
Regioselective [2 + 2] cycloaddition reaction within a pair of polymorphic co-crystals based upon halogen bonding interactions†
Eric
Bosch
 *a,
Samantha J.
Kruse
b,
Eric W.
Reinheimer
c,
Nigam P.
Rath
d and
Ryan H.
Groeneman
*b
*a,
Samantha J.
Kruse
b,
Eric W.
Reinheimer
c,
Nigam P.
Rath
d and
Ryan H.
Groeneman
*b
aDepartment of Chemistry, Missouri State University, 901 South National Ave., Springfield, MO 65897, USA. E-mail: ericbosch@missouristate.edu; Tel: +1 417 836 4277
bDepartment of Biological Sciences, Webster University, 470 East Lockwood Ave., St. Louis, MO 63119, USA. E-mail: ryangroeneman19@webster.edu; Tel: +1 314 246 7466
cRigaku Corporation, 9009 New Trails Dr., The Woodlands, TX 77381, USA
dDepartment of Chemistry and Biochemistry and the Center for Nanoscience, University of Missouri-St. Louis, 1 University Blvd., St. Louis, MO 63121, USA
First published on 14th October 2019
Abstract
The realization of a pair of photoreactive polymorphic co-crystals that are held together by the combination of I⋯N halogen bonding interactions and C–H⋯Cl contacts is reported. The reactant molecule within these co-crystals is based upon an unsymmetrical olefin, namely 4-stilbazole, that results in a regioselective solid-state [2 + 2] cycloaddition reaction in both polymorphic forms. Each solid undergoes a quantitative photoreaction which yields exclusively the head-to-tail photoproduct.
Introduction
Halogen bonding continues to be a reliable and highly investigated non-covalent interaction employed in the formation of numerous molecular solids.1 In particular, halogen bonding continues to be an important interaction in the area of co-crystallization where it has been used to control various physical and chemical properties of these multi-component materials.2 The solid-state photoreactivity, in terms of the [2 + 2] cycloaddition reaction, of halogen-bonded co-crystals is still surprisingly limited in the chemical literature. To the best of our knowledge, all of the reported examples are based on the alignment of a symmetric olefin-containing reactant molecule. For example, Metrangolo and Resnati used a tetratopic halogen-bond donor to align a pair of trans-1,2-bis(4-pyridyl)ethylene3 (4,4-BPE) molecules to undergo a cycloaddition reaction. Similarly, MacGillivray then reported the use of a ditopic halogen-bond acceptor to align a pair of trans-1,2-bis(2,3,5,6-tetrafluoro-4-iodophenyl)ethylene molecules to achieve a photoreaction.4 Lastly, the resulting photoproduct subsequently aligned a pair of 4,4-BPE molecules in a photoreactive configuration.5Recently, we reported the ability to achieve a [2 + 2] cycloaddition reaction within a co-crystal based upon a new halogen-bond donor 1,4-diiodoperchlorobenzene (C6I2Cl4).6 Thus the co-crystal with 4,4-BPE, namely (C6I2Cl4)·(4,4-BPE), underwent a solid-state photoreaction since molecules of C6I2Cl4 π–π stacked in a homogeneous manner along with a face-to-face configuration. As a consequence of this type of stacking and crystal symmetry, a pair of carbon–carbon double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) on neighbouring 4,4-BPE molecules are found at a distance of 4.08 Å which meets the distance criteria for a photoreaction developed by Schmidt.7 The preferred homogeneous π–π stacking pattern for C6I2Cl4 was confirmed by utilizing density functional theory calculations that determined a lower energy for this configuration when compared to a hypothetical heterogeneous stacking pattern of the aromatic rings.
C) on neighbouring 4,4-BPE molecules are found at a distance of 4.08 Å which meets the distance criteria for a photoreaction developed by Schmidt.7 The preferred homogeneous π–π stacking pattern for C6I2Cl4 was confirmed by utilizing density functional theory calculations that determined a lower energy for this configuration when compared to a hypothetical heterogeneous stacking pattern of the aromatic rings.
Using this as inspiration, we report here the ability to form a pair of polymorphic co-crystals based upon C6I2Cl4 with an unsymmetrical reactant molecule, namely 4-stilbazole (4-SB) (Scheme 1). Each polymorph is held together by the combination of I⋯N halogen bonds along with C–H⋯Cl contacts to yield an infinite one-dimensional chain. The different polymorphic forms come about due to the stacking pattern of the C6I2Cl4 molecules in the various solids. In particular, form I has a homogeneous offset π–π stacking arrangement of C6I2Cl4 while form II has a heterogeneous pattern (Scheme 1).
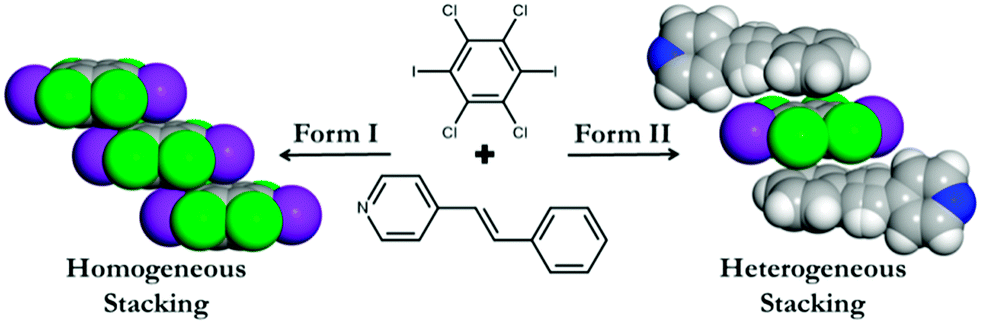 | ||
| Scheme 1 The π–π stacking arrangement within the two polymorphs of (C6I2Cl4)·2(4-SB). | ||
As a consequence of both I⋯N halogen bonds and C–H⋯Cl contacts, each polymorph forms an infinite assembly where nearest neighbouring 4-SB molecules are aligned parallel and in an anti-orientation. The two polymorphs have different separation distance between the pair of CC within the solid, but after exposure to light both polymorphs undergo a solid-state [2 + 2] cycloaddition reaction. The anti-orientation of the pair of 4-SB reactant molecules as well as the resulting 1H NMR of the photoproduct are in agreement that the head-to-tail photoproduct, namely 1,3-bis(4-pyridyl)-2,4-bis(phenyl)cyclobutane (ht-PP), was realized (Scheme 2). This is the first reported regioselective [2 + 2] cycloaddition reaction of an unsymmetrical alkene based upon halogen bonding interactions.
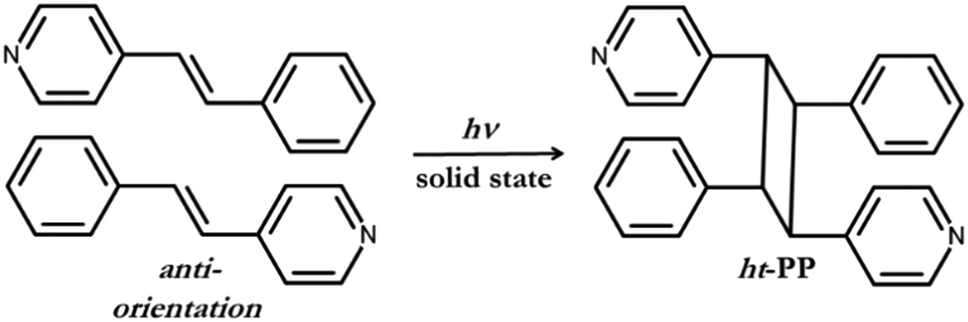 | ||
| Scheme 2 The anti-orientation of 4-SB before photoreaction and the resulting regiochemistry of the head-to-tail photoproduct, namely ht-PP. | ||
Experimental
Materials
4-Stilbazole (4-SB) as well as the solvent toluene were purchased from Sigma-Aldrich Chemical (St. Louis, MO, USA) and used as received. The halogen bond donor 1,4-diiodoperchlorobenzene (C6I2Cl4) was synthesized by a previous reported method.8General methods
Photoreactions were conducted using UV-radiation from a 450 W medium-pressure mercury lamp in an ACE Glass photochemistry cabinet. Each polymorph of (C6I2Cl4)·2(4-SB) were placed between a pair of Pyrex glass plates for irradiation. The photoreactivity and the overall yield for the photoreaction were determined by using 1H NMR Spectroscopy. 1H NMR spectra were collected using a Bruker Avance 400 MHz spectrometer using DMSO-d6 as the solvent. X-ray powder diffraction data was collected at room temperature on a Rigaku Ultima IV X-ray diffractometer using Cu Kα1 radiation (λ = 1.54056 Å) between 5° to 40° two-theta.Synthesis of form I and form II
Co-crystals of (C6I2Cl4)·2(4-SB) in both forms were synthesized by dissolving 25.0 mg of C6I2Cl4 in 2.0 mL of toluene, which was then combined with a separate 2.0 mL toluene solution containing 19.4 mg of 4-SB (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar equivalent). Form I was achieved by removing the cap of the 20 mL scintillation vial while form II was formed by covering the vial with either a folded Kimwipe or placing cotton in the opening. The rate of solvent evaporation is attributed to the formation of the particular polymorph. In both cases, after most of the solvent was evaporated plate-like single crystals suitable for X-ray diffraction were realized ranging from two days for form I and up to three days for form II.
:2 molar equivalent). Form I was achieved by removing the cap of the 20 mL scintillation vial while form II was formed by covering the vial with either a folded Kimwipe or placing cotton in the opening. The rate of solvent evaporation is attributed to the formation of the particular polymorph. In both cases, after most of the solvent was evaporated plate-like single crystals suitable for X-ray diffraction were realized ranging from two days for form I and up to three days for form II.
Single crystal X-ray diffraction data collection
Crystals were mounted on a MiTeGen cryoloop in random orientations for data collection. Data collections were performed using a Bruker Venture Duo Photon-II single crystal X-ray diffractometer equipped with an Oxford Cryostream device. Apex II and SAINT software packages were used for data collection and integration. All of the data were corrected for systematic errors using SADABS based on the Laue symmetry using equivalent reflections. Structure solution and refinement were carried out using the SHELXTL-PLUS software package. All structures were solved by direct methods with a full matrix least-squares refinement. Non-hydrogen atoms were refined anisotropically to convergence. All hydrogen atoms were treated using an appropriate riding model (AFIX m3). Unit cell parameters for both polymorphs and the partial photoproduct co-crystal are found within Table 1. X-ray diffraction and refinement data are also found in Tables S1 and S2.†| Polymorphic form | I | Partial photoproduct | II |
|---|---|---|---|
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
| a/Å | 5.7320(3) | 5.6832(2) | 10.9356(2) |
| b/Å | 11.2839(6) | 11.0865(4) | 16.4835(3) |
| c/Å | 12.3090(7) | 12.2076(5) | 8.6541(2) |
| α/° | 83.794(2) | 82.9822(14) | 90 |
| β/° | 82.313(2) | 82.3948(12) | 96.0154(8) |
| γ/° | 77.034(2) | 77.3857(12) | 90 |
| V/Å3 | 766.35(7) | 740.53(5) | 1551.37(5) |
Results and discussion
Structure and photoreactivity of form I
Single crystal analysis revealed that form I crystallizes in the centrosymmetric triclinic space group P. The asymmetric unit contains a half of C6I2Cl4 and a single molecule of 4-SB. The co-crystal is sustained by I⋯N halogen bonds [I⋯N 2.9047(1) Å] with a C–I⋯N bond angle of 175.0519(2)° at 290 K (Fig. 1). The aromatic rings within 4-SB are nearly coplanar with an angle of 5.96°. In contrast, the angle between C6I2Cl4 and the pyridine ring within 4-SB is found to be 75.29°. The ethylene bridge in 4-SB is found to be disordered over two position which is common in other similar shaped molecules.9 After a free variable refinement of the diffraction data the ratios of the two components were determined to be 0.89/0.11 at 290 K.‡ In order to determine the type of disorder, a multivariable crystallographic study was undertaken. The same crystal was then cooled to 250 K and a second complete data set was collected. Again, only the disorder in the ethylene bridge within 4-SB was able to be modelled. After a free variable refinement the ratios of the ethylene group changed to 0.92/0.08 which is indicative of dynamic motion (i.e. pedal motion)9 within form I. To determine the overall purity of the bulk material a powder X-ray diffraction study was performed on the resulting solid after complete evaporation of the solvent when the vial is completely open. The resulting diffractogram is in good agreement with the calculated powder pattern of form I (Fig. S4†).
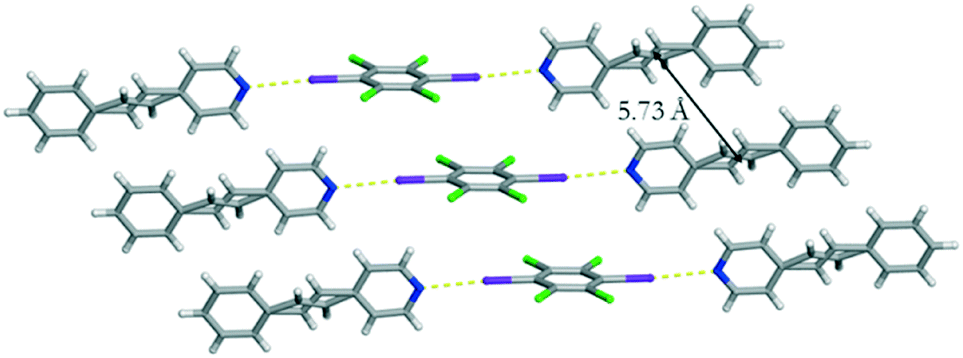 | ||
| Fig. 1 X-ray structure of form I illustrating the I⋯N halogen bonds as well as the offset homogeneous π–π stacking arrangement of C6I2Cl4. The I⋯N halogen bonds are shown with yellow dashed lines. | ||
Unlike our previous photoreactive solid (C6I2Cl4)·(4,4-BPE),6 molecules of C6I2Cl4 within form I are found to be offset with a centroid-to-centroid distance of 5.73 Å. Due to crystal symmetry, a pair of CC are also found to be at a distance of 5.73 Å which is beyond the accepted limit for a photoreaction (Fig. 1). In addition, a pair of 4-SB molecules between nearest neighbouring halogen bonded assemblies is found to be in a syn-orientation.
Beside the I⋯N halogen bonds, there are C–H⋯Cl contacts10 [C⋯Cl 3.8357(15) Å] within form I where the chlorine atoms interact with the para hydrogen atom on the benzene ring producing a one-dimensional chain (Fig. 2). This combination of non-covalent interactions positions a pair of 4-SB molecules in an anti-orientation with the shortest CC separation distance of 4.16 Å, which is within the limit for a photoreaction (Fig. 2). All four chlorine atoms accept C–H⋯Cl contacts, in particular the other crystallographically unique chlorine is interacting with both a α-hydrogen atom on the pyridine ring [C⋯Cl 3.8525(2) Å] as well as the disordered ethylene hydrogen atom [C⋯Cl 3.8574(2) Å] ultimately yielding a three dimensional solid (Fig. 3). Lastly, form I also contains some weak Cl⋯π interactions between the chlorine and the pyridine ring with an atom-to-centroid distance of 3.37 Å.
 | ||
| Fig. 2 X-ray structure of form I illustrating the anti-orientation within the one-dimensional chain. The I⋯N halogen bonds and C–H⋯Cl contacts are shown with yellow dashed lines. | ||
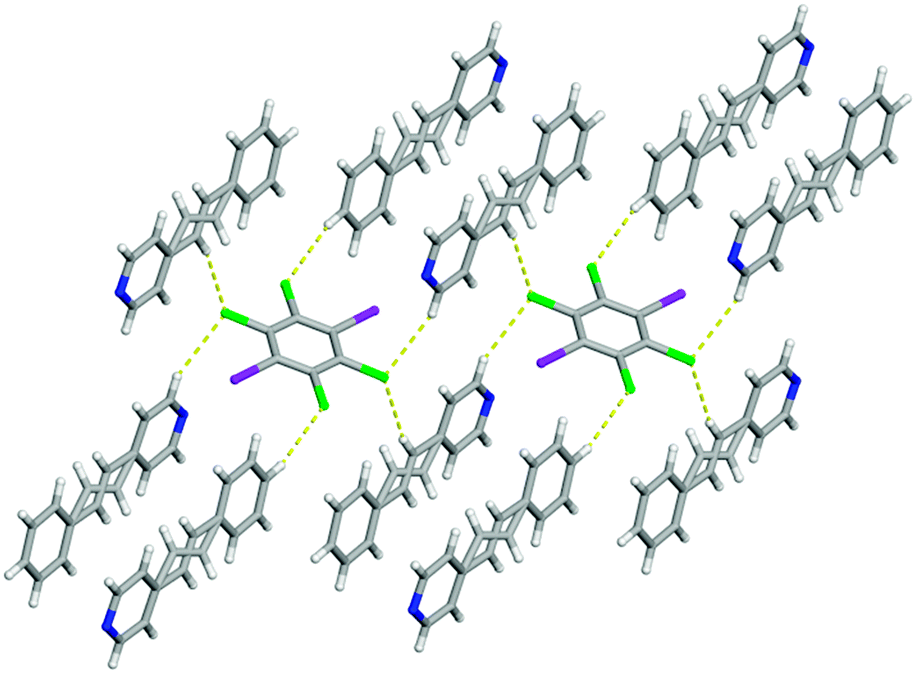 | ||
| Fig. 3 X-ray structure of form I illustrating all of the different types of C–H⋯Cl contacts. The C–H⋯Cl contacts are shown with yellow dashed lines. | ||
To determine if a solid-state photoreaction would occur, a powdered sample of form I was placed between glass plates and put in a photoreactor cabinet to be exposed to broadband UV radiation from a 450 W medium-pressure mercury lamp. A photoreaction was detected as evidenced by the loss of the olefinic peak on 4-SB at 7.57 ppm (Fig. S1†) along with the appearance of the cyclobutane peak at 4.59 ppm (Fig. S2†) in the 1H NMR spectrum. The position and the particular shape of the 1H NMR signal for the cyclobutane ring confirms the photoproduct to be the ht-PP regioisomer.11 The yield for the [2 + 2] cycloaddition reaction was determined to be quantitative after 30 hours of irradiation.
During X-ray data collection, an additional crystal of form I was investigated via single crystal X-ray diffraction. Again, the solid crystallizes in the centrosymmetric triclinic space group P; however, new atoms appeared in the difference map. After refinement, it was determined that the crystal was partially photoreacted forming the ht-PPvia a single-crystal-to-single-crystal reaction12 with an overall yield of 8.3% (Fig. 4). The unit cell parameters are similar to the unreacted form I (Table 1) making these two structures isomorphic in nature. The solid is again held together by the combination of both I⋯N halogen bonds [I⋯N 2.9910(1) Å; C–I⋯N 172.2938(2)°] and C–H⋯Cl contacts [C⋯Cl 3.7864(1) Å] at 100 K. It must be noted that the crystal underwent the [2 + 2] cycloaddition reaction from ambient light and not from UV light in the photoreactor. The partial photoproduct crystal structure is in agreement with the regiochemistry determined by 1H NMR for form I to be the ht-PP.
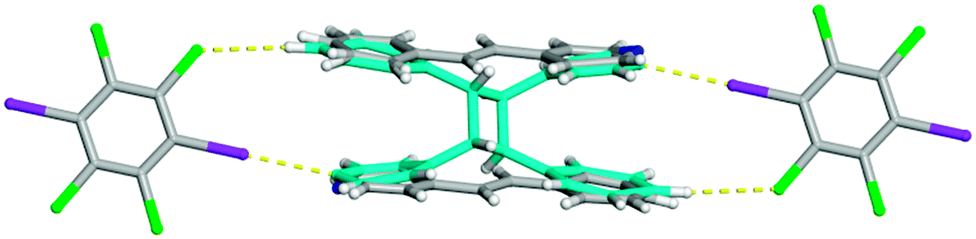 | ||
| Fig. 4 X-ray structure of the partially photoreacted co-crystal of form I forming the ht-PP shown in turquoise. The I⋯N halogen bonds and C–H⋯Cl contacts are shown with yellow dashed lines. | ||
Structure and photoreactivity of form II
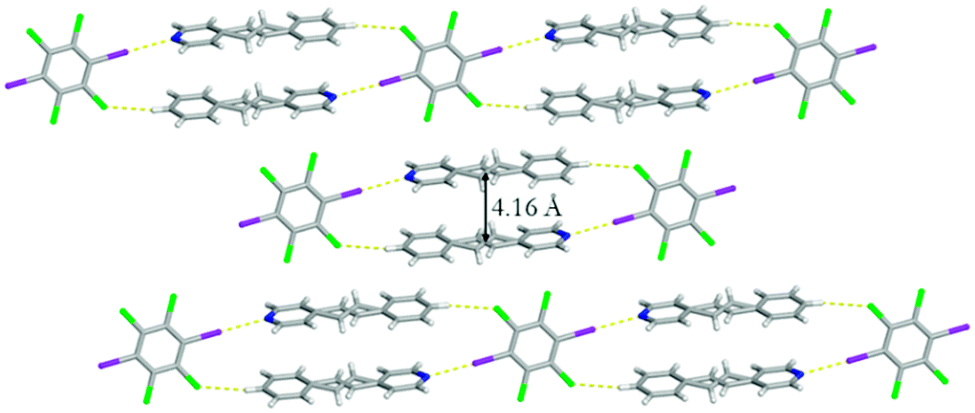
Changing the evaporation rate of toluene led to the formation of a second polymorph of (C6I2Cl4)·2(4-SB), namely form II. Unlike before, form II crystallizes in the centrosymmetric monoclinic space group P21/c. Again, the asymmetric unit contains a half of C6I2Cl4 and a whole molecule of 4-SB. A similar one-dimensional chain held together by a combination of I⋯N halogen bonds [I⋯N 2.9292(1) Å; C–I⋯N 174.9740(1)°] along with C–H⋯Cl contacts [C⋯Cl 3.7445(1) Å] at 290 K was observed for form II (Fig. 5). The aromatic rings within 4-SB are twisted at a much larger angle than observed in form I with a value of 54.91°. The ethylene bridge in 4-SB is again found to be disordered over two positions and after a free variable refinement the ratios were determined to be 0.55/0.45 at 290 K.‡ A second data set at 250 K, on the same crystal, resulted in a final free variable refinement of 0.56/0.44 which is inconclusive on the presence of pedal motion within form II.9 A second type of disorder was observed in the form of rotational disorder of the C6I2Cl4 template where after refinement the ratios were determined to be 0.98/0.02. As before, the 4-SB molecules within the one-dimensional chain are found to be in an anti-orientation with a slightly greater CC separation distance of 4.58 Å (Fig. 5). Again, the bulk material was studied via powder X-ray diffraction to determine the purity after complete evaporation of the solvent when the opening of the vial is covered with cotton. The diffractogram partially agrees with the calculated powder pattern for form II, but a mixture of products is likely (Fig. S5†).
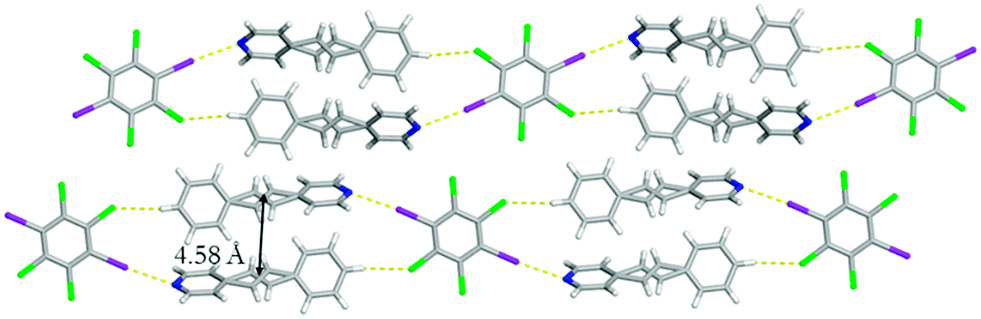 | ||
| Fig. 5 X-ray structure of form II illustrating the anti-orientation within the one-dimensional chain. The I⋯N halogen bonds and C–H⋯Cl contacts are shown with yellow dashed lines. The disorder observed in C6I2Cl4 was removed for clarity. | ||
The difference in the π–π stacking arrangement of C6I2Cl4 gave rise to the different polymorphs. In form I, C6I2Cl4 are found to stack in a homogeneous offset pattern which is drastically different than form II where C6I2Cl4 π–π stacks with benzene rings in a heterogeneous configuration (Fig. 6). This stacking arrangement does not place any other pair of CC at a closer distance than was observed within the one-dimensional chain.
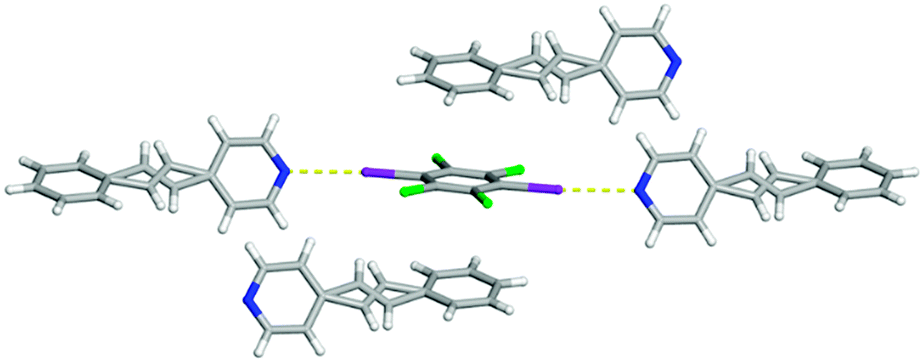 | ||
| Fig. 6 X-ray structure of form II illustrating the I⋯N halogen bonds as well as the heterogeneous π–π stacking arrangement of C6I2Cl4. The I⋯N halogen bonds are shown with yellow dashed lines. The disorder observed in C6I2Cl4 was removed for clarity. | ||
To determine if form II would undergo a solid-state [2 + 2] cycloaddition reaction a powdered sample was again placed between glass plates and put in a photoreactor. As before, a photoreaction was observed by the loss of the olefinic peak on 4-SB at 7.57 ppm in the 1H NMR spectrum along with the presence of a peak at 4.59 ppm which belongs to the cyclobutane ring on ht-PP (Fig. S1 and S3†). The closest CC separation distance is found when the 4-SB molecules are in an anti-orientation which is in agreement with the observed 1H NMR spectrum that only the ht-PP was formed within form II. Again, a quantitative yield for the photoreaction was reached after 30 hours of irradiation. The disordered olefin within form II must undergo molecular pedal motion in order to reach the observed quantitative yield for the photoreaction.13 Even though the olefin–olefin distance within form II is beyond the accepted value for a photoreaction, there are numerous examples in the literature that have reported photoreactions at even greater distances. In particular, a solid-state photoreaction was observed for p-formylcinnamic acid which has a separation distance of 4.825 Å between neighbouring olefins.14
Conclusions
In this contribution, we report a pair of regioselective [2 + 2] cycloaddition reactions based upon halogen bonding interactions. The combination of both I⋯N halogen bonds and C–H⋯Cl contacts position a pair of unsymmetrical 4-SB reactant molecules in the correct orientation to undergo a solid-state photoreaction in both polymorphs resulting in the head-to-tail photoproduct. Currently, we are investigating functionalized olefin-based pyridines to determine if C6I2Cl4 can act as a template to achieve additional solid-state photoreactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. H. G. gratefully acknowledges financial support from Webster University in the form of various Faculty Research Grants. Funding from the National Science Foundation (MRI, CHE-1827756) for the purchase of the Bruker Venture Duo Photon-II diffractometer is also acknowledged.Notes and references
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- (a) M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426 CrossRef CAS; (b) C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047 RSC; (c) J. W. Steed, Chem. Commun., 2018, 54, 13175 RSC; (d) P. Metrangolo and G. Resnati, Halogen Bonding II: Impact on Materials Chemistry and Life Sciences, Springer, Berlin-Heidelberg, 2008 CrossRef.
- T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500 CrossRef CAS PubMed.
- M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477 CrossRef CAS.
- M. A. Sinnwell, J. N. Blad, L. R. Thomas and L. R. MacGillivray, IUCrJ, 2018, 5, 491 CrossRef CAS PubMed.
- E. Bosch, S. J. Kruse, H. R. Krueger Jr. and R. H. Groeneman, Cryst. Growth Des., 2019, 19, 3092 CrossRef CAS.
- G. J. M. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS.
- C. M. Reddy, M. T. Kirchner, R. C. Gundakaram, K. A. Padmanabhan and G. R. Desiraju, Chem. – Eur. J., 2006, 12, 2222 CrossRef CAS PubMed.
- (a) J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244 RSC; (b) J. Harada and K. Ogawa, J. Am. Chem. Soc., 2001, 123, 10884 CrossRef CAS PubMed.
- V. R. Hathwar, S. M. Roopan, R. Subashini, F. N. Khan and T. N. Guru Row, J. Chem. Sci., 2010, 122, 677 CrossRef CAS.
- (a) A. L. Grobelny, N. P. Rath and R. H. Groeneman, CrystEngComm, 2018, 20, 3951 RSC; (b) A. L. Grobelny, N. P. Rath and R. H. Groeneman, Photochem. Photobiol. Sci., 2019, 18, 989 RSC; (c) D. Liu and J.-P. Lang, CrystEngComm, 2014, 16, 76 RSC.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893 CrossRef CAS.
- (a) E. Elacqua, P. Kaushik, R. H. Groeneman, J. C. Sumrak, D.-K. Bučar and L. R. MacGillivray, Angew. Chem., Int. Ed., 2012, 51, 1037 CrossRef CAS PubMed; (b) A. M. P. Peedikakkal and J. J. Vital, Chem. – Eur. J., 2008, 14, 5329 CrossRef CAS PubMed.
- F. Nakanishi, H. Nakanishi, M. Tsuchiya and M. Hasegawa, Bull. Chem. Soc. Jpn., 1976, 49, 3096 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, single crystal X-ray data, 1H NMR spectra, and powder X-ray diffractograms. CCDC 1947988, 1947989, 1947991–1947993. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01379b |
| ‡ Modelling the observed disorder over the entire molecule of 4-SB was attempted but did not result in a better overall model of the structure. |
| This journal is © The Royal Society of Chemistry 2019 |