
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetranuclear oxido-bridged thorium(IV) clusters obtained using tridentate Schiff bases†‡
Sokratis T.
Tsantis
 a,
Aimilia
Lagou-Rekka
a,
Konstantis F.
Konidaris
ab,
Catherine P.
Raptopoulou
c,
Vlasoula
Bekiari
*b,
Vassilis
Psycharis
*c and
Spyros P.
Perlepes
*ad
a,
Aimilia
Lagou-Rekka
a,
Konstantis F.
Konidaris
ab,
Catherine P.
Raptopoulou
c,
Vlasoula
Bekiari
*b,
Vassilis
Psycharis
*c and
Spyros P.
Perlepes
*ad
aDepartment of Chemistry, University of Patras, 26504, Patras, Greece. E-mail: perlepes@patreas.upatras.gr; Tel: +30 2610 996730
bSchool of Agriculture Sciences, University of Patras, 30200 Messolonghi, Greece. E-mail: mpekiari@teimes.gr; Tel: +30 26310 58260
cInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, 15310 Aghia Paraskevi Attikis, Greece. E-mail: v.psycharis@inn.demokritos.gr; Tel: +30 210 6503346
dFoundation for Research and Technology-Hellas (FORTH), Institute of Chemical Engineering Sciences (ICE-HT), Platani, P.O. Box 144, 26504 Patras, Greece
First published on 2nd September 2019
Abstract
Thorium(IV) complexes are currently attracting intense attention from inorganic chemists due to the development of liquid-fluoride thorium reactors and the fact that thorium(IV) is often used as a model system for the study of the more radioactive Np(IV) and Pu(IV). Schiff-base complexes of tetravalent actinides are useful for the development of new separation strategies in nuclear fuel processing and nuclear waste management. Thorium(IV)–Schiff base complexes find applications in the colorimetric detection of this toxic metal ion and the construction of fluorescent on/off sensors for Th(IV) exploiting the ligand-based light emission of its complexes. Clusters of Th(IV) with hydroxide, oxide or peroxide bridges are also relevant to the environmental and geological chemistry of this metal ion. The reactions between Th(NO3)4·5H2O and N-salicylidene-o-aminophenol (LH2) and N-salicylidene-o-amino-4-methylphenol (L′H2) in MeCN have provided access to complexes [Th4O(NO3)2(LH)2(L)5] (1) and [Th4O(NO3)2(L′H)2(L′)5] (2) in moderate yields. The structures of 1·4MeCN and 2·2.4 MeCN have been determined by single-crystal X-ray crystallography. The complexes have similar molecular structures possessing the {Th4(μ4-O)(μ-OR′)8} core that contains the extremely rare {Th4(μ4-O)} unit. The four ThIV atoms are arranged at the vertexes of a distorted tetrahedron with a central μ4-O2− ion bonded to each metal ion. The H atom of one of the acidic –OH groups of each 3.21 LH− or L′H− ligand is located on the imine nitrogen atom, thus blocking its coordination. The ThIV centres are also held together by one 3.221 L2− or (L′)2− group and four 2.211 L2− or (L′)2− ligands. The metal ions adopt three different coordination numbers (8, 9, and 10) with a total of four coordination geometries (triangular dodecahedral, muffin, biaugmented trigonal prismatic, and sphenocorona). A variety of H-bonding interactions create 1D chains and 2D layers in the crystal structures of 1·4 MeCN and 2·2.4 MeCN, respectively. The structures of the complexes are compared with those of the uranyl complexes with the same or similar ligands. Solid-state and IR data are discussed in terms of the coordination mode of the organic ligands and the nitrato groups. 1H NMR data suggest that solid-state structures are not retained in DMSO. The solid complexes emit green light at room temperature upon excitation at 400 nm, the emission being ligand-centered.
Introduction
The ions of early actinide (An) elements are unique in inorganic chemistry in that they combine large radii and lanthanide (Ln) ions’ Lewis acidity with the potential for remarkable covalency (due to greater f-orbital radial extension for An vs. Ln ions), and due to the fact that they can achieve a broad range of oxidation states (mainly for U, Np and Pu).1–3These characteristics provide An complexes with some special reactivity profiles. Thorium was discovered in 1829 by the famous chemist Jöns Jacob Berzelius and owes its name to Thor, the Scandinavian god of thunder and war.4 Natural Th is radioactive, but many of its current uses exploit its chemical, rather than its nuclear, features. Thorium is the first true An element and has an empty 5f orbital, its outer electronic configuration being 6d25f2; therefore, Th(IV) is the most stable and almost exclusive oxidation state.5
There has been a renaissance in the coordination and material chemistry of Th(IV) for the last 5 years or so.1,6–21 There are several reasons for this. The main reason is that Th is widely considered to be the next generation nuclear fuel as the development of Liquid-Fluoride Thorium Reactors (LFTRs) comes closer to commercialization.5,6,22,23 India is keen on using thorium as the nuclear fuel (this country has approximately 30% of the world's thorium reserves) and plans to produce one third of its electricity from thorium by 2050.5 The production of thorium is usually achieved by the separation of Th4+ from Ln3+ ions, since the main thorium production mineral is monazite which contains the ions of 4f metals; mining is based on liquid–liquid extraction with organic ligands,24,25 hence the importance of the Th(IV) coordination chemistry. There is also intense interest for the development of new efficient solid absorbents for the selective extraction of Th4+ ions from 4f-metal ions.16
Other research areas of Th(IV) chemistry that are currently attracting attention from inorganic chemists include the incorporation of this diamagnetic ion into coordination clusters instead of the paramagnetic U(IV) to elucidate the magnetic exchange interactions between the d-metal ions in d/UIV complexes,26 its interaction with ionic liquids to model the aspects of the extraction of An ions from radioactive feeds (a process of great relevance in nuclear fuel cycle activities),13 the understanding of Th(IV) peroxide chemistry,8,14 the study of the solution and solid-state structural chemistry of Th(IV) hydrolysis and condensation products,9,11 the stabilization of novel secondary building units in Th(IV)-based Metal–Organic Frameworks (MOFs),15,19 the characterization of complexes with very high coordination numbers (up to 14) and unusual coordination geometry,7 the in-depth investigation of the thermodynamic and electronic properties of heterometallic Anz/ThIV (z = various) and An/ThIV/3d-metal ion MOFs with “structural memory”20 and the progress in compounds with Th(IV)-ligand multiple bonds.12
Schiff bases have been used extensively as ligands with metals across the Periodic table.27 These molecules are attractive for their easy and high-yield preparation, their solubility in a variety of organic solvents, and their ability to stabilize metals in various oxidation states and control behaviour of metal ions during catalysis;28–33 in addition, their steric and electronic properties are highly tunable. The coordination chemistry of the trans-UO22+ ion (uranyl ion) with Schiff bases is a well explored area.34–38 However, tetravalent actinides, An(IV), have been little studied. Schiff-base complexes of An(IV) are of great utility for the development of new An separation strategies in nuclear fuel processing and nuclear waste management.39 Restricting the discussion to Th(IV), its Schiff-base complexes (which are scarce in the literature40–42 are closely related to the development of colorimetric detection of this metal ion and applications in real-time samples,43 to the construction of on/off sensors for Th(IV) exploiting the ligand-based light emission of its complexes,44 and to the study of Th(IV) reactions with the derivatives of vitamin B6 as a means to understand the assimilation of this metal ion and other heavy nuclei by living organisms and its acute toxicological effects.45 A drawback associated with tetradentate dianionic O,N,N,O ligands of the salen2−-type is their planarity and charge. The valence completion of An(IV), e.g. Th(IV), requires often two ligands per metal ion and this may result in hindered reactivity due to steric effects. Once the bis-Schiff-base complex is isolated, no further ligand-substitution is possible and the redox chemistry of redox-active AnIV centres, e.g. UVI, is rather poor.41 Therefore, the interest in this area has shifted to potentially dianionic, tridentate Schiff bases. Two such ligands are N-salicylidene-o-aminophenol [or N-(2-hydroxyphenyl)salicylaldimine; LH2 in Scheme 1] and N-salicylidene-o-amino-4-methylphenol [or N-(2-hydroxy-5-methylphenyl)-salicylaldimine; L′H2 in Scheme 1]. The number 4 in the first (empirical) name of L′H2 (which is in disagreement with the numbering code in Scheme 1) is used because its origin is from the name of the starting material, i.e., 4-methyl-2-aminophenol used for the synthesis of L′H2.46
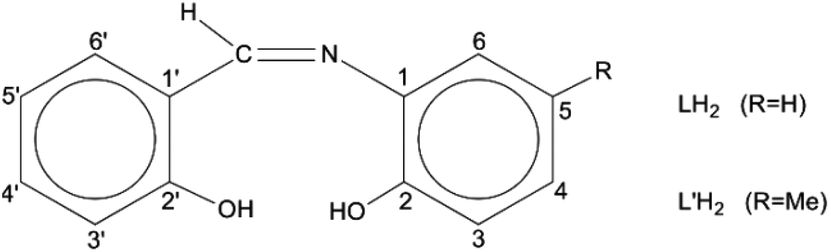 | ||
| Scheme 1 General structural formula of the tridentate ONO Schiff bases used in the present work and their abbreviations. | ||
Upon deprotonation of one or both –OH groups of LH2 and L′H2, each negatively charged phenoxido atom can bridge two or three metal ions favouring cluster formation, while the position of the imino N atom simultaneously assures the formation of chelating rings that can lead to thermodynamic stability of the products in solution. The anions of the ligands have been widely used in main group-,47 3d-,48–51 4f-52,53 and mixed 3d/4f-metal54 chemistry by our group and other scientists, with emphasis on the magnetic properties of the complexes. The use of LH2 and L′H2 in 5f-metal chemistry is negligible (vide infra). No Th(IV) complexes have been prepared with these ligands.
Having all the above mentioned in mind, and given our interest in the chemistry of An ions55–57 and in the coordination chemistry of LH2 and its derivatives,46,48–52 we report herein the reactions of Th(NO3)4·5H2O with LH2 and L′H2, and the full characterization of the interesting products.
Experimental section
Materials, physical and spectroscopic measurements
All manipulations were performed under aerobic conditions using materials (reagent grade) and solvents as received. The free ligands LH2 and L′H2 were synthesized by the reactions of salicylaldehyde, and 2-aminophenol and 4-methyl-2-aminophenol, respectively, in refluxing MeOH or EtOH using published procedures;58–60 the yields were in the 75–80% range. Their purity was checked by microanalyses and 1H NMR spectra. Caution! Natural thorium (primary isotope 232Th) is a weak α-emitter (4.012 MeV) with a half-life of 1.41 × 1010 years. Manipulations and reactions should be carried out in monitored fume hoods in a radiation laboratory which has α- and β-counting equipment; the laboratory atmosphere is inspected monthly for contamination. Elemental analyses (C, H, and N) were performed by the University of Patras microanalytical service. FT-IR spectra (4000–400 cm−1) were recorded using a PerkinElmer 16PC spectrometer with samples prepared as KBr pellets and as Nujol or hexachlorobutadiene mulls between CsI disks. Solid-state emission and excitation spectra were recorded using a Cary Eclipse fluorescence spectrophotometer. 1H NMR spectra in DMSO-d6 were recorded using a 400 MHz Bruker Avance DPX spectrometer with TMS as an internal standard. FT-Raman spectra were recorded using a Bruker (D) FRA-106/S component attached to an Equinox 55 spectrometer. An R510 diode-pumped Nd:YAG laser at 1064 nm was used for Raman excitation with a laser power of 250 mW on the sample, utilizing an average of 100 scans at 4 cm−1 resolution. Conductivity measurements were carried out at 25 °C with a Metrohm-Herisau E-527 bridge and a cell of standard constant.Synthetic details
Single-crystal X-ray crystallography
Yellowish orange (0.06 × 0.11 × 0.43 mm) and orange (0.17 × 0.17 × 0.36 mm) crystals of 1·4MeCN and 2·2.4MecN, respectively, were taken from the mother liquor and immediately cooled to −103 °C (1·4MeCN) and −93 °C (2·2.4MecN). X-ray diffraction data were collected using a Rigaku R-AXIS SPIDER Image Plate diffractometer using graphite-monochromated Mo Kα radiation. Data collection (ω-scans) and processing (cell refinement, data reduction and empirical absorption correction) were performed using the CrystalClear program package.61 The structures were solved by direct methods using SHELXS-97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 62 and refined by full-matrix least-squares techniques on F2 with the latest version of SHELXL (2014/7).63 The H atoms were either located by difference maps and refined isotropically or were introduced at calculated positions as riding on their respective bonded atoms. All non-H atoms were refined anisotropically. Most structural plots were drawn using the Diamond 3 program package.64
62 and refined by full-matrix least-squares techniques on F2 with the latest version of SHELXL (2014/7).63 The H atoms were either located by difference maps and refined isotropically or were introduced at calculated positions as riding on their respective bonded atoms. All non-H atoms were refined anisotropically. Most structural plots were drawn using the Diamond 3 program package.64
Important crystallographic data are listed in Table 1. Full details can be found in the CIF files.
| Parameter | 1·4MeCN | 2·2.4 MeCN |
|---|---|---|
| Formula | C99H77N13Th4O21 | C102.8H86.2N11.4Th4O21 |
| F w | 2712.89 | 2745.39 |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
| a/Å | 13.5738(3) | 16.3097(3) |
| b/Å | 14.9072(3) | 26.3838(5) |
| c/Å | 26.8572(5) | 22.6412(4) |
| α/° | 81.513(1) | 90.0 |
| β/° | 76.826(1) | 94.057(1) |
| γ/° | 61.585(1) | 90.0 |
| V/Å3 | 4649.11 (17) | 9718.4(3) |
| Z | 2 | 4 |
| ρ calcd/g cm−3 | 1.938 | 1.876 |
| T/K | 170 | 180 |
| Radiation/μ (mm−1) | Mo Kα/6.46 | Mo Kα/6.18 |
| Reflections collected/unique (Rint) | 86266/20248 (0.049) |
91224/21120 (0.045) |
| Reflections with I > 2σ(I) | 16985 |
18560 |
| No. of parameters | 1462 | 1279 |
| R 1[I > 2σ(I)], wR2 (all data) | 0.0295, 0.0576 | 0.0325/0.073 |
| GOF (F2) | 1.02 | 1.05 |
| Δρmax/Δρmin (e Å−3) | 1.561/−1.15 | 2.13/−1.43 |
| CCDC number | 1945475 | 1945476 |
Results and discussion
Synthetic comments
In the present study, we have investigated the reactions between the tridentate Schiff bases LH2 and L′H2 and thorium sources; Th(IV) complexes with these ligands were unknown in the literature. A variety of reaction systems involving Th(NO3)4·5H2O and ThCl4, solvent media, reaction ratios, the absence/presence of external bases and crystallization techniques were systematically studied for arriving at the optimized procedures described in the Experimental section. Working with ThCl4, we obtained powders which could not be crystallized; IR spectroscopy indicated that the powders were products.Treatment of Th(NO3)4·5H2O and LH2 (1:2) in MeCN at room temperature gave a yellow solution from which were subsequently isolated yellowish crystal of [Th4O(NO3)2(LH)2(L)5]·4MeCN (1·4MeCN) in rather low yields (∼30%), eqn (1). Efforts to prepare a compound with all the ligands in their doubly deprotonated form (L2−), e.g., [Th4O(L)7] were not successful, even when we used an external base (Et3N, Bun4NOH) to LH2 ratios up to 2:1. The obtained product was again 1; somewhat to our surprise the yield of the reaction increased only by 10%, reaching 40–45% (the procedure with the addition of the base is not described in the Experimental section).
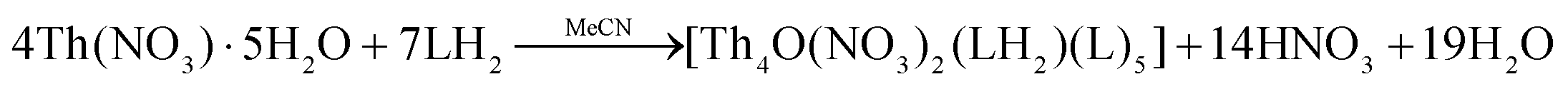 | (1) |
A completely analogous reaction between Th(NO3)4·5H2O and L′H2 in MeCN, using the same concentrations of the reactants as in the case of 1, resulted only in a few orange crystals (yield lower than 10%) of [Th4O(NO3)2(L′H)2(L′)5]·2.4MeCN (2·2.4MeCN). The addition of an external base was deemed necessary to increase the yield. Thus, the Th(NO3)4·5H2O/L′H2/Bun4NOH (1:2:2) reaction system in MeCN gave a light orange solution, from which the product was isolated in a ca. 40% yield, eqn (2). Increase of the L′H2 to OH− ratio from 2:2 to 2:4, to further improve the yield, resulted in the precipitate of amorphous hydroxide thorium(IV) species.
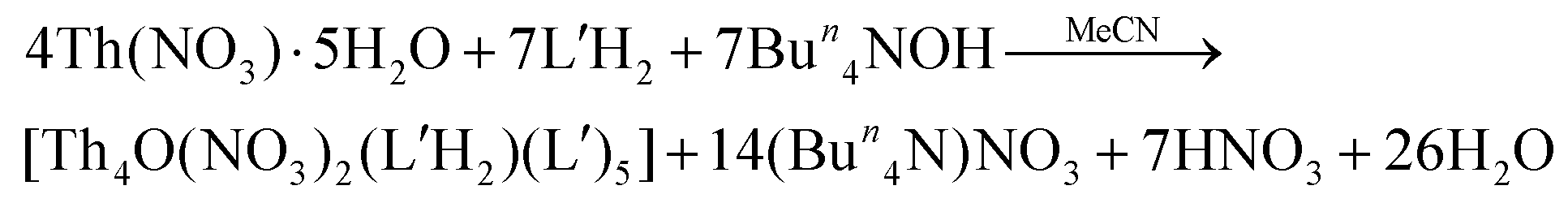 | (2) |
Description of the structures
Aspects of the molecular and crystal structures of complexes 1·4MeCN and 2·2.4MeCN are shown in Fig. 1–6 and Fig S1–S3.‡ Selected numerical data are listed in Tables 2, 3 and S1–S5.‡ The molecular structures of the two complexes are very similar. Thus, only the molecular structure of 1·4MeCN will be described in detail.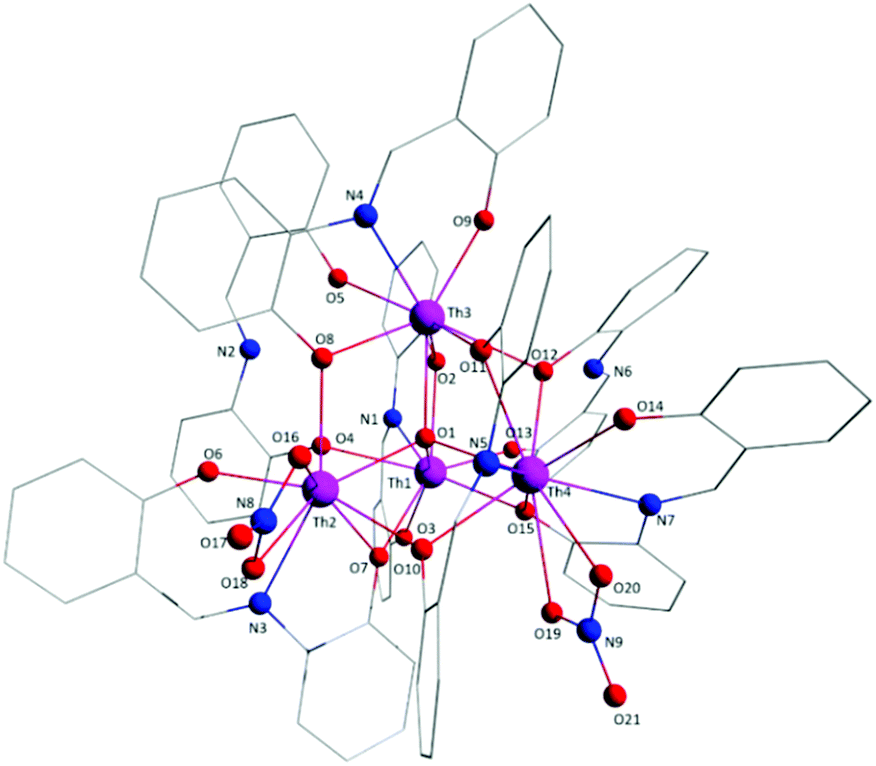 | ||
| Fig. 1 Partially labelled plot of the tetranuclear molecule that is present in the structure of 1·4MeCN. Colour scheme: Th, pink; O, red; N, blue; C, grey. All H atoms have been omitted for clarity. | ||
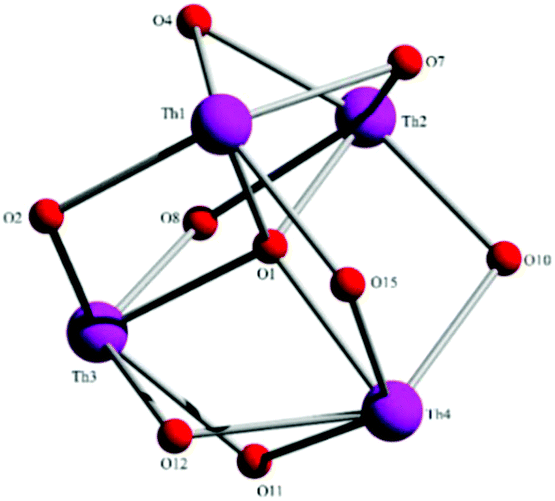 | ||
| Fig. 2 The {ThIV4(μ4-O)(μ-OR′)8} core of molecules 1 and 2. O1 is the μ4-O2− group in 1, O4 and O12 are the bridging oxygen atoms of the iminiumphenolate parts of the LH− in 1 ligands, while the remaining oxygen atoms belong to the L2− groups. | ||
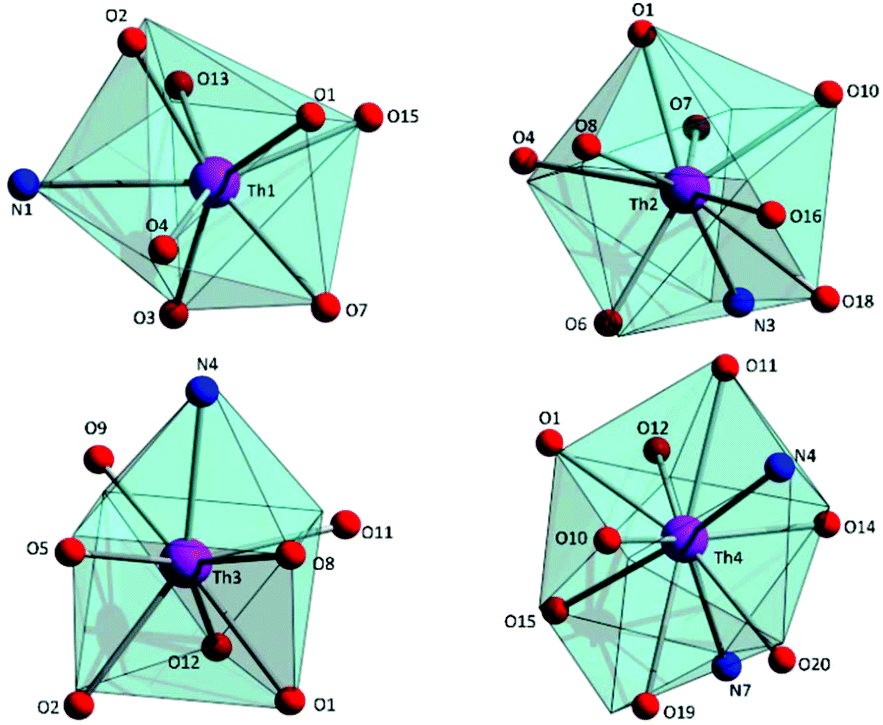 | ||
| Fig. 3 The triangular dodecahedral, muffin-type, biaugmented trigonal prismatic and sphenocorona coordination polyhedra for Th1, Th2, Th3 and Th4, respectively, in the structure of 1·4MeCN. The very small spheres define the vertices of the ideal polyhedra. | ||
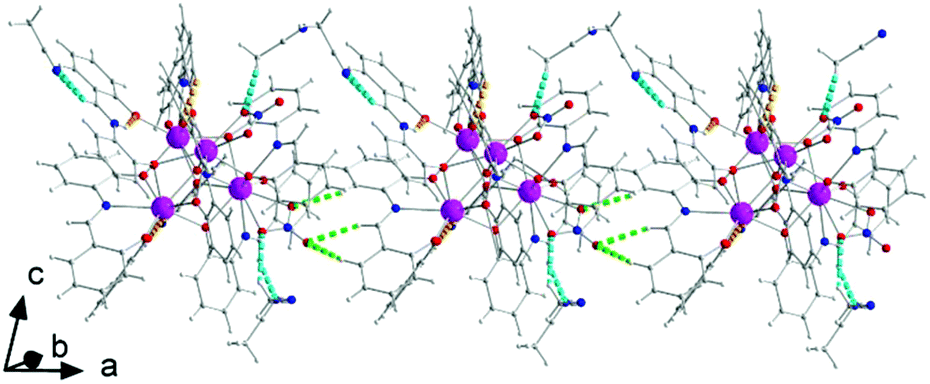 | ||
| Fig. 4 A portion of a chain of tetranuclear cluster and MeCN molecules parallel to the α crystallographic axis in the crystal structure of 1·4MeCN. Dashed orange lines indicate intramolecular N–H⋯O bonds within the LH− ligands and dashed cyan lines indicate H bonds involving the lattice MeCN molecules; dashed light green lines represent the H bonds that are responsible for the formation of chains. | ||
 | ||
| Fig. 5 Partially labelled plot of the tetranuclear molecule that is present in the structure of 2·2.4MeCN. Colour scheme: Th, pink; O, red; N, blue; C, grey. All H atoms have been omitted for clarity. | ||
 | ||
| Fig. 6 A part of a layer parallel to the (−101) plane formed as a result of C–H⋯O H bonds (dashed light green lines) between the tetranuclear cluster molecules in the crystal structure of 2·2.4MeCN, see the text for details. Only the H atoms involved in H bonds that contribute to the layer formation are shown. | ||
| Distances | |||
| Th1⋯Th2 | 3.776(1) | Th2⋯Th3 | 3.992(1) |
| Th1⋯Th3 | 3.935(1) | Th2⋯Th4 | 4.001(1) |
| Th1⋯Th4 | 4.039(1) | Th3⋯Th4 | 3.816(1) |
| Th1–O1 | 2.434(3) | Th3–O2 | 2.447(3) |
| Th1–O2 | 2.410(3) | Th3–O5 | 2.372(3) |
| Th1–O3 | 2.205(3) | Th3–O8 | 2.436(3) |
| Th1–O4 | 2.486(3) | Th3–O9 | 2.228(3) |
| Th1–O7 | 2.463(3) | Th3–O11 | 2.379(3) |
| Th1–O13 | 2.335(3) | Th3–O12 | 2.574(3) |
| Th1–O15 | 2.437(3) | Th3–N4 | 2.601(4) |
| Th1–N1 | 2.650(3) | Th4–O1 | 2.440(3) |
| Th2–O1 | 2.369(3) | Th4–O10 | 2.493(4) |
| Th2–O4 | 2.566(3) | Th4–O11 | 2.598(3) |
| Th2–O6 | 2.235(3) | Th4–O12 | 2.579(3) |
| Th2–O7 | 2.459(3) | Th4–O14 | 2.213(3) |
| Th2–O8 | 2.511(3) | Th4–O15 | 2.479(3) |
| Th2–O10 | 2.445(3) | Th4–O19 | 2.652(3) |
| Th2–O16 | 2.573(3) | Th4–O20 | 2.574(3) |
| Th2–O18 | 2.612(3) | Th4–N5 | 2.741(4) |
| Th2–N3 | 2.636(4) | Th4–N7 | 2.660(4) |
| Th3–O1 | 2.380(3) | ||
| Angles | |||
| Th1–O1–Th2 | 103.7(1) | Th2–O1–Th3 | 114.4(1) |
| Th1–O1–Th3 | 109.7(1) | Th2–O1–Th4 | 112.6(1) |
| Th1–O1–Th4 | 111.9(1) | Th3–O1–Th4 | 104.7(1) |
| O4–Th1–O13 | 148.7(1) | Th1–O7–Th2 | 100.2(1) |
| O6–Th2–O10 | 153.6(1) | Th2–O8–Th3 | 107.6(1) |
| O1–Th3–O9 | 150.3(1) | Th3–O11–Th4 | 100.0(1) |
| O11–Th4–O19 | 161.2(1) | Th1–O15–Th4 | 110.5(1) |
| Distances | |||
| Th1⋯Th2 | 4.029(1) | Th2⋯Th3 | 3.988(1) |
| Th1⋯Th3 | 3.823(1) | Th2⋯Th4 | 3.763(1) |
| Th1⋯Th4 | 4.029(1) | Th3⋯Th4 | 3.942(1) |
| Th1–O | 2.438(3) | Th2–N2 | 2.633(4) |
| Th1–O1 | 2.483(3) | Th3–O | 2.379(3) |
| Th1–O2 | 2.660(3) | Th3–O2 | 2.379(3) |
| Th1–O5 | 2.195(3) | Th3–O14 | 2.539(3) |
| Th1–O6 | 2.467(3) | Th3–O16 | 2.435(3) |
| Th1–O7 | 2.642(3) | Th3–O17 | 2.386(3) |
| Th1–O8 | 2.642(3) | Th3–O19 | 2.241(3) |
| Th1–O14 | 2.585(3) | Th3–O20 | 2.413(3) |
| Th1–N1 | 2.725(4) | Th3–N9 | 2.618(4) |
| Th1–N3 | 2.693(3) | Th4–O | 2.427(3) |
| Th2–O | 2.384(3) | Th4–O4 | 2.440(3) |
| Th2–O1 | 2.504(3) | Th4–O6 | 2.421(3) |
| Th2–O3 | 2.230(3) | Th4–O13 | 2.321(3) |
| Th2–O4 | 2.462(3) | Th4–O15 | 2.235(3) |
| Th2–O10 | 2.561(3) | Th4–O16 | 2.406(3) |
| Th2–O11 | 2.621(3) | Th4–O18 | 2.534(3) |
| Th2–O18 | 2.497(3) | Th4–N7 | 2.644(4) |
| Th2–O20 | 2.529(3) | ||
| Angles | |||
| Th1–O–Th2 | 113.3(1) | Th2–O–Th3 | 113.7(1) |
| Th1–O–Th3 | 105.1(1) | Th2–O–Th4 | 102.9(1) |
| Th1–O–Th4 | 111.8(1) | Th3–O–Th4 | 110.2(1) |
| O6–Th1–N1 | 156.5(1) | Th1–O1–Th2 | 107.8(1) |
| O20–Th2–N2 | 154.0(1) | Th2–O4–Th4 | 100.3(1) |
| O2–Th3–O17 | 151.9(1) | Th1–O14–Th3 | 96.5(1) |
| O–Th4–O15 | 148.0(1) | Th2–O18–Th4 | 96.8(1) |
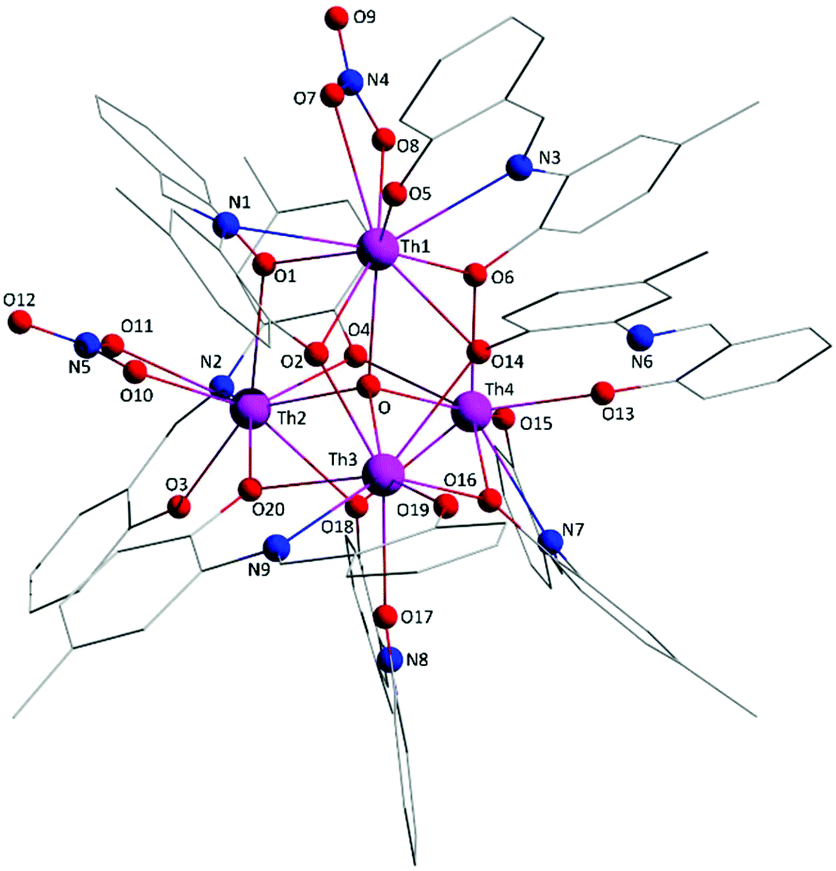
The crystal structure of 1·4MeCN consists of molecules [Th4O(NO3)2(LH)2(L)5] and MeCN in an 1:4 ratio. The tetranuclear molecule (Fig. 1) contains four ThIV atoms arranged at the vertexes of a distorted tetrahedron, with a central μ4-O2− ion (O1) bonded to each metal ion. The ThIV centres are also held together by two 3.21 (Harris notation65) LH− groups, one 3.221 L2− and four 2.211 L2− ligands (Scheme 2). Two slightly anisobidentate chelating nitrato groups (donor atoms O16/O18 and O19/O20) complete the coordination spheres of Th2 and Th4. The core of the molecule is {ThIV4(μ4-O)(μ-OR′)8} (Fig. 2). Of interest is the fact that the H atom of one of the acidic –OH groups of each LH− ligand is clearly located on the imine nitrogen atom (N2 and N6), thus blocking the coordination of the latter.
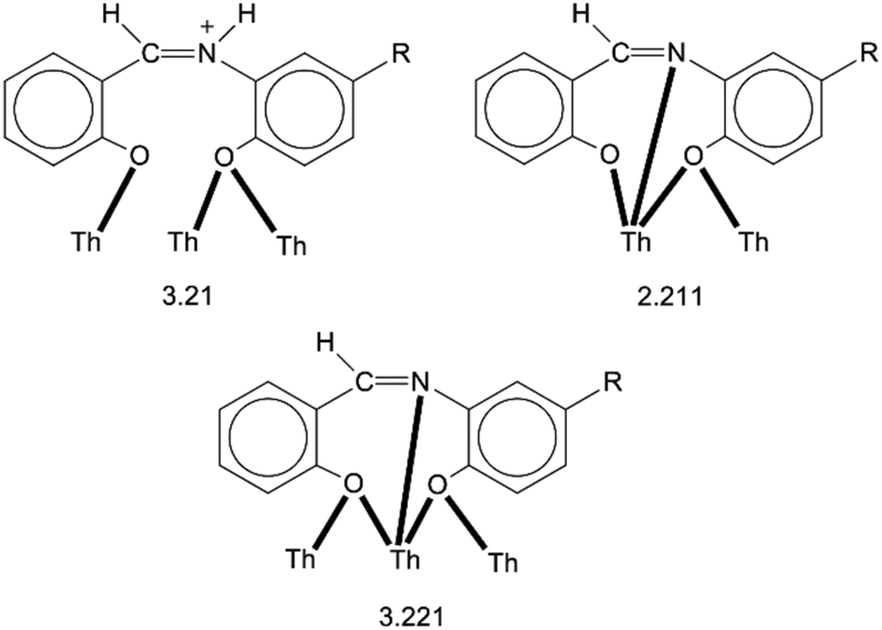 | ||
| Scheme 2 The crystallographically established coordination modes of LH− (R = H), L2− (R = H), L′H− (R = Me) and (L′)2− (R = Me) in complexes 1·4MeCN and 2·2.4MeCN, and the Harris notation that describes these modes. | ||
The ThIV⋯ThIV separations range from 3.776(1) to 4.039(1) Å. The Th1⋯Th2 [3.776(1) Å] and Th3⋯Th4 [3.816(1) Å] distances are shorter than the other four ThIV⋯ThIV distances [3.935(1)–4.039(1) Å], because the Th1/Th2 and Th3/Th4 pairs are linked by three monoatomic O bridges (O1, O4, and O7 for the Th1/Th2 pair; O1, O11, and O12 for the Th3/Th4 pairs), whereas the Th1/Th3, Th1/Th4, Th2/Th3 and Th2/Th4 pairs are each connected by two monoatomic O bridges (O1 and one phenoxido O atom). The ThIV–(μ4-O2−)–ThIV angles are in the 103.7(1)–112.6(1)° range, in agreement with the distorted tetrahedral arrangement of the metal centres. Th1 is 8 coordinate; it is coordinated by seven oxygen atoms, with the Th1–O bond lengths ranging from 2.205(3) to 2.486(3) Å and one nitrogen atom. Two oxygen atoms are terminal from one LH− (O13) and one L2− (O3) ligands, four are bridging from a LH− (O4) and three L2− (O2, O7, and O15) ligands and the seventh oxygen atom is the oxido group. Th2 is 9-coordinate; it is surrounded by eight oxygen atoms, with the Th2–O bond lengths ranging from 2.235(3) to 2.612(3) Å, and by one nitrogen atom. One oxygen atom is bridging from a LH− (O4) ligand, four oxygen atoms (one terminal, O6; three bridging, O7/O8/O10) belong to three L2− ligands, two oxygen atoms are from a chelating nitrato group (O16 and O18) and the eighth oxygen is the oxido group. Th3 is 8-coordinate; it is bonded to seven oxygen atoms, with the Th3–O bond length ranging from 2.228(3) to 2.574(3) Å, and to one nitrogen atom. Two oxygen atoms are terminal from one LH− (O5) and one L2− (O9) ligands, four are bridging from a LH− (O12) and three L2− (O2, O8, and O11) ligands and the seventh oxygen atom is the oxido group. Finally, Th4 is surrounded by eight oxygen and two nitrogen atoms being 10-coordinate. The Th4–O bond lengths range from 2.213(3) to 2.652(3) Å. One oxygen atom is bridging from a LH− (O12) ligand, four oxygen atoms (one terminal, O14; three bridging, O10/O11/O15) belong to two L2− ligands, two oxygen atoms are from a chelating nitrato group (O19 and O20) and the eighth oxygen atom is the oxido group. The Th–N bond distances are in the range of 2.601(4)–2.741(4) Å and are typical for 8-, 9- and 10-coordinate species with Schiff bases as ligands.40–45
To estimate the closer coordination polyhedra defined by the donor atoms around the ThIV centres in 1·4MeCN, a comparison of the experimental structural data with the theoretical values for the most common polyhedral shapes with 8, 9 and 10 vertices was performed using the SHAPE program.66 The best fit was obtained for the triangular dodecahedron (Th1), the muffin (Th2), the biaugmented trigonal prism (Th3) and sphenocorona (Th4) (Fig. 3 and Tables S3–S5‡).
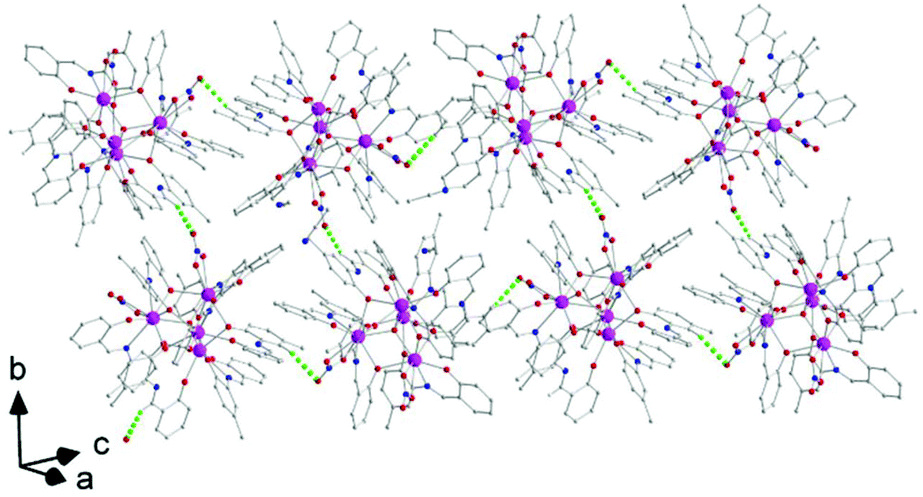
There are two classical, relatively strong, intraligand H bonds within the tetranuclear molecule (Fig. S1, Table S1‡), with the protonated nitrogen atoms (N2 and N6) of the LH− ligands as donors and their terminal, formerly salicylaldehyde, phenolato oxygen atoms (O5 and O13) as acceptors. A variety of intermolecular H-bonding interactions are present in the crystal structure. Three lattice MeCN molecules, three coordinated nitrato oxygen atoms (O16, O19, and O20) and one non-coordinated nitrato oxygen atom (O21) act as acceptors, while the donors are aromatic and aliphatic carbon atoms from the ligands and MeCN. The overall result is the formation of chains parallel to the α axis (Fig. 4).
Molecule 2 has a very similar structure (Fig. 5 and S2‡) to that of 1 (Fig. 1). The core, the metal topology, the coordination polyhedra (Fig. S3, Tables S3–S5‡) and the coordination modes of the organic and nitrato ligands are almost identical in the two structures. In the numbering schemes of the metal centres, Th1, Th2, Th3 and Th4 in 2 are equivalent to Th4, Th2, Th3 and Th1, respectively, in 1. The ThIV⋯ThIV distances and the ThIV–(μ4-O2−)–ThIV angles are in the ranges 3.763(1)–4.029(1) Å [3.776(1)–4.039(1) Å in 1·4MeCN] and 102.9(1)–113.7(1)° [103.7(1)–112.6(1)° in 1·4MeCN], respectively. At the supramolecular level, the tetranuclear [Th4O(NO3)2(L′H)2(L′)5] molecules are connected through H bonds forming layers parallel to the (−101) plane (Fig. 6). The acceptors are the non-coordinated oxygen atoms O9 and O12 of the nitrato groups, and the donors are the imine carbon atom one L′H− ligand and one aromatic carbon atom of the iminium phenolato part of the other L′H− ligand.
Complexes 1·4MeCN and 2·2.4MeCN are the third and fourth, respectively, tetranuclear thorium(IV) complexes with the tetrahedral {ThIV4(μ4-O)} unit. The previously structurally characterized examples are complexes [Th4OCl8(EO4)3],67 where EO4 is the tetraethylene glycolate(−2) ligand, and [Th4OCl2I6{O(CH2)2OCH3}6].68 Interestingly, the authors note that the oxido bridge in the former complex is the result of an oxide impurity from the ThCl4 starting material.67 In the latter complex, the source of the central μ4-O2− unit was verified to be68 dimethoxyethane (DME) present in the starting [Th4Cl4−xIx(DME)2] material, and not adventitious H2O (the reaction and the manipulation were performed in a strictly inert atmosphere (N2) under vacuum). In the case of 1 and 2, the source of the oxido group seems to be H2O from the solvent (MeCN) and the hydrated thorium(IV) nitrate. Such μ4-O2− groups are extremely rare in An(IV) cluster chemistry despite their occurrence in the fluorite-type structures.8 Generally, the tetravalent actinides Th–Pu represent some of the hardest cations of the Periodic table; because of their high charge density and acidity, these metal ions are particularly prone to hydrolysis and condensation.9,18
Compounds 1 and 2 are new members of a large family of complexes of 3d-, 4f- and mixed 3d/4f-metal ions (ref. 47–54 are only representative) containing anionic forms of LH2 and L′H2. However, they join a handful of structurally characterized An complexes which are listed in Table 4. Unfortunately, no U(IV), Np(IV) and Pu(IV) complexes with any forms of LH2 and L′H2 have been reported, so no direct comparison of An complexes with the metal in the IV oxidation state are possible. The previous An examples were mononuclear (neutral69 and anionic34,70) and dinuclear55 uranyl (UO22+) complexes. The completely different structures between the uranyl and thorium(IV) complexes (Table 4) are attributed to the completely different chemical nature of the two metal species. In a general sense, as compared to the hexavalent actinides, which almost invariably form the well-established actinyl cation (AnVIO22+), the tetravalent actinides adopt more spherical or isotropic coordination geometries, thus leading to structural units and types that are quite distinct from those of the hexavalent An ions.18 Restricting this discussion to Th(IV) and UVIO22+, Th(IV) exhibits an effective charge that matches its formal oxidation state, whereas U(VI), in the form of the uranyl species, behaves as a metal ion with an effective charge of +3.3.10
| Complexa | Coordination mode of the ligand | Ref. |
|---|---|---|
| a The lattice solvent molecules have been omitted. b A similar complex [(UO2)2(L′′)2(EtOH)2], where (L′′)2− is the dianion of N-salicylidene-o-amino-4-chlorophenol (L′′H2; R = Cl in Scheme 1), has also been structurally characterized.55 c For the monoanionic ligands. d For the dianionic ligands. | ||
| [UO2(L)2(DMSO)2] | 1.111 | 69 |
| (Bun4N)2[UO2(L)(H2PO4)2] | 1.111 | 70 |
| [(UO2)2(L′)2(EτOH)2]b | 2.211 | 55 |
| [Th4O(NO3)2(LH)2(L)5] | 3.21,c 2.211,d 3.221d | This work |
| [Th4O(NO3)2(L′H)2(L′)5] | 3.21,c 2.211,d 3.221d | |
A final interesting point is the fact that the 3.210 ligation mode of LH− and L′H− established in the structures of 1·4MeCN and 2·2.4MeCN, respectively, is observed for the first time in the coordination chemistry of LH2 and L′H2.
Spectroscopic studies
In the IR spectra of 1 and 2 (Fig. S4 and S5‡), the medium-intensity and broad band at ∼3450 cm−1 is attributed to the ν(NH+) vibration of the LH− or L′H− ligands (vide supra);71 the broadness of the band is indicative of H bonding. The spectrum of 1 exhibits a medium-intensity and a strong band at 1622 and 1604 cm−1, respectively, which is assigned46,55,71 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration, ν(CN), of the Schiff base linkage. These bands have been shifted to lower wavenumbers compared with the corresponding band in the spectrum of the free LH2 ligand (1630 cm−1, Fig. S6‡), in agreement with the protonation or coordination of the imine nitrogen. The appearance of the two bands reflects the presence of two types (protonated and coordinated) of CN groups in the complex. The two bands appear at 1628 and 1602 cm−1 in the spectrum of 2. The band at 1602 cm−1 has been shifted to a lower wavenumber compared with the band in the spectrum of the free L′H2 ligand (1628 cm−1, Fig. S7‡), whereas the band at 1628 cm−1 has remained at the same wavenumber. The latter experimental fact is not unusual.72,73 Extensive experimental and theoretical studies on complexes with Schiff bases containing a CN bond (with the carbon atom attached to an aromatic ring) have revealed that a change in the s character of the N lone pair occurs upon coordination or protonation and the s character of the N orbital involved in the CN bond increases; this change in hybridization leads to a greater CN stretching constant relative to the free neutral ligands, thus shifting the ν(CN) vibrational mode to slightly higher wavenumbers. The IR bands at ∼1470, ∼1270 and ∼1030 cm−1 in the spectra of both complexes are assigned74 to the ν1(A1)[ν(NO)], ν5(B2)[νas(NO2)] and ν2(A1)[νs(NO2)] vibrations of the bidentate chelating (C2v) nitrato group; the separation of the two highest-wavenumber stretching bands is ∼200 cm−1, in agreement with the bidentate character of the coordinated nitrato groups.52,74
N stretching vibration, ν(CN), of the Schiff base linkage. These bands have been shifted to lower wavenumbers compared with the corresponding band in the spectrum of the free LH2 ligand (1630 cm−1, Fig. S6‡), in agreement with the protonation or coordination of the imine nitrogen. The appearance of the two bands reflects the presence of two types (protonated and coordinated) of CN groups in the complex. The two bands appear at 1628 and 1602 cm−1 in the spectrum of 2. The band at 1602 cm−1 has been shifted to a lower wavenumber compared with the band in the spectrum of the free L′H2 ligand (1628 cm−1, Fig. S7‡), whereas the band at 1628 cm−1 has remained at the same wavenumber. The latter experimental fact is not unusual.72,73 Extensive experimental and theoretical studies on complexes with Schiff bases containing a CN bond (with the carbon atom attached to an aromatic ring) have revealed that a change in the s character of the N lone pair occurs upon coordination or protonation and the s character of the N orbital involved in the CN bond increases; this change in hybridization leads to a greater CN stretching constant relative to the free neutral ligands, thus shifting the ν(CN) vibrational mode to slightly higher wavenumbers. The IR bands at ∼1470, ∼1270 and ∼1030 cm−1 in the spectra of both complexes are assigned74 to the ν1(A1)[ν(NO)], ν5(B2)[νas(NO2)] and ν2(A1)[νs(NO2)] vibrations of the bidentate chelating (C2v) nitrato group; the separation of the two highest-wavenumber stretching bands is ∼200 cm−1, in agreement with the bidentate character of the coordinated nitrato groups.52,74
The Raman spectra of the well dried samples of complexes 2 and 1 are shown in Fig. 7 and S8,‡ respectively. The characteristic ν(Caromatic–H) peak appears at ∼3050 cm−1 in both spectra;11,46,72,75,76 extra peaks at 2918 and 2860 cm−1 in the spectrum of 2 are due to the νas(CH3) and νs(CH3) modes, respectively.46,75 The peak at ∼1620 cm−1 in both spectra is due to the ν(CN) mode of the Schiff-base linkage;46,76 the clear splitting of this peak reflects the presence of two types of imine linkages (protonated and coordinated) in the tetranuclear complexes. The two highest-frequency stretching nitrogen–oxygen vibrations of the bidentate chelating nitrato groups are located74,77 at 1477 and 1266 cm−1 for 1, and at 1500 and 1284 cm−1 for 2.
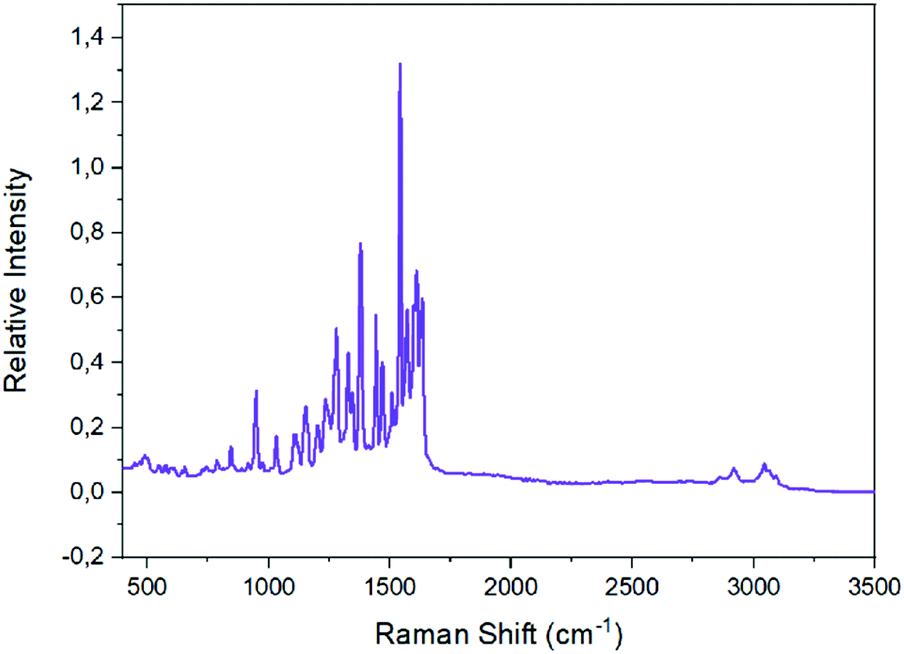 | ||
| Fig. 7 The Raman spectrum of 2. | ||
The 1H NMR spectra of 1 and 2 in DMSO-d6 (Fig. S9 and S10‡) are extremely complicated, suggesting the presence of several different species in solution and indicating that the structures of the complexes are not retained in solution. For example, the spectrum of 2 shows four signals between δ 2.00 and 2.30 ppm, with different integration ratios, attributed11,46 to the protons of the methyl groups. The imine protons appear around δ 9 ppm in the form of multiple signals.43 Of particular interest is the appearance of signals at δ values in the region 14.08–13.76 ppm and at ∼9.7 ppm. The former can be assigned to the very acidic protons of –CNH+ groups from the LH− and L′H− ligands; the latter most probably indicates the presence of –OH groups arising from protonation of the deprotonated phenolato oxygen atoms, the most possible proton sources being some of the –CNH+ groups and/or hydrolysis of [Th(H2O)x]4+ species; a strong evidence of hydrolytic processes comes from the appearance of signals in the δ 4.8–6.2 ppm region assigned11 to coordinated OH− groups. Our proposal for the decomposition of the complexes in solution is reinforced by the molar conductivity values, ΛM (10−3 M, 25 °C), in DMSO, which are 86 (1) and 69 (2) S cm2 mol−1; these values indicate the presence of ionic species in this solvent.78
Solid-state, room-temperature emission spectra of the free ligands LH2 and L′H2 (Fig. S11 and S12,‡ respectively), and complexes 1 and 2 (Fig. 8 and S13‡) were recorded. The optical behavior of the two complexes is similar. Upon maximum excitation at 400 nm, the solid complexes show a rather sharp emission band with a maximum at ∼ 540 nm (emission of green light). As Th(IV) is non-emissive, the emission can be assigned to a charge transfer state within the coordinated organic ligands.44,79–84 Tetravalent thorium has a 5f0 configuration. All of the electrons are spin paired in this electronic state and emission is not expected.81 It is also hardly oxidizing and consequently low-energy metal-centered charge-transfer excited states do not exist.80 Additional evidence for the intraligand character of the green emission in the complexes comes from the study of the emission properties of the free ligands. Upon maximum excitation at 497 nm (LH2) or 465 nm (L′H2), the ligands emit at 540 nm (LH2) and 610 nm (L′H2). The development of “turn-on” or “turn-off” fluorescent sensors for Th(IV) is an area of intense interest due to their potential use for thorium analysis/identification in nuclear waste investigation.44,82–84
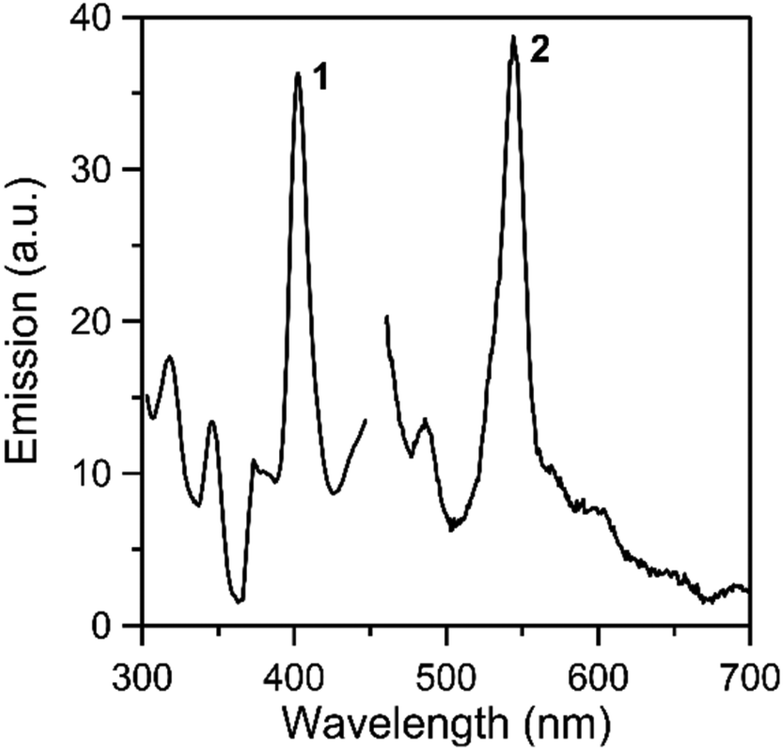 | ||
| Fig. 8 Solid-state, room-temperature excitation (curve 1; maximum emission at 540 nm) and emission (curve 2; maximum excitation at 400 nm) spectra of complex 1. | ||
Conclusions and perspectives
Even at the time of submission of the present work, thorium chemistry continues to surprise the inorganic chemistry community.85–91 For example, the number of the crystallographically characterised Th(III) complexes (mainly with cyclopentadienyl ancillary ligands) increases and the first example of a square planar thorium complex has been just reported.89 Many scientists believe that with the harmful effects of global climate change becoming more evident with every passing year, more research activity and allocated resources will help the Z = 90 element realize its great potential and become a truly valuable source of materials in our energy economy.5As far as the present work is concerned, it is rather difficult to conclude on a research topic, i.e., Th(IV) complexes with Schiff-base ligands, which is still at its infancy. In this report, we believe that we have contributed to some extent into the chemistry of thorium(IV) clusters, and into the coordination chemistry of LH2 and L′H2 and related tridentate Schiff bases. Complexes 1 and 2 have interesting molecular structures and contain the extremely rare {ThIV4(μ4-O)}14+ core. They also join a handful of fluorescent Th(IV) complexes. The observed ligand-based green emission might indicate that LH2 and L′H2 and related tridentate Schiff bases containing naptholate (instead of phenolate) aromatic rings, can be considered to be fluorescent sensors for toxic Th(IV) analysis. The development of such a fluorescence detection in solution might provide a simple, rapid, selective and low-cost method for thorium ion determination. Several fluorogenic optodes for Th(IV), with a variety of organic molecules as sensing materials, have been developed in the last 10 years or so. However, some limitations such as a poor sensitivity, a limited working pH range and minor or major interference from other metal ions (e.g. Al3+, UO22+, Ti4+, Zr4+ and trivalent lanthanides), due to their similar chemical properties, need to be addressed. Very recently Kumar and Kumar designed, synthesized, fabricated and used an excellent 8-aminoquinoline-based fluorescent optode for the quantification of Th4+ in various aqueous, monazite sand and gas mantle samples.92 In the present case, the ligand L′H2 could be, in principle, used as the sensor of Th(IV) in the presence of UO22+, since the complex of the latter with (L′)2− does not emit light at room temperature under several excitation wavelengths.55
Future work will be focused on the study of the coordination chemistry of tetradentate Schiff bases containing one –OH and one –COOH group (instead of two –OH groups) on different aromatic rings towards Th(IV) and the investigation of the emission properties of Th(IV) complexes in solution. Work is also in progress to develop the Th(IV)-oxime/oximate chemistry (oximes also contain a CN group) and synthesize the almost elusive thorium η2-(O,N)-oximate complexes.93 These efforts, already well advanced, will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Research Director George Voyiatzis (ICE-HT/FORTH) for helpful discussion concerning the Raman spectra of the complexes and Dr Zoi G. Lada for recording these spectra.Notes and references
- N. R. Andreychuk, T. Dickie, D. J. H. Emslie and H. A. Jenkins, Dalton Trans., 2018, 47, 4866–4876 RSC.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999, pp. 1130–1164 Search PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Harlow, UK, 5th edn, 2018, pp. 1033–1064 Search PubMed.
- M. S. Wickleder, B. Fourest and P. K. Dorhout, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, The Netherlands, 4th edn, 2010, vol. 1, ch. 3, pp. 52–133 Search PubMed.
- J. Arnold, T. L. Gianetti and Y. Kashtan, Nat. Chem., 2014, 6, 554 CrossRef CAS.
- L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Coord. Chem. Rev., 2014, 266–267, 171–193 CrossRef CAS.
- C. D. Tutson and A. E. V. Gorden, Coord. Chem. Rev., 2017, 333, 27–43 CrossRef CAS.
- J. Qiu and P. C. Burns, Chem. Rev., 2013, 113, 1097–1120 CrossRef CAS.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef CAS.
- P. O. Adelani and T. E. Albrecht-Schmitt, Inorg. Chem., 2010, 49, 5701–5705 CrossRef CAS.
- M. Vasiliu, K. E. Knope, L. Soderholm and D. A. Dixon, J. Phys. Chem. A, 2012, 116, 6917–6926 CrossRef CAS.
- E. P. Wildman, G. Balázs, A. J. Wooles, M. Scheer and S. T. Liddle, Nat. Commun., 2016, 7, 12884 CrossRef CAS.
- P. K. Mohapatra, Dalton Trans., 2017, 46, 1730–1747 RSC (perspective).
- S. S. Galley, C. E. Van Alstine, L. Maron and T. E. Albrecht-Schmitt, Inorg. Chem., 2017, 56, 12692–12694 CrossRef CAS.
- C. Falaise, K. Kozma and M. Nyman, Chem. – Eur. J., 2018, 24, 14226–14232 CrossRef CAS.
- Z. Wang, A. T. Brown, K. Tan, Y. J. Chabal and K. J. Balkus Jr., J. Am. Chem. Soc., 2018, 140, 14735–14739 CrossRef.
- A. McSkimming, J. Su, T. Cheisson, M. R. Gau, P. J. Carroll, E. R. Batista, P. Yang and E. J. Schelter, Inorg. Chem., 2018, 57, 4387–4394 CrossRef CAS.
- N. A. Vanagas, J. N. Wacker, C. L. Rom, E. N. Glass, I. Colliard, Y. Qiao, J. A. Bertke, E. Van Keuren, E. J. Schelter, M. Nyman and K. E. Knope, Inorg. Chem., 2018, 57, 7259–7269 CrossRef CAS.
- P. Li, S. Goswami, K.-i. Otake, X. Wang, Z. Chen, S. L. Hanna and O. K. Farha, Inorg. Chem., 2019, 58, 3586–3590 CrossRef CAS.
- O. A. Ejegbavwo, C. R. Martin, O. A. Olorunfemi, G. A. Leith, R. T. Ly, A. M. Rice, E. A. Dolgopolova, M. D. Smith, S. G. Karakalos, N. Birkner, B. A. Powell, S. Pandey, R. J. Koch, S. T. Misture, H.-C. zur Loye, S. R. Phillpot, K. S. Brinkman and N. B. Shustova, J. Am. Chem. Soc., 2019, 141, 11628–11640 CrossRef CAS.
- M. Nakase, T. Yamamura, K. Shirasaki, M. Nagai and K. Takeshita, Sep. Sci. Technol., 2019, 54, 1952–1959 CrossRef CAS.
- M. Lung and O. Gremm, Nucl. Eng. Des., 1998, 180, 133–146 CrossRef CAS.
- M. Follows, Educ. Chem., Is thorium the perfect fuel? | Feature | Education in Chemistry, https://eic.rsc.org/feature/is-thorium-the-perfect-fuel/2000092.article, (accessed 26 July 2019).
- Y. Dong, S. Li, X. Su, Y. Wang, Y. Shen and X. Sun, Hydrometallurgy, 2017, 171, 387–393 CrossRef CAS.
- Z. Zhu, Y. Pranolo and C. Y. Cheng, Miner. Eng., 2015, 77, 185–196 CrossRef CAS.
- L. Salmon, P. Thuéry, E. Rivière and M. Ephritikhine, Inorg. Chem., 2006, 45, 83–93 CrossRef CAS.
- T. Tidwell, Angew. Chem., Int. Ed., 2008, 47, 1016–1020 CrossRef CAS.
- J. Costamagna, J. Vargas, R. Latorre, A. Alvarado and G. Mena, Coord. Chem. Rev., 1992, 119, 67–88 CrossRef CAS.
- S. Yamada, Coord. Chem. Rev., 1999, 190–192, 537–555 CrossRef CAS.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- D. M. Rudkevich, W. Verboom, Z. Brzozka, M. J. Palys, W. P. R. V. Stauthamer, G. J. van Hummel, S. M. Franken, S. Harkema, J. F. J. Engbersen and D. N. Reinhoudt, J. Am. Chem. Soc., 1994, 116, 4341–4351 CrossRef CAS.
- J. L. Sessler, T. D. Mody and V. Lynch, Inorg. Chem., 1992, 31, 529–531 CrossRef CAS.
- P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206–8208 CrossRef CAS.
- M. S. Bharara, S. A. Tonks and A. E. V. Gorden, Chem. Commun., 2007, 4006–4008 RSC.
- K. Takao, M. Kato, S. Takao, A. Nagasawa, G. Bernhard, C. Hennig and Y. Ikeda, Inorg. Chem., 2010, 49, 2349–2359 CrossRef CAS.
- C. A. Hawkins, C. G. Bustillos, R. Copping, I. May and M. Nilsson, Actinide and Fission Product Partitioning and Transmutation, NEA/NSC/R, 2015, vol. 2, pp. 315–323 Search PubMed.
- For example, see: A. Bino and R. Chayat, Inorg. Chim. Acta, 1987, 129, 273–276 CrossRef CAS.
- For example, see: A. N. Dame, M. S. Bharara, C. L. Barnes and J. R. Walensky, Eur. J. Inorg. Chem., 2015, 2996–3005 CrossRef CAS.
- For example, see: B. E. Klamm, C. J. Windorff, C. Celis-Barros, M. L. Marsh, D. S. Meeker and T. E. Albrecht-Schmitt, Inorg. Chem., 2018, 57, 15389–15398 CrossRef CAS.
- R. Selva Kumar, S. K. Ashok Kumar, K. Vijayakrishna, A. Sivaramakrishna, C. V. S. Brahmmananda Rao, N. Sivaraman and S. K. Sahoo, Inorg. Chem., 2018, 57, 15270–15279 CrossRef.
- E. E. Hardy, K. M. Wyss, J. D. Gorden, I. R. Ariyarathna, E. Miliordos and A. E. V. Gorden, Chem. Commun., 2017, 53, 11984–11987 RSC.
- D. F. Back, É. Bonfada, G. M. de Oliveira and E. S. Lang, J. Inorg. Biochem., 2007, 101, 709–714 CrossRef CAS.
- K. I. Alexopoulou, E. Zagoraiou, T. F. Zafiropoulos, C. P. Raptopoulou, V. Psycharis, A. Terzis and S. P. Perlepes, Spectrochim. Acta, Part A, 2015, 136, 122–130 CrossRef CAS.
- A. Kagkelari, G. S. Papaefstathiou, C. P. Raptopoulou and T. F. Zafiropoulos, Polyhedron, 2009, 28, 3279–3283 CrossRef CAS.
- K. I. Alexopoulou, A. Terzis, C. P. Raptopoulou, V. Psycharis, A. Escuer and S. P. Perlepes, Inorg. Chem., 2015, 54, 5615–5617 CrossRef CAS.
- P. S. Perlepe, A. A. Athanasopoulou, K. I. Alexopoulou, C. P. Raptopoulou, V. Psycharis, A. Escuer, S. P. Perlepes and T. C. Stamatatos, Dalton Trans., 2014, 43, 16605–16609 RSC.
- For example, see: S. Dutta, P. Basu and A. Chakravorty, Inorg. Chem., 1991, 30, 4031–4037 CrossRef CAS.
- For example, see: R. Herchel, I. Nemec, M. Machata and Z. Trávniček, Dalton Trans., 2016, 45, 18622–18634 RSC.
- N. C. Anastasiadis, D. A. Kalofolias, A. Philippidis, S. Tzani, C. P. Raptopoulou, V. Psycharis, C. J. Milios, A. Escuer and S. P. Perlepes, Dalton Trans., 2015, 44, 10200–10209 RSC.
- E. C. Mazarakioti, L. Cunha-Silva, V. Bekiari, A. Escuer and T. C. Stamatatos, RSC Adv., 2015, 5, 92534–92538 RSC.
- D. I. Alexandropoulos, T. N. Nguyen, L. Cunha-Silva, T. F. Zafiropoulos, A. Escuer, G. Christou and T. C. Stamatatos, Inorg. Chem., 2013, 52, 1179–1181 CrossRef CAS.
- S. T. Tsantis, V. Bekiari, C. P. Raptopoulou, D. I. Tzimopoulos, V. Psycharis and S. P. Perlepes, Polyhedron, 2018, 152, 172–178 CrossRef CAS.
- S. T. Tsantis, M. Mouzakitis, A. Savvidou, C. P. Raptopoulou, V. Psycharis and S. P. Perlepes, Inorg. Chem. Commun., 2015, 59, 57–60 CrossRef CAS.
- S. T. Tsantis, E. Zagoraiou, A. Savvidou, C. P. Raptopoulou, V. Psycharis, L. Szyrwiel, M. Hołyńska and S. P. Perlepes, Dalton Trans., 2016, 45, 9307–9319 RSC.
- E. Labisbal, L. Rodríguez, A. Vizoso, M. Alonso, J. Romero, J.-A. García-Vázquez, A. Sousa-Pedrares and A. Sousa, Z. Anorg. Allg. Chem., 2005, 631, 2107–2114 CrossRef CAS.
- Y. Elerman, A. Elmali, O. Atakol and I. Svoboda, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 2344–2346 CrossRef.
- D. Gürbüz, A. Cinarli, A. Tavman and A. S. Birteksz, Chin. J. Chem., 2012, 30, 970–978 CrossRef.
- CrystalClear, Rigaku/MSC Inc., The Woodlands, TX, USA, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- DIAMOND: Crystal and Molecular Structure Visualization, Ver. 3.1, Crystal Impact GbR, Bonn, Germany, 2011 Search PubMed.
- R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349–2356 RSC.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE, version 2.0, Barcelona, Spain, 2010 Search PubMed.
- R. D. Rogers, A. H. Bond and M. M. Witt, Inorg. Chim. Acta, 1991, 182, 9–17 CrossRef CAS.
- N. E. Travia, B. L. Scott and J. L. Kiplinger, Chem. – Eur. J., 2014, 20, 16846–16852 CrossRef CAS.
- R. Graziani, U. Casellato, P. A. Vigato, O. A. Rajan and A. Chakravorty, Inorg. Chim. Acta, 1981, 49, 129–134 CrossRef CAS.
- S. T. Tsantis, C. P. Raptopoulou, D. I. Tzimopoulos, V. Psycharis and S. P. Perlepes, manuscript in preparation.
- D. Maniaki, I. Mylonas-Margaritis, J. Mayans, A. Savvidou, C. P. Raptopoulou, V. Bekiari, V. Psycharis, A. Escuer and S. P. Perlepes, Dalton Trans., 2018, 47, 11859–11872 RSC.
- E. C. Mazarakioti, A. S. Beobide, V. Angelidou, C. G. Efthymiou, A. Terzis, V. Psycharis, G. A. Voyiatzis and S. P. Perlepes, Molecules, 2019, 24, 2219 CrossRef.
- J. J. Lopez-Garriga, G. T. Babcock and J. F. Harrison, J. Am. Chem. Soc., 1986, 108, 7241–7251 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 4th edn, 1986, pp. 254–257 Search PubMed.
- Z. G. Lada, A. S. Beobide, A. Savvidou, C. P. Raptopoulou, V. Psycharis, G. A. Voyiatzis, M. M. Turnbull and S. P. Perlepes, Dalton Trans., 2017, 46, 260–274 RSC.
- F. R. Dollish, W. G. Fateley and F. F. Bentley, Characteristic Raman Frequencies of Organic Compounds, Wiley, New York, 1974, pp. 1–11, 134–142, 162–190 Search PubMed.
- W. G. Fateley, F. R. Dollish, N. T. McDerrit and F. F. Bentley, Infrared and Raman Selection Rules for Molecular and Lattice Vibrations: The Corellation Method, Wiley, New York, 1972, pp. 1–51 Search PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- G. Knör and A. Strasser, Inorg. Chem. Commun., 2002, 5, 993–995 CrossRef.
- H. Kunkely and A. Vogler, Chem. Phys. Lett., 1999, 304, 187–190 CrossRef CAS.
- B. A. Maynard, R. E. Sykora, J. T. Mague and A. E. V. Gorden, Chem. Commun., 2010, 46, 4944–4946 RSC.
- J. Wen, L. Dong, J. Tian, T. Jiang, Y.-Q. Yang, Z. Huang, X.-Q. Yu, C.-W. Hu, S. Hu, T.-Z. Yang and X.-L. Wang, J. Hazard. Mater., 2013, 263, 638–642 CrossRef CAS.
- R. Gao, J. Hu, K. Zhang, Y. He, P. Liu, S. Luo, Y. Yang, L. Yang, W. Feng and L. Yuan, Chin. J. Chem., 2013, 31, 689–694 CrossRef CAS.
- J. Wen, L. Dong, S. Hu, W. Li, S. Li and X. Wang, Chem. – Asian J., 2016, 11, 49–53 CrossRef CAS.
- J. Du, D. M. King, L. Chatelain, E. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- P. Farger, B. Haidon, P. Roussel, B. Arab-Chapelet and M. Rivenet, Inorg. Chem., 2019, 58, 1267–1277 CrossRef CAS PubMed.
- T. Cheisson, K. D. Kersey, N. Mahieu, A. McSkimming, M. R. Gau, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2019, 141, 9185–9190 CrossRef PubMed.
- C. Zhang, G. Hou, G. Zi and M. D. Walter, Dalton Trans., 2019, 48, 2377–2387 RSC.
- D. N. Huh, S. Roy, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2019, 141, 12458–12463 CrossRef CAS.
- J. N. Wacker, M. Vasiliu, I. Colliard, R. L. Ayscue, III, S. Y. Han, J. A. Bertke, M. Nyman, D. A. Dixon and K. E. Knope, Inorg. Chem., 2019, 58, 10871–10882 CrossRef CAS PubMed.
- C. J. Inman and F. G. N. Cloke, Dalton Trans., 2019, 48, 10782–10784 RSC.
- R. Selva Kumar and S. K. Ashok Kumar, Dalton Trans., 2019, 48, 12607–12614 RSC , and references cited therein.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2005, 2591–2593 RSC.
Footnotes |
| † Dedicated to Professor Annie K. Powell on the occasion of her 60th birthday: a great scientist, an excellent mentor for the Patras group, a fantastic personality and a precious friend. |
| ‡ Electronic supplementary information (ESI) available: Structural plots and spectroscopic data. Cifs and checkcifs for the two complexes. CCDC 1945475 and 1945476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03189h |
| This journal is © The Royal Society of Chemistry 2019 |