
Theory-guided materials design: two-dimensional MXenes in electro- and photocatalysis
Albertus D.
Handoko
 a,
Stephan N.
Steinmann
b and
Zhi Wei
Seh
*a
a,
Stephan N.
Steinmann
b and
Zhi Wei
Seh
*a
aInstitute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis, Singapore 138634, Singapore. E-mail: sehzw@imre.a-star.edu.sg
bUniv Lyon, ENS de Lyon, CNRS, Université Lyon 1, Laboratoire de Chimie UMR 5182, F-69342, Lyon, France
First published on 3rd April 2019
Abstract
Two-dimensional transition metal carbides and nitrides (MXenes) have made a significant impact on sustainable energy research in the fields of energy storage and conversion. Unlike short-term energy storage strategies (e.g. batteries and supercapacitors), catalytic conversion of simple molecules to value-added chemicals using renewable energy represents a more long-term solution to the world's energy crisis. Significant advances in density functional theory and low-cost computing in the past decade have enabled the generation of reliable materials data from fundamental physics equations. The paradigm shift towards theory-guided materials design is expected to enhance the catalyst discovery and development process by providing rational guidance to screen viable MXene catalysts more rapidly than an experimental-only approach. In this review, we aim to provide a critical appraisal of the latest theoretical and experimental work on MXenes in the fields of electro- and photocatalytic energy conversion, including relevant reactions involving hydrogen, oxygen, carbon dioxide and nitrogen molecules. In the process, we will also be pointing out current limitations in theoretical models, existing scientific gaps and future research directions for this field.
 Albertus D. Handoko | Albertus Denny Handoko is a Scientist at the Institute of Materials Research and Engineering, A*STAR. He attained his core competency in solvothermal/hydrothermal synthesis methods and X-ray characterisation during his PhD studies at Nanyang Technological University (Singapore). He went for postdoctoral research fellowships at the University College London (UK) and National University of Singapore where he gained research experience and developed keen interest in photo- and electrochemistry. His current research effort is dedicated to understanding advanced electrocatalytic systems for renewable energy conversion reactions such as CO2 reduction, water splitting and purification. |
 Stephan N. Steinmann | Dr Stephan N. Steinmann is a CNRS researcher at the Ecole Normale Superieure de Lyon (France). After his PhD thesis on the development of dispersion corrections to standard density functional approximations in 2012 at the Ecole Polytechnique Federale de Lausanne (Switzerland), he went for a post-doctoral stay to Duke University (USA), developing electronic structure methods. Back in Europe since 2014, he has been developing advanced methods to treat heterogeneous electro-catalysis and the metal/liquid interface in general. Concomitantly, his current research is devoted to strong collaborations with experimental groups from academia and industry. |
 Zhi Wei Seh | Zhi Wei Seh is a scientist and NRF fellow at the Institute of Materials Research and Engineering, A*STAR. He received his BS and PhD degrees in Materials Science and Engineering from Cornell University and Stanford University, respectively. His research interests lie in the design of new materials for energy storage and conversion, including advanced battery and electrocatalyst systems. He has published in top journals such as Science, Nature Communications, Nature Energy and Nature Catalysis, and has also received numerous awards including the Highly Cited Researchers (Clarivate Analytics), Emerging Investigators (Journal of Materials Chemistry A), and Innovators under 35 Asia (MIT Technology Review) awards. |
1. Introduction
The burgeoning energy demand linked to rapid global population growth and expanding industrialization calls for new sources of sustainable and renewable energy. The U.S. Energy Information Administration estimates that close to 80% of the world's primary energy consumption is derived from fossil fuels, including coal, natural gas, petroleum, and related products.1 Fossil fuel is not a renewable source of energy, and its depletion within the next century could pose grave consequences if suitable alternatives are not found on time.2 In addition, fossil fuel combustion is also a major source of CO2, along with other noxious pollutants like soot, SOx, and NOx that further exacerbate environmental issues.Electro- and photocatalytic energy conversion presents an attractive pathway to convert earth abundant molecules (e.g. H2O, CO2, or N2) into high-value products and fuels (e.g. H2, hydrocarbons, oxygenates, or NH3). While sustainable conversion can be achieved by tapping into renewable energy sources, the viability of this process is contingent on the development of selective, robust and affordable catalysts that allow faster reaction rates to occur at lower overpotentials. The search for high performance catalysts, especially those without precious metal content, has attracted intensive research interest.3 MXenes, a contemporary family of 2D materials based on transition metal carbides or nitrides, have shown interesting properties and promise in catalytic energy conversion.4 So far, of the 54 MXenes proposed to exist in silico, only 22 of them have been experimentally realised (Fig. 1). One of the challenges in MXene synthesis is the instability of some of its constituents in strong HF etchants, particularly nitrogen and transition metals reactive to fluorine.5 More recently, milder synthesis routes using in situ HF generated from fluoride salts in acids have been demonstrated.6,7
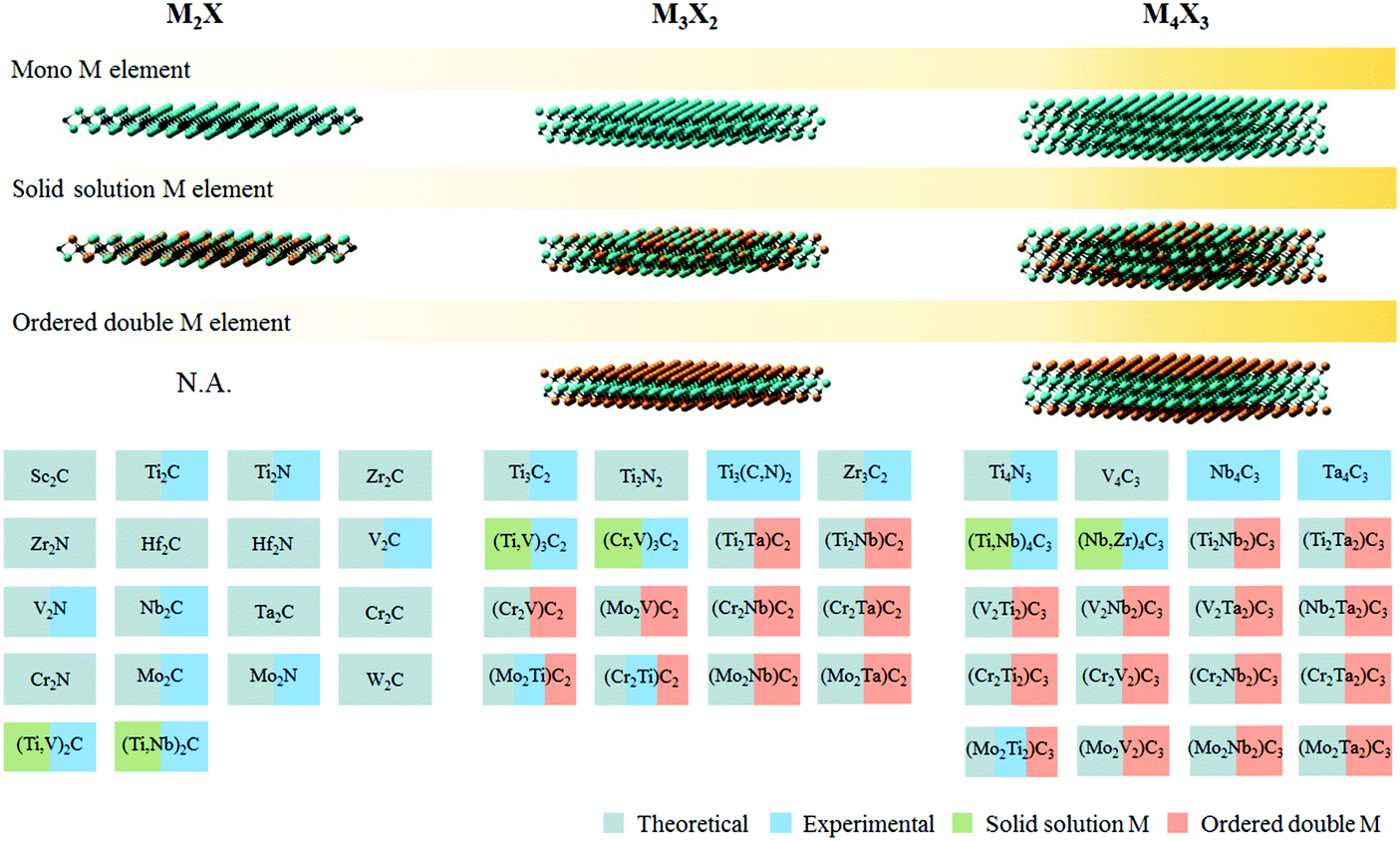 | ||
| Fig. 1 Different types of MXene discovered so far, both theoretically and experimentally. Adapted with permission from ref. 4 Springer-Nature, Copyright 2017. Updated with newly synthesised MXenes from ref. 7 and 8. | ||
In this review, we will discuss the latest promising research on MXenes in the field of electro- and photocatalytic energy conversion, based on the strategy of theory-guided materials design. First, the fundamentals of catalytic energy conversion will be elaborated, including the H2 evolution reaction (HER), O2 evolution reaction (OER), O2 reduction reaction (ORR), CO2 reduction reaction (CO2RR) and N2 reduction reaction (N2RR). The key properties of MXenes relevant to electro- and photocatalysis will also be discussed. A critical appraisal of the state-of-the-art research using MXenes as catalysts in the above reactions will follow, including both theoretical and experimental aspects. Using computational chemistry to guide materials design has the potential to accelerate catalyst development by uncovering broader governing principles and providing rational design guidelines. In the process, we will also be pointing out current limitations in theoretical models, existing scientific gaps and future research directions for this field.
2. Fundamentals
2.1 Basic concepts
Electrocatalysis exploits the thermodynamic and kinetic tuning of electrochemical reactions occurring on electrode surfaces as a function of the applied external voltage. An electrocatalyst modifies the catalytic reaction rate compared to solution electrochemistry or an inert electrode, by providing favourable reaction sites, altering adsorption energies and/or introducing alternative reaction pathways. Therefore, faster product formation can be achieved at lower overpotentials. Modifications in exposed facets,9 surface functionalisation,10 size11–13 and doping/co-catalyst addition14,15 are common strategies to enhance electrochemical activity and selectivity. Depending on the target reaction, MXenes can be suitable electrocatalysts due to their high electronic conductivity and optimal adsorption strength for key reaction intermediates.In photocatalysis, the driving force arises from electron/hole pairs generated in semiconducting materials upon light irradiation. The additional step of photocarrier generation makes photocatalysis generally less efficient than electrocatalysis due to the associated energy losses. Important strategies to improve photocatalyst performance usually involve bandgap alteration,16 addition of co-catalysts,17–19 or the formation of a cascading junction (Z-scheme) structure20–22 aimed at enhancing light absorption and promoting photogenerated carrier separation.23 MXenes can be a prime choice for co-catalysts as their Fermi level is compatible with the Fermi levels of popular wide-bandgap photocatalysts like TiO2 and ZnO, thus encouraging electron migration to MXene surfaces and simultaneous formation of a Schottky barrier at the interface to prevent back-flow.18
Before going into the details of electro- and photocatalytic processes, there are several fundamental terms that need to be defined.
Overpotential describes how much additional voltage above the thermodynamic requirement is needed to drive an electrochemical reaction. Expressed as a potential difference, overpotential is considered as one of the primary metrics in evaluating the catalytic activity as it is strongly related to the redox reaction kinetics and the overall energy efficiency of the process. Experimentally, it is determined by the onset potential at which a redox event is observed, usually marked by a significant rise of the current density compared to the baseline (non-redox) level. Electro- and photocatalytic reactions such as CO2RR can be complicated, with multiple electron transfer steps involving different intermediates and overpotentials.24 It is common to take the largest overpotential (i.e. the most difficult) of those individual steps to be the overall redox overpotential. An excellent catalyst should display an overall overpotential value close to zero.
Faradaic efficiency (FE) describes how efficient a catalytic system is in using electron input to facilitate a particular chemical reaction. In catalysis, FE is usually calculated by comparing the eoutput, estimated based on the amount of product generated (or reactant consumed), with the actual einput as measured by a coulombmeter or a potentiostat. A real catalytic reaction usually shows FE less than unity, as Faradaic losses can be incurred when electrons or ions participate in unmonitored side reactions. Comparison of FE among different products can serve as an insightful indicator for both energy efficiency and product selectivity, especially important for CO2RR where product selectivity is a critical challenge.
Turnover number (TON) describes the intrinsic activity of a catalyst. It is usually defined as the total number of reactant molecules converted (or the total number of product molecules generated) per active site. TON is also often used to describe the “recyclability” of a catalyst, which is calculated by the number of molecules of the reagent that a molecule of catalyst can convert before it becomes inactive. In both cases, an ideal catalyst should have a very large TON, approaching infinity. Sometimes, the term turnover frequency (TOF) is used, which is simply TON per unit time. TOF describes the number of catalytic conversions that occur at the active centre per unit time, which also gives information on the overall kinetics. Thus, the higher the intrinsic reaction rate, the higher the TOF value.
Tafel plot/equation relates the rate of electrochemical reaction on a particular catalyst to the applied overpotential:
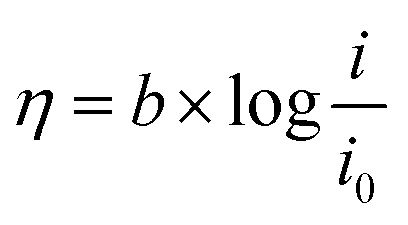 | (1) |
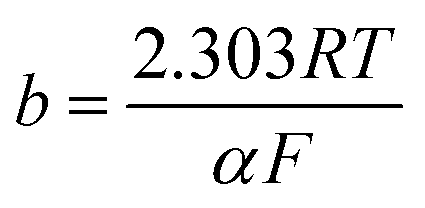 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i” axis at zero η. Smaller b and larger i0 values are indicators of a good catalyst, implying a larger reaction transfer coefficient and better binding between the reactants/electrolyte and catalyst. Tafel slope values, subject to limitations, can also be used as indicators of reaction pathways and rate determining steps in electrochemical reactions. For example, a multi-step reaction with the rate determining chemical step preceded by two 1-electron electrochemical steps should display a Tafel slope of around 30 mV dec−1.25
i” axis at zero η. Smaller b and larger i0 values are indicators of a good catalyst, implying a larger reaction transfer coefficient and better binding between the reactants/electrolyte and catalyst. Tafel slope values, subject to limitations, can also be used as indicators of reaction pathways and rate determining steps in electrochemical reactions. For example, a multi-step reaction with the rate determining chemical step preceded by two 1-electron electrochemical steps should display a Tafel slope of around 30 mV dec−1.25
The Sabatier principle states that the adsorption between the catalysts and reactants should be neither too weak nor too strong.26 When the adsorption is too weak, it becomes difficult to bind the reactants onto the catalyst surface, and no reaction would take place. On the other hand, very strong adsorption causes the reactant to be too strongly bound, slowing down the product desorption step, and sometimes leading to rapid catalyst deactivation.27 Hence, an optimal catalyst should bind the reactants just strong enough to allow surface adsorption of the reactant, while still allowing facile desorption of the intended products. Taking a relatively simple HER as an example, the catalytic activity (expressed as i0) of the elementary steps (Volmer, Tafel or Heyrovský) is exponentially proportional to the partial pressure of hydrogen (pH2) and the standard Gibbs free energy of adsorption of atomic hydrogen (ΔGH) by:28
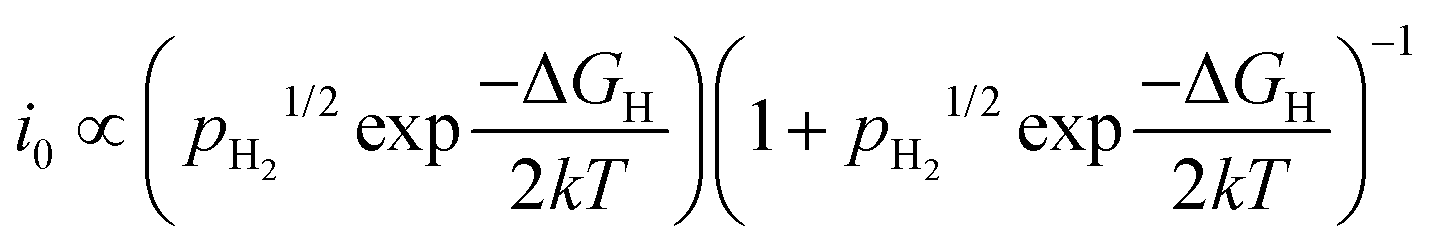 | (3) |
Plotting logi0 against ΔGH results in a triangle that yields a maximum value of i0 when ΔGH is close to 0, and rising and falling logi0 when 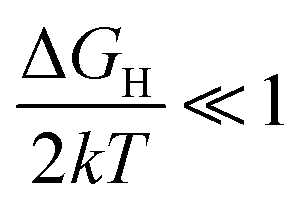 (very strong *H adsorption, * denotes an adsorption site on the electrode) and
(very strong *H adsorption, * denotes an adsorption site on the electrode) and 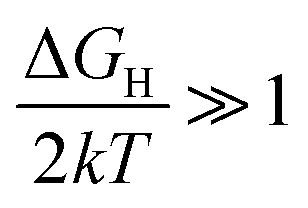 (i.e. very weak *H adsorption), respectively, giving rise to a ‘volcano plot’.
(i.e. very weak *H adsorption), respectively, giving rise to a ‘volcano plot’.
2.2 Computational methods
Density functional theory (DFT) is frequently exploited to calculate the adsorption energy based on the differences between the energy of a “clean” atomic surface model plus half the energy of a H2 molecule, representing a proton/electron pair. This model is known as the computational hydrogen electrode (CHE).29 In combination with DFT computations for the intermediates (e.g. the energy of hydrogen bound onto the same surface model), thermodynamic overpotentials can be estimated at low computational cost. The thermodynamic overpotential is defined as the potential at which each electrochemical step (coupled proton/electron transfer) is thermo-neutral. These overpotentials can then be plotted as a function of a descriptor (e.g., the d-band centre or the adsorption energy of a particular intermediate) to obtain a volcano plot. Though simple, volcano plots have proven to be a powerful tool that can predict the activity of different materials in catalysing various reactions.3,30,31In its most commonly used CHE formulation, neither electrolyte presence nor electrode polarization due to the applied electrochemical potential is taken into account. It also assumes that no additional kinetic barriers exist, i.e. if a reaction is thermodynamically favourable at a given potential, it will also occur at a high rate. These assumptions, which simplify the computational work significantly, often provide correct trends,32–34 although they cannot capture the details of the electrochemical processes.35–37 Furthermore, for complex reactions, where chemical and electrochemical steps have to be considered, microkinetic simulations are necessary to estimate the rate of the electrochemical reactions, rather than taking the thermodynamic overpotential as provided by CHE as a proxy for the activity of a given catalyst.38,39
In order to lift the approximations of CHE, transition states for all processes need to be determined, which is particularly challenging for electrochemical reactions.40,41 Second, the electrochemical potential needs to be included explicitly, which is equivalent to simulating electrode surfaces in the presence of a surface charge. Third, the solvent needs to be modelled as well. The computationally most efficient approach is to rely on an implicit solvent/electrolyte,42–44 which comes at the cost of neglecting “direct” (i.e. chemical) effects of the solvent, such as the competition of the adsorption between reactants and the solvent or the assistance of solvent molecules in breaking bonds.45 Alternatively, aqueous electrolytes are often approximated as ice-like layers,46,47 which is very cumbersome for studying various reaction intermediates, as the (arbitrary) placement of adsorbates into or under these ice-like layers is associated with uncontrolled effects on their properties. Furthermore, for non-aqueous solvents, no obvious organization for placing explicit solvent molecules is currently available. All these challenges explain why the simplistic CHE model, with its success in predicting activity trends, continues to be highly popular.
3. Properties of MXenes
In order to elaborate about MXenes, one has to understand a special family of the layered MAX phases. The acronym MAX comes from the materials’ composition, Mn+1AXn (n = 1, 2 and 3), where ‘M’ stands for early transition metals such as Sc, Ti, Cr, Nb, V, Mo and so on, ‘A’ is a group IIIA or IVA element, and ‘X’ represents C and/or N. MAX is composed of Mn+1Xn face-sharing octahedral layers and interlayer cation A, resulting in a hexagonal P63/mmc structure, the same symmetry as graphite (Fig. 2A). By etching out the interlayer cations A, a graphene-like 2D transition metal carbide/nitride Mn+1XnTx (here T represents the surface termination groups such as fluorine, oxygen or hydroxyl) is obtained, and the name MXene was given due to their remarkable similarity to graphene and the relation to the MAX phase.5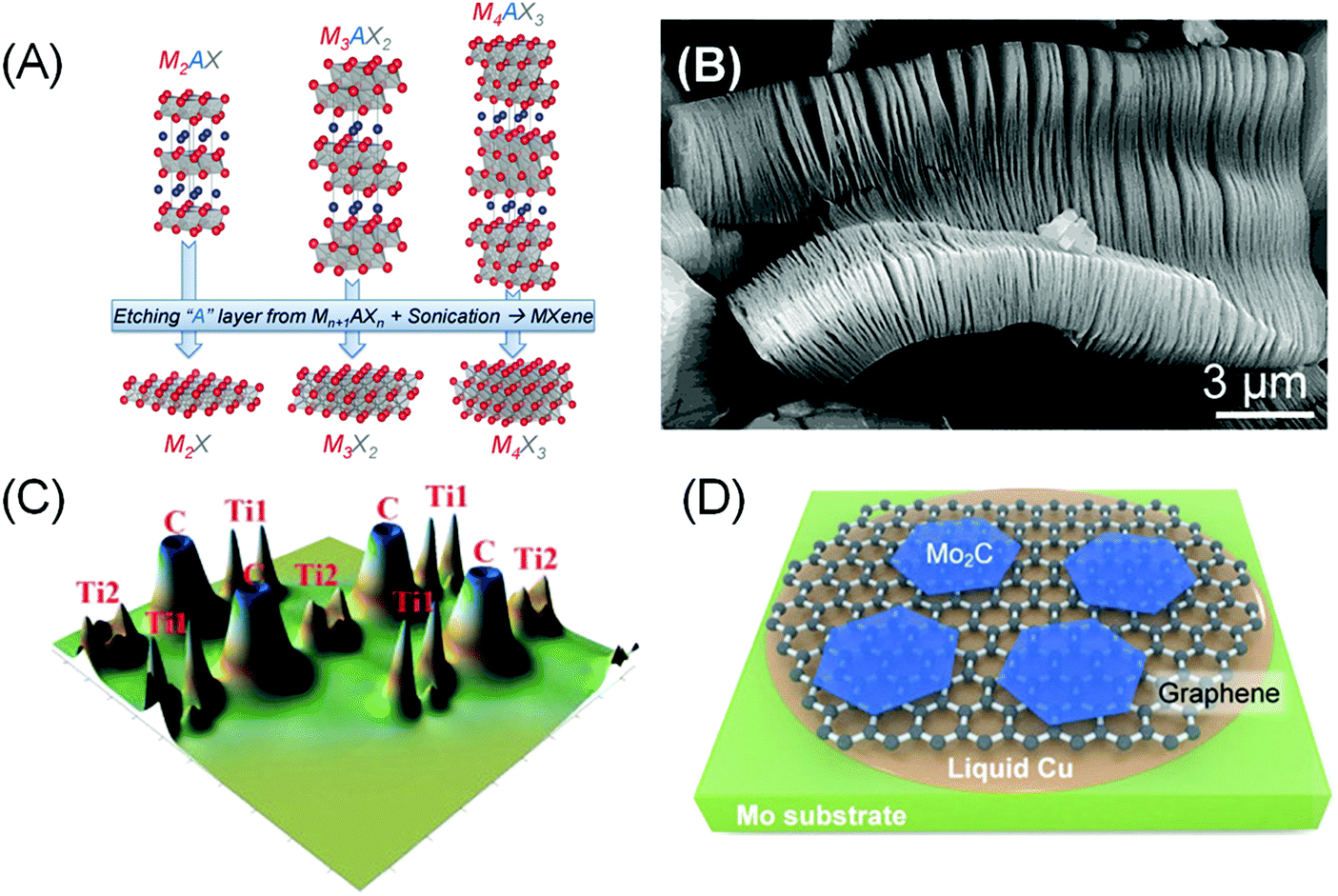 | ||
| Fig. 2 (A) Structure of MAX and the corresponding MXenes after etching an “A” layer and sonication. Reprinted with permission from ref. 58 Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. (B) Scanning electron micrograph of Ti3C2Tx after HF etching of Ti3AlC2. Reprinted with permission from ref. 59 Copyright 2012 American Chemical Society. (C) Calculated valence electron density of bare Ti2C. Ti1 refers to the outermost Ti atoms that have direct bonds with ‘A’ atoms, while Ti2 represents inner Ti atoms that interact only with X atoms. Reprinted from ref. 49 Copyright 2012, with permission from Elsevier. (D) Schematic showing CVD growth of bare Mo2C MXenes on a molten Cu on Mo substrate from a CH4 precursor. A graphene underlayer was formed in situ at a high CH4 flow rate. Reprinted with permission from ref. 52 Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The relatively strong metallic M–A bonds in MAX phases suggest that the mechanical method is not suitable to exfoliate the Mn+1Xn layers. However, the distinct chemical reactivity between M–A and M–X bonds make selective etching plausible. Thus the first reported carbide MXene, Ti3C2Tx, was obtained via aqueous hydrofluoric acid (HF) etching of Ti3AlC2 at room temperature (Fig. 2B).5 The majority of other MXene phases were synthesized with a similar recipe, using either direct addition of HF or via in situ formation of HF (e.g. by adding LiF to HCl), sometimes at elevated temperature and/or prolonged heating.4,6
Most pristine (bare) MXenes show metallic characteristics.48 The high conductivity can be linked to the redistribution of the outer M(1) atoms’ 3d states from the broken M(1)–A bonds into delocalized M(1)–M(1) metallic-like bonding states placed in the window around the Fermi level (Fig. 2C).49 The M(1) atoms are distinct from the inner M(2) atoms that interact more closely with the X atoms.
Further delamination of the MXene layers can be achieved by intercalation strategies, using sonication in moieties such as dimethyl sulfoxide, tetrabutylammonium hydroxide or amines.4,6,50,51 Other synthetic methods, such as etching MAX powders in molten fluoride salt at high temperature and in an inert environment, or chemical vapour deposition (CVD), have so far been demonstrated only on limited types of MXenes.52–56 We note that, while requiring high vacuum and high temperature, the CVD growth method is particularly promising for extra-large grain growth of bare MXenes that may be of interest in energy storage application.52,53 Nitride variant of MXenes is also obtainable via ammoniation of the carbide MXene.8
So far, among the 70 odd MAX phases, only Al-containing ones have shown successful conversion to MXenes.4 This led to the exploration of other, non-MAX layered materials, like Zr3Al3C5 that have been shown to form Zr3C2Tx after HF etching.57 Unlike the case for the MAX precursor, where only a single layer of ‘A’ element is removed, multiple layers of atoms are removed for the non-MAX precursors (e.g. 3 Al–C layers for Zr3Al3C5).
To date, 22 MXenes have been successfully synthesized and dozens more theoretically predicted.4 However, producing single phase MXene monolayers remains highly challenging for many MXene compositions, and less hazardous synthesis procedures are still under investigation.
3.1 Surface terminations
To maintain charge balance, surface terminations quickly form on the “dangling” M(1) atoms during the etching of A atoms in aqueous HF solutions. –OH, –O, and –F are the most common species that can be observed filling the Tx surface termination sites in wet-etched MXenes. An exception is MXenes produced via the CVD method, of which bare Mo2C has been grown on a molten Cu/Mo substrate from a CH4 precursor.52 Larger and thinner Mo2C grains were achieved by increasing the CH4 flow rate, which results in in situ formation of a graphene underlayer that improves the crystallinity of Mo2C (Fig. 2D).Precise characterization on the surface coverage and ratios of termination groups using X-ray techniques remains challenging due to the intrinsic low-scattering strength of the light elements. Solid state nuclear magnetic resonance (NMR) spectroscopy is one potential solution, as it has been successfully used to quantify the surface termination composition on Ti3C2Tx MXene.60 More importantly, the study indicates that the ratio of Tx surface termination species can be tuned by adjusting the synthesis method. DFT calculations also indicate that the tuneability of the Tx population is also related to the different metal species in MXenes, which governs the stability of the M–Tx interaction.61,62
Being the outer-most layer, the composition of Tx significantly influences MXene's physical and electronic properties. For example, –O terminated MXenes have been calculated to have smaller lattice parameters and thinner interlayer thickness than –F and –OH terminated variants, leading to higher mechanical strength.63 The presence of Tx termination also makes MXenes more semiconducting. The source of this electronic property alteration is linked to the distinct electron orbital hybridization that yields bandgap variations. For example, the bandgap of Sc2CTx MXene was found to be dependent on the Tx species in the order of Sc2CO2 > Sc2CF2 > Sc2C(OH)2.63 Generally –F and –OH have similar effects on the electronic structures because both can only receive one electron, but –O differs significantly because it readily demands two electrons.63 It is also shown that –O functionalisation is most favourable for high Li-ion capacity, superior to those with –F and –OH terminations, as Li prefers to adsorb on O.64,65 In some other application areas, such as heavy metal ion adsorption, –OH terminated MXenes are preferred.66
The Tx composition was also found to affect surface adsorption and magnetisation behaviour. For example, –O terminated Ti3C2Tx exhibits a near zero GH of 0.003 eV while bare and –F terminated Ti3C2Tx give high values of −0.927 and 1.995 eV, respectively.18 Moreover, –OH terminated Ti3C2Tx is calculated to show stronger CO2 adsorption than –F terminated ones.19 In general, the experimentally hardly avoidable surface termination by Tx suppresses the magnetism and electronic conductivity in MXenes. For example, the semi-metallic ferromagnetic behaviour of bare Cr2C has been shown to become semiconducting antiferromagnetic upon –F, –OH and –H functionalization.67 Similarly, other properties such as optical, thermal and charge transport are effectively modified by the presence of surface functionalizations.68
3.2 Carbide vs. nitride MXenes
The relatively more electronegative nitrogen compared to carbon at the MXene X sites gives stronger bonding to the electropositive M, thus variations in physical and electronic properties related to the shorter M–M and M–X interatomic distances are expected. For example, bare Tin+1NnTx has been calculated to have smaller lattice constant and monolayer thickness compared to Tin+1CnTx, accompanied by larger in-plane Young's modulus.69 It is interesting that Tin+1NnTx MXenes, including –O terminated ones, have higher density of states (DOS) at the Fermi level, as opposed to carbide MXenes.70 These findings lead to predictions of higher electronic conductivity and more catalytically active surface on nitride MXenes.Despite their immense promise, nitride MXenes are generally more difficult to obtain due to less stable M–N layers in the etching solution and higher formation energy. Nevertheless, an etching strategy to produce Ti2NTx by exploiting the K+ ions that can intercalate and weaken the M–X layers has recently been proposed.7 An alternative strategy is to obtain nitride MXene via ammoniation of the carbide MXene, for example Mo2NTx synthesized from Mo2CTx is reported to exhibit 3 orders of magnitude higher electrical conductivity than the corresponding carbide.8 Although limited by difficulties in synthesis, we expect that nitride MXenes will continue to be the subject of intense experimentation to exploit their superior electronic and physical properties.
3.3 Effect of metal species
Many interesting properties of MXenes arise from the open d orbitals in the M transition metal. Thus, modification of M with different metal species, either as solid solutions, ordered layers or ad-atoms, is expected to bring a dramatic change in their properties. The strong interaction between M and the adjacent Tx suggests that MXenes with different metals may energetically favour different functional groups. For example, –F and –O species are more commonly detected in Ti3C2Tx, whereas –F and –OH are more common for V2CTx.61,62 Consequently, alteration of M species composition will bring about a significant change in the electronic properties of MXenes.48 A more subtle change in the electronic structure has been predicted on MXenes with varying M but the same Tx preference, as seen in the increased bandgap in the order of Ti2CO2 < Zr2CO2 < Hf2CO2.68 Addition of transition metal ad-atoms over the MXene layer has also been reported to yield interesting HER catalytic properties.71The discovery of double-transition metal MXenes,72 containing a sandwiched configuration of different M′ and M′′ transition metal species, has enriched the family further. The metals can be either randomly distributed (e.g. (Nb,Zr)4C3Tx, solid solution73), or form ordered layers of M′–X–M”–X–M′ (e.g. Mo2TiC2Tx) or M′–X–M′′–X–M′′–X–M′ (e.g. Mo2Ti2C3Tx74). The overall electronic properties will be highly dependent on the overall DOS near the Fermi level which contains both M′ and M′′ partially filled d orbitals due to the degeneracy effect. This means double-transition metal MXene can be intrinsic semiconductors (like the ordered Mo2TiC2 and Cr2TiC274,75) or a semi-metal (like Ti2MnC2Tx76). It is interesting that Ti2MnC2Tx is still found to be ferromagnetic in the ground state regardless of its surface terminations,76 while surface terminations have been shown to quench magnetism in single MXenes like Ti2CTx.
Understandably, the outer M′ atoms still play a more important role in MXene's surface behaviour compared to inner M′′. For example, Mo2TiC2Tx shows Li adsorption behaviour closer to Mo3C2Tx, rather than the isostructural Ti3C2Tx, thus the former shows larger capacity when applied as a Li-battery electrode.74 The current investigation on double transition metal MXenes is very limited, but it is expected to expand rapidly. Khazaei et al. proposed that many compositions are topological insulators due to their large spin orbital coupling between metal elements that can be widely applied in many energy applications such as thermoelectrics.77
3.4 Effect of layer thickness
Due to the higher volume to surface ratio, the stability of MXenes generally increases with increasing layer thickness, such that M2XTx < M3X2Tx < M4X3Tx, in agreement with the experimental observation on the better stability of Ti3C2Tx compared to Ti2CTx.48 The effect of layer thickness on mechanical properties is investigated by first-principles calculations, and highest Young's moduli for the thinnest MXene are obtained from a comparison of Ti2C, Ti3C2 and Ti4C3.78 The equilibrium bond strength calculations indicate that Ti2C has the shortest Ti–Ti distance but the longest Ti–C bond among the three.49 As surface properties mainly depend on the outer layer atoms, different layer thicknesses do not induce a significant effect on their preferred surface functionalization, e.g. Tin+1CnTx show similar Tx preference of –O > –F > –OH, regardless of the value of n.78 However, distinct metal d band degeneracy was observed on MXenes with the same atomic constituent but different n, thus displaying significantly different electronic structures.79 DFT calculations found that the DOS near the Fermi level generally reduces with increasing n for pristine Tin+1Xn.49 Interestingly, bulk Ti3C2Tx (n = 2) displays much smaller electrical resistivity than Ti2CTx (n = 1) possessing similar surface functionalization, probably due to the increasing difficulty for electron hopping in between the layers when n is smaller.59 Due to the varying accessibility and tuneability of the active sites, different values of n have also been shown to cause variations in the HER activity.11,80With appropriate delamination, MXenes can be obtained as a monolayer or multiple layers; the inter-layer spacing can be tuned by intercalation using multivalent cations, water or organic molecules. The transition metals with partially filled d-orbitals can have different oxidation and spin states, bringing up a vast range of interesting properties. The hydrophilic nature of Tx leads to stable water-based solutions or suspensions, which facilitates applications requiring coating or thin film fabrication.
While it has been established that most MXenes show metallic-like behaviours, the actual bulk conductivity depends greatly on the preparation method. Milder etching and delamination conditions generally result in higher bulk conductivity,81 and so is more thorough removal of intercalated species.82 In summary, the flexibility and tuneability of atomic composition and surface functionalization are key to the excellent electronic, physical and surface properties of MXenes, making them unique among the 2D materials reported so far.
In the next few sections, we will discuss how the excellent properties of MXenes can be exploited to enhance catalytic conversion to various value-added chemicals and fuels.
4. MXene catalysts for hydrogen evolution
4.1 Electrocatalytic hydrogen evolution reaction (HER)
As one of the two half-reactions of electrochemical water splitting, HER provides a cornerstone in the exploration of other, more complex electrocatalytic processes that involve multiple electron transfer. In brief, the mechanism for HER can be separated into three distinct elementary reactions under acidic conditions:Volmer (adsorption/discharge reaction)
| H+ + e− + *→ *H | (4) |
| 2*H→ H2 + 2* | (5) |
Heyrovský (ion + adsorbed *H reaction)
| H+ + *H + e− → H2 + * | (6) |
| H2O + e− + * → *H + OH− | (7) |
| H2O + *H + e− → H2 + * + OH− | (8) |
Nørskov's finding opened a new dimension in catalytic research and now DFT is routinely employed to predict the reactivity of catalysts, based on the calculated binding strength to a particular surface structure. In the quest to search for a prominent HER electrocatalyst amongst MXenes, a holistic consideration of intrinsic metal orbital hybridization, surface terminations as well as layer thickness is required. MXene's high DOS near the Fermi level, constructed from the partially filled d orbitals of M(1) and M(2), is expected to favourably interact with the hydrogen 1s orbital. This is advantageous compared to other, rather inert, 2D materials such as MoS2,86 as the entire basal plane of MXene's highly anisotropic monolayered structure has the potential to act as active sites for hydrogen adsorption.10,87
DFT is an ideal tool to assess MXene's potential for HER electrocatalysis, as it provides a framework for rapid, precise, systematic and yet qualitatively correct predictions of the catalytic performance. The primary metric is GH, and volcano plots based on calculated GH values are then constructed to aid in material screening (Fig. 3A).80,87 In the case of MXenes, the composition and role of Tx surface functional groups for *H adsorption sites need to be clarified and taken into account for a more realistic approximation. In M2XTx type MXenes, –O or –OH was found to be the preferred Tx species, with five MXenes with theoretical HER overpotential under 100 mV identified: Sc2C(OH)2, Mo2CO1.75(OH)0.25, Hf2NO2, Nb2NO2 and V2NO2 (Fig. 3B).87 Ling et al. further observed that the HER overpotential calculation may be simplified as it is directly related to the electron gain of the –O Tx group from various transition metals in M2CO2 systems.88 These theoretical works enable the rational selection of promising MXene catalysts for further experimental testing.
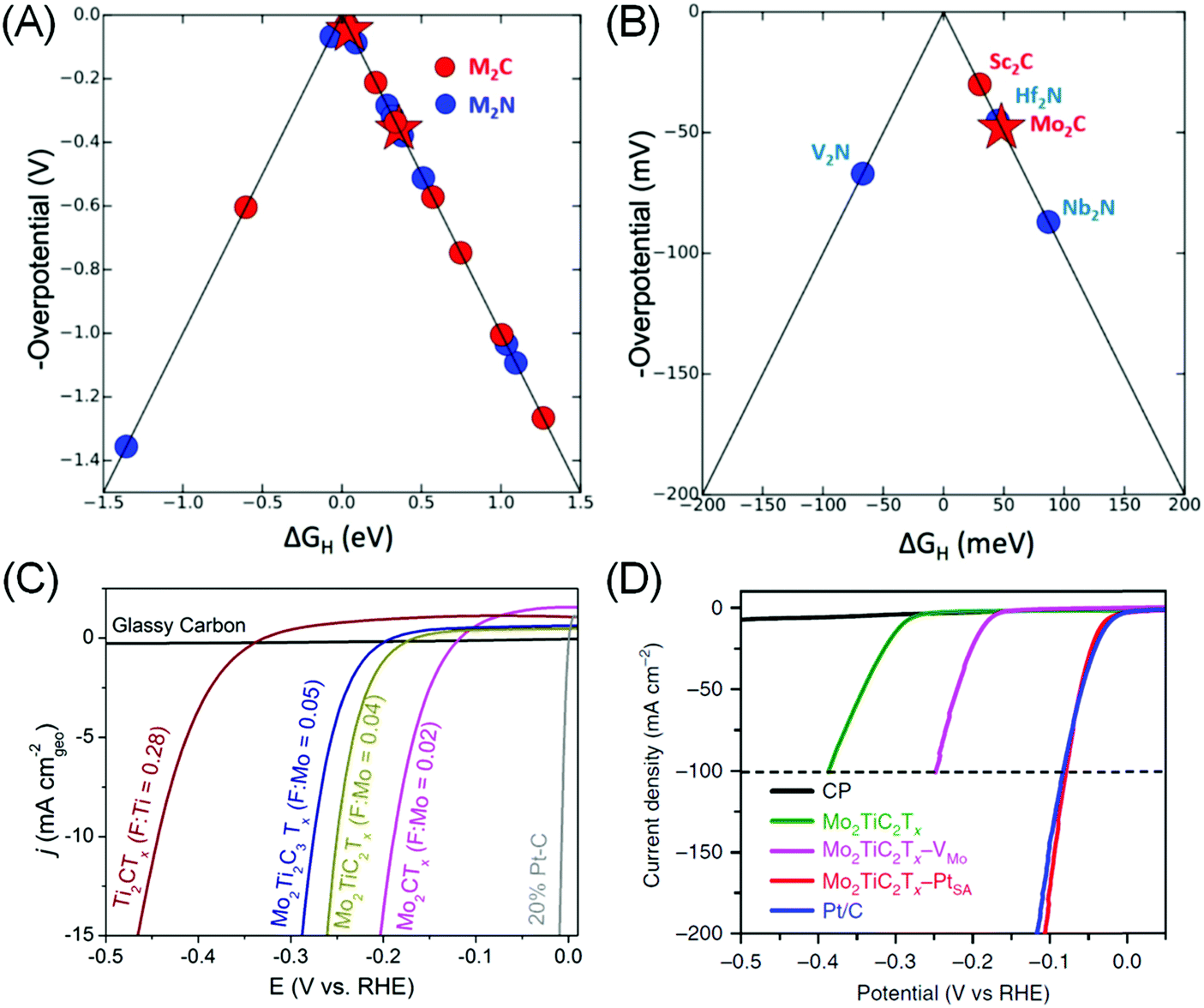 | ||
| Fig. 3 (A) Volcano plot showing calculated ΔGH and theoretical overpotentials for different M2XTx MXenes and (B) zoom-in to the top of the volcano plot. (a and b) Reprinted with permission from ref. 87 Copyright 2012 American Chemical Society. (C) Linear scanning voltammetry (LSV) of various MXenes containing different –F termination contents in 0.5 M H2SO4 electrolyte. From ref. 10. (D) HER polarization curves of carbon paper (CP), Mo2TiC2Tx MXene, Mo2TiC2Tx with Mo vacancy (–VMo), Mo2TiC2Tx with embedded Pt single atom (–PtSA) and Pt/C (40%) in 0.5 M H2SO4 solution. Reprinted by permission from ref. 15 Springer-Nature, Copyright 2018. | ||
Using theory-guided materials design, Handoko et al. demonstrated Mo2CTx MXenes that can achieve a HER current density of 10 mA cm−2 at 190 mV overpotential (Fig. 3C). It was found that the HER overpotential and activity of MXenes vary significantly with changing Tx species composition.10 In particular, –F species was found to be particularly detrimental to MXene's HER activity: Ti3C2Tx obtained from etching in 50% HF (with 70% –F coverage on the basal plane) exhibited >350 mV worse HER overpotential compared to the mild LiF–HCl etched ones (with 18% –F coverage). This observation is consistent with separate DFT investigations of Ti, V and Nb-based MXenes that suggest that –O functionalization weakens *H adsorption of bare MXenes towards an ideal GH value.79 This is a strong indicator that the Tx species plays a dominant role in the catalytic activity of MXenes. For example, the HER activity variation of Mo2CTx > Mo2TiC2Tx > Mo2Ti2C3Tx is more closely correlated with the extent of –F coverage on their surfaces than the outer metallic species (Fig. 3C), as Mo is the outer M′ species in all three cases above.10
Several other promising theoretical and experimental approaches have been explored to enhance the HER activity of MXenes, such as:
(1) Controlling the surface functionalization. In particular, replacing –F termination with other moieties such as –O or –OH with suitable *H adsorption strength as described in the case of Ti3C2Tx.10
(2) Interlayer delamination. Delamination of MXene interlayers increases the basal plane accessible for HER. This effect has been demonstrated on Mo2CTx, where the overpotential to achieve 10 mA cm−2 current density can be reduced by 30 mV after delamination.10 The improved overpotential indicates that the basal planes of Mo2CTx are catalytically active towards HER, unlike in the case of 2H-phase MoS2, where the edge sites have been shown to be more active.86 Alternatively, the interlayer delamination may have also improved the electron hopping to the basal plane.89 A similar improvement in HER overpotential has also been demonstrated on Ti2CTx after delamination treatment in N-methyl-2-pyrrolidone.87,90
(3) Morphology modification and interfacial coupling. For example, specially shaped Ti3C2Tx nanofibres etched from Ti3AlC2 fibres show a much lower HER overpotential of 169 mV and a smaller Tafel slope of 97 mV dec−1 compared to Ti3C2Tx sheets.91 Interfacial coupling between MXene and carbon or other 2D materials, like reduced graphene (rGO) or MoS2, has also shown enhanced HER activity. A lower HER overpotential of 236 mV was observed on Mo2C/graphene compared to 320 mV for bulk Mo2C.52 Separately, nanohybrids of MoS2/Ti3C2/C composites show a lower HER overpotential of 135 mV compared to 352 mV for MoS2/rGO without MXene.92 The reasoning for the improved HER activity has been linked to the lowered Schottky barrier contact.93,94
(4) Chemical modification such as addition of ad-atoms or vacancies. Ling et al. proposed to introduce an electron donor onto the surface of V2CO2, to weaken its high hydrogen bonding strength to the surface O, so as to enhance the HER activity.95 Addition of Fe, Co and Ni transition metal promoters was found to induce significant charge transfer between the promoters to the surface O, leading to less charge transfer between O and H and increased GH closer to 0 eV. The most electropositive Fe was found to induce the strongest charge transfer to O, thus increase the GH with the largest magnitude, followed by Co and then Ni.95 In this way, it is possible to optimise the hydrogen adsorption strength by adding different metal ad-atoms. DFT is also an ideal tool in this case, as high-throughput screening combination of various MXenes and ad-atoms can be performed. Several combinations with GH close to ideal were identified using this approach: Os–Ta2CO2, Ir–Sc2CO2, Ag–Nb2NO2, Re–Nb2NO2, and W–Nb2NO2.71 Investigations into MXenes with metallic vacancies such as Mo1.33CTx and W1.33CTx were also reported to show promising HER activity.96 Very recently, electrochemical treatment of Mo2TiC2Tx MXene has resulted in Mo vacancy creation that increases its HER activity.15 A similar treatment in the presence of Pt resulted in atomic Pt embedment in the Mo2TiC2Tx lattice, achieving similar HER performance to the Pt/C catalyst, but with 40 times the mass activity (Fig. 3D).
Other approaches such as introducing structural strain on monolayer MXenes can also be promising. A strained surface has been shown to increase the energy of metal d-states and increase surface reactivity.97 Strasser et al. demonstrated that the catalytic activity of Pt–Cu alloys can be tuned by introducing surface strain through de-alloying.98 Similar concepts may be applicable to MXenes, as strain has also been predicted to change the covalent character of the M–X bond and release d-electrons.99 These newly formed states may also show favourable interactions with adsorbed *H and increase HER activity.
4.2 MXenes in photocatalytic HER
Although most bare and terminated MXenes have no or very small bandgaps, they have found their way as co-catalysts for photocatalytic HER due to their electrocatalytic HER performance. Additionally, MXenes are relatively water stable with a hydrophilic surface and metallic conductivity, making them a good substitute to noble metal co-catalysts. For example, a composite of 5 wt% MXene and a classic photocatalyst, TiO2 (Fig. 4A), was reported to exhibit up to 940% higher H2 production rate under visible light compared to bare TiO2.17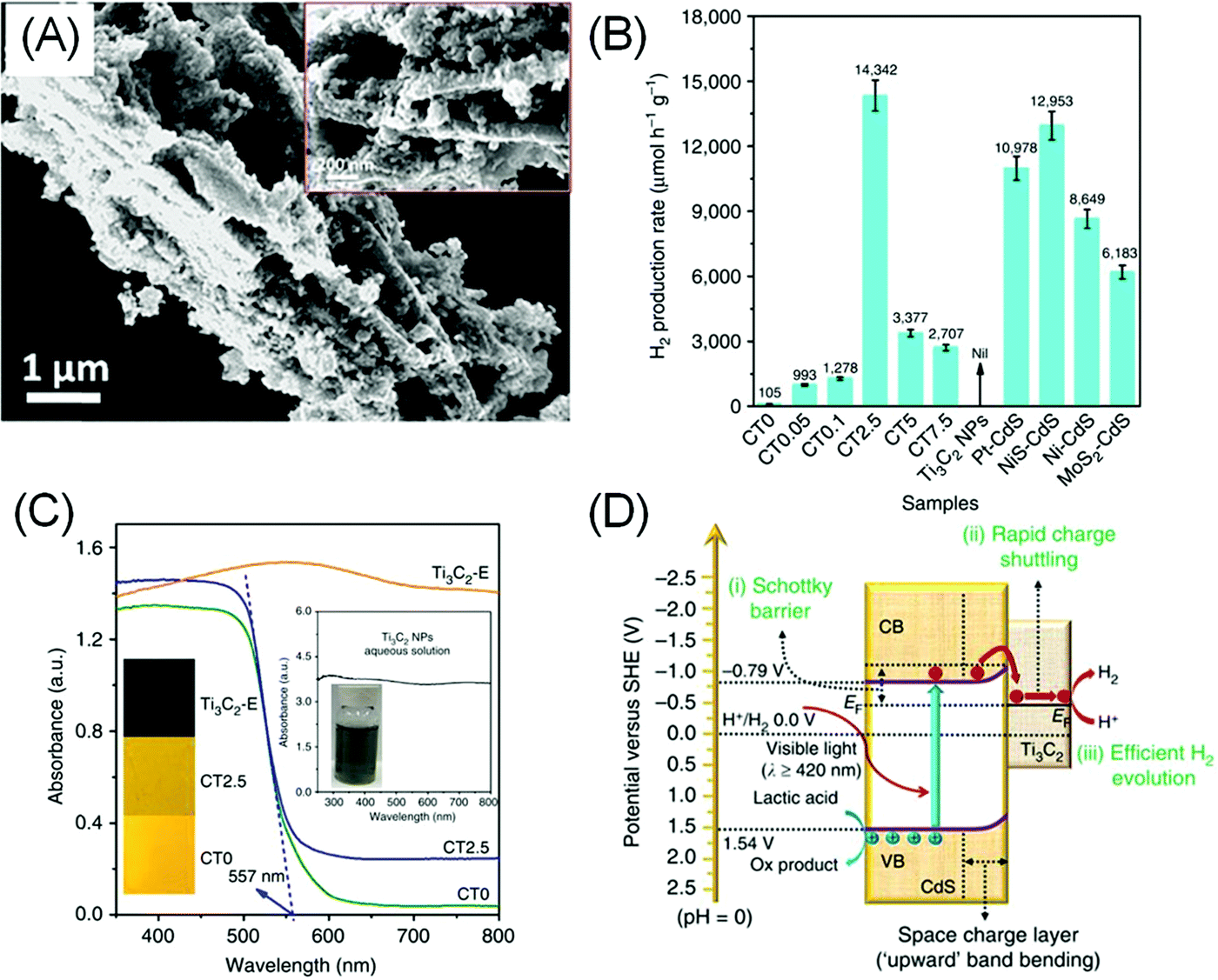 | ||
| Fig. 4 (A) Electron micrographs of the TiO2/Ti3C2Tx (50 wt%) composite. Reprinted with permission from ref. 17 Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA (B) photocatalytic H2 evolution performance of the CdS/Ti3C2 composite (CT) with varying Ti3C2Tx/CdS mass loading ratio (0 to 7.5%) compared to bare Ti3C2Tx and other composites of CdS with different catalysts. (C) UV-Vis diffuse reflectance spectra of the composite. (D) Proposed charge separation and transfer mechanism in the CdS/Ti3C2 system under visible light irradiation. (B and C) Reprinted from ref. 18 (CC BY 4.0). | ||
Similarly, 2.5 wt% loading of –O/–OH terminated Ti3C2Tx on CdS nanoparticles yields a 136.6 times higher H2 production rate, superior to Pt/CdS under the same condition (Fig. 4B), presumably due to the better interfacial contact between MXenes and CdS.18 Understandably, higher MXene loading does not necessarily result in better HER, as too much MXenes will block the light from the CdS semiconductor (Fig. 4C). In another example, Ti3C2Tx and Ti2CTx were also shown to exhibit activity enhancement on g-C3N4.100,101
As metallic-like MXene could not have generated photo-carriers, the enhanced photocatalytic HER was attributed to the inherent catalytic activity of MXene in facilitating HER. Additionally, the formation of a Schottky barrier between MXene and the semiconductor encourages electron pooling and ameliorates charge recombination within the semiconductor band structure (Fig. 4D). The height of the Schottky barrier is proportional to the work function of the outer M′ transition metals, therefore higher photocatalytic activity can be further optimised by selecting a MXene–semiconductor composite pair with a suitable Fermi level.
We note that some MXenes, especially Sc and Y based ones like Sc2C(OH)2, are predicted to be good HER photocatalysts, as they have an indirect bandgap of around 2 eV and a conduction band minimum above the HER theoretical potential, thus opening up further opportunities.102,103
5. MXene catalysts for oxygen evolution and reduction
5.1 Oxygen evolution reaction (OER)
OER is the anodic half-reaction for overall water splitting. However, the substantial overpotentials required to overcome the kinetic barrier, even with the most efficient precious metal catalyst, make this process particularly challenging.3 Additionally, OER (along with ORR) is also the major bottleneck step for rechargeable metal–air batteries.104 Electrochemical OER is proposed to be a four-electron transfer process with the overall reaction as follows:In acidic electrolyte:
| 2H2O → O2 + 4H+ + 4e− | (9) |
| 4OH− → O2 + 2H2O + 4e− | (10) |
| H2O + * → *OH + H+ + e− | (11) |
| *OH → *O + H+ + e− | (12) |
| 2*O → O2 + 2* | (13) |
| H2O + *→ *OH + H+ + e− | (14) |
| *OH → *O + H+ + e− | (15) |
| *O + H2O → *OOH + H+ + e− | (16) |
| *OOH → O2 + * + H+ + e− | (17) |
Admittedly, OER is a more complex process than HER, and a universal “activity descriptor” for its activity is elusive, as different rate determining steps exist for different catalysts and reaction conditions. Seminal works by Trasatti suggested that the OER reactivity of simple oxides can be correlated with the standard enthalpy of transition from lower to higher oxides,114 while Bockris and Ottagawa suggested that the catalytic OER activity on perovskites is inversely correlated with *OH binding energy.110 Using DFT calculation, Rossmeisl et al. suggest that *O binding energy is a more suitable descriptor, putting RuO2 at the apex of the volcano plot.115 More recently Suntivich et al. proposed, through molecular orbital theory (MOT) calculation, that the level of surface cation (eg) occupancy in perovskite type AMO3 is a suitable descriptor, as it determines the binding of intermediates to the transition metal M (Fig. 5A).31,116
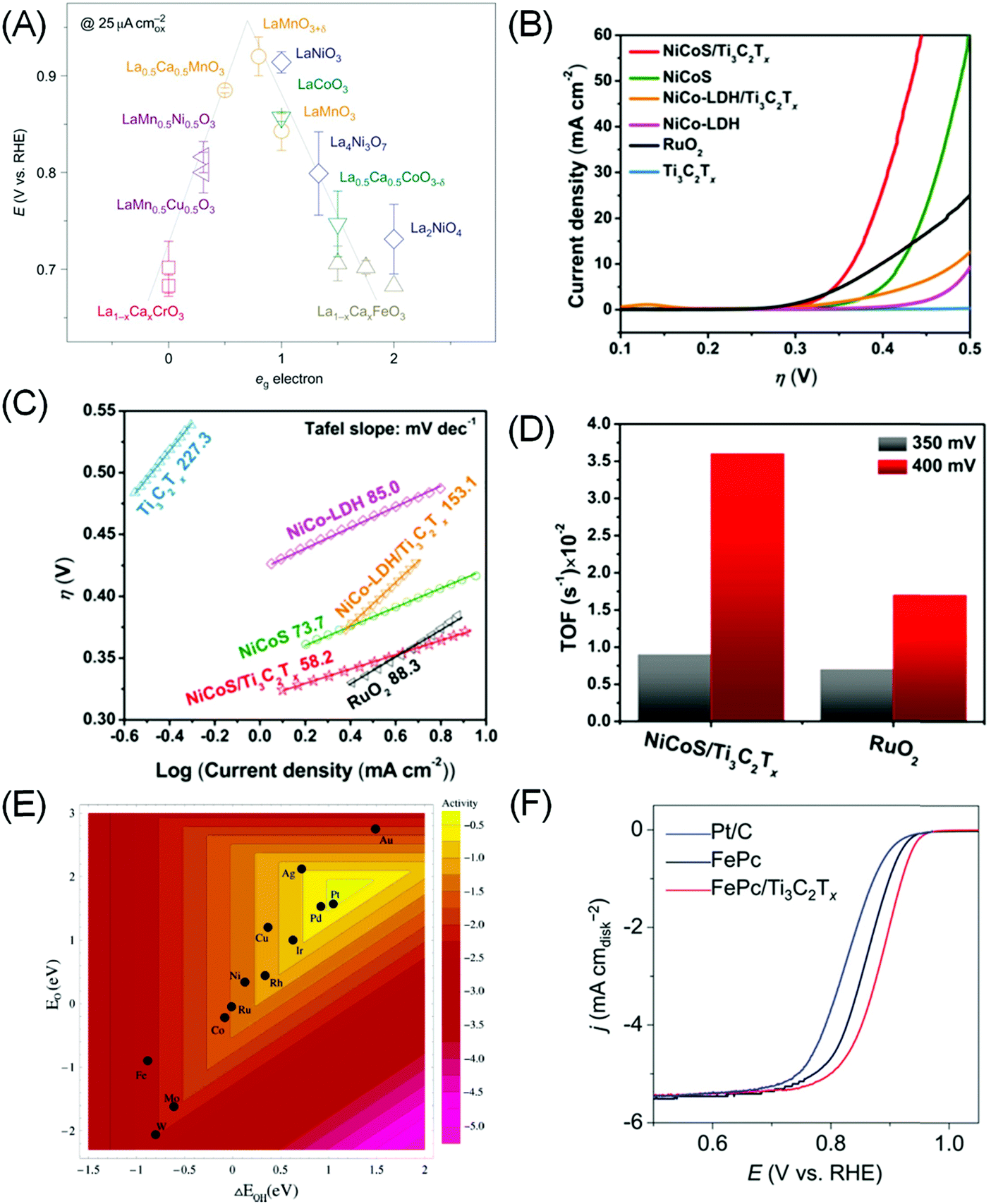 | ||
| Fig. 5 (A) The relation between the measured OER catalytic activity and the occupancy of the eg-symmetry electron of the transition metal (M in AMO3 perovskite). Reprinted by permission from ref. 116 Springer-Nature, Copyright 2011. (B) Linear sweep voltammetry curves and (C) Tafel plot of NiCoS/Ti3C2Tx and NiCoS-LDH/Ti3C2Tx composites compared to the baseline compound and RuO2. (D) TOF comparison of the NiCoS/Ti3C2Tx composite with RuO2. (B–D) Reprinted with permission from ref. 117 Copyright 2018 American Chemical Society. (E) Trends in ORR activity of single transition metals plotted as a function of both *O and *OH binding energy. Reprinted with permission from ref. 29 Copyright 2004 American Chemical Society. (F) ORR polarization curves in O2-saturated 0.1 M KOH showing superior activity of the Fe-phthalocyanine (FePc)/Ti3C2Tx hybrid compared to bare FePc and Pt/C benchmark. Reprinted with permission from ref. 120 Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
By themselves MXenes have not shown efficient OER electrocatalytic activity to date. However, when coupled with other catalysts, a synergistic effect has been shown to significantly accelerate the oxygen evolution process.117–119 As most OER catalysts are based on metal oxides, the associated low electrical conductivity of oxides (except for ruthenates) is a major bottleneck. The hybrid systems assembled with MXenes would result in unique nanostructures, as the metallic conductivity and hydrophilic surfaces of MXenes, as well as enhanced surface area of the hybrids, can effectively promote the charge transfer. Yu et al. created a 2D hierarchy of 80 wt% FeNi–LDH (i.e. layered double hydroxide)–Ti3C2Tx achieving an onset overpotential of 240 mV and a low overpotential of 298 mV at 10 mA cm−2.118 This is superior to the 372 and 407 mV displayed by bare FeNi–LDH and Ni(OH)2 respectively, and also to that of the benchmark RuO2 (397 mV). Similarly, Ni–Co mixed metal sulphide (NiCoS) incorporated with Ti3C2Tx also yields a more competitive OER rate than RuO2 in terms of smaller overpotential and Tafel slope, and larger TOF (Fig. 5B–D). XPS measurement suggested that the NiCoOOH originated from NiCoS during OER acted as the active sites.117
5.2 Oxygen reduction reaction (ORR)
ORR is a fundamental reaction for many processes in biology, energy conversion or materials dissolution, including fuel cells and metal–air batteries.104 Under acidic conditions, ORR can be described as a simple dissociation of adsorbed oxygen on the catalyst surface, followed by the formation of hydroxyl species via electron transfer and hydrogenation:29| O2 + 2* → 2*O | (18) |
| *O + H+ + e− → *OH | (19) |
| *OH + H+ + e− → H2O + * | (20) |
| O2 + * → *O2 | (21) |
| *O2 + H+ + e− → *OOH | (22) |
| *OOH + H+ + e− → *O + H2O | (23) |
| *O + H+ + e− → *OH | (24) |
| *OH + H+ + e−→ H2O + * | (25) |
In alkaline electrolytes, another indirect mechanism involves electron transfer (tunnelling) to the solvated molecular oxygen cluster in the outer Helmholtz plane.123 This mechanism explains the high ORR activity of non-noble metal catalysts in alkaline electrolytes, typically yielding peroxide (HO2−)aq that is detectable in the rotating ring disc electrode experiment.
| *OH + [O2(H2O)n]aq + e− → *OH + (HO2˙)ads + OH− + (H2O)n−1 | (26) |
| (HO2˙)ads + e− → (HO2−)ads | (27) |
| (HO2−)ads → (HO2−)aq | (28) |
Some research has been dedicated to search for a “bifunctional” oxygen catalyst (i.e. both OER and ORR active), as it is of great interest to metal–air batteries. However, finding such a material turns out to be challenging, simply because the requirement of variable oxidation states and stable structures at very different working potentials is difficult to fulfill.104 Perovskites126 and compounds of Mn that can accommodate multiple stable oxidation states127 are the few prominent candidates that demonstrate this bifunctional activity.
As in the case of OER, there are only sparse reports on the use of MXenes as ORR catalysts. Lin et al. found that Ti3C2Tx only becomes reasonably ORR active (onset potential ∼0.8 V vs. RHE) after delamination into a single layer with tetrapropylammonium hydroxide.128 The most striking ORR performance is displayed when MXene is combined with other catalysts with intrinsic ORR activity. Notably, Li et al. showed that combining Ti3C2Tx with Fe-phthalocyanine (FePc) can result in excellent ORR activity surpassing Pt/C in alkaline electrolytes (Fig. 5F).120 While FePc is known to be ORR active,129 combining it with Ti3C2Tx doubles its activity. The source of activity enhancement is proposed to be Fe 3d electron delocalisation and spin-state transition of Fe(II) ions upon coupling with Ti3C2Tx.
In another example, Xue et al. prepared Mn3O4/Ti3C2Tx nanocomposites using the hydrothermal method, and found a dominant four electron transfer ORR mechanism and an onset overpotential as low as that of Pt/C.130 The enhancement mechanism is fundamentally the same as that for HER and OER as discussed earlier. The Mn3O4 nanoparticles were detected to be well dispersed on MXene nanosheets, and they increased the surface area by inhibiting aggregation.130 Interestingly, the Ti in Ti3C2Tx MXene can form strong hybridization with Ag when the alkaline-treated MXene was stirred with AgNO3 solution in the presence of poly(vinylpyrrolidone) and deionized water for 60 mins.131 The existence of reductive low valence Ti2+ and Ti3+, confirmed by XPS, results in the formation of metallic Ag, therefore a hybrid MXene with bimetallic Ag/Ti was obtained. The MXene/Ag0.9Ti0.1 nanowire exhibited an onset potential and a half-wave potential of 0.921 and 0.782 V vs. RHE at 1600 rpm, respectively, which were significantly more positive than those of the 20 wt% Ag/C catalyst or even pure Ag nanowire.131 The MXene served not only as a structural support, but also as an efficient charge carrier conductor. The Ti doping induced vacancies or defects also provided more active sites for O2 adsorption.131
While by itself MXene has not been considered as an active ORR catalyst, the synergistic coupling between MXenes and other catalytic materials has opened up a new approach for the development of efficient electrocatalysts. Of particular interest is the fact that MXene coupling can induce a significant change and delocalisation in the catalyst's d-band electronic structure. We note that there is still much to be explored in this area as only one MXene, Ti3C2Tx, has been investigated. We speculate that, with realisation of new types of MXenes, many interesting properties can be discovered by coupling existing catalyst materials with MXenes.
6. MXene catalysts for CO2 and N2 reduction
6.1 CO2 capture and electrocatalytic CO2 reduction reaction (CO2RR)
Electrocatalytic reduction of CO2 to value-added products has attracted a lot of attention due to its value proposition of creating a closed-loop anthropogenic carbon cycle.132 Depending on the nature of the catalyst surface, CO2RR has been proposed to begin with the formation of either *COOH, *CO2− radicals or *OCHO.133–135 From there, a maze of multi-step processes continue towards one or more of the 16 distinct molecular products that have been reported on the Cu surface.136 With so many viable reaction pathway permutations, finding an excellent catalyst for CO2RR goes beyond lowering the required overpotential, but also tailoring the catalyst to enhance reaction selectivity towards a certain product. The complexity of CO2RR has called for advanced DFT approaches beyond computational hydrogen electrode (CHE) models that incorporate explicit solvent layers44,137 and surface charge.35The “universal” linear dependence of Ea and ΔE on the surface structure of the reaction site proposed by Nørskov and co-workers83,84 has an overreaching consequence for CO2RR, especially upon the revelation that the adsorption energies of similarly bound intermediates (e.g. *C and *CH; *O and *OH; etc.) are linearly correlated.122,138 These unfavourable scaling relations suggest that the CO2RR limiting potential is not expected to surpass that of Cu(211),139 unless a new catalyst surface structure with differently coordinated intermediates is found.
In this regard, MXenes, with delocalized M(1)–M(1) metallic-like bonding states and tuneable surface termination Tx, present a viable solution. Interactions between CO2 and MXenes have been subjected to theoretical investigations due to their potential use in CO2 sequestration and activation.140,141 On bare M2C MXenes, CO2 adsorption was found to be relatively strong, with binding strength ranging between −1.13 and −3.69 eV on W2C and Ti2C respectively.140 Like on many transition metal surfaces, the adsorbed CO2 molecule remains intact but bent, reminding of an activated CO2δ− anionic species (on Pt and Ni(111)).142 It should be noted that DFT calculations performed on bare (non-terminated) MXenes may be unrealistic as most MXenes are quickly functionalized with Tx termination species during synthesis and/or catalytic testing in aqueous solutions.
Li et al. screened the interaction between various bare M3C2 MXenes (M = group IV, V and VI transition metals) and CO2 using DFT calculations.143 Similar to Morales-Garcia et al.140 they found that MXenes can adsorb and activate CO2, and proceed along the reaction pathway towards CH4. What's interesting in Li et al.'s work is that some of the intermediate molecules appear to be coordinated to the MXene's surface through –O,143 different from pure transition metal surfaces. Cr3C2 and Mo3C2 were identified to be the most promising M3C2 MXenes for CO2RR based on the least endergonic overall reaction free energy towards CH4.
More recently, Handoko et al. found that CO2RR to CH4 can proceed through a more favourable *HCOOH pathway on the majority of M2XTx MXenes.144 This work takes into account the most favourable terminating species Tx on each M2XTx MXene, which is a more accurate representation of actual MXenes. On M2XTx MXenes, the intermediates were bound to the MXene surface either through the –C coordination (such as *COOH) or through the –H coordination (such as *HCOOH). Linear scaling relations were established using the binding energies of *COOH and *HCOOH as important descriptors. Fig. 6A and B depict the reaction coordinate and adsorption configuration on Ti2CO2 MXene. W2CO2 and Ti2CO2 are identified to be promising CO2RR catalysts, based on the highest CO2 limiting potential of −0.35 and −0.52 V vs. RHE respectively (close to the red apex of the 2D volcano plot shown in Fig. 6C) and best selectivity against competing HER (Fig. 6D). This limiting potential corresponds to the theoretical CO2RR to CH4 overpotential of 0.52 and 0.69 V, significantly lower than that of Cu (0.91 to 1.10 V).145,146
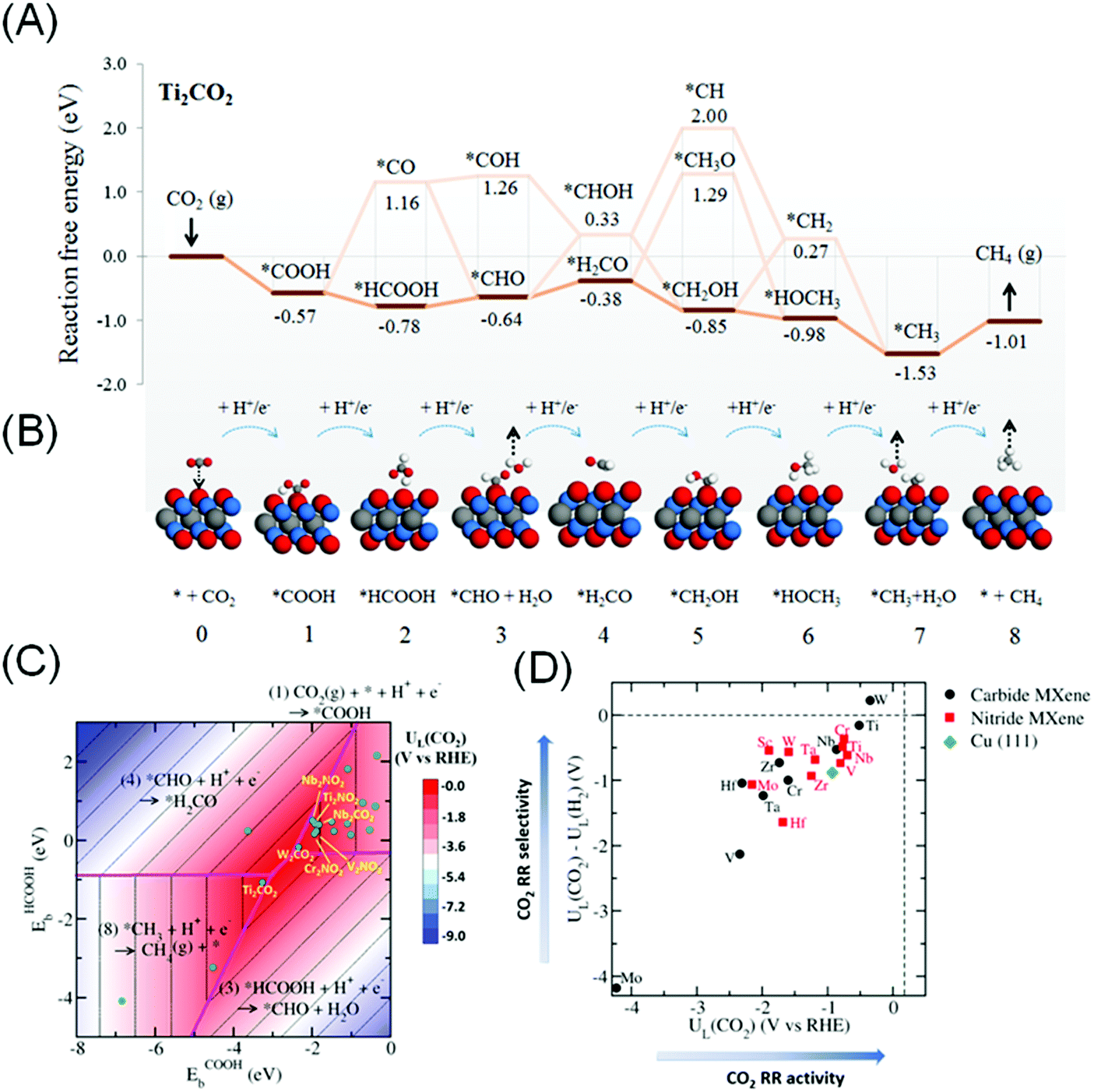 | ||
| Fig. 6 (A) The lowest energy path and (B) schematics of the molecular adsorption geometry for CO2RR to CH4 on Ti2CO2 MXene. (C) Contour volcano plot of the limiting potential (UL) for CO2RR and (D) plot of UL(CO2)–UL(H2) against UL(CO2), signifying selectivity of the MXene catalyst towards CO2RR relative to HER. Reprinted from ref. 144 with permission from The Royal Society of Chemistry. | ||
6.2 Photocatalytic CO2RR
Research into photocatalytic CO2RR is largely inspired by the natural photosynthesis process, where complex carbohydrate molecules and oxygen can be obtained from CO2, water and sunlight.22 Arguably, photocatalytic CO2RR is even more challenging than electrocatalytic CO2RR due to the additional processes of photocarrier generation and charge transfer.147 These processes are generally inefficient and the quantum yield of photocatalytic CO2RR is typically less than 1%.148As elaborated previously, most MXenes are not able to generate photocarriers due to their metallic characteristics. Only some –O and –OH terminated MXenes like Ti2CO2 and Sc2C(OH)2 with a narrow bandgap (around 0.9 to 2.0 eV) may be photocatalytically active. DFT calculations suggest that Ti2CO2 could be active for photocatalytic CO2RR to HCOOH.149 The oxygen vacancy (Ov) on –O terminated Ti2CO2 was found to play an essential role in enhancing CO2 adsorption since the adsorption on a perfect –O terminated surface was too weak. It is interesting that the energy barrier for the rate limiting step was much lower than that of anatase TiO2.150
Instead of being the main photocatalyst, MXenes can also take the role of a co-catalyst or conductive support when combined with an efficient semiconductor photoabsorber during CO2RR. One of the sources of inefficiency is the relatively poor CO2 adsorption and activation. Ye et al. proposed that the –OH functional groups on the MXene surface may serve as basic sites that facilitate the adsorption of relatively acidic CO2.19 The group employed KOH treatment to replace majority –F termination with –OH on Ti3C2Tx and combined it as a co-catalyst with the commercial P25 TiO2 (Fig. 7A). The resulting photocatalytic CO2RR to CO rate was increased by 3 times and the CH4 production rate was boosted by 277 times.19 In another example, a hybrid Ti3C2/Bi2WO6 composite shows a higher yield of CH4 and CH3OH by 4.6 times compared to bare Bi2WO6.151
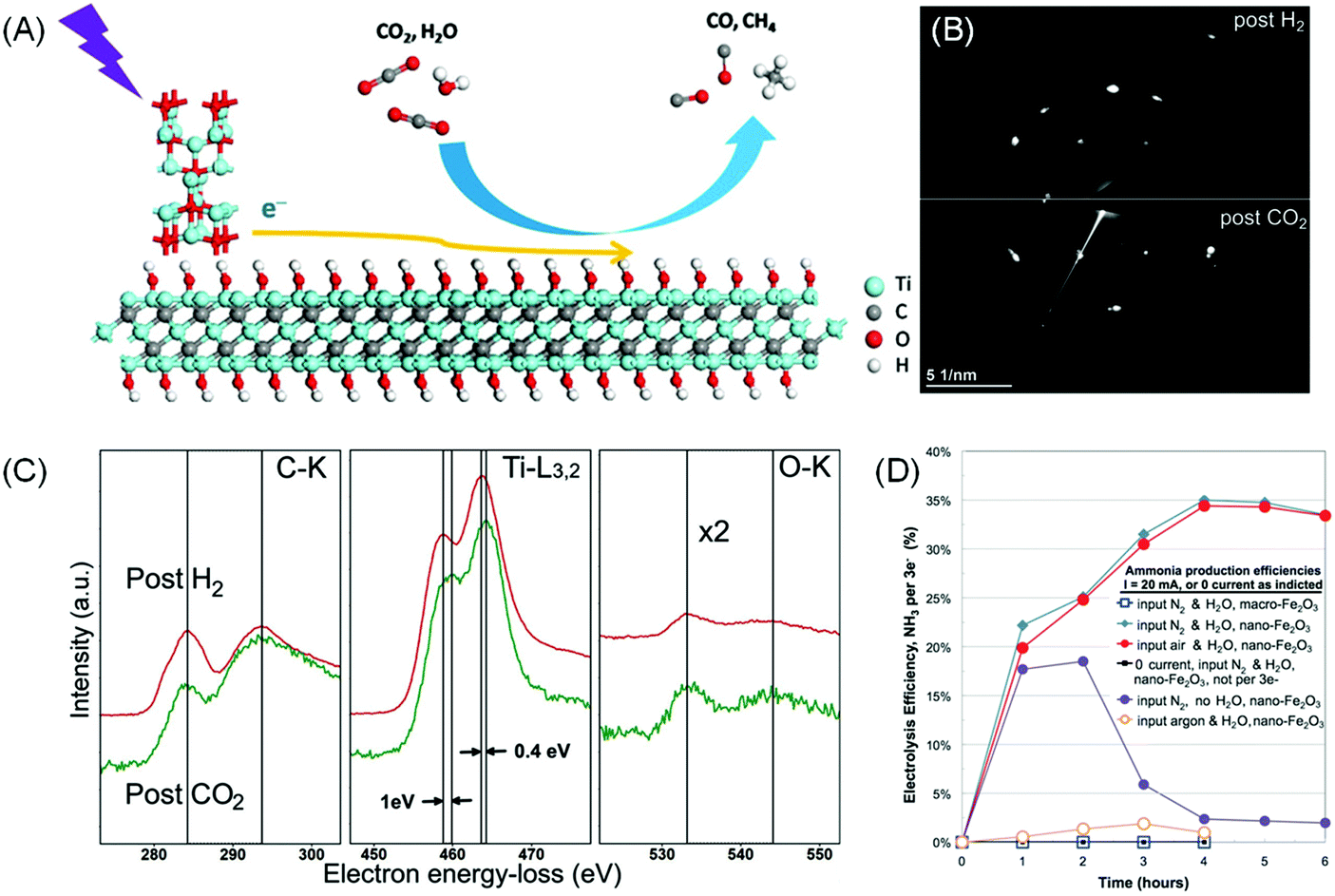 | ||
| Fig. 7 (A) Proposed photocatalytic CO2 reduction mechanism on the Ti3C2(OH)2/P25 composite under light irradiation. Reprinted with permission from ref. 19 Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA. (B) Electron diffraction pattern and (C) EELS spectra on Ti3C2 acquired after exposure to 3.5 mbar CO2 gas at 100 °C for 0.5 h preceded by H2 exposure for 1 h at 650 °C. From ref. 152 (D) Faradaic efficiency of electrocatalytic N2 reduction in a molten NaOH/KOH mixture at 200 °C at a constant current of 10 mA cm−2. Different reactions and controls are as described. Reprinted with permission from ref. 153 Copyright 2014 American Association for the Advancement of Science. | ||
Recently Persson et al. devised a sequential heating treatment in an H2 environment during in situ TEM experiment to realise O-deficient Ti3C2 MXene.152In situ CO2 exposure to this fresh Ti3C2 results in immediate CO2 saturation on the basal plane while maintaining the 2D structure (Fig. 7B and C). This result shows that careful modification of the MXene surface may also be a viable approach to improve MXene's CO2 uptake. Introducing MXenes as co-catalysts, with high conductivity, proper band structure alignment (i.e. favourable Fermi level) to extract photo-induced electrons, and enhanced active sites for CO2 adsorption, provides a realistic solution to improve the reaction efficiency. With proper surface engineering, MXenes can be tuned into a semiconductor with a suitable bandgap for photocatalytic CO2RR.
6.3 N2 reduction reaction (N2RR)
Ammonia is an important molecule, widely used for fertilizers, and is an important precursor to most nitrogen containing products, including amines for polymers and basic chemical compounds like nitric acid.154 Currently, 3–5% of natural gas worldwide is used to make ammonia through successive processes of reverse gas shift, Haber–Bosch and purification, releasing large amounts of CO2 to the atmosphere.155 Although the Haber–Bosch process (N2 + 3H2 → 2NH3) is exothermic, the kinetics are very slow under ambient conditions. These kinetic limitations are overcome by the combination of an iron-based catalyst with high pressure, high temperature conditions.153Electrochemical N2 reduction (N2 + 6H+ + 6e− → 2NH3) aims at forming ammonia under ambient conditions, replacing H2 as a hydrogen source by proton/electron pairs, therefore reducing the number of process steps, cost and CO2 emission related to natural gas reforming.156 Pickett and Talarmin demonstrated the possibility of electrochemically synthesising ammonia from N2via controlled hydrogenation of a dinitrogen complex (trans-[W(N2)2(Ph2PCH2CH2PPh2)2]) over a Hg-pool electrode.157 While promising, the reaction rate is uncompetitive compared to the high temperature process using H2. Two approaches are common in electrochemical N2 reduction: moderate temperature (400–600 °C) using a solid oxide proton conductor electrolyte158 and an aqueous alkaline electrolyte using a Nafion membrane.159 Generally the aqueous electrolyte approach is attracting more interest due to higher ammonia TOF, but the overall FE is generally below 1% due to competition from HER. Biological N2RR using extracted or synthesised Fe-nitrogenases has also been proposed;160 however, such a process is very sensitive to O2.
Recently, Licht et al. demonstrated that FE up to 35% can be achieved by conducting N2RR on a dispersed nano-Fe2O3 catalyst in molten hydroxide at 200 °C (Fig. 7D).153 The cell voltage required to drive 10 mA cm−2 current density was 1.2 V. However, the proposed contraption was not stable as electrostatics would eventually coagulate the nano-Fe2O3 catalyst and halt the electrochemical process.
Herein, MXene has the potential to step into the foray as the state-of-the-art N2RR catalyst is still based on the same Fe2O3 that was used in 1910. Current investigation for MXenes to be applied as N2RR electrocatalysts is led by theoretical DFT calculations.161,162 Although N2 is relatively inert, chemisorption of N2 on bare M3C2 MXenes was found to be spontaneous.162 Importantly, the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond appears to be stretched >20% upon adsorption compared to the gas phase, indicating weakening/activation. The first hydrogenation step *N2 + *H → N–NH˙ on V3C2 was found to proceed with activation barriers of 0.64 eV,162 which compares favourably with the 1.98 eV estimated for isolated N2 to N2˙.163
N bond appears to be stretched >20% upon adsorption compared to the gas phase, indicating weakening/activation. The first hydrogenation step *N2 + *H → N–NH˙ on V3C2 was found to proceed with activation barriers of 0.64 eV,162 which compares favourably with the 1.98 eV estimated for isolated N2 to N2˙.163
Therefore, MXenes are predicted theoretically to be promising N2 capture and reduction electrocatalysts. In order to further reduce the energy input, the effect of metal doping in MXenes has been explored, and a low overpotential of 0.47 V was obtained for Fe-doped Mo2N where Fe-dopants were found to weaken the metal–nitrogen interaction and promote ammonia formation.161
7. Future prospects and outlook
7.1 Computations
To date, computational studies of MXenes have been largely dominated by (semi-local) DFT. Since these functionals are notoriously ill adapted for band-gap predictions,164,165 applications that aim at exploiting the (small) band-gap of Tx terminated MXenes can be especially problematic. In analogy to other 2D materials such as MoS2166 or graphitic carbon-nitrides,167 we encourage the community to rely on the more robust many-body perturbation treatments, also known as the GW approximation. We note that first steps in this direction have already been taken.168 Furthermore, systematic studies of MXenes’ topological electronic structure169 might reveal particularly intriguing applications.For the exploitation of MXenes in catalysis, more in-depth studies of the evolution of the surface state as a function of the reactive conditions are needed, i.e. reactive gases, ionic species, solvent, electrochemical potential and so on. As in the case of transition metal alloys,170,171 the surface of MXenes might restructure considerably under reaction conditions, even if the initial or bare surface resembles a randomised alloy. In order to accelerate the development of these catalysts, theoretical insight into the bonding mechanism and the difference compared to transition metal surfaces can be obtained via energy decomposition analysis, recently extended to metallic surfaces.172
To obtain an atomistic understanding of the working catalyst and promising performance of MXenes in electrocatalysis, efforts towards the inclusion of the electrochemical potential and the influence of the solvent need to be made.36,37,46,173,174 These efforts are not specific to MXenes, but generally necessary for metallic catalysts. Both aspects are highly challenging and not always fully separable; to account for the electrochemical potential, the electrolyte also needs to be modelled. On the other hand, in the context of bio-mass valorization,175 aqueous solution-based catalytic processes over metal catalysts are on the rise.176 In these processes, the chemisorption of water molecules at the solid/liquid interface has been shown to significantly modify the reactivity of the catalyst.177 Although highly challenging to model even for mildly reactive surfaces such as Pt(111),178 the structuration of the metal/liquid interface is expected to depend on the nature of the MXenes and their surface termination, which may open the door to more active and selective catalysts compared to known transition metal based nanostructures.
7.2 Applications
As an important family of post-graphene 2D materials, MXenes have made a significant impact on sustainable energy storage and conversion. But most of the research on MXenes for electro- and photocatalysis is still at its early stage. Although theoretical calculations predict that MXenes can be promising efficient catalysts for many energy conversion reactions, their experimental investigation is still limited to a few compositions, and their roles in OER, ORR, CO2RR and N2RR are still waiting to be further explored. We could expect remarkable performances for other MXene compositions, e.g. nitride-based MXenes are predicted to exhibit better CO2 adsorption than their carbide analogues.Besides acting as an individual electrocatalyst, MXenes found their important role by coupling with other materials to form hybrid catalysts for HER, OER, ORR, CO2RR and N2RR, though most of the investigations were based on Ti3C2Tx MXene. Since there are 22 different MXenes reported until now, the exploration of other compositions and their coupling effects with various existing catalysts could certainly bring many interesting results. We expect that the coupling of the unique properties of MXenes with other CO2RR catalysts such as Cu and careful composition and structural engineering, will boost the reaction activity and selectivity. Similar to the synergistic effects observed in electrocatalysis, we also expect new combinations of MXenes and other semiconducting materials to significantly improve photocatalysis reactions. Overall, MXenes as catalysts or co-catalysts are still a relatively under-explored field, and theory-guided materials design is expected to bring about significant breakthroughs in clean energy research in the near future.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Institute of Materials Research and Engineering, A*STAR (IMRE/17-1R1211).References
- Monthly Energy Review July 2018, U.S. Energy Information Administration, 2018.
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633–7644 CrossRef CAS.
- B. Soundiraraju and B. K. George, ACS Nano, 2017, 11, 8892–8900 CrossRef CAS PubMed.
- P. Urbankowski, B. Anasori, K. Hantanasirisakul, L. Yang, L. Zhang, B. Haines, S. J. May, S. J. L. Billinge and Y. Gogotsi, Nanoscale, 2017, 9, 17722–17730 RSC.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2018, 1, 173–180 CrossRef CAS.
- J. Di, C. Yan, A. D. Handoko, Z. W. Seh, H. Li and Z. Liu, Mater. Today, 2018, 21, 749–770 CrossRef CAS.
- A. D. Handoko, C. W. Ong, Y. Huang, Z. G. Lee, L. Lin, G. B. Panetti and B. S. Yeo, J. Phys. Chem. C, 2016, 120, 20058–20067 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10, 10876–10883 RSC.
- H. Wang, R. Peng, Z. D. Hood, M. Naguib, S. P. Adhikari and Z. Wu, ChemSusChem, 2016, 9, 1490–1497 CrossRef CAS PubMed.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- M. Ye, X. Wang, E. Liu, J. Ye and D. Wang, ChemSusChem, 2018, 11, 1606–1611 CrossRef CAS PubMed.
- S. Ho-Kimura, S. J. A. Moniz, A. D. Handoko and J. Tang, J. Mater. Chem. A, 2014, 2, 3948–3953 RSC.
- E. Pastor, F. M. Pesci, A. Reynal, A. D. Handoko, M. Guo, X. An, A. J. Cowan, D. Klug, J. Durrant and J. Tang, Phys. Chem. Chem. Phys., 2014, 16, 5922–5926 RSC.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- A. J. Cowan, J. Tang, W. Leng, J. R. Durrant and D. R. Klug, J. Phys. Chem. C, 2010, 114, 4208–4214 CrossRef CAS.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef.
- S. Fletcher, J. Solid State Electrochem., 2009, 13, 537–549 CrossRef CAS.
- P. Sabatier, La catalyse en chimie organique, C. Béranger, Paris, 1913 Search PubMed.
- P. Trens, R. Durand, B. Coq, C. Coutanceau, S. Rousseau and C. Lamy, Appl. Catal., B, 2009, 92, 280–284 CrossRef CAS.
- R. Parsons, in Synchrotron Techniques in Interfacial Electrochemistry, ed. C. A. Melendres and A. Tadjeddine, Springer, Netherlands, Dordrecht, 1994, pp. 21–32 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS PubMed.
- S. N. Steinmann, C. Michel, R. Schwiedernoch, J.-S. Filhol and P. Sautet, ChemPhysChem, 2015, 16, 2307–2311 CrossRef CAS PubMed.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- T. Cheng, L. Wang, B. V. Merinov and W. A. Goddard, J. Am. Chem. Soc., 2018, 140, 7787–7790 CrossRef CAS PubMed.
- K. S. Exner and H. Over, Acc. Chem. Res., 2017, 50, 1240–1247 CrossRef CAS PubMed.
- K. S. Exner, I. Sohrabnejad-Eskan, J. Anton, T. Jacob and H. Over, ChemElectroChem, 2017, 4, 2902–2908 CrossRef CAS.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2016, 7, 1686–1690 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., 2013, 125, 2519–2522 CrossRef.
- Y.-H. Fang and Z.-P. Liu, J. Am. Chem. Soc., 2010, 132, 18214–18222 CrossRef CAS PubMed.
- K. Letchworth-Weaver and T. A. Arias, Phys. Rev. B, 2012, 86, 075140 CrossRef.
- S. N. Steinmann, C. Michel, R. Schwiedernoch and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 13949–13963 RSC.
- B. Schweitzer, S. N. Steinmann and C. Michel, Phys. Chem. Chem. Phys., 2019, 21, 5368–5377 RSC.
- C. D. Taylor, S. A. Wasileski, J.-S. Filhol and M. Neurock, Phys. Rev. B, 2006, 73, 165402 CrossRef.
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- I. R. Shein and A. L. Ivanovskii, Comput. Mater. Sci., 2012, 65, 104–114 CrossRef CAS.
- M. Naguib, R. R. Unocic, B. L. Armstrong and J. Nanda, Dalton Trans., 2015, 44, 9353–9358 RSC.
- O. Mashtalir, M. R. Lukatskaya, M. Q. Zhao, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2015, 27, 3501–3506 CrossRef CAS PubMed.
- D. Geng, X. Zhao, Z. Chen, W. Sun, W. Fu, J. Chen, W. Liu, W. Zhou and K. P. Loh, Adv. Mater., 2017, 29, 1700072 CrossRef PubMed.
- D. Geng, X. Zhao, L. Li, P. Song, B. Tian, W. Liu, J. Chen, D. Shi, M. Lin, W. Zhou and K. P. Loh, 2D Mater., 2017, 4, 011012 CrossRef.
- B. Wang, A. Zhou, Q. Hu and L. Wang, Int. J. Appl. Ceram. Technol., 2017, 14, 873–879 CrossRef CAS.
- C. Cui, M. Hu, C. Zhang, R. Cheng, J. Yang and X. Wang, Chem. Commun., 2018, 54, 8132–8135 RSC.
- P. Urbankowski, B. Anasori, T. Makaryan, D. Er, S. Kota, P. L. Walsh, M. Zhao, V. B. Shenoy, M. W. Barsoum and Y. Gogotsi, Nanoscale, 2016, 8, 11385–11391 RSC.
- J. Zhou, X. Zha, F. Y. Chen, Q. Ye, P. Eklund, S. Du and Q. Huang, Angew. Chem., Int. Ed., 2016, 55, 5008–5013 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS.
- M. A. Hope, A. C. Forse, K. J. Griffith, M. R. Lukatskaya, M. Ghidiu, Y. Gogotsi and C. P. Grey, Phys. Chem. Chem. Phys., 2016, 18, 5099–5102 RSC.
- T. Hu, Z. Li, M. Hu, J. Wang, Q. Hu, Q. Li and X. Wang, J. Phys. Chem. C, 2017, 121, 19254–19261 CrossRef CAS.
- A. Champagne, L. Shi, T. Ouisse, B. Hackens and J.-C. Charlier, Phys. Rev. B, 2018, 97, 115439 CrossRef CAS.
- X.-H. Zha, K. Luo, Q. Li, Q. Huang, J. He, X. Wen and S. Du, EPL, 2015, 111, 26007 CrossRef.
- Y. Xie, M. Naguib, V. N. Mochalin, M. W. Barsoum, Y. Gogotsi, X. Yu, K. W. Nam, X. Q. Yang, A. I. Kolesnikov and P. R. Kent, J. Am. Chem. Soc., 2014, 136, 6385–6394 CrossRef CAS PubMed.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- C. Si, J. Zhou and Z. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 17510–17515 CrossRef CAS PubMed.
- M. Khazaei, M. Arai, T. Sasaki, C.-Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- N. Zhang, Y. Hong, S. Yazdanparast and M. A. Zaeem, 2D Mater., 2018, 5, 045004 CrossRef.
- G. R. Berdiyorov, EPL, 2015, 111, 67002 CrossRef.
- P. Li, J. Zhu, A. D. Handoko, R. Zhang, H. Wang, D. Legut, X. Wen, Z. Fu, Z. W. Seh and Q. Zhang, J. Mater. Chem. A, 2018, 6, 4271–4278 RSC.
- B. Anasori, J. Halim, J. Lu, C. A. Voigt, L. Hultman and M. W. Barsoum, Scr. Mater., 2015, 101, 5–7 CrossRef CAS.
- J. Yang, M. Naguib, M. Ghidiu, L.-M. Pan, J. Gu, J. Nanda, J. Halim, Y. Gogotsi and M. W. Barsoum, J. Am. Ceram. Soc., 2016, 99, 660–666 CrossRef CAS.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- W. Sun, Y. Xie and P. R. C. Kent, Nanoscale, 2018, 10, 11962–11968 RSC.
- L. Dong, H. Kumar, B. Anasori, Y. Gogotsi and V. B. Shenoy, J. Phys. Chem. Lett., 2017, 8, 422–428 CrossRef CAS.
- M. Khazaei, A. Ranjbar, M. Arai and S. Yunoki, Phys. Rev. B, 2016, 94, 125152 CrossRef.
- V. N. Borysiuk, V. N. Mochalin and Y. Gogotsi, Nanotechnology, 2015, 26, 265705 CrossRef PubMed.
- G. Gao, A. P. O’Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- M. Pandey and K. S. Thygesen, J. Phys. Chem. C, 2017, 121, 13593–13598 CrossRef CAS.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. Man Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 CrossRef CAS PubMed.
- H. Wang, Y. Wu, J. Zhang, G. Li, H. Huang, X. Zhang and Q. Jiang, Mater. Lett., 2015, 160, 537–540 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 209, 275–278 CrossRef.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- Y. Yu, S.-Y. Huang, Y. Li, S. N. Steinmann, W. Yang and L. Cao, Nano Lett., 2014, 14, 553–558 CrossRef CAS PubMed.
- S. Li, P. Tuo, J. Xie, X. Zhang, J. Xu, J. Bao, B. Pan and Y. Xie, Nano Energy, 2018, 47, 512–518 CrossRef CAS.
- W. Yuan, L. Cheng, Y. An, H. Wu, N. Yao, X. Fan and X. Guo, ACS Sustainable Chem. Eng., 2018, 6, 8976–8982 CrossRef CAS.
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed.
- Y. Liu, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2016, 138, 15853–15856 CrossRef CAS PubMed.
- J. You, C. Si, J. Zhou and Z. Sun, J. Phys. Chem. C, 2019, 123, 3719–3726 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang, Q. Chen and J. Wang, Adv. Sci., 2016, 3, 1600180 CrossRef PubMed.
- R. Meshkian, M. Dahlqvist, J. Lu, B. Wickman, J. Halim, J. Thörnberg, Q. Tao, S. Li, S. Intikhab, J. Snyder, M. W. Barsoum, M. Yildizhan, J. Palisaitis, L. Hultman, P. O. Å. Persson and J. Rosen, Adv. Mater., 2018, 30, 1706409 CrossRef PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- G. Gao, G. Ding, J. Li, K. Yao, M. Wu and M. Qian, Nanoscale, 2016, 8, 8986–8994 RSC.
- M. Shao, Y. Shao, J. Chai, Y. Qu, M. Yang, Z. Wang, M. Yang, W. F. Ip, C. T. Kwok, X. Shi, Z. Lu, S. Wang, X. Wang and H. Pan, J. Mater. Chem. A, 2017, 5, 16748–16756 RSC.
- Y. Sun, D. Jin, Y. Sun, X. Meng, Y. Gao, Y. Dall’Agnese, G. Chen and X.-F. Wang, J. Mater. Chem. A, 2018, 6, 9124–9131 RSC.
- A. C. Rajan, A. Mishra, S. Satsangi, R. Vaish, H. Mizuseki, K.-R. Lee and A. K. Singh, Chem. Mater., 2018, 30, 4031–4038 CrossRef CAS.
- K. Xiong, P. Wang, G. Yang, Z. Liu, H. Zhang, S. Jin and X. Xu, Sci. Rep., 2017, 7, 15095 CrossRef PubMed.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- H. Dau, L. Iuzzolino and J. Dittmer, Biochim. Biophys. Acta, 2001, 1503, 24–39 CrossRef CAS.
- A. D. Handoko, S. Deng, Y. Deng, A. W. F. Cheng, K. W. Chan, H. R. Tan, Y. Pan, E. S. Tok, C. H. Sow and B. S. Yeo, Catal. Sci. Technol., 2016, 6, 269–274 RSC.
- Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef CAS.
- J. O. M. Bockris, J. Chem. Phys., 1956, 24, 817–827 CrossRef CAS.
- J. O. Bockris and T. Otagawa, J. Phys. Chem., 1983, 87, 2960–2971 CrossRef CAS.
- R. L. Doyle and M. E. G. Lyons, in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Cham, 2016, pp. 41–104 Search PubMed.
- C.-H. Lee, A. Riga and E. Yeager, in Mass Transport Phenomena in Ceramics, ed. A. R. Cooper and A. H. Heuer, Springer US, Boston, MA, 1975, pp. 489–499 Search PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 125–131 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- H. Zou, B. He, P. Kuang, J. Yu and K. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 22311–22319 CrossRef CAS PubMed.
- M. Yu, S. Zhou, Z. Wang, J. Zhao and J. Qiu, Nano Energy, 2018, 44, 181–190 CrossRef CAS.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS PubMed.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng, W. Chen, L. Mai and S. Guo, Adv. Mater., 2018, 30, 1803220 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. Munter, P. Moses, E. Skúlason, T. Bligaard and J. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CrossRef CAS.
- J. Rossmeisl, J. K. Nørskov, C. D. Taylor, M. J. Janik and M. Neurock, J. Phys. Chem. B, 2006, 110, 21833–21839 CrossRef CAS PubMed.
- S. Liu, M. G. White and P. Liu, J. Phys. Chem. C, 2016, 120, 15288–15298 CrossRef CAS.
- J.-I. Jung, M. Risch, S. Park, M. G. Kim, G. Nam, H.-Y. Jeong, Y. Shao-Horn and J. Cho, Energy Environ. Sci., 2016, 9, 176–183 RSC.
- G. Fu, X. Jiang, Y. Chen, L. Xu, D. Sun, J.-M. Lee and Y. Tang, NPG Asia Mater., 2018, 10, 618–629 CrossRef CAS.
- H. Lin, L. Chen, X. Lu, H. Yao, Y. Chen and J. Shi, Sci. China Mater., 2018, 62, 662–670 CrossRef.
- W. Li, A. Yu, D. C. Higgins, B. G. Llanos and Z. Chen, J. Am. Chem. Soc., 2010, 132, 17056–17058 CrossRef CAS PubMed.
- Q. Xue, Z. Pei, Y. Huang, M. Zhu, Z. Tang, H. Li, Y. Huang, N. Li, H. Zhang and C. Zhi, J. Mater. Chem. A, 2017, 5, 20818–20823 RSC.
- Z. Zhang, H. Li, G. Zou, C. Fernandez, B. Liu, Q. Zhang, J. Hu and Q. Peng, ACS Sustainable Chem. Eng., 2016, 4, 6763–6771 CrossRef CAS.
- G. A. Olah, G. K. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Martínez, J. García-Lastra, J. Rossmeisl and M. Koper, Phys. Rev. Lett., 2012, 108, 116103 CrossRef CAS PubMed.
- C. Shi, H. A. Hansen, A. C. Lausche and J. K. Norskov, Phys. Chem. Chem. Phys., 2014, 16, 4720–4727 RSC.
- Á. Morales-García, A. Fernández-Fernández, F. Viñes and F. Illas, J. Mater. Chem. A, 2018, 6, 3381–3385 RSC.
- R. Morales-Salvador, Á. Morales-García, F. Viñes and F. Illas, Phys. Chem. Chem. Phys., 2018, 20, 17117–17124 RSC.
- S. N. Steinmann, C. Michel, R. Schwiedernoch, M. Wu and P. Sautet, J. Catal., 2016, 343, 240–247 CrossRef CAS.
- N. Li, X. Chen, W.-J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- A. D. Handoko, K. H. Khoo, T. L. Tan, H. Jin and Z. W. Seh, J. Mater. Chem. A, 2018, 6, 21885–21890 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- X. Hong, K. Chan, C. Tsai and J. K. Nørskov, ACS Catal., 2016, 6, 4428–4437 CrossRef CAS.
- A. D. Handoko, K. Li and J. Tang, Curr. Opin. Chem. Eng., 2013, 2, 200–206 CrossRef.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- X. Zhang, Z. Zhang, J. Li, X. Zhao, D. Wu and Z. Zhou, J. Mater. Chem. A, 2017, 5, 12899–12903 RSC.
- Y. Ji and Y. Luo, ACS Catal., 2016, 6, 2018–2025 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- I. Persson, J. Halim, H. Lind, T. W. Hansen, J. B. Wagner, L.-Å. Näslund, V. Darakchieva, J. Palisaitis, J. Rosen and P. O. Å. Persson, Adv. Mater., 2019, 31, 1805472 CrossRef PubMed.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- L. Lassaletta, G. Billen, B. Grizzetti, J. Anglade and J. Garnier, Environ. Res. Lett., 2014, 9, 105011 CrossRef.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press, Cambridge, 2004 Search PubMed.
- I. A. Amar, R. Lan, C. T. G. Petit and S. Tao, J. Solid State Electrochem., 2011, 15, 1845 CrossRef CAS.
- C. J. Pickett and J. Talarmin, Nature, 1985, 317, 652–653 CrossRef CAS.
- F. Kosaka, T. Nakamura, A. Oikawa and J. Otomo, ACS Sustainable Chem. Eng., 2017, 5, 10439–10446 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- D. F. Harris, D. A. Lukoyanov, S. Shaw, P. Compton, M. Tokmina-Lukaszewska, B. Bothner, N. Kelleher, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, Biochemistry, 2018, 57, 701–710 CrossRef CAS PubMed.
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 121, 27563–27568 CrossRef CAS.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed.
- H. Xiao, J. Tahir-Kheli and W. A. Goddard, J. Phys. Chem. Lett., 2011, 2, 212–217 CrossRef CAS.
- T. Le Bahers, M. Rérat and P. Sautet, J. Phys. Chem. C, 2014, 118, 5997–6008 CrossRef.
- D. Y. Qiu, F. H. da Jornada and S. G. Louie, Phys. Rev. B, 2016, 93, 235435 CrossRef.
- S. N. Steinmann, S. T. A. G. Melissen, T. Le Bahers and P. Sautet, J. Mater. Chem. A, 2017, 5, 5115–5122 RSC.
- Y. Zhang, W. Xia, Y. Wu and P. Zhang, Nanoscale, 2019, 11, 3993–4000 RSC.
- B. Bradlyn, L. Elcoro, J. Cano, M. G. Vergniory, Z. Wang, C. Felser, M. I. Aroyo and B. A. Bernevig, Nature, 2017, 547, 298 CrossRef CAS PubMed.
- E. Vignola, S. N. Steinmann, K. Le Mapihan, B. D. Vandegehuchte, D. Curulla and P. Sautet, J. Phys. Chem. C, 2018, 122, 15456–15463 CrossRef CAS.
- F. Tao and M. Salmeron, Science, 2011, 331, 171–174 CrossRef CAS PubMed.
- R. Staub, M. Iannuzzi, R. Z. Khaliullin and S. N. Steinmann, J. Chem. Theory Comput., 2019, 15, 265–275 CrossRef CAS PubMed.
- S. N. Steinmann and P. Sautet, J. Phys. Chem. C, 2016, 120, 5619–5623 CrossRef CAS.
- R. Sundararaman, W. A. GoddardIII and T. A. Arias, J. Chem. Phys., 2017, 146, 114104 CrossRef PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- R. Réocreux and C. Michel, Curr. Opin. Green Sustain. Chem., 2018, 10, 51–59 CrossRef.
- C. Michel, J. Zaffran, A. M. Ruppert, J. Matras-Michalska, M. Jędrzejczyk, J. Grams and P. Sautet, Chem. Commun., 2014, 50, 12450–12453 RSC.
- S. N. Steinmann, R. Ferreira De Morais, A. W. Götz, P. Fleurat-Lessard, M. Iannuzzi, P. Sautet and C. Michel, J. Chem. Theory Comput., 2018, 14, 3238–3251 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |