
Quantification of liquid phase faecal odourants to evaluate membrane technology for wastewater reuse from decentralised sanitation facilities†
E.
Mercer
a,
C. J.
Davey
 a,
P.
Campo
a,
D.
Fowler
b,
L.
Williams
a,
A.
Kolios
c,
A.
Parker
a,
S.
Tyrrel
a,
C.
Walton
d,
E.
Cartmell
e,
M.
Pidou
a and
E. J.
McAdam
*a
a,
P.
Campo
a,
D.
Fowler
b,
L.
Williams
a,
A.
Kolios
c,
A.
Parker
a,
S.
Tyrrel
a,
C.
Walton
d,
E.
Cartmell
e,
M.
Pidou
a and
E. J.
McAdam
*a
aCranfield Water Science Institute, Cranfield University, Vincent Building, Bedfordshire, UK. E-mail: e.mcadam@cranfield.ac.uk
bEnvironmental Analytical Facility, Cranfield University, Vincent Building, Bedfordshire, UK
cNaval Architecture, Ocean and Marine Engineering, University of Strathclyde, Glasgow, UK
dCentre for Environmental and Agricultural Informatics, Cranfield University, Building 146, Bedfordshire, UK
eScottish Water, Castle House, Carnegie Campus, Dunfermline, UK
First published on 29th November 2018
Abstract
Public willingness to use decentralised sanitation facilities or arising water products is discouraged due to malodour, preventing improved sanitation practices or water reuse opportunities in low income countries. Whilst odour is characterised in the gas phase, it originates in the liquid phase. Consequently, controlling odour at source could prevent gas-phase partitioning and limit produced water contamination. This study therefore developed an analytical method for the quantitation of a range of liquid phase volatile organic compounds (VOCs) classified into eight chemical groups, known to be primary indicators of faecal odour, to provide characterisation of real fluids and to permit evaluation of several potential membrane separation technologies for liquid phase odourant separation. The gas chromatography mass spectrometry method provided quantitation in the range of 0.005 mg L−1 to 100 mg L−1 with instrument detection limits ranging from 0.005 mg L−1 to 0.124 mg L−1. Linear calibration curves were achieved (r2 > 0.99) with acceptable accuracy (77–115%) and precision (<15%) for quantitation in the calibration range below 1 mg L−1, and good accuracy (98–104%) and precision (<2%) determined for calibration in the range 1–100 mg L−1. Pre-concentration of real samples was facilitated via solid phase extraction. Subsequent application of the method to the evaluation of two thermally driven membranes based on hydrophilic (polyvinyl alcohol) and hydrophobic (polydimethylsiloxane) polymers evidenced contrasting separation profiles. Importantly, this study demonstrates the method's utility for liquid phase VOC determination which is of use to a range of disciplines, including healthcare professionals, taste and odour specialists and public health engineers.
Water impactIn order to meet proposed sustainable development goals, advanced decentralised sanitation facilities are being developed for low income countries which allow for improved sanitation practices and water reuse opportunities. However, malodour discourages public acceptance for such technologies. This article introduces an analytical method which quantifies liquid phase faecal odour for the development of treatment technologies to change public perception. |
1. Introduction
Large scale centralised wastewater treatment is not economically practicable for implementation in many low income country contexts. Local communities are therefore instead dependent upon decentralised sanitation solutions such as pit latrines which may not provide a safe barrier to discharge of faecal material into local water resources. Malodour associated with these sanitation facilities has also been shown to exacerbate discharge of faecal material into the environment with users preferring open defecation to foul-smelling pit latrines.1,2 The odour profile associated with decentralised sanitation can be considered distinct from that of centralised treatment facilities since the absence of flush water and other water sources limit the primary composition to urine and faeces. Whilst analytical determination has determined around 279 and 381 volatile organic compounds (VOCs) associated with urine and faeces respectively from healthy individuals,3 the faecal-borne VOCs indole, skatole (3-methyl-1H-indole) and p-cresol (4-methylphenol) amongst others, are considered key contributors to malodour arising from pit latrines.4 Previous research has demonstrated that VOCs originate in the liquid phase as microbial metabolites, with factors such as diet and health influencing composition and concentration of VOCs, and the physico-chemical environmental conditions (e.g. pH and temperature) encouraging partitioning into the gas phase where odour is finally perceived by the olfactory system.5Recent technological innovations seek to deliver alternative sustainable sanitation solutions that can facilitate sufficient water quality for safe discharge to the environment or to promote local water reuse.6,7 As water supplies often arise from sources of unknown provenance, the local production of water to reuse standards can be considered an attractive proposition. However, a major limiting criterion that governs willingness to use reclaimed water is odour.8 Odour abatement technologies presently provide elimination or neutralisation of malodourous compounds already partitioned into the gas phase.2 Through introducing barrier technology into this new genre of sanitation solutions for liquid phase treatment, the partitioning of odorous VOCs from the liquid phase into the gas phase could be mediated at source and potentially averted, therefore enhancing the potential willingness of users to use locally engineered sanitation solutions and the arising water product for a range of reuse applications.8 Pervaporation fosters water transport through application of a vapour pressure gradient and permeation through a polymeric membrane. The availability of waste heat, coupled with characteristically low water volumes from these new decentralised sanitation solutions, make thermally driven membrane separation a practicable solution for water recovery.7 For non-porous (or dense) membranes, the polymer chemistry can favour permeation of water over VOCs thereby imparting selectivity into the separation that will exert an influence on the final odour profile of the treated water.
Whilst the management of odourants in the liquid phase is an attractive proposition, there is presently not an analytical solution of sufficient resolution to characterise the separation performance of membrane technology for this application. The conventional analytical route that has been previously exploited for liquid phase VOC odourant determination is headspace sampling with pre-concentration onto a sorbent (e.g. Tenax) before introduction into gas chromatography mass spectrometry (GC-MS).2,9 Such indirect techniques introduce temporal and sample volume restrictions in addition to limitations with respect to recovery which do not guarantee accurate quantitation of the liquid phase VOC profile. Lin et al.2 recently introduced a direct method for liquid phase VOC odourant characterisation of pit latrine faecal sludge using solid phase extraction (SPE) for pre-concentration from the liquid phase before determination by GC-MS. The authors used the method to successfully identify a discrete range of VOCs in the liquid phase representative of faecal odour. Pre-concentration by SPE was also selected for study by Chappuis et al.4 to extract compounds from pit latrine air in which the equilibrium was shifted to the liquid phase to trap and concentrate the compounds, enabling quantitation close to the odour detection thresholds (ODTs) to be achieved.
Although SPE-GC-MS has been demonstrated as a suitable method for liquid phase VOC quantitation, only a discrete range of VOCs has been determined, representing a limited range of chemical structures that is not sufficiently definitive to aid in the characterisation and development of membrane technology for the selective separation of liquid phase odourants. This study therefore seeks to develop an analytical method for the determination of liquid phase odourants sufficient to characterise a broad range of VOC chemistries including organo-sulphurs, aromatics, phenols, alcohols, aldehydes, ketones, esters and hydrocarbons, that are known contributors to faecal odour,3,9 and within a single elution to simplify the analytical procedure. Specific objectives are therefore to: (i) develop a method for the quantitation of liquid phase VOCs within a single elution, which present a broad range of chemistries, representative of those commonly associated with faeces and urine; (ii) develop and validate solid phase extraction for the liquid-phase pre-concentration stage; (iii) apply the method for VOC quantitation in urine and faecally contaminated urine; and (iv) confirm the methods validity through application to pervaporative membranes of differing polarity that should engender distinct differences in liquid phase VOC separation.
2. Materials and methods
2.1. Chemicals and reagents
All chemicals were sourced from Fisher Scientific (Loughborough, UK) or Sigma Aldrich (Dorset, UK). The VOCs analytes (1-butanol, 1-propanol, benzaldehyde, indole, skatole, ethyl butyrate, ethyl propionate, limonene, 2-butanone, p-cresol, dimethyl disulfide and dimethyl trisulfide) had a purity of at least 98%. Diethyl ether, propylene glycol, and the methyl octanoate internal standard (IS) were of extra pure grade (≥99%) and the methanol used for SPE conditioning and acetone used for glassware cleaning was laboratory grade.2.2. Standards preparation
Stock solutions and working standards were prepared in class A volumetric glassware which was cleaned to remove residual contaminants by soaking glassware in deionised water, acetone and methanol for 10 min. Respectively, within a sonicator and then dried overnight at 50 °C. For calibration purposes, a 1000 mg L−1 stock solution of all VOCs was prepared in diethyl ether and working standards were subsequently diluted according to the calibration concentration. Three calibration curves, 0.005–1, 1–10 and 10–100 mg L−1 were generated to cover a wide concentration range (Fig. S1–S3†). A 10 mg L−1 stock solution of methyl octanoate (IS) in diethyl ether was prepared for the lower calibration curve (<1 mg L−1) and spiked in the low range standards for a final concentration of 1 mg L−1. High range standards (1–100 mg L−1) were spiked with the IS for a final concentration of 10 mg L−1. Internal calibration curves were obtained for each VOC with the mean response factor used to determine unknown concentrations.2.3. Gas chromatography mass spectrometry
Compound identification and quantification were performed by a Shimazdu-TQ8040 GC-MS (Shimadzu, Milton Keynes, UK), equipped with a semi polar ZB-624 fused silica GC column 60 m × 0.25 mm, 1.4 μm (Phenomenex, Macclesfield, UK). The initial oven temperature was held at 35 °C for 5 min then increased to 170 °C at a rate of 10 °C min−1 in order to elute 1-propanol, 2-butanone, 1-butanol, ethyl propionate, dimethyl disulfide, and ethyl butyrate. This temperature was sustained for 2 min to provide separation between dimethyl trisulfide, benzaldehyde and limonene. Then the temperature was ramped at 30 °C min−1 up to 240 °C for the detection of the internal standard (methyl octanoate) and p-cresol and further increased to 250 °C at 5 °C min−1, which was maintained for 5 min, allowing for the separation of indole and skatole. The total runtime was 29.83 min. Helium was used as the carrier gas (236.1 kPa) at a linear column flow rate of 2.47 mL min−1 to maintain a velocity of 40 cm s−1. The mass spectrometer was operated in single quad mode with a detector voltage relative to the tuning result (0.2 kV), ionisation energy of −70 eV at an ion source temperature of 200 °C and interface temperature of 250 °C. A solvent cut time was applied until 8.95 min. Initially, the MS was operated in scan mode in order to identify the retention times and target ions through in house MS libraries and NIST MS search with a scan range of 30–500 m/z. Compounds of interest were then detected in single ion monitoring (SIM) mode by the principal ion and two reference ions (Table 2).2.4. Determining SPE recovery factors
A synthetic solution was prepared in order to determine SPE recovery factors. A 1000 mg L−1 stock solution containing all VOCs was prepared in propylene glycol to completely dissolve all compounds. Aliquots were subsequently added to three individual buffered solutions (potassium chloride buffer pH 2, potassium phosphate monobasic 6.5 and tris(hydroxymethyl)aminomethane pH 9) according to Robinson and Stokes,10 within a volumetric flask for a GC-MS injection concentration of 100 mg L−1. Multiple pH levels were studied to identify the influence of natural pH variations within faecally contaminated urine, on compound recovery, as similarly practiced by Lin et al.2Oasis® HLB cartridges (1 g), sourced from Waters (Milford, USA), were used and attached to an Agilent VacElut20 manifold (Agilent Technologies, Stockport, UK). The cartridges were first conditioned by subsequently passing 10 mL of diethyl ether, methanol and deionised water, facilitated by a vacuum pump (N 022 AN.18, KNF Neuberger, Whitney, UK). Samples (20 mL) were then loaded onto the cartridges. The VOCs were eluted with 1 mL of methyl octanoate (IS) in diethyl ether (0.057 μg mL−1) followed by 5 mL of pure diethyl ether. The residual sample water which collected at the bottom of the beaker was removed carefully using a glass Pasteur pipette (Fisher Scientific, Loughborough, UK). Diethyl ether extracts were concentrated to 0.5 mL under nitrogen gas and then analysed by GC-MS. The response ratios were compared between the calibration standard and the sample in order to calculate the recovery factors of the compounds. All trials were triplicated at pH 2, 6.5 and 9. The method detection limit (MDL) was determined by:
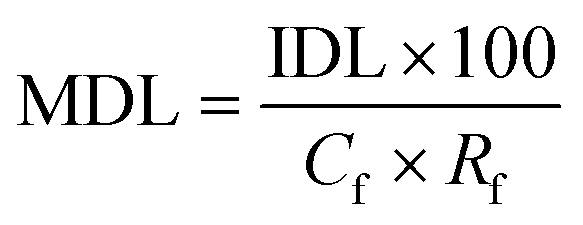 | (1) |
 | (2) |
2.5. Characterisation of urine and faecally contaminated urine
Fresh urine and faeces samples were collected and analysed within 12 hours of collection.Informed consent of real samples was obtained from anonymous volunteers through a collection regime approved by the Cranfield University Research Ethics System (CURES, project ID 3022).
Faecally contaminated urine was prepared by producing a composite sample containing a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 urine-to-faeces ratio, which represents the typical proportions produced by an individual per day.11 With this purpose, 5 g of fresh faeces along with 50 g of fresh urine were combined in a 50 mL centrifuge tube and vortexed for 30 seconds. The supernatant was then filtered through cotton wool and sand (50 mL) and a 20 mL aliquot was processed by SPE. Fresh urine samples (20 mL) were also processed with SPE. All samples were eluted with 0.2 mL IS solution (0.057 μg mL−1) and 10 mL diethyl ether and concentrated down to 100 μL. Duplicate samples were also prepared with a concentration factor of five was also processed to capture p-cresol concentrations exceeding the calibration range i.e. (2.5 mL sample, 1 mL IS solution, 10 mL diethyl ether, concentrated down to 500 μL).
:1 urine-to-faeces ratio, which represents the typical proportions produced by an individual per day.11 With this purpose, 5 g of fresh faeces along with 50 g of fresh urine were combined in a 50 mL centrifuge tube and vortexed for 30 seconds. The supernatant was then filtered through cotton wool and sand (50 mL) and a 20 mL aliquot was processed by SPE. Fresh urine samples (20 mL) were also processed with SPE. All samples were eluted with 0.2 mL IS solution (0.057 μg mL−1) and 10 mL diethyl ether and concentrated down to 100 μL. Duplicate samples were also prepared with a concentration factor of five was also processed to capture p-cresol concentrations exceeding the calibration range i.e. (2.5 mL sample, 1 mL IS solution, 10 mL diethyl ether, concentrated down to 500 μL).
2.6. Membrane technology set-up
Commercially available polydimethylsiloxane (PDMS) and polyvinyl alcohol (PVA) membranes were evaluated (Fig. S4 and Table S1†). The PDMS and PVA membranes exhibited contact angles of 116 ± 1.4° and 43 ± 1.1°, indicating them to be hydrophobic and hydrophilic polymers respectively. Vapour pressure gradient was established using a diaphragm vacuum pump (MD 4CNT, Vacuubrand, Brackley) operating at 0.05 bar on the permeate side. Permeate samples were collected (20 mL) within a liquid nitrogen cold trap (−196 °C). The permeate, feed and retentate samples were analysed using the SPE-GC-MS method to establish a mass balance. The feed reservoir was submerged within a thermostatic bath at 50 °C (Grant TC120, Cambridge, UK) with a feed flowrate of 0.2 L min−1 applied (520 s, Watson Marlow, Falmouth, UK). Separation efficiency of the PVA and PDMS membranes was expressed through removal efficiency (%):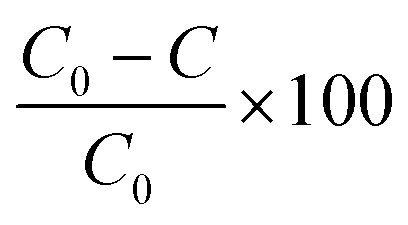 | (3) |
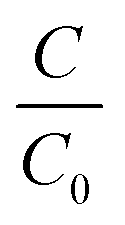 | (4) |
3. Results and discussion
3.1. Method development
The VOC analytes comprised of alcohols (1-butanol, 1-propanol), aldehydes (benzaldehyde), aromatics (indole, skatole), esters (ethyl butyrate, ethyl propionate), hydrocarbons (limonene), ketones (2-butanone), phenols (p-cresol) and organo-sulphur containing compounds (dimethyl disulfide, dimethyl trisulfide) (Table 1). The compounds represent a broad range of physico-chemical properties such as acid dissociation constant (pKa, −7 to 16.1), octanol–water partitioning coefficient (logKow 0.25 to 4.57), water solubility (0.013 to 1000 g L−1) and volatility (0.00048 to 19.1 mol m−3 Pa−1),12–15 which confer a challenging separation for any barrier technology, and is representative of the chemistries frequently associated with faecal odour.2,4,9
| Compound | Chemical group | Chemical composition | Chemical structure | Molecular weight | pKa | logKow at 20 °C |
Water solubility at 25 °C | Henry's volatility constant at 25 °C | Boiling point | Vapour pressure at 25 °C |
|---|---|---|---|---|---|---|---|---|---|---|
| (g mol−1) | (g L−1) | (mol m−3 Pa−1) | (°C) | (mm Hg) | ||||||
| a Pubchem (2018).12 b YMDB (2018).13 c Gu and Berry (1991).14 d Sander (2015).15 | ||||||||||
| 1-Butanol | Alcohol | C4H9OH |
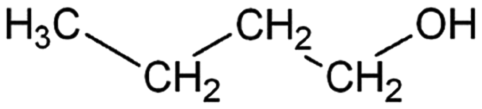
|
74.12 | 16.1a | 0.88a | 63.2a | 1.2d | 111.7a | 7a |
| 1-Propanol | Alcohol | C3H8O |
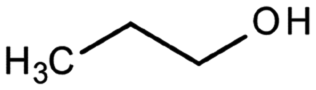
|
60.1 | 16.1a | 0.25a | 1000a | 1.5d | 97a | 14.9a |
| Benzaldehyde | Aldehyde | C7H6O |
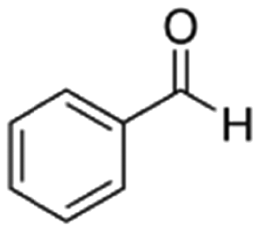
|
106.12 | 14.9a | 1.48a | 6.95a | 0.38d | 178.1a | 1.27a |
| Indole | Aromatic heterocycle | C8H7N |
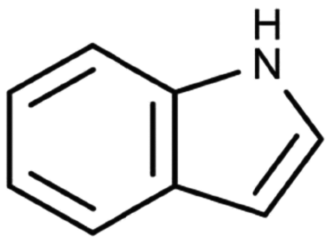
|
117.15 | −3.6c | 2.14a | 3.56a | 19.1d | 254a | 0.0122a |
| Skatole | Aromatic heterocycle | C9H9N |
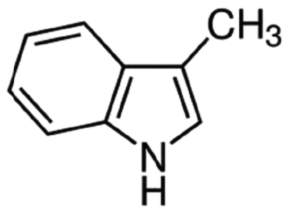
|
131.17 | −4.6c | 2.6a | 0.498a | 4.7d | 265a | 0.0055a |
| Ethyl butyrate | Ester | C6H12O2 |
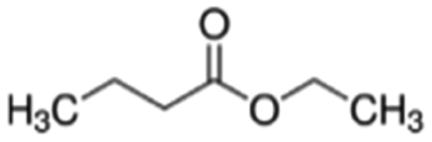
|
116.16 | −7b | 1.85a | 2.7b | 0.029d | 121a | 14a |
| Ethyl propionate | Ester | C5H10O2 |
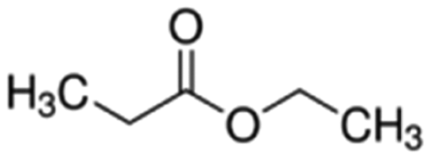
|
102.13 | −7b | 1.21a | 19.2a | 0.041d | 98.9a | 35.8a |
| Limonene | Hydrocarbon | C10H16 |
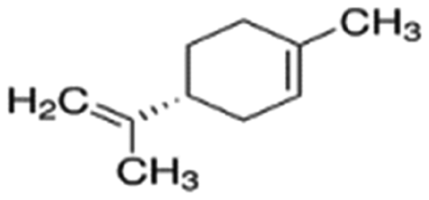
|
136.24 | −4.2b | 4.57a | 0.013a | 0.00048d | 177a | 1.98a |
| 2-Butanone | Ketone | C4H8O |
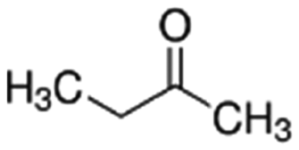
|
72.11 | 14.7a | 0.29a | 223a | 8.1d | 79.7a | 90.6a |
| p-Cresol | Phenol | C7H8O |
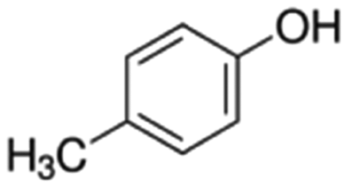
|
108.14 | 10.26a | 1.94a | 21.5a | 10d | 201.9a | 0.11a |
| Dimethyl disulfide | Sulphur containing | C2HS2 |
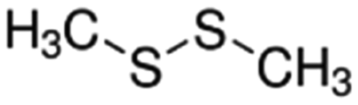
|
94.19 | — | 1.77a | 3a | 0.0065d | 110a | 28.7a |
| Dimethyl trisulfide | Sulphur containing | C2H6S3 |
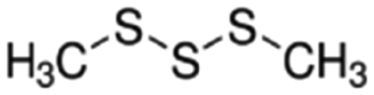
|
126.25 | — | 1.926a | 2.39a | 0.021d | 170a | 1.06a |
In order to identify a method capable of detecting each of the 9 selected VOCs in this range within a single elution, various injection split ratios were trialled in scan mode. The optimum split ratios were selected according to the upper limit of detector saturation which was associated to the later emerging higher boiling point compounds (aromatics) and a signal to noise ratio of >10 for the lower boiling point compounds (alcohols). The injection port was operated at a split of 1:5, 1:12.5 and 1:100 for the low calibration range (0.005–1 mg L−1), medium calibration range (1–10 mg L−1) and high calibration range (10–100 mg L−1) respectively; three calibration ranges were adopted to ensure that the ‘natural’ concentration of faecally contaminated urine as well as sample concentrations post-separation could be determined. The respective injection volumes were 2.5, 1 and 1 μL. The split ratio conditions were then applied to SIM mode to increase selectivity and sensitivity (Table 2). The final peak of the elution (Fig. 1a and b) represents butylated hydrocarbon (BHT), the stabilisation agent within the diethyl ether solvent. All compounds were detected within a 27 minute runtime. Peaks generally had good tailing factors close to one which was within the recommended analytical range of ≤2 (Fig. S5 and S6 and Table S2†).16,17
| Compound | Retention time (minutes) | Principal ion (m/z) | Reference ion 1 (m/z) | Reference ion 2 (m/z) |
|---|---|---|---|---|
| 1-Propanol | 9.455 | 31 | 42 | 59 |
| 2-Butanone | 10.213 | 43 | 72 | 57 |
| 1-Butanol | 12.437 | 56 | 41 | 43 |
| Ethyl propionate | 12.903 | 57 | 74 | 75 |
| Dimethyl disulfide | 14.087 | 94 | 79 | 45 |
| Ethyl butyrate | 15.087 | 71 | 43 | 88 |
| Dimethyl trisulfide | 19.478 | 126 | 79 | 45 |
| Benzaldehyde | 19.653 | 106 | 105 | 77 |
| Limonene | 19.862 | 68 | 93 | 67 |
| p-Cresol | 22.498 | 107 | 108 | 77 |
| Indole | 25.688 | 117 | 90 | 89 |
| Skatole | 26.76 | 130 | 131 | 77 |
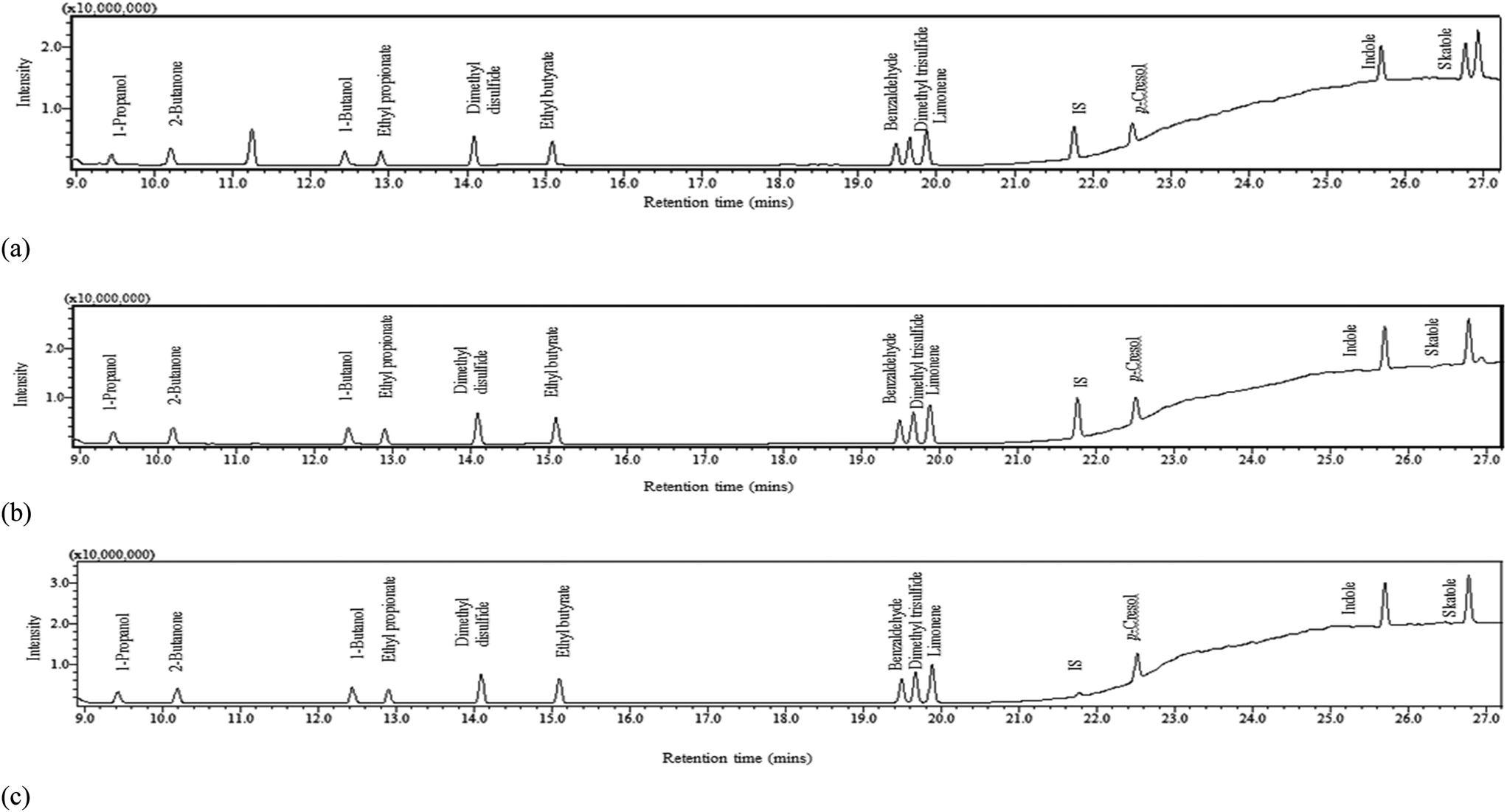 | ||
| Fig. 1 Chromatograms in single ion monitoring mode (SIM) at (a) 1 mg L−1 volatile organic compound (VOC) concentration with 1 mg L−1 IS concentration, (b) 10 mg L−1 VOC concentration with 10 mg L−1 IS concentration and (c) 100 mg L−1 VOC concentration with 10 mg L−1 IS concentration. | ||
3.2. GC-MS calibration
Calibration was based on a linear regression analysis of the mean response factor fit (Table 3).18 A good correlation coefficient was obtained for each of the three calibration curves (r2 > 0.99). Residual standard deviations (RSD) of the response factors of all calibration curves were within the acceptance criteria of <20%.19 The instrument limit of detection (LD) was calculated as 3.3 σ/slope, and limit of quantification (LQ) as 10 σ/slope where σ is standard deviation of seven trace (0.005 mg L−1) replicates.18 The LD ranged from 0.005 mg L−1 (p-cresol) to 0.124 mg L−1 (2-butanone) and the LQ from 0.014 mg L−1 to 0.351 mg L−1.| Calibration range | Slope | Intercept | r 2 | LDa | LQb | RF | Mean RF | RF SD | |
|---|---|---|---|---|---|---|---|---|---|
| (mg L−1) | (mg L−1) | (mg L−1) | (% RSD) | ||||||
| a LD calculated as 3.3σ/slope, where σ is standard deviation of seven 0.005 mg L−1 replicates (Currie, 1999).18 b LQ calculated as 10σ/slope, where, σ is standard deviation of seven 0.005 mg L−1 replicates (Currie, 1999).18 Note: RSD is acceptable when <20% (EPA, 2003).19 | |||||||||
| 1-Propanol | 10–100 | 0.531 | 0.00479 | 1.000 | 3.266 | 0.52 | 0.017 | ||
| 1–10 | 0.456 | 0.00382 | 0.992 | 10.48 | 0.455 | 0.047 | |||
| 0.005–1 | 0.674 | 0.00677 | 1.000 | 0.019 | 0.077 | 11.66 | 0.75 | 0.087 | |
| 2-Butanone | 10–100 | 0.874 | 0.07026 | 0.999 | 2.57 | 0.89 | 0.023 | ||
| 1–10 | 0.817 | 0.01183 | 0.994 | 8.429 | 0.84 | 0.07 | |||
| 0.005–1 | 1.39 | 0.194 | 0.991 | 0.124 | 0.351 | 14.53 | 1.87 | 0.27 | |
| 1-Butanol | 10–100 | 0.4144 | 0.01558 | 1.000 | 2.71 | 0.63 | 0.017 | ||
| 1–10 | 0.381 | −0.00232 | 0.996 | 9.54 | 0.365 | 0.034 | |||
| 0.005–1 | 0.468 | −0.00213 | 0.999 | 0.036 | 0.099 | 17.93 | 0.436 | 0.078 | |
| Ethyl propionate | 10–100 | 0.614 | 0.0693 | 0.999 | 2.68 | 0.99 | 0.027 | ||
| 1–10 | 0.588 | 0.00609 | 0.995 | 0.914 | 0.59 | 0.054 | |||
| 0.005–1 | 0.753 | 0.00412 | 0.999 | 0.011 | 0.045 | 4.975 | 0.78 | 0.039 | |
| Dimethyl disulfide | 10–100 | 0.976 | 0.0939 | 0.999 | 1.81 | 0.468 | 0.0085 | ||
| 1–10 | 1.08 | 0.00586 | 0.998 | 9.16 | 1.07 | 0.098 | |||
| 0.005–1 | 1.2003 | 0.00316 | 1.000 | 0.005 | 0.019 | 7.25 | 1.17 | 0.085 | |
| Ethyl butyrate | 10–100 | 0.462 | 0.0301 | 1.000 | 2.5 | 0.729 | 0.018 | ||
| 1–10 | 0.49 | 0.00362 | 0.997 | 8.82 | 0.49 | 0.043 | |||
| 0.005–1 | 0.561 | 0.00184 | 1.000 | 0.006 | 0.026 | 16.54 | 0.545 | 0.09 | |
| Dimethyl trisulfide | 10–100 | 0.718 | 0.047 | 0.999 | 2.03 | 0.71 | 0.014 | ||
| 1–10 | 0.797 | −0.000266 | 0.997 | 8.86 | 0.78 | 0.069 | |||
| 0.005–1 | 0.8114 | 0.000512 | 1.000 | 0.010 | 0.031 | 13.01 | 0.769 | 0.1 | |
| Benzaldehyde | 10–100 | 0.685 | 0.0823 | 0.999 | 3.09 | 0.5 | 0.015 | ||
| 1–10 | 0.731 | 0.004297 | 0.997 | 8.03 | 0.73 | 0.0587 | |||
| 0.005–1 | 0.76 | 0.00259 | 1.000 | 0.005 | 0.021 | 5.38 | 0.76 | 0.041 | |
| Limonene | 10–100 | 0.479 | 0.0741 | 1.000 | 3.09 | 0.503 | 0.0156 | ||
| 1–10 | 0.474 | 0.00898 | 0.997 | 5.7 | 0.495 | 0.028 | |||
| 0.005–1 | 0.529 | 0.0105 | 0.999 | 0.041 | 0.165 | 8.85 | 0.568 | 0.05 | |
| p-Cresol | 10–100 | 0.69 | 0.0809 | 1.000 | 2.42 | 0.741 | 0.0173 | ||
| 1–10 | 0.71 | 0.00152 | 0.997 | 9.87 | 0.7 | 0.069 | |||
| 0.005–1 | 0.681 | −0.000495 | 0.999 | 0.019 | 0.057 | 18.46 | 0.698 | 0.129 | |
| Indole | 10–100 | 1.39 | 0.545 | 0.996 | 5.74 | 1.56 | 0.0896 | ||
| 1–10 | 1.49 | 0.0121 | 0.997 | 7.57 | 1.5 | 0.113 | |||
| 0.005–1 | 1.433 | 0.00895 | 1.000 | 0.005 | 0.027 | 12.523 | 1.56 | 0.195 | |
| Skatole | 10–100 | 1.509 | 0.558 | 0.994 | 5.3 | 1.67 | 0.089 | ||
| 1–10 | 1.6625 | 0.00763 | 0.998 | 7.51 | 1.66 | 0.12 | |||
| 0.005–1 | 1.519 | 0.00755 | 1.000 | 0.005 | 0.014 | 13.1862 | 1.6 | 0.211 | |
Accuracy and precision for each calibration range was determined by analysis of the mid-point concentration (Table 4; 0.5 mg L−1, 5 mg L−1 and 50 mg L−1). Accuracy was calculated as the ratio between measured and theoretical concentrations of 6 replicate solutions in different vials and precision was calculated as the RSD of 6 replicate injections from the same vial. According to the EPA method 8000C19 and Little,20 accuracy and precision was classed as acceptable for all compounds at all calibration levels which was ≤30%. This also demonstrates sample stability after standing time which then permits repeat injections from the same vial.
| 0.5 mg L−1 | 5 mg L−1 | 50 mg L−1 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mid-point mean | Accuracya | Precisionb | Mid-point mean | Accuracya | Precisionb | Mid-point mean | Accuracya | Precisionb | |
| (mg L−1) | (%) | (RSD) | (mg L−1) | (%) | (RSD) | (mg L−1) | (%) | (RSD) | |
| a Accuracy calculated as the percentage ratio between measured and theoretical concentrations of 6 replicate solutions in different vials. b Precision calculated as the RSD of 6 replicated injections from the same vial. Note: 1. Accuracy acceptance: ≤30% (EPA, 2003).19 2. Criteria for precision: ≤25% is excellent, less than or equal to 30% is acceptable (Little, 2016).20 | |||||||||
| 1-Propanol | 0.46 ± 0.08 | 92.6 | 3.7 | 5.25 ± 0.04 | 104.9 | 0.3 | 50.70 ± 1.50 | 101.4 | 1.6 |
| 2-Butanone | 0.45 ± 0.07 | 89.6 | 15.6 | 5.16 ± 0.08 | 103.2 | 0.8 | 49.14 ± 3.69 | 98.3 | 1.7 |
| 1-Butanol | 0.50 ± 0.02 | 100.6 | 5.34 | 5.15 ± 0.05 | 103.1 | 1.4 | 49.59 ± 5.05 | 99.2 | 2.4 |
| Ethyl propionate | 0.51 ± 0.07 | 102.7 | 3.6 | 5.19 ± 0.05 | 103.7 | 0.7 | 50.13 ± 1.89 | 100.3 | 1.1 |
| Dimethyl disulfide | 0.54 ± 0.06 | 107.6 | 3.6 | 5.15 ± 0.03 | 103.1 | 0.6 | 50.27 ± 1.24 | 100.5 | 1.5 |
| Ethyl butyrate | 0.55 ± 0.06 | 109.3 | 3.6 | 5.18 ± 0.03 | 103.6 | 0.7 | 50.04 ± 2.41 | 100.1 | 1.6 |
| Dimethyl trisulfide | 0.58 ± 0.05 | 115.3 | 2.5 | 5.12 ± 0.01 | 102.4 | 1.1 | 50.96 ± 1.82 | 101.9 | 1.7 |
| Benzaldehyde | 0.53 ± 0.07 | 106.7 | 0.9 | 5.16 ± 0.02 | 103.1 | 0.2 | 49.68 ± 1.44 | 99.4 | 1.4 |
| Limonene | 0.39 ± 0.12 | 77.3 | 0.9 | 5.17 ± 0.03 | 103.4 | 0.4 | 49.28 ± 1.02 | 98.6 | 1.6 |
| p-Cresol | 0.50 ± 0.07 | 100.4 | 4.6 | 5.13 ± 0.01 | 102.7 | 2.3 | 50.39 ± 1.53 | 100.8 | 2.7 |
| Indole | 0.47 ± 0.08 | 93.8 | 2.5 | 5.17 ± 0.02 | 103.4 | 0.7 | 49.55 ± 1.89 | 99.1 | 1.0 |
| Skatole | 0.49 ± 0.07 | 98.4 | 7.0 | 5.13 ± 0.03 | 102.7 | 0.4 | 49.55 ± 2.80 | 99.1 | 1.9 |
3.3. Solid phase extraction
Solids phase extraction recovery efficiency was evaluated to permit calculation of recovery factors. Recoveries for p-cresol (90%), indole (81%) and skatole (88%) are comparable to those stated by Lin et al. (Table 5).2 Further analytes with recoveries deemed to be either ‘recommended’ or ‘acceptable’ in accordance with EPA guidelines21 were 2-butanone (56%), dimethyl disulfide (63%), 1-butanol (100%), benzaldehyde (77%) ethyl propionate (82%) and ethyl butyrate (89%). However, poor recoveries were identified for compounds including 1-propanol and limonene. We suggest that the poor extraction efficiency of 1-propanol can be ascribed to its strong affinity for water, which limits the probability for partitioning onto the solid phase (Table 1, logKow 0.25). Conversely, the poor extraction efficiency for limonene can be attributed to its high volatility, which increases the probability for sample losses at the vacuum and evaporation stages of sample preparation, coupled with its significant hydrophobicity (logKow 4.57) which can initiate strong interactions with the sorbent that are known to inhibit SPE recovery.22 Wells recommended inclusion of an organic modifier for compounds with logKow exceeding 4,23 along with the addition of methanol to increase eluotropic strength: this is recommended for improving SPE recovery for this compound for future research. Importantly, an RSD below 10% was recorded for each compound, which evidenced that SPE can achieve consistent recovery to within the acceptance criteria specified in the SPE EPA method 3535A (SW-846),21 which demonstrates that correction factors could be applied (Table 5) to determine method detection limits (MDL). For illustration, method detection limits for p-cresol (Cf 200, Rf 0.9) and indole (Cf 200, Rf 0.81) were 0.1 and 0.03 μg L−1. These values are several orders of magnitude lower than identified by De Preter et al.24 by using purge and trap with GC-MS to determine faecal fermentation, which suggests direct determination from the liquid phase may enhance method sensitivity.
| SPE recoverya (% ± RSD) in this study | Average SPE recovery (%) | SPE recovery (%) Lin et al., (2013)2 | |||||
|---|---|---|---|---|---|---|---|
| pH 2 | pH 6.5 | pH 9 | All trials | pH 5 | pH 6 | pH 7 | |
| a SPE recovery calculated as the percentage ratio between SPE measured and theoretical concentrations (100 mg L−1 injection concentration representing the upper calibration limit). Note: 1. SPE recovery recommended as: 70–130% (EPA, 2007).21 2. RSD acceptance: ≤30% (EPA, 2007).21 | |||||||
| 1-Propanol | 21 ± 1 | 26 ± 4 | 21 ± 5 | 22 ± 3 | |||
| 2-Butanone | 64 ± 4 | 52 ± 2 | 53 ± 3 | 56 ± 7 | |||
| 1-Butanol | 106 ± 5 | 106 ± 2 | 100 ± 6 | 100 ± 4 | |||
| Ethyl propionate | 85 ± 2 | 79 ± 4 | 83 ± 3 | 82 ± 3 | |||
| Dimethyl disulfide | 69 ± 4 | 54 ± 3 | 66 ± 2 | 63 ± 8 | |||
| Ethyl butyrate | 84 ± 4 | 95 ± 4 | 89 ± 3 | 89 ± 6 | |||
| Dimethyl trisulfide | 55 ± 2 | 44 ± 2 | 51 ± 2 | 50 ± 6 | |||
| Benzaldehyde | 76 ± 2 | 77 ± 3 | 79 ± 2 | 77 ± 2 | |||
| Limonene | 23 ± 2 | 24 ± 2 | 21 ± 1 | 22 ± 2 | |||
| p-Cresol | 96 ± 6 | 90 ± 6 | 83 ± 4 | 89 ± 7 | 103 ± 5 | 97 ± 0.5 | 103 ± 11 |
| Indole | 80 ± 7 | 82 ± 6 | 81 ± 6 | 81 ± 1 | 89 ± 2 | 90 ± 16 | 96 ± 2 |
| Skatole | 87 ± 5 | 89 ± 5 | 89 ± 5 | 88 ± 2 | 96 ± 5 | 97 ± 9 | 100 ± 2 |
3.4. Characterisation of faecally contaminated urine
Liquid phase concentrations in urine and faecally contaminated urine samples from eleven volunteers were determined for the full-suite of VOCs except those which exhibited poor SPE recoveries (Table 6). In general, concentrations ranged between the MDL and 1 mg kg−1 in urine samples, which is anticipated for fresh urine samples such as those measured in this study, which generally produce little odour when compared to aged urine.25 The presence of indole and skatole in fresh urine is also evident in the literature, though concentrations were considered sufficiently low not to be impactful as an odorant.9 However, p-cresol, was present at a considerable concentration (max. 13.01 mg kg−1). para-Cresol arises in urine from the breakdown of tyrosine by cresol producing bacteria in the intestine.26 Seigfried and Zimmerman27 reported an average p-cresol concentration in urine of 18 mg L−1. This is similar to the maximum value, the broader variation potentially arising from various factors such as protein intake,28 and the presence of specific urease positive isolates (e.g. Enterobacteriaceae)25 which are known contributors to raised p-cresol concentration. The use of ‘mid-stream’ urine collection techniques commonly used in medical studies (and not employed in this study) will also expectedly increase average concentration. Importantly, comparable values to the literature provide confirmation of the suitability of the method to real samples. Bacteria constitute 60% of faecal solids dry mass,29 with Escherichia coli (Table S3†) representing the dominant bacterial species that is primarily responsible for the oxidation of fatty acids to alcohols, and the conversion of the amino acids tyrosine and tryptophan to p-cresol and indole and skatole respectively.28,30,31 Faecally contaminated urine samples therefore generally exhibited higher VOC concentrations, and specifically for 1-butanol (alcohol) and indole (aromatic) which accords with the literature data on faecally contaminated urine.2 The analytical data was compared to thresholds compiled from literature by van Gemert32 used simply as a reference in order to contextualise the data (Table 6). At the background concentrations provided in urine, ethyl propionate, dimethyl disulfide, ethyl butyrate, p-cresol, indole and skatole were greater than the lower detection threshold for odour in water. The same VOC range was also above the taste threshold for water as was benzaldehyde. Significantly, each of the identified VOCs was determined in urine and faecally contaminated urine samples, with several at elevated concentrations, which suggests that the VOC range selected is pertinent for the development of membrane technology for liquid phase odourant abatement.| Odour descriptor32 | Urine | Faecally contaminated urine. 10:1 urine to faeces ratio |
Faeces2 | Detection threshold32 | ||||
|---|---|---|---|---|---|---|---|---|
| N = 11 | N = 11 | N = 2 | Air (odour) | Water (odour) | Water (taste) | |||
| Range | Range | Range | Range | Range | Range | Range | ||
| (mg kg−1 urine) | (mg kg −1 urine) | (mg kg−1 faeces) | (mg kg−1 faeces) | (mg m−3) | (mg kg−1) | (mg kg−1) | ||
| 2-Butanone | Acetone like | <LD-1.323 | 0.014–0.315 | 0.140–3.146 | 0.21–1000 | 7–100 | 3–60 | |
| 1-Butanol | Alcohol like | <LD-0.016 | <LD-0.185 | <LD-1.846 | 0.015–3000 | 0.27–511 | 2–100 | |
| Ethyl propionate | Fruity, rum | <LD-0.008 | <LD-0.02 | <LD-0.198 | 0.3–1 | 0.0001–0.067 | 0.00049–0.004 | |
| Dimethyl disulfide | Rotten cabbage | <LD-0.013 | <LD-0.014 | <LD-0.142 | 0.0011–3.5 | 0.00016–0.09 | 0.03–0.068 | |
| Ethyl butyrate | Pineapple | <LD-0.006 | <LD-0.02 | <LD-0.197 | 0.000016–0.1 | 0.000001–0.4 | 0.0001–0.45 | |
| Benzaldehyde | Bitter almond | <LD-0.060 | 0.0009–0.012 | 0.009–0.107 | 0.01–3400 | 0.32–4.6 | 0.05–1.5 | |
| p-Cresol | Sweet, tar-like | 0.003–13.01 | 0.214–2.67 | 2.139–26.683 | 20–25 | 0.00002 | 0.055–0.2 | 0.002–0.018 |
| Indole | Feacal | <LD-0.514 | 0.012–1.001 | 0.113–10.015 | 5–8 | 0.00035–0.0071 | 0.13–0.59 | 0.5 |
| Skatole | Faecal, nauseating | <LD-0.045 | 0.007–0.162 | 0.074–1.619 | 2–6 | 0.00035–0.00078 | 0.0002–0.052 | 0.05 |
3.5. Pervaporative membranes govern odour transport in faecally contaminated urine
An initial mass balance was conducted across the membrane experimental set-up to confirm minimum VOC losses. A 10 mg L−1 synthetic solution (comprising each VOC) was introduced to the feed-side and the mass balance constructed at the end of permeation was found to be 100 ± 10% (PVA used for assessment), which demonstrates the developed methods capability for technology assessment. An RSD of ≤13% was identified for replicate samples from membrane experimental studies. The membranes were subsequently challenged with the 10 ppm synthetic faecally contaminated urine. For the PVA membrane, removal efficiency ranged between 60 ± 5% (benzaldehyde) and 85 ± 0.5% (dimethyl disulfide, p-cresol) (Fig. 2a). The separation efficiency can be accounted for by the selectivity of the polymer toward water, the intrinsic polarity increasing the solubility parameter of the polymer for water, whilst the lower molecular weight of water increases the diffusivity parameter for the polymer, the product of these two parameters providing an enhanced water permeability.33 Whilst the presence of alcohols or carbonyl groups (e.g. benzaldehyde) are generally thought to influence the solubility parameter, a trend between VOC physico-chemical or structural properties (Table 1) was not evident.34 This can be accounted for by the comparatively low partial pressure exhibited by the VOCs relative to water, which limits the associative driving force for separation. Van Baelen et al.34 also observed that polyvinyl alcohol is soluble in water and prone to swelling above 20% wt water. This results in an open membrane structure which decreases selectivity, and is exacerbated at elevated temperatures. In this study, the PVA membrane was used for illustrative purposes and the material used is recommended for separations comprising 50% wt water solutions. Increasing crosslinking of the PVA polymer will increase membrane stability in the presence of water.35 Therefore whilst good VOC separation was facilitated by the PVA membrane, optimisation of cross-linking is recommended for future investigation into PVA for liquid phase odourant abatement. | ||
| Fig. 2 Assessment of pervaporative membrane processes as a liquid phase treatment to manage odourants at source. Performance expressed as (a) removal efficiency for a hydrophilic membrane material and (b) enrichment factor for a hydrophobic membrane. PVA (polyvinyl alcohol) and PDMS (polydimethylsiloxane). Error bars represent the standard deviation of a triplicate at pH 6.5. | ||
For the hydrophobic PDMS membrane, permeate was enriched for all VOCs with enrichment factors (β) ranging 6.1 ± 0.8 to 35.9 ± 0.2 (eqn (3), Fig. 2b). The selectivity toward VOCs can be ascribed to the enhanced affinity of PDMS toward non-polar compounds.36 A broad trend between the octanol–water coefficient, which corresponds to compound hydrophobicity, and enrichment factor was identified from benzaldehyde (logKow = 1.48, β = 36) to ethyl propionate (logKow = 1.21, β = 27), 1-butanol (logKow = 0.88, β = 26) and 2-butanone (logKow = 0.29, β = 23). However, although p-cresol, indole, and skatole presented a stronger hydrophobic contribution (Table 1), β factors of 6–17 were identified for these compounds. In addition to compound mobility and solubility within PDMS, vapour pressure difference also governs separation.37 The relatively lower permeability of these compounds can thus be accounted for by their vapour pressure which is around an order of magnitude lower than the other compounds. Since the PDMS polymer promotes VOC enrichment of the permeate, it is rational to expect an intensification of the ‘repulsive’ or ‘nauseating’ perception ordinarily associated with faecally contaminated urine (Table 6). However, the resulting permeate odour could be described as sweet, chemical, earthy and floral, with little perceivable evidence of faecal odour, and was hedonically more pleasant than the PVA permeate (Table 7). The range of physico-chemical characteristics represented with these compounds therefore illustrates the mechanisms which determine enrichment/rejection and can be used to suggest the behaviour of related compounds. Selectivity is governed by vapour pressure (low vapour pressures resulting in concentration polarisation at the downstream interface), volatility (liquid phase stability) and hydrophobicity (by inclusion of highly hydrophobic groups i.e. benzene or length of hydrocarbon chain). For example, we can infer that vanillin, which contains a hydrophobic aromatic ring (logKow 1.21) but low vapour pressure (0.00047 mm Hg at 25 °C), could be enriched similarly to p-cresol, indole and skatole. Importantly, the arising data suggests that thermally driven barrier technology could be engineered to change perception through modification of the odour profile rather than developed simply for elimination. This is analogous to the perfumery industry in which indole, one of the core constituents of odour arising from faecally contaminated urine is also a critical ingredient in jasmine perfume.9
| Membrane material | Permeate odour descriptor |
|---|---|
| Polyvinyl alcohol | Sweaty, chemical, sweet, onion |
| Polydimethylsiloxane | Sweet, chemical, earthy, floral |
4. Conclusions
In this study, an analytical method for the detection of liquid phase VOCs responsible for faecal odour has been developed and verified. The following conclusions have been drawn:• A quantitative method has been developed to enable co-elution of a range of VOCs comprised of a broad spectrum of physicochemical properties in a single elution.
• Sample concentration by SPE permit low method detection limits sufficient to measure liquid phase concentrations within and below the detection threshold range reported for odour and taste. The utility of this method extends to a broad range of stakeholders including healthcare professionals, taste and odour specialists and public health engineers.
• Consistent recovery was identified for solid phase extraction, while acceptable recoveries were also determined for nine VOCs, which were subsequently analysed in real matrices.
• Comparison of VOC data determined in urine and faecally contaminated urine samples to literature data, provided confirmation of the appropriateness of this method for evaluation of real samples, and also that the VOCs determined are relevant and appropriate for the quantitation of faecal odourants in the liquid phase.
• The method was successfully applied for the evaluation of pervaporative membranes, where SPE coupled with the lower calibration range, was capable of quantification within PVA membrane permeate which presents an analytical challenge due to the polymers capability for separation.
• The method holds immediate value for public health engineers, medical and taste and odour scientists. However, through development of a GC-MS method, the accessibility of the technique extends beyond those prescribed sectors to a wide range of institutions/laboratories thanks to the inclusion of such equipment as ‘standard’.
• Dense hydrophilic polymeric membranes offer the greatest selective separation of liquid phase VOCs, yet the more concentrated permeate produced from PDMS presented the more hedonically pleasant permeate, which suggests there is more than one route to challenging perception of faecal odour in reuse product water.
• Further research on the combination of VOC and non-VOC odourants, building from this method, would be beneficial to develop a holistic odour management approach.
• Whilst further membrane development is warranted for this application, the method was capable of facilitating diagnostic investigation of VOC separation and further demonstrated that the combination of hedonic characterisation coupled with quantitative methods are demanded to develop a technical solution for liquid phase odourant separation, which offers significant potential for the advancement of decentralised sanitation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based on research funded by the Bill & Melinda Gates Foundation (OPP1111272). The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the funders. Enquiries for access to the data referred to in this article should be directed to: E-mail: researchdata@cranfield.ac.uk. The authors give special thanks to Charles Chappuis and Christian Starkenmann at Firmenich for their assistance with the development of the SPE protocol.References
- UNICEF and WHO, Progress on Sanitation and Drinking Water: 2015 Update and MDG Assessment, WHO Press, NY, USA, 2015 Search PubMed.
- J. Lin, J. Aoll, Y. Niclass, M. I. Velazco, L. Wunsche, J. Pika and C. Starkenmann, Qualitative and quantitative analysis of volatile constituents from latrines, Environ. Sci. Technol., 2013, 47, 7876–7882 CrossRef CAS PubMed.
- B. de Lacy Costello, A. Amann, H. Al-Kateb, C. Flynn, W. Filipiak, T. Khalid, D. Osborne and N. M. Ratcliffe, A review of the volatiles from the healthy human body, J. Breath Res., 2014, 8, 014001 CrossRef CAS PubMed.
- C. J.-F. Chappuis, Y. Niclass, C. Vuilleumier and C. Starkenmann, Quantitative headspace analysis of selected odorants from latrines in africa and india, Environ. Sci. Technol., 2015, 49, 6134–6140 CrossRef CAS PubMed.
- M. Brattoli, G. de Gennaro, V. de Pinto, A. D. Loiotile, S. Lovascio and M. Penza, Odour detection methods: olfactometry and chemical sensors, Sensors, 2011, 11, 5290–5322 CrossRef CAS PubMed.
- R. Otterpohl, U. Braun and M. Oldenburg, Innovative technologies for decentralised wastewater management in urban and peri-urban areas, in 5th Specialised Conference on Small Water and Wastewater Treatment Systems, Istanbul, Turkey, 2002 Search PubMed.
- F. Kamranvand, C. J. Davey, H. Sakar, O. Autin, E. Mercer, M. Collins, L. Williams, A. Kolios, A. Parker, S. Tyrrel, E. Cartmell and E. J. McAdam, Impact of fouling, cleaning and faecal contamination on the separation of water from urine using thermally driven membrane separation, Sep. Sci. Technol., 2018, 53, 1372–1382 CrossRef CAS.
- P. H. Cruddas, A. Parker and A. Gormley, User perspectives to direct water reuse from the Nano Membrane Toilet, in 38th WEDC International Conference, Loughborough, UK, 2015 Search PubMed.
- C. Starkenmann, in Springer Handbook of Odor, ed. A. Buettner, Springer, Berlin, 2017, pp. 921–936 Search PubMed.
- R. Robinson and R. Stokes, Electrolyte solutions, Dover Publicatoins, Mineola, 2nd edn, 2002 Search PubMed.
- C. Rose, A. Parker, B. Jefferson and E. Cartmell, The characterization of feces and urine: a review of the literature to inform advanced treatment technology, Crit. Rev. Environ. Sci. Technol., 2015, 45, 1827–1879 CrossRef CAS PubMed.
- PubChem, https://pubchem.ncbi.nlm.nih.gov/, (accessed 10 February 2018).
- Yeast Metabolome database (YMDB), http://www.ymdb.ca/, (accessed 10 February 2018).
- J. D. Gu and D. F. Berry, Degradation of substituted indoles by an indole-degrading methanogenic consortium, Appl. Environ. Microbiol., 1991, 57, 2622–2627 CAS.
- R. Sander, Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- A. Technologies, The Secrets of Good Peak Shape in HPLC, https://www.agilent.com/cs/library/eseminars/public/secretsofgoodpeakshapeinhplc.pdf, (accessed 10 February 2018).
- M. F. Wahab, D. C. Patel and D. W. Armstrong, Peak shapes and their measurements: the need and the concept behind total peak shape analysis, LCGC North Am., 2017, 35, 846–853 CAS.
- L. Currie, Detection and quantification limits: origins and historical overview, Anal. Chim. Acta, 1999, 391, 127–134 CrossRef CAS.
- U. E. P. A. (Epa), EPA Method 8000C, Gas Chromatography, 2003 Search PubMed.
- T. Little, Establishing Acceptance Criteria for Analytical Methods, BioPharm Int., 2016, 29(10), 44–48 Search PubMed.
- U. E. P. A. (Epa), Method 3535A (SW-846): Solid-Phase Extraction (SPE), 2007, vol. 1 Search PubMed.
- M. Nakamura, M. Nakamura and S. Yamada, Conditions for solid-phase extraction of agricultural chemicals in waters by using n-octanol-water partition coefficients, Analyst, 1996, 121, 469–475 RSC.
- M. Wells, in Solid-Phase Extraction: Principles, Techniques and Applications, ed. N. Simpson, Marcel Dekker, New York, 2000 Search PubMed.
- V. De Preter, G. Van Staeyen, D. Esser, P. Rutgeerts and K. Verbeke, Development of a screening method to determine the pattern of fermentation metabolites in faecal samples using on-line purge-and-trap gas chromatographic–mass spectrometric analysis, J. Chromatogr. A, 2009, 1216, 1476–1483 CrossRef CAS PubMed.
- M. Troccaz, Y. Niclass, P. Anziani and C. Starkenmann, The influence of thermal reaction and microbial transformation on the odour of human urine, Flavour Fragrance J., 2013, 28, 200–211 CrossRef CAS.
- S. Gabriele, R. Sacco, L. Altieri, C. Neri, A. Urbani, C. Bravaccio, M. P. Riccio, M. R. Iovene, F. Bombace, L. De Magistris and A. M. Persico, Slow intestinal transit contributes to elevate urinary p-cresol level in Italian autistic children, Autism Res., 2016, 9, 752–759 CrossRef PubMed.
- M. Siegfried and R. Zimmermann, Biochem. Z., 1911, 34, 471 Search PubMed.
- B. Geypens, D. Claus, P. Evenepoel, M. Hiele, B. Maes, M. Peeters, P. Rutgeerts and Y. Ghoos, Influence of dietary protein supplements on the formation of bacterial metabolites in the colon, Gut, 1997, 41, 70–76 CrossRef CAS PubMed.
- F. Guarner and J.-R. Malagelada, Gut flora in health and disease, Lancet, 2003, 361, 512–519 CrossRef.
- L. D. J. Bos, P. J. Sterk and M. J. Schultz, Volatile Metabolites of Pathogens: A Systematic Review, PLoS Pathog., 2013, 9, e1003311 CrossRef CAS PubMed.
- R. Vanholder, R. De Smet and G. Lesaffer, p-Cresol: a toxin revealing many neglected but relevant aspects of uraemic toxicity, Nephrol., Dial., Transplant., 1999, 14, 2813–2815 CrossRef CAS.
- L. van Gemert, Odour thresholds. Compilations in air water and other media, Oliemans Punter & Partners BV, Utrecht, 2nd edn, 2011 Search PubMed.
- L. Liu and S. E. Kentish, Pervaporation performance of crosslinked PVA membranes in the vicinity of the glass transition temperature, J. Membr. Sci., 2018, 553, 63–69 CrossRef CAS.
- D. Van Baelen, B. Van der Bruggen, K. Van den Dungen, J. Degreve and C. Vandecasteele, Pervaporation of water–alcohol mixtures and acetic acid–water mixtures, Chem. Eng. Sci., 2005, 60, 1583–1590 CrossRef CAS.
- J. M. Gohil, A. Bhattacharya and P. Ray, Studies On The Crosslinking Of Poly (Vinyl Alcohol), J. Polym. Res., 2006, 13, 161–169 CrossRef CAS.
- R. Baker, Membrane Technology and Applications, John Wiley and Sons, Chichester, 3rd edn, 2012 Search PubMed.
- Pervaporation, Vapour Permeation and Membrane Distillation: Principles and Applications, ed. A. Basile, A. Figoli and M. Khayet, Woodhead Publishing, Cambridge, UK, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ew00693h |
| This journal is © The Royal Society of Chemistry 2019 |