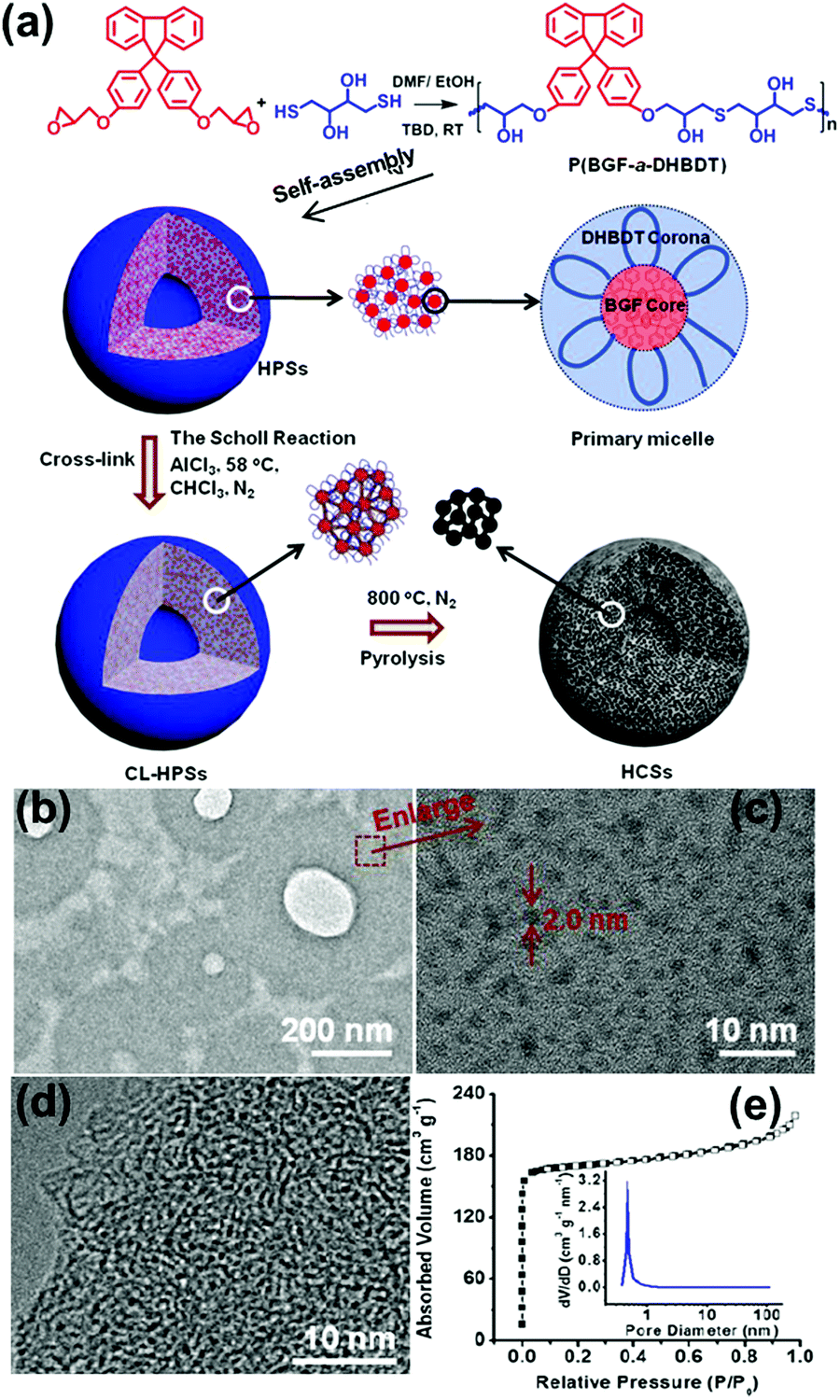 | ||
| Fig. 8 (a) Schematic of the preparation of HCSs and the self-assembly mechanism. (b) TEM image and (c) HR-TEM image of the ultrathin slices of the HPSs embedded in epoxy resin. (d) HR-TEM image of the ultrathin slices of the HCSs embedded in epoxy resin. (e) N2 adsorption/desorption isotherms of the HCSs at 77K. The insert shows pore size distribution calculated using the DFT method. Reproduced with permission from ref. 57. Copyright 2019, American Chemical Society. | ||
3.4 MMAs obtained from H-shaped polymers
H-shaped polymers, as a kind of non-linear polymers, can be described as a central linear polymer that are terminated by two other polymer chains on both ends.34 Tian and coworkers synthesized (AC)B(AC)-type H-shaped terpolymers using PEG for A, polytetrahydrofuran (PTHF) for B and poly(N,N-dimethylaminoethyl methacrylate) (PDMA) for C ((PEG)(PDMA)-PTHF-(PDMA)(PEG)) (Fig. 9a). The results showed that this H-shaped copolymer could self-assemble into dot-like micelles of around 10–30 nm in an aqueous solution at pH 5.0 and 7.4; in addition, when the pH was increased to 9.2, these micelles aggregated into large MMAs of around 100 nm.34 Moreover, when ultrasonic vibration was employed to offer sufficient energy to accelerate the movement and increase the entanglement of the PDMA chains, this H-shaped copolymer could self-assemble into large MMAs with a size of about 270–500 nm in an aqueous solution (Fig. 9b).58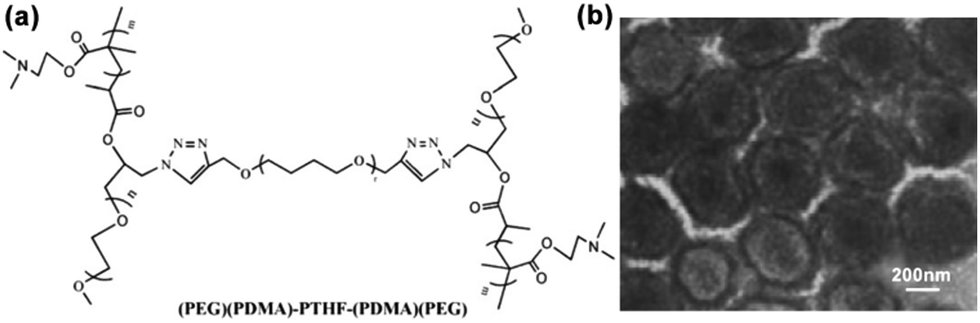 | ||
| Fig. 9 (a) Structure of the H-shaped (PEG)(PDMA)-PTHF-(PDMA)(PEG) copolymer. (b) TEM image of the MMAs obtained under ultrasonic vibration. Reproduced with permission from ref. 58, Copyright 2013, Elsevier. | ||
3.5 MMAs obtained from star polymers
Star polymers consist of several linear polymer chains connected at one point. Our group synthesized a four-arm star polymer with a porphyrin core and four poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) arms (THPD) through anionic polymerization (Fig. 10a). At pH 6.2, this star copolymer with four hydrophilic PDMAEMA arms and a hydrophobic core could undergo self-assembly in an aqueous solution.59 The results showed that when the concentration of this star copolymer was below 0.05 mg mL−1, it formed UMs with the average diameter of about 3.8 nm. In contrast, when the copolymer concentration was above 0.05 mg mL−1, it self-assembled into large UMAs with the average diameter of 76 nm and about 200 UMs inside, determined by counting the particles in the TEM image (Fig. 10b). These UMAs can further be designed to construct an artificial light-harvesting nanosystem by introducing a natural protein; this has been discussed in Section 4.1. | ||
| Fig. 10 Schematic for the self-assembly of the pomegranate-like porphyrin-phycocyanin light-harvesting nanosystem (Ex Light = excitation light; Em Light = emitted light). (b) TEM image of the THPD MMAs. (c) TEM image of the THPE-PC micelles. (d) The fluorescence decay profiles of the THPD MMAs and THPE-PC MMAs. Reproduced with permission from ref. 59, Copyright 2016, Wiley-VCH. | ||
3.6 MMAs obtained from comb copolymers
Comb polymers are a kind of special grafting polymers formed by the simultaneous grafting of several linear chains on one main chain and resemble a comb-shaped polymer. We synthesized a comb-shaped amphiphilic multiarm copolymer of PEHO-star-PEO with a linear PEHO core and PEO arms through sequential cationic ring-opening polymerization at low temperatures (Fig. 11). The resulting micelles were around 154 nm, which were also a kind of SMAs with the basic building units of small micelles.60 Huang and coworkers utilized a “grafting from” method to synthesize the comb copolymer ethyl cellulose-graft-PS (EG-g-PS) through ATRP with the EC 2-bromo-isobutyryl ester as the macro-initiator. Their results showed that this comb copolymer could self-assemble into small UMs at low copolymer concentrations, and large MMAs could be formed at relatively high concentrations. Moreover, the micelle size could be increased by increasing the copolymer concentration and the PS chain length.61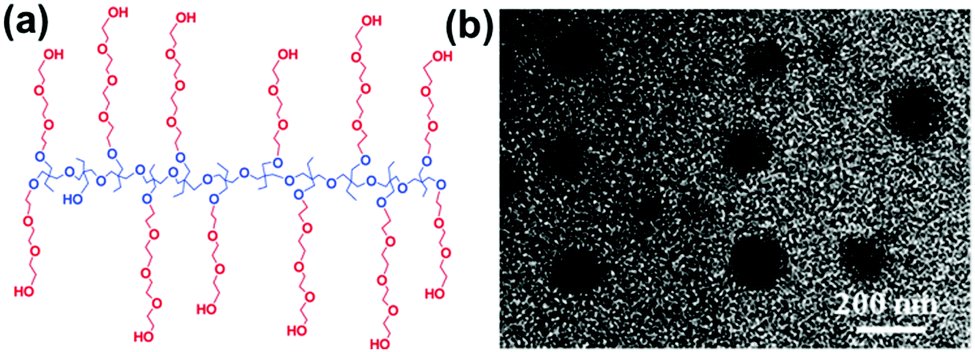 | ||
| Fig. 11 (a) Structure of the comb-shaped PEHO-star-PEO. (b) TEM image of large micelles. Reproduced with permission from ref. 60, Copyright 2010, American Chemical Society. | ||
3.7 MMAs obtained from dendritic polymers
Dendritic polymers are a class of cascade-branched molecules with a three-dimensional (3D) highly branched architecture and exhibit the special properties of a large population of terminal functional groups, lower viscosity and better solubility.62–64 Generally, dendritic copolymers that self-assemble into MMAs in solution may be classified into dendrimer-based multiarm copolymers, hyperbranched multiarm copolymers and arborescent (dendrigraft) copolymers.65,66 Hereinafter, we have provided a brief example for each type of dendritic precursors for MMAs. | ||
| Fig. 12 Chemical structures of the hyperbranched blocks incorporated into hyperbranched polymers for the preparation of MMAs. | ||
Through reversible addition–fragmentation chain transfer polymerization, we synthesized a hyperbranched multiarm copolymer with Boltorn H20 as the core and poly (acrylic acid) as the arms (H20-star-PAA). In the dilution solution, the H20-star-PAA always exhibited UMs (around 5 nm), and with a decrease in pH, the PAA shell of the UMs gradually collapsed to minimize the size of UMs. However, in the concentrated solution, when the pH of the solution was low, the UMs furthered aggregated into UMAs with the size of around 80 nm.80
Thermosensitive polymer micelles also obey the MMA mechanism during the phase transition process. We synthesized thermosensitive amphiphilic star copolymers with a hydrophobic HBPO core and many hydrophilic poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) arms. At room temperature (below LCST), the HBPO-star-PDMAEMAs synchronously formed small UMs (<10 nm) and large UMAs (around 100 nm) above the CMC.29 Furthermore, when the temperature was increased to LCST, the PDMAEMA shells dehydrated; this led to the shrinking of the micelles, and then, the micelles underwent secondary aggregation to form larger MMAs (around 1000 nm) via the increasing intermicellar hydrophobic interaction. Finally, this aggregation turned the solution from transparent to turbid, which showed a sharp thermosensitive phase transition at the LCST.81 Jiang and coworkers synthesized amphiphilic azobenzene-containing hyperbranched poly(ether amine)s (hPEA-AZOs). The results showed a sharp transformation between UMs to UMAs in response to temperature, pH and ionic strength.82
There are also some precursors in the formation of SMAs. The thermosensitive large spherical micelles obtained from adamantly-terminated hyperbranched polyglycerol (HPG-AD) are a kind of SMAs (around 200 nm) with the building blocks of SMs of around 20 nm at 20 °C. With an increase in solution temperature, the SMAs underwent secondary aggregation into larger micelles of around 550 nm.31 Recently, we synthesized a hyperbranched multiarm copolymer with an HBPO core and many poly(acrylamide-co-acrylonitrile) (P(AAm-co-AN)) arms, which showed sharp thermoresponsive phase transition at the upper critical solution temperature (UCST). The HBPO-star-P(AAm-co-AN) copolymers obeyed the SMA mechanism during the UCST transition. Above the UCST, the HBPO-star-P(AAm-co-AN)s formed UMs of around 10 nm. When cooled down to the UCST, the hydrophilic/hydrophobic balance of the copolymer micelles was broken up, which led to the formation of SMs of around 20 nm. With a further decrease in temperature, the SMs further shrunk and aggregated into large SMAs of around 400 nm.83 Upon heating, the large SMA micelles disassembled into unimolecular micelles (Fig. 13). However, there should be two steps of disassembly: from SMA to small micelles and then to unimolecular micelles, which is a slow disassembly process. It is the slow disassembly dynamics that leads to the slow UCST transition during the heating cycle as well as the obvious hysteresis between the cooling and the heating cycles.
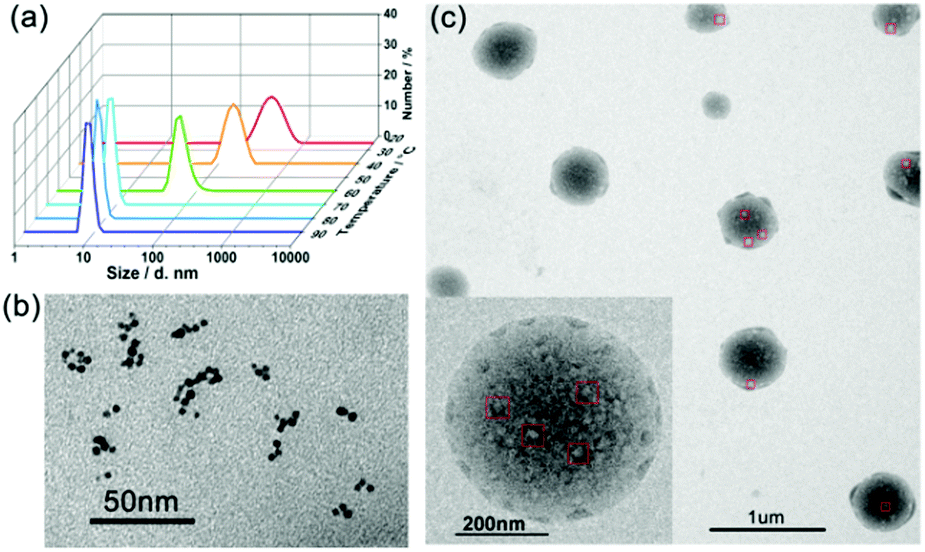 | ||
| Fig. 13 (a) DLS plots of the micellar solution of HBPO-star-P(AAm-co-AN)s upon cooling. (b) TEM images of the UMs at 80 °C. (c) TEM images of the SMAs at 25 °C. Reproduced with permission from ref. 83. Copyright 2018, American Chemical Society. | ||
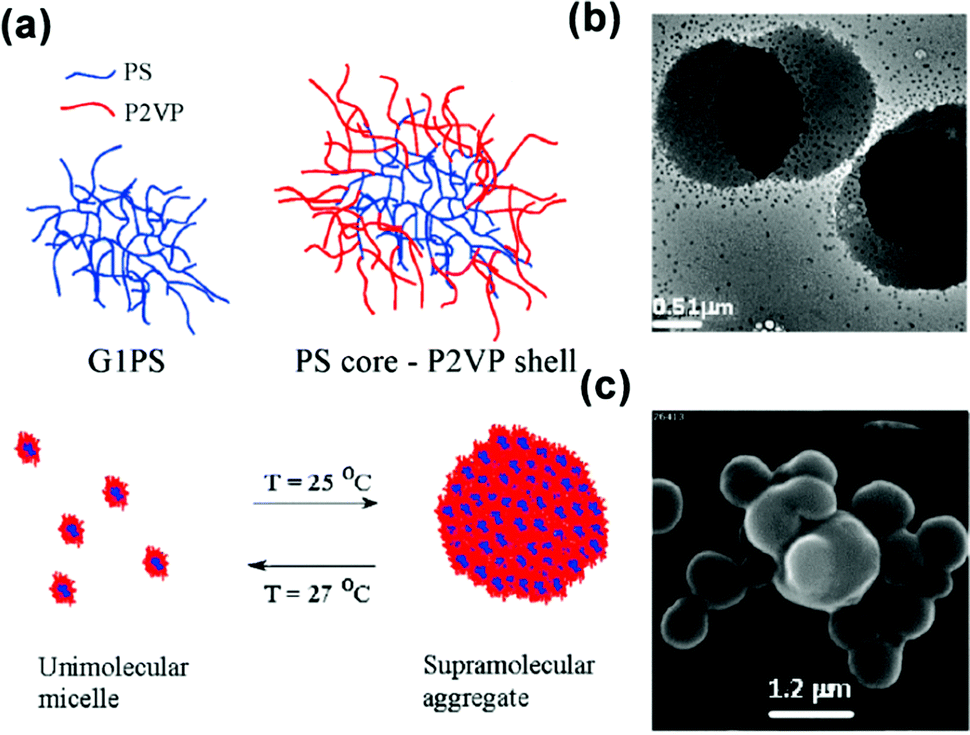 | ||
| Fig. 14 (a) Schematic for the temperature-responsive self-assembly of arborescent PS–P2VP copolymers in toluene. (b) TEM image and (c) SEM image of arborescent PS–P2VP copolymers in toluene at 20 °C. Reproduced with permission from ref. 86. Copyright 2008, American Chemical Society. | ||
4. Applications
The MMA mechanism plays an important role in the understanding of the micellization processes of amphiphilic copolymers, which further provides a clear and definite guide for the design and development of novel micelle structures and functional materials with micelles as building units. Supramolecular interactions, such as electrostatic interactions, hydrogen bonding and coordination bonds, have been introduced between the micellar building units of MMA, which expand the MMA applications. Their globular structure, adjustable size and diverse design qualify MMAs for various applications. In this section, we have summarized their applications in a wide range of fields including light-harvesting antennas,59,88 porous nanomaterials,89 controlled drug/gene/protein delivery, cancer diagnosis and bioimaging.4.1 Light-harvesting nanosystems
As abovementioned in Section 3.5, the four-arm star polymer THPD could assemble into UMAs above CMC at the pH of 6.2. Through the electrostatic self-assembly, we successfully introduced the phycocyanin protein into the THPD UMAs; this led to the formation of a novel micelle-like hybrid natural-artificial light-harvesting nanosystem;59 the resulting nanosystem showed a special structure of pomegranate-like UMAs, which consisted of one phycocyanin acceptor in the centre and multiple porphyrin donors in the shell. The TEM result clearly proved the presence of this special structure (Fig. 10c). In addition, this system could display an efficient transfer of excitation energy (about 80.1%) in water by avoiding self-quenching among the porphyrin donors and afford efficient energy transfer from up to 179 porphyrin donors to one phycocyanin acceptor (Fig. 10d). In the same way, we employed multi-graphene quantum dots as donors and the above-mentioned porphyrin unimolecular micelle as the acceptor to construct a srikaya-like light-harvesting antenna. The high energy-transfer efficiency of the constructed antenna in an aqueous solution was as high as 93.6% with the antenna effect of 7.3.884.2 Nanoporous carbon spheres
Nanoporous carbon spheres integrate the advantages of carbon materials and a spherical structure, which allows them to exhibit unique properties of regular geometry, good liquidity and controlled size distribution, and they have therefore been applied in a wide range of research fields, such as catalysis, adsorption and energy conversion and storage.90–93 The MMAs show unique advantages in the preparation of microporous carbon spheres (pore size <2 nm) and mesoporous carbon spheres (pore size = 2–50 nm).As mentioned in Section 3.3, we prepared HPSs of P(BGF-a-DHBDT) and the alternating polymer was stacked in the form of MMAs in the hollow sphere wall. Through the Scholl reaction, the BGF segments were cross-linked and stabilized hollow polymer spheres (CL-HPSs) were obtained (Fig. 8a). Benefiting from the uniform DHBDT segment length, hollow carbon nanospheres (HCSs) with uniform micropores (0.5–1.0 nm) of a narrow size distribution were prepared successfully through carbonization at 800 °C under N2 protection and KOH activation (Fig. 8d and e).57 This was the first reported template-free method for HCSs with uniform micropores, which showed highly superior electrochemical performance. This method ingeniously utilized the microphase separation of the hydrophilic and hydrophobic segments in MMAs and the characteristic spherical structure of MMAs. Using the hydrophilic segments as pore sources, the hydrophobic cross-linking segments as carbon sources and the spherical structure of MMAs as the integral framework, HCSs were prepared successfully by a template-free method, which provides a new idea for the fabrication of microporous carbon spheres.
Soft templating synthesis is a preferred way to prepare mesoporous carbon spheres. Actually, it can be categorized into the composite MMAs, obtained by the co-assembly of block copolymer micelles and carbon precursors. Different from the above template-free method, this method uses the integral micelles as the thermally decomposable soft templates, and a thermosetting polymer as the carbon precursor. Through controlling the micelle size (hydrophobic/hydrophilic volume ratio, block length, etc.) and the interaction between the micelles and carbon precursors (pH, ionic strength, temperature), microporous carbon spheres with a controllable pore size and element doping could be easily obtained.89,93,94 Up to now, significant progress has been made in the composite MMA method to synthesize microporous carbon spheres.90,95,96 In the following, we briefly report the recent advances in the mesoporous spheres with non-Pluronic and Pluronic amphiphilic block copolymers as templates, respectively.
Yamauchi and coworkers97 obtained highly nitrogen-doped mesoporous carbon spheres with extra-large pores (16 nm) through the co-assembly of polystyrene-block-poly(ethylene oxide) (PS-b-PEO) micelles and the self-polymerization of dopamine (DA). First, they prepared the stable DA/PS-b-PEO composite MMAs through the hydrogen bonding between the catechol and the N–H groups in DA and an –OH group in the PEO block. Then, PDA/PS-b-PEO composite spheres were obtained under alkaline conditions by the self-polymerization of the DA. Finally, the PS-b-PEO micelles, as pore-forming agent, were removed from the spheres during the carbonization process and PDA was retained as both carbon and nitrogen sources, which allowed synthesizing the nitrogen-doped mesoporous carbon spheres (Fig. 15). The resulting mesoporous carbon spheres showed high electrocatalysis activity and excellent long-term stability towards the oxygen reduction reaction. Mai and coworkers98 took the same approach to synthesize nitrogen-doped mesoporous carbon nanospheres with well-controlled pore sizes ranging from 8 to 38 nm. A linear equation was also first achieved for the quantitative control of the average pore size of the mesoporous carbon nanospheres by simply adjusting the PS length of PS-b-PEO. In addition, the MCNSs exhibited electrochemical performance with high specific capacitances, superior rate capability and outstanding cycling stability.
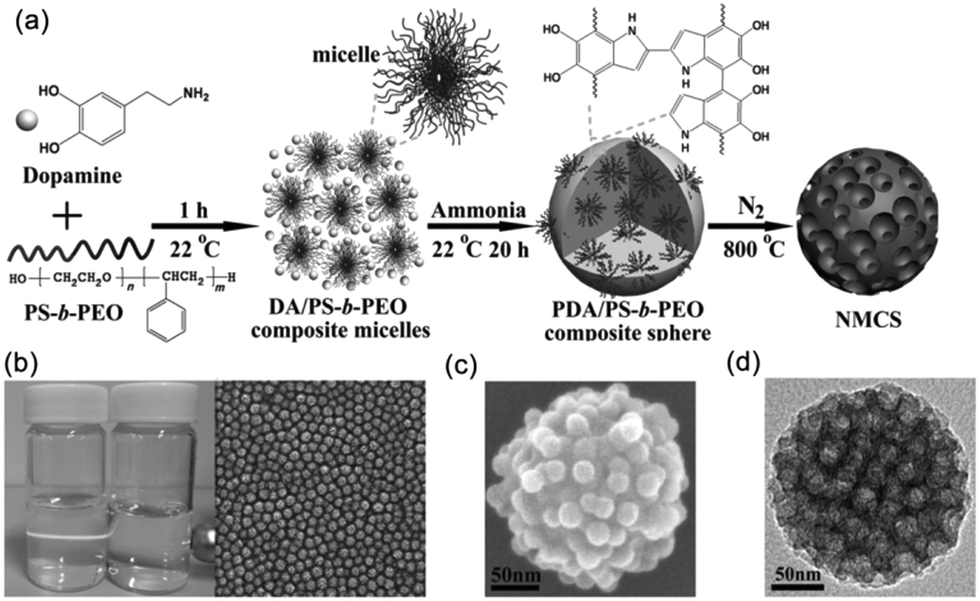 | ||
| Fig. 15 (a) Formation process of the N-doped mesoporous carbon nanospheres. (b) Characterizations of the DA/PS-b-PEO composite micelles. (c) SEM image of a PDA/PS-b-PEO composite sphere. (d) TEM image of NMCS. Reproduced with permission from ref. 97. Copyright 2014, Wiley-VCH. | ||
Su and coworkers99 prepared size-controlled nitrogen-containing mesoporous carbon nanospheres through an aqueous self-assembly process employing Pluronic F127 micelles as the template and spherical control agent and 3-aminophenol as the carbon and nitrogen sources. The resulting nanospheres exhibited good catalytic performance for the direct dehydrogenation of ethylbenzene. Li and coworkers100 employed hexadecyl trimethyl ammonium bromide and F127 as a co-template, resorcinol as a polymer precursor and tetraethyl orthosilicate as an inorganic precursor to synthesize mesoporous carbon nanospheres with a dual mesoporous texture (4.1 and 24.1 nm), which showed a highly efficient absorption of bulky dye molecules.
4.3 Biological applications
In recent years, micelles have been applied in a wide range of biological fields, such as drug delivery, bioimaging and diagnosis.71,101–103 MMAs have many unique advantages in the above biological applications. First, MMAs possess more hydrophobic cores than UMs, which favour the enhancement of drug encapsulation and controllable drug delivery. Second, the shortening of the hydrophilic arms results in the increase in aggregate size.5,28 Thus, a wide range of sizes of MMAs could be obtained by adjusting the length of the hydrophilic segments, which further favours the optimum size selection for efficient endocytosis. Third, functional mixed MMAs could be easily obtained by the co-assembly of UMs with varying functionalities, which provides micelles the capability of being designed to realize tumour-coordinated therapy technology in an efficient manner. Fourth, MMAs exhibit an abundant structure with a lot of cores and shells from the building block micelles and the cores are separated by the shells. This special structure provides the chance to use the cores and shells as the design units to build more versatile micelles. Fifth, the MMAs usually have a low critical micelle concentration (CMC) in aqueous solution, which exhibits high stability, prolonged plasma half-lives and reduced cytotoxicity. Consequently, inspired by MMAs’ unique and outstanding properties, considerable work has been done in an effort to explore their applications in the biological field. In this section, we give a brief overview of the typical achievements in the biological applications by our group in recent years.Through the “bottom-up” strategy, we constructed a multi-polyprodrug-arm hyperbranched amphiphilic copolymer with controlled drug loading for cancer therapy.104 This copolymer assembled into about 150 nm-sized MMAs, which provided a suitable size for effective cellular internalization, extended blood retention time and excellent anticancer activity both in vitro and in vivo. We developed a facile supramolecular approach for the preparation of size-controllable copolymer MMAs via adenine–uracil recognition between adenine-functionalized HBPO-star-PCL and uracil-modified PEG (PEG-U).105 Through decreasing the molar ratio of the PEG-U, the size of the resulting MMAs could be regulated from 10 nm to 200 nm, which provides the optimized size for drug delivery. The above work fully verified the properties of the adjustable particle size of the MMAs.
In addition to the controllability of the MMA size, multifunctional MMAs could be obtained by a simple co-assembly method. By using UMs with different functions as the building blocks, we presented a mixed MMA for targeted cancer imaging by the simple co-assembly of carboxyfluorescein-functionalized HBP (HSPFAM) and aptamer-functionalized HBP (HSPDNA).106 Among these, HSPFAM is for imaging and HSPDNA is for targeting, respectively. This MMAs showed bright green fluorescence in MCF-7 cells and no significant fluorescence could be seen from cells (Fig. 16).
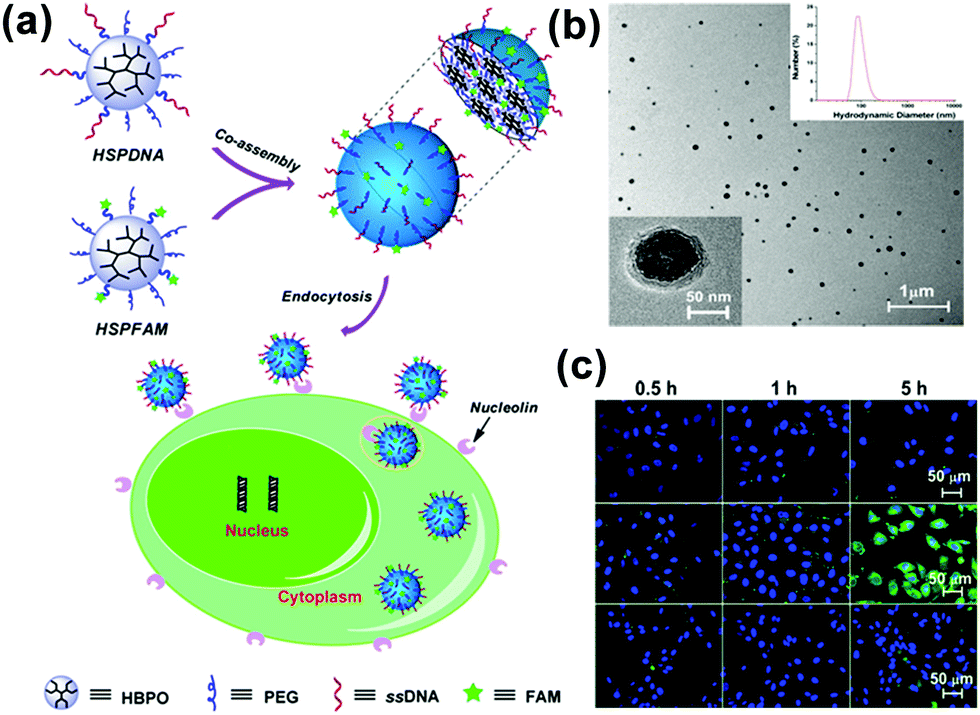 | ||
| Fig. 16 (a) Schematic for the co-assembly of HSPFAM and HSPDNA and their delivery to cancer cells through an endocytosis-based internalization mechanism. (b) TEM images of the co-assembled MMAs. (c) Confocal laser scanning microscopy images of the cells incubated with multifunctional MMAs. Reproduced with permission from ref. 106. Copyright 2014, American Chemical Society. | ||
Moreover, phase separation between the hydrophilic and hydrophobic segments in the MMAs endows them with special optical properties and certain applications. By employing a co-solvent self-assembly approach, we presented a emission enhancement process of star conjugated copolymers (HCP-star-PEG) during the process of UMs aggregating into UMAs.107 Importantly, this study proved that the hydrophilic PEG shell of the HCP-star-PEG UMs could efficiently restrict the concentration self-quenching and intermolecular aggregation of the conjugated polymers core, which realized emission enhancement and exhibited excellent biological application potential (Fig. 17). Furthermore, we constructed a combing two-photo-activated (2P) fluorescence resonance energy transfer (FRET) and near-infrared (NIR) photothermal effect of micelles for enhanced photodynamic therapy.108 Thermoresponsive hyperbranched polyether (HPE) and photosensitizer Chlorin e6 (Ce6) were successively grafted onto hyperbranched conjugated polymers (HCP) (HCP@HPE-Ce6). Under irradiation with NIR light, the heating temperature was over the LCST of HCP@HPE, shortening the distance between HCP and Ce6 by the collapse of the HPE cell, which further switched “on” the FRET process from the HCP core to Ce6s. In NIR fluorescence imaging, the fluorescence increase of PSs of HCP@HPE-Ce6 was observed under excitation at 800 nm along with the fluorescence quenching of HCP. Consequently, the required Ce6 content of HCP@HPE-Ce6 for IC50 was only a quarter of the dosage used in traditional free Ce6 treatment.
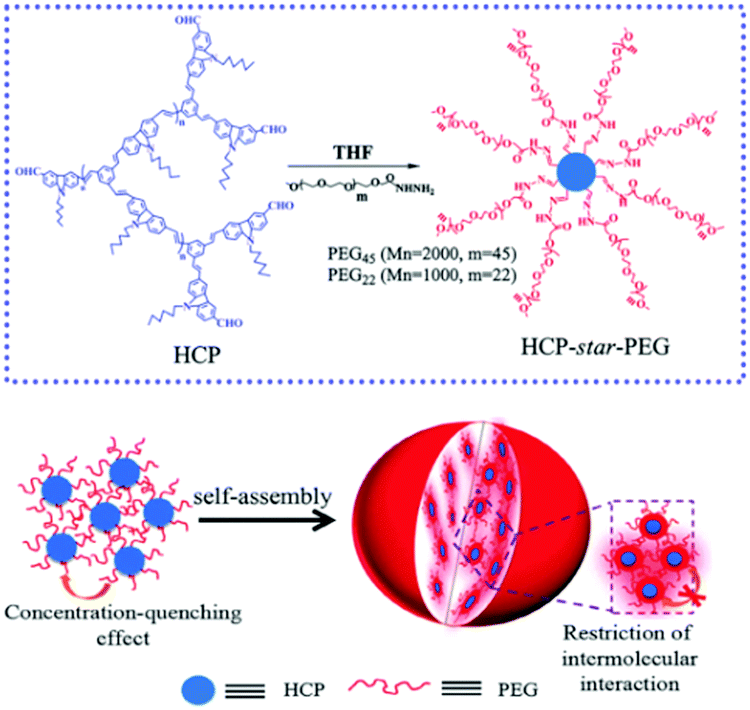 | ||
| Fig. 17 Schematic for the mechanism of HCP-star-PEG45 MMAs and the emission enhancement. Reproduced with permission from ref. 107. Copyright 2011, Royal Society of Chemistry. | ||
4.4 Smart separation
Micelles formed by the self-assembly of amphiphilic molecules have the ability of achieving the selective encapsulation of hydrophobic guest molecules in water. As mentioned in Section 3.5, during the phase transition process (temperature or pH responsive), micelles aggregate from UMs/SMAs to MMAs. Integrating the encapsulation ability and responsive aggregation, smart separation could be realized by adding external stimulus. Yin and coworkers first synthesized micelles with hydrophobic PPO and TMS as the core and hydrophilic Jeffamine L100 as the shell. Then, hybrid core-cross-linked micelles were obtained through hydrolysis. These micelles had the ability to selectively encapsulate Rose Bengal (RB) and rhodamine 6G (R6G). Upon heating, the core-linked micelles aggregated and settled to the bottom of the centrifugal tube, which realized the separation of RB and R6G from the aqueous solution.109,1104.5 Multidimensional ordered porous materials
In addition to nanoporous carbon sphered mentioned above, MMAs can actually be extended to form multidimensional ordered porous materials, such as ordered nanoporous films and ordered porous inorganic materials.Cochen and coworkers reported the preparation of the two-dimensional porous films through an MMA mechanism (Fig. 18a). They first prepared the stable reverse micelles solution of the PS-b-PAA diblock copolymer in toluene with segregated PAA cores and PS shells. Then, through casting the micellar solution onto silicon nitride substrates, it formed a micellar monolayer with lateral hexagonal arrays of spherical PAA microdomains in a PS matrix. Finally, upon exposure to the solution of a monovalent base, due to hydration and swelling of the PAA cores, cavitation of the individual PAA micelles cores occurred, leading to the formation of cavities in the centre of the micelles and exposing the PAA domains to the film's surface.111 Further, the cavitated PAA domains could serve effectively as open nanoreactors for nanocluster synthesis, like Ag and PbS nanoclusters. Park and coworkers fabricated a reversible ordered nanoporous film of PS-b-poly(2-vinylpyridine) (PS-b-P2VP) micelles and used such films as templates to control the spatial location of metal precursors and nanoparticles. In addition, the sizes of the nanoparticles could be adjusted by controlling the concentration of the metal precursor solutions.112 Chai and coworkers utilized the PS-b-P2VP micellar monolayer as structural elements to produce parallel metal nanowires and modulated the pattern morphology by controlling the solution concentration, metal deposition time and molecular weight of PS-b-P2VP.113 Mai and coworkers114–117 also used the bilayers or graphene oxide (GO) as 2D templates to accommodate the assembly of single layer polymer micelles on the surface. Then, they also polymerized some percursors, like pyrrole and aniline, on the surface of the micelle monolayer. After the removal of the micelles thorough dissolution and hydrothermal treatment, 2D nanoplates with highly ordered spherical (Fig. 18b) or cylindrical mesopores were obtained. These nanomaterials show great potential in energy storage and conversion.
 | ||
| Fig. 18 (a) Schematic of the fabrication of a nanoporous PS-b-PAA micellar film and AFM height image of the corresponding micellar film. (b) TEM image and fast Fourier transform image (inset) showing the ordered 2D mesostructure of a polyaniline nanosheet with a hexagonal lattic. (c) SEM image of the silica scaffold. Reproduced with permission from ref. 111. Copyright 2002, American Chemical Society. Reproduced with permission from ref. 117 and 118. Copyright 2016 and 2017, Wiley-VCH. | ||
The MMA mechanism can also be applied to prepare more complex multidimensional ordered structures. Che and coworkers employed the co-assembly of polymer micelles and silica source to prepare complex mesoporous silica materials. They used poly(tert-butyl acrylate)117(PtBA)-b-PS77-b-PEO191 micelles as templates and tetraethyl orthosilicate as silica sources to obtain a silica scaffold with a shifted Schoen gyroid (P) network and the interconversion of the P and the Schwarz diamond (D) networks.118 The small-angle X-ray scattering and SEM results showed that the calcined materials had a highly ordered structure (Fig. 18c). Besides, through varying the degree of polymerization of the hydrophobic blocks, eight different structures could be formed in the mesoporous materials, including normal-phase cage-type, normal-phase 2D hexagonal, lamellar, shifted double-diamond, single-gyroid, shifted double-primitive, inverse-phase 2D hexagonal and inverse-phase micellar structures.119 Herein, the composite MMAs and inorganic source exhibited a more stacking pattern and complicated structure due to the cooperative formation effect.
5. Conclusions
The MMA mechanism, including UMA and SMA, has been widely accepted to explain the formation of large spherical multimolecular micelles. For the UMA mechanism, the copolymers first form UMs, and then, the UMs further aggregate into large micelles without microphase separation. For the SMA mechanism, the copolymers first self-assemble into small micelles through microphase separation, and then, the small micelles further aggregate into large micelles. The UMA is more suitable for highly branched polymers, such as dendritic polymers, star polymers and graft polymers, that can form UMs in solution. The SMA is suitable not only for amphiphilic highly branched polymers, but also for amphiphilic comb copolymers, linear copolymers and even small molecules. Therefore, the MMA mechanism can be regarded as a universal mechanism for the formation of large spherical multimolecular micelles from many kinds of topological precursors, ranging from small molecules to globular macromolecules.The MMAs possess a special hierarchical structure, which endows them with unique advantages in terms of structural controllability and design diversity. The self-limiting growth mechanism during the secondary aggregation process favours the formation of MMAs with an adjustable size and narrow size distribution. Then, each building block of primary micelles inside the MMA can be considered as a design unit. Through the co-assembly of primary micelles, MMAs with multiple functions can be obtained directly. Furthermore, the MMAs can be regarded as a special composite structure, which has an integral spherical framework and many hydrophobic cores and hydrophilic shells inside; moreover, the cores are separated from each other by shells, and the shells are interconnected. This special structure provides the chance to use the cores and shells as the design units to build more versatile micelles.
The special structure of MMAs makes them very useful in some applications such as light-emitting materials, light-harvesting systems, nanoporous materials and biomedical areas. For light-emitting materials, since the building blocks of UMAs are UMs with chromophores, all the chromophores are separated from each other to avoid π–π stacking effectively, and as a result, the final UMAs emit very strong light. Thus, UMAs are very useful in light-emitting materials and devices used in bioimaging. Due to the same advantage, the UMA structure also shows good potential to funnel solar energy due to the high energy-transfer efficiency inside the micelle in an aqueous solution. It can even generate multi-donors to only one acceptor structure in the micelle through a delicate sequential self-assembly process of donors and acceptors; this is very useful for highly efficient light-harvesting antennae and photosynthesis. For nanoporous materials, the cores and shells of the UMs or small micelles inside the MMAs can be respectively used as a pore source and carbon source to fabricate microporous or mesoporous materials. Moreover, the pore size can be controlled through changing the size of the primary micelles by adjusting the molecular weight and hydrophilic fractions of the constituent polymers. For example, porous nanoparticles with uniform micropores can be obtained from MMAs of alternating copolymers. For biomedical applications, MMAs can be easily uptaken by tumour cells or tissues through efficient endocytosis. Most importantly, they can quickly disassociate into the primary micelles of UMs or small micelles under a mild environmental stimulus and then release the encapsulated guest molecules or drugs. Thus, MMAs have potential in controlled drug delivery or chemosensors.
In short, MMAs widely exist in the self-assembly of amphiphiles, and they also show some unique structural characteristics and property advantages in their applications. However, the studies on MMAs are just at the very early stage, and the application potential of MMAs has not been explored clearly. For example, MMAs have been used to fabricate nanoporous particles, and thus, their application can also be extended to the formation of separation membranes with delicate pore structures; the light-emitting property of MMAs is really advantageous; however, to date, their applications are still limited in fluorescent materials, and more light-emitting materials and devices can be expected from MMAs; MMAs are useful in energy transfer for light harvesting; however, their applications in photosynthesis are still greatly limited. We envisage that this review can further stimulate new ideas and inspire continued endeavours in this emerging research area in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Jiangsu Collaborative Innovation Center of Biomedical Functional Materials. We acknowledge the National Natural Science Foundation of China (21890733, 51773115), the Program for Basic Research of Shanghai Science and Technology Commission (17JC1403400) for the financial support.Notes and references
- M. Jones and J. Leroux, Eur. J. Pharm. Biopharm., 1999, 48, 101–111 CrossRef CAS PubMed
.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS PubMed
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS
- A. Halperin, Macromolecules, 1987, 20, 2943–2946 CrossRef CAS
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed
- M. Moffitt, H. Vali and A. Eisenberg, Chem. Mater., 1998, 10, 1021–1028 CrossRef CAS
- Z. Li, K. Ellina, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed
- J. F. Lutz and A. Laschewsky, Macromol. Chem. Phys., 2005, 206, 813–817 CrossRef CAS
- J. Du and R. K. O’Reilly, Chem. Soc. Rev., 2011, 40, 2402–2416 RSC
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2009, 25, 13718–13725 CrossRef CAS PubMed
- C. A. Fustin, V. Abetz and J. F. Gohy, Eur. Phys. J. E: Soft Matter Biol. Phys., 2005, 16, 291–302 CrossRef CAS PubMed
- R. Erhardt, M. Zhang, A. Bo, H. Zettl, C. Abetz, P. Frederik, G. Krausch, V. Abetz and A. H. E. Mu, J. Am. Chem. Soc., 2003, 125, 3260–3267 CrossRef CAS PubMed
- B. Fang, A. Walther, A. Wolf, Y. Xu, J. Yuan and A. H. E. Müller, Angew. Chem., Int. Ed., 2009, 48, 2877–2880 CrossRef CAS PubMed
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841–11876 RSC
- F. Wurm, H. M. König, S. Hilf and A. F. M. Kilbinger, J. Am. Chem. Soc., 2008, 130, 5876–5877 CrossRef CAS PubMed
- L. Nie, S. Liu, W. Shen, D. Chen and M. Jiang, Angew. Chem., Int. Ed., 2007, 46, 6321–6324 CrossRef CAS PubMed
- L. Cheng, G. Zhang, L. Zhu, D. Chen and M. Jiang, Angew. Chem., Int. Ed., 2008, 47, 10171–10174 CrossRef CAS PubMed
- A. Walther and A. H. E. Müller, Soft Matter, 2008, 4, 663–668 RSC
- R. Erhardt, A. Bo, H. Zettl, H. Kaya, W. Pyckhout-hintzen, G. Krausch, V. Abetz, A. H. E. Müller and F. Ju, Macromolecules, 2001, 34, 1069–1075 CrossRef CAS
- H. Xu, R. Erhardt, V. Abetz, A. H. E. Müller and W. A. Goedel, Langmuir, 2001, 17, 6787–6793 CrossRef CAS
- A. Walther, C. Barner-Kowollik and A. H. E. Müller, Langmuir, 2010, 26, 12237–12246 CrossRef CAS PubMed
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed
- H. Qiu, G. Russo, P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 11882–11885 CrossRef CAS PubMed
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS PubMed
- X. Li, Y. Gao, C. E. Boott, D. W. Hayward, R. Harniman, G. R. Whittell, R. M. Richardson, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2016, 138, 4087–4095 CrossRef CAS PubMed
- X. Li, Y. Gao, C. E. Boott, M. A. Winnik and I. Manners, Nat. Commun., 2015, 6, 8127 CrossRef PubMed
- O. E. C. Gould, H. Qiu, D. J. Lunn, J. Rowden, R. L. Harniman, Z. M. Hudson, M. A. Winnik, M. J. Miles and I. Manners, Nat. Commun., 2015, 6, 10009 CrossRef PubMed
- Y. Mai, Y. Zhou and D. Yan, Macromolecules, 2005, 38, 8679–8686 CrossRef CAS
- H. Hong, Y. Mai, Y. Zhou, D. Yan and J. Cui, Macromol. Rapid Commun., 2007, 28, 591–596 CrossRef CAS
- M. R. Radowski, A. Shukla, H. Von Berlepsch, C. Böttcher, G. Pickaert, H. Rehage and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 1265–1269 CrossRef CAS PubMed
- X. Sun, Y. Zhou and D. Yan, Macromol. Chem. Phys., 2010, 211, 1940–1946 CrossRef CAS
- Y. Wang, B. Li, H. Jin, Y. Zhou, Z. Lu and D. Yan, Chem. – Asian J., 2014, 9, 2281–2288 CrossRef CAS PubMed
- B. N. S. Thota, L. H. Urner and R. Haag, Chem. Rev., 2016, 116, 2079–2102 CrossRef CAS PubMed
- P. Chem, C. Mu, X. Fan, W. Tian, Y. Bai, Z. Yang, W. Fan and H. Chen, Polym. Chem., 2012, 3, 3330–3339 RSC
- P. Huang, J. Ao, L. Zhou, Y. Su, W. Huang, X. Zhu and D. Yan, Bioconjugate Chem., 2016, 27, 1564–1568 CrossRef CAS PubMed
- Z. Hou, Y. Zhou and D. Yan, Chin. Polym. Bull., 2015, 5–20 CAS
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS
- H. Jin, W. Huang, X. Zhu, Y. Zhou and D. Yan, Chem. Soc. Rev., 2012, 41, 5986 RSC
- W. Dong, Y. Zhou, D. Yan, H. Li and Y. Liu, Phys. Chem. Chem. Phys., 2007, 9, 1255 RSC
- Y. Zhang, H. Xu, X. Ma, Z. Shi and J. Yin, Macromol. Rapid Commun., 2016, 37, 998–1004 CrossRef CAS PubMed
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef CAS PubMed
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed
- P. Huang, M. Hu, L. Zhou, Y. Wang, Y. Pang, G. Tong, W. Huang, Y. Su and X. Zhu, RSC Adv., 2015, 5, 86254–86264 RSC
- M. Hu, P. Huang, Y. Wang, Y. Su, L. Zhou, X. Zhu and D. Yan, Bioconjugate Chem., 2015, 26, 2497–2506 CrossRef CAS PubMed
- T. Zhang, P. Huang, L. Shi, Y. Su, L. Zhou, X. Zhu and D. Yan, Mol. Pharmaceutics, 2015, 12, 2328–2336 CrossRef CAS PubMed
- Y. Wang, P. Huang, M. Hu, W. Huang, X. Zhu and D. Yan, Bioconjugate Chem., 2016, 27, 2722–2733 CrossRef CAS PubMed
- J. Kwak, S. S. Nam, J. Cho, E. Sim and S.-Y. Lee, Phys. Chem. Chem. Phys., 2017, 19, 10274–10281 RSC
- W. Zhang, L. Shi, K. Wu and Y. An, Macromolecules, 2005, 38, 5743–5747 CrossRef CAS
- X. Zhang, J. Ma, S. Yang and J. Xu, Soft Mater., 2013, 11, 394–402 CrossRef CAS
- T. K. Paira, S. Banerjee, M. Raula, A. Kotal, S. Si and T. K. Mandal, Macromolecules, 2010, 43, 4050–4061 CrossRef CAS
- L. Huang, J. Hu, L. Lang, X. Chen, Y. Wei and X. Jing, Macromol. Rapid Commun., 2007, 28, 1559–1566 CrossRef CAS
- Q. Xu, T. Huang, S. Li, K. Li, C. Li, Y. Liu, Y. Wang, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2018, 57, 8043–8047 CrossRef CAS PubMed
- J. Chen, C. Yu, Z. Shi, S. Yu, Z. Lu, W. Jiang, M. Zhang, W. He, Y. Zhou and D. Yan, Angew. Chem., Int. Ed., 2015, 54, 3621–3625 CrossRef CAS PubMed
- C. Li, C. Chen, S. Li, T. Rasheed, P. Huang, T. Huang, Y. Zhang, W. Huang and Y. Zhou, Polym. Chem., 2017, 8, 4688–4695 RSC
- Y. Zhang, C. Li, T. Rasheed, P. Huang and Y. Zhou, Chin. J. Polym. Sci., 2018, 36, 897–904 CrossRef CAS
- S. Li, C. Yu and Y. Zhou, Sci. China: Chem., 2019, 62, 226–237 CrossRef CAS
- C. Li, T. Rasheed, H. Tian, P. Huang, Y. Mai, W. Huang and Y. Zhou, ACS Macro Lett., 2019, 8, 331–336 CrossRef CAS
- Y. Bai, X. Fan, W. Tian, C. Mu, Z. Yang, W. Fan, H. Zhang and W. Zhang, Polymer, 2013, 54, 1734–1738 CrossRef CAS
- Y. Liu, J. Jin, H. Deng, K. Li, Y. Zheng, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7952–7957 CrossRef CAS PubMed
- H. Cheng, X. Yuan, X. Sun, K. Li, Y. Zhou and D. Yan, Macromolecules, 2010, 43, 1143–1147 CrossRef CAS
- W. Liu, R. Liu, Y. Li, H. Kang, D. Shen and M. Wu, Polymer, 2009, 50, 211–217 CrossRef CAS
- B. I. Voit, Acta Polym., 1995, 46, 87–99 CrossRef CAS
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed
- S. Stiriba, H. Frey and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 1329–1334 CrossRef CAS PubMed
- S. J. Teertstra and M. Gauthier, Prog. Polym. Sci., 2004, 29, 277–327 CrossRef CAS
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC
- M. Liu and J. M. J. Fréchet, Pharm. Sci. Technol. Today, 1999, 2, 393–401 CrossRef CAS PubMed
- A. D’Emanuele and D. Attwood, Adv. Drug Delivery Rev., 2005, 57, 2147–2162 CrossRef PubMed
- Z. Zhang, F. Wang, Z. Zhang and F. Wang, J. Dispersion Sci. Technol., 2008, 29, 1092–1097 CrossRef CAS
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS
- J. Liu, W. Huang, Y. Pang and D. Yan, Chem. Soc. Rev., 2015, 44, 3942–3953 RSC
- Z. Hou, T. Huang, C. Cai, T. Resheed and C. Yu, Chin. J. Polym. Sci., 2017, 35, 602–610 CrossRef CAS
- M. Qi, W. Huang, G. Xiao, X. Zhu, C. Gao and Y. Zhou, Acta Polym. Sin., 2017, 2, 214–228 Search PubMed
- H. Cheng, S. Wang, J. Yang, Y. Zhou and D. Yan, J. Colloid Interface Sci., 2009, 337, 278–284 CrossRef CAS PubMed
- J. Liu, W. Huang, Y. Pang, P. Huang, X. Zhu, Y. Zhou and D. Yan, Angew. Chem., Int. Ed., 2011, 50, 9162–9166 CrossRef CAS PubMed
- J. Liu, Y. Pang, J. Chen, P. Huang, W. Huang, X. Zhu and D. Yan, Biomaterials, 2012, 33, 7765–7774 CrossRef CAS PubMed
- L. Yang, S. Fu, X. Zhu, L. M. Zhang, Y. Yang, X. Yang and H. Liu, Biomacromolecules, 2010, 11, 3395–3405 CrossRef CAS PubMed
- C. Gao and X. Zheng, Soft Matter, 2009, 5, 4788 RSC
- M. Jiang, Y. Wu, Y. He and J. Nie, Polym. Int., 2009, 58, 31–39 CrossRef CAS
- L. Zhong, X. He, Y. Zhou, D. Yan and C. Pan, Acta Polym. Sin., 2007, 986–991 CAS
- H. Hong, Y. Mai, Y. Zhou, D. Yan and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 668–681 CrossRef CAS
- B. Yu, X. Jiang, R. Wang and J. Yin, Macromolecules, 2010, 3, 10457–10465 CrossRef
- M. Qi, K. Li, Y. Zheng, T. Rasheed and Y. Zhou, Langmuir, 2018, 34, 3058–3067 CrossRef CAS PubMed
- M. Gauthier, J. Li and J. Dockendorff, Macromolecules, 2003, 36, 2642–2648 CrossRef CAS
- A. V. Vivek, K. Babu and R. Dhamodharan, Macromolecules, 2009, 42, 2300–2303 CrossRef CAS
- S. Il Yun, G. E. Gadd, V. Lo, M. Gauthier and A. Munam, Macromolecules, 2008, 41, 7166–7172 CrossRef
- C. Xie, Z. Ju, C. Zhang, Y. Yang and J. He, Macromolecules, 2013, 46, 1437–1446 CrossRef CAS
- Y. Liu, S. Li, K. Li, Y. Zheng, M. Zhang, C. Cai, C. Yu, Y. Zhou and D. Yan, Chem. Commun., 2016, 52, 9394–9397 RSC
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS PubMed
- A. Lu, G. Hao, Q. Sun, X. Zhang and W. Li, Chem. Synth. Appl. Graphene Carbon Mater., 2012, 213, 1107–1131 CAS
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed
- M. Kruk, Acc. Chem. Res., 2012, 45, 1678–1687 CrossRef CAS PubMed
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 RSC
- W. Sun, M. Chen, S. Zhou and L. Wu, Langmuir, 2014, 30, 12011–12017 CrossRef CAS PubMed
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2014, 54, 588–593 Search PubMed
- H. Tian, Z. Lin, F. Xu, J. Zheng, X. Zhuang, Y. Mai and X. Feng, Small, 2016, 12, 3155–3163 CrossRef CAS PubMed
- J. Wang, H. Liu, J. Diao, X. Gu, H. Wang, J. Rong, B. Zong and D. S. Su, J. Mater. Chem. A, 2015, 3, 2305–2313 RSC
- A. Chen, Y. Li, Y. Yu, Y. Li, K. Xia, Y. Wang and S. Li, J. Mater. Sci., 2016, 51, 7016–7028 CrossRef CAS
- D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023–4071 RSC
- Y. Huang, R. Dong, X. Zhu and D. Yan, Soft Matter, 2014, 10, 6121 RSC
- Y. Huang, D. Wang, X. Zhu, D. Yan and R. Chen, Polym. Chem., 2015, 6, 2794–2812 RSC
- P. Sun, D. Chen, H. Deng, N. Wang, P. Huang, X. Jin and X. Zhu, Bioconjugate Chem., 2017, 28, 1470–1480 CrossRef CAS PubMed
- D. Wang, H. Chen, Y. Su, F. Qiu, L. Zhu, X. Huan, B. Zhu, D. Yan, F. Guo and X. Zhu, Polym. Chem., 2013, 4, 85–94 RSC
- S. Yu, R. Dong, J. Chen, F. Chen, W. Jiang, Y. Zhou, X. Zhu and D. Yan, Biomacromolecules, 2014, 15, 1828–1836 CrossRef CAS PubMed
- F. Qiu, C. Tu, R. Wang, L. Zhu, Y. Chen, G. Tong, B. Zhu, L. He, D. Yan and X. Zhu, Chem. Commun., 2011, 47, 9678–9680 RSC
- Y. Huang, F. Qiu, L. Shen, D. Chen, Y. Su, C. Yang, B. Li, D. Yan and X. Zhu, ACS Nano, 2016, 10, 10489–10499 CrossRef CAS PubMed
- R. Wang, X. Jiang, C. Di and J. Yin, Macromolecules, 2010, 43, 10628–10635 CrossRef CAS
- R. Wang, B. Yu, X. Jiang and J. Yin, Adv. Funct. Mater., 2012, 22, 2606–2616 CrossRef CAS
- Y. Boontongkong and R. E. Cohen, Macromolecules, 2002, 35, 3647–3652 CrossRef CAS
- H. Cho, H. Park, T. P. Russell and S. Park, J. Mater. Chem., 2010, 20, 5047–5051 RSC
- J. Chai and J. M. Buriak, ACS Nano, 2008, 2, 489–501 CrossRef CAS PubMed
- H. Tian, J. Qin, D. Hou, Q. Li, C. Li, Z.-S. Wu and Y. Mai, Angew. Chem., Int. Ed., 2019, 58, 10173–10178 CrossRef CAS PubMed
- S. Liu, P. Gordiichuk, Z. Wu, Z. Liu, W. Wei, M. Wagner, M.-N. Nasser, D. Wu, Y. Mai, A. Herrmann, M. Klaus and X. Feng, Nat. Commun., 2015, 6, 8817 CrossRef CAS PubMed
- S. Liu, F. Wang, R. Dong, T. Zhang, J. Zhang and X. Zhuang, Adv. Mater., 2016, 28, 8365–8370 CrossRef CAS PubMed
- S. Liu, J. Zhang, R. Dong, P. Gordiichuk, T. Zhang, X. Zhuang, Y. Mai, F. Liu, A. Herrmann and X. Feng, Angew. Chem., Int. Ed., 2016, 128, 12704–12709 CrossRef
- W. Mao, X. Cao, Q. Sheng, L. Han and S. Che, Angew. Chem., Int. Ed., 2017, 56, 10670–10675 CrossRef CAS PubMed
- X. Cao, W. Mao, Y. Mai, L. Han and S. Che, Macromolecules, 2018, 51, 4381–4396 CrossRef CAS
| This journal is © the Partner Organisations 2019 |