
Straightforward chemo- and stereoselective fluorocyclopropanation of allylic alcohols: exploiting the electrophilic nature of the not so elusive fluoroiodomethyllithium†
Marco
Colella‡
a,
Arianna
Tota‡
a,
Angela
Großjohann
a,
Claudia
Carlucci
a,
Andrea
Aramini
b,
Nadeem S.
Sheikh
*c,
Leonardo
Degennaro
*a and
Renzo
Luisi
 *a
*a
aDepartment of Pharmacy – Drug Sciences, University of Bari, “A. Moro” Via E. Orabona 4, 70125 Bari, Italy. E-mail: renzo.luisi@uniba.it; leonardo.degennaro@uniba.it
bDepartment of Discovery Dompé Farmaceutici S.p.A., Via Campo di Pile, L’Aquila 67100, Italy
cDepartment of Chemistry, College of Science, King Faisal University, P. O. Box 380, Al-Ahsa 31982, Saudi Arabia. E-mail: nsheikh@kfu.edu.sa
First published on 24th June 2019
Abstract
An unprecedented direct fluorocyclopropanation of allylic alcohols is reported. This simple method involves the not so elusive fluoroiodomethyllithium, a carbenoidic intermediate that under the developed conditions discloses its electrophilic nature. Gratifyingly, the reaction turned out to be highly chemo- and stereoselective, and DFT calculations provided insights into the structure and nature of this new type of carbenoid.
Fluorinated compounds are of paramount importance in modern synthetic chemistry. The presence of a fluorine atom in an organic molecule could profoundly affect some molecular properties, and this is particularly significant in medicinal chemistry.1 With regard to the preparation of fluorinated molecules, several strategies have been invented in the past few decades.2 Among the fluorinated molecules of interest in medicinal chemistry, monofluorinated cyclopropanes are attractive scaffolds found in several biologically active compounds (Fig. 1).3
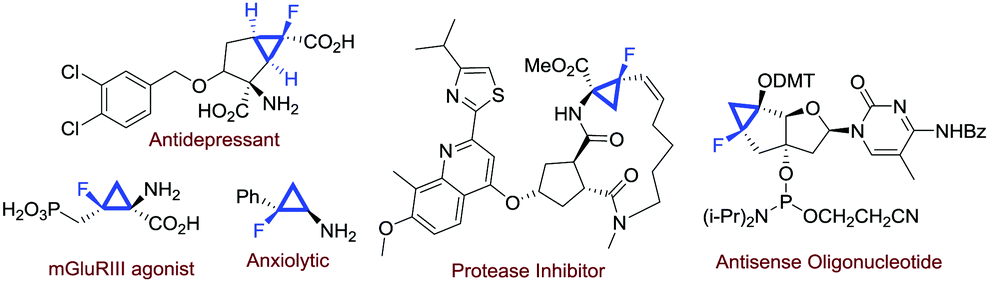 | ||
| Fig. 1 Bioactive entities bearing a monofluorocyclopropane unit. | ||
Notwithstanding the potential of the fluorocyclopropane unit, the synthesis of these molecular motifs is rather difficult. The available strategies for the preparation of mono-fluorocyclopropanes rely on the Michael initiated ring closure,4 nucleophilic fluorination,5 addition of carbene to fluoroalkenes6 and the addition of fluorocarbene or fluorocarbenoids to alkenes.6 Every protocol has pros and cons mainly related to the availability and the ease of preparation of the starting materials, or the stability of the intermediates involved in the process. A rather underexploited strategy, for making fluorinated molecules, is based on the use of fluorinated organometallics such as fluorinated carbenoids where both the fluorine and the metal atoms (i.e. Li, Zn or Mg) are bound to the same carbon. The intrinsic nature of fluorinated carbenoids would make them suitable electrophilic reagents for the cyclopropanation of alkenes. In fact, it has been demonstrated that halo carbenoids could react as nucleophiles or electrophiles depending on the reaction conditions and the nature of the coupling partners.7 Concerning the direct fluorocyclopropanation of alkenes, Schlosser reported that the reactions of mono-fluorocarbenoids with alkenes furnished, albeit in low yields, the expected fluorocyclopropanes.8 Similarly, Burton succeeded in generating fluorochloro-, and fluorobromocarbenoids, which were subsequently employed for the preparation of 1,1′-halofluorocyclopropanes.9 However, these strategies found limited synthetic applicability, and noticeably, required very low temperatures (i.e. −116 °C) for controlling the reactivity of such unstable intermediates. A more synthetically useful strategy for the preparation of mono-fluorocyclopropanes was introduced by Terashima, who exploited a zinc fluorocarbenoid generated from fluorodiiodomethane in the presence of diethylzinc (Scheme 1). The strategy was found to be successful for the cyclopropanation of enamines.10 More recently, Charette reported the use of difluoroiodomethane as the starting material for the formation of the same zinc fluorocarbenoids reported by Terashima but using an unusual F/I exchange reaction (Scheme 1). The Charette's methodology allowed for a direct and effective fluorocyclopropanation of allylic alcohols with high stereocontrol, nevertheless a long reaction time was required (15 hours stirring at −40 °C), and the use of dichloromethane as the solvent.11 In continuation of a research plan on the chemistry of fluorinated organometallics, and in particular fluorocarbenoids,12 we recently reported that very unstable fluoroiodomethyllithium could be generated by deprotonation of fluoroiodomethane with a suitable lithium amide.13 According to Moss and Burton's studies, such fluorinated carbenoid was considered highly unstable and difficult to generate for synthetic purposes.14 We have successfully demonstrated that, under internal quenching conditions, fluoroiodomethyllithium could react, as a nucleophile, with carbonyls or imines to provide unusual and synthetically useful fluoroepoxides and fluoroaziridines (Scheme 1).13 Herein, we report an efficient direct fluorocyclopropanation of allylic alcohols, exploiting the unprecedented electrophilic behavior of the same fluoroiodomethyllithium carbenoid (Scheme 1). Pleasingly, our approach exhibited an excellent chemo- and stereoselectivity, adopting a fast and easy procedure for the formation of monofluorocyclopropanes.
 | ||
| Scheme 1 Strategies for accessing fluorocyclopropanes. | ||
This investigation began while studying the reactivity of fluoroiodomethyllithium 1-Li with α,β-unsaturated carbonyls. With the aim to prepare unsaturated fluoroepoxides, traces of compound 4a were detected during the analysis of the crude reaction mixture by 19F NMR (δ = −218 ppm) (Scheme 2). Inspired by the Charette’ reports,11 we hypothesized that LDA would have reduced ketone 2, providing lithiated alkoxide 2-Li that smoothly underwent cyclopropanation in the presence of fluorocarbenoid 1-Li.15
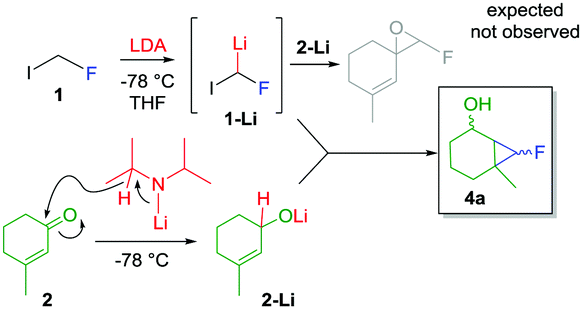 | ||
| Scheme 2 LDA mediated fluorocyclopropanation. | ||
In order to verify our hypothesis and demonstrate the electrophilic nature of the fluorocarbenoid 1-Li, fluoroiodomethane 1 (4 equiv.) was reacted with LDA (4 equiv.) in the presence of cinnamyl alcohol 2b (1 equiv.) in a 1/1 mixture of THF/Et2O at −78 °C.16 To our delight, the expected fluorocyclopropane 4b was found in the reaction mixture in 28% yield. Surprisingly, only one main diastereoisomer was detected by 19F NMR analysis (dr 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). The stereochemistry of the fluorinated cyclopropane was ascertained on the basis of the values of the 3JH–H and 3JH–F coupling constants, and by comparison with already reported molecules (see ESI†). The optimization was continued, searching for the optimal conditions to maximize the yield of the desired fluorocyclopropane. Parameters such as the stoichiometry of the reagents, the nature of the solvent, and temperature were judiciously screened (see ESI†). As a result of this study, a dramatic role of the solvent was observed – ethereal solvents such as THF likely exalted the electrophilic nature of the carbenoid – along with the effect of the concentration and temperature. The optimal conditions provided fluorocyclopropane 4b up to 80% yield using 1/LDA/2b in a 1:3:2 molar ratio, respectively, and running the reaction at −50 °C (see ESI†). With the determination of optimal conditions, the role of the unsaturated acceptor was evaluated (Scheme 3). The use of a preformed lithium alcoholate 5 was not beneficial leading to 4b in only 20% yield. The use of allylic ether 6 resulted in an unfruitful reaction as well as in the case of cynammic acid 7, homoallylic alcohol 8, styrene 9, and propargyl alcohol 10 (Scheme 3). These experiments would rule out the involvement of a carbolithiation sequence. In addition, although the alcohol is expected to be incompatible with the organolithium species, these data strongly suggested that the hydroxyl group at the allylic position is an essential requirement for the process.17
:10). The stereochemistry of the fluorinated cyclopropane was ascertained on the basis of the values of the 3JH–H and 3JH–F coupling constants, and by comparison with already reported molecules (see ESI†). The optimization was continued, searching for the optimal conditions to maximize the yield of the desired fluorocyclopropane. Parameters such as the stoichiometry of the reagents, the nature of the solvent, and temperature were judiciously screened (see ESI†). As a result of this study, a dramatic role of the solvent was observed – ethereal solvents such as THF likely exalted the electrophilic nature of the carbenoid – along with the effect of the concentration and temperature. The optimal conditions provided fluorocyclopropane 4b up to 80% yield using 1/LDA/2b in a 1:3:2 molar ratio, respectively, and running the reaction at −50 °C (see ESI†). With the determination of optimal conditions, the role of the unsaturated acceptor was evaluated (Scheme 3). The use of a preformed lithium alcoholate 5 was not beneficial leading to 4b in only 20% yield. The use of allylic ether 6 resulted in an unfruitful reaction as well as in the case of cynammic acid 7, homoallylic alcohol 8, styrene 9, and propargyl alcohol 10 (Scheme 3). These experiments would rule out the involvement of a carbolithiation sequence. In addition, although the alcohol is expected to be incompatible with the organolithium species, these data strongly suggested that the hydroxyl group at the allylic position is an essential requirement for the process.17
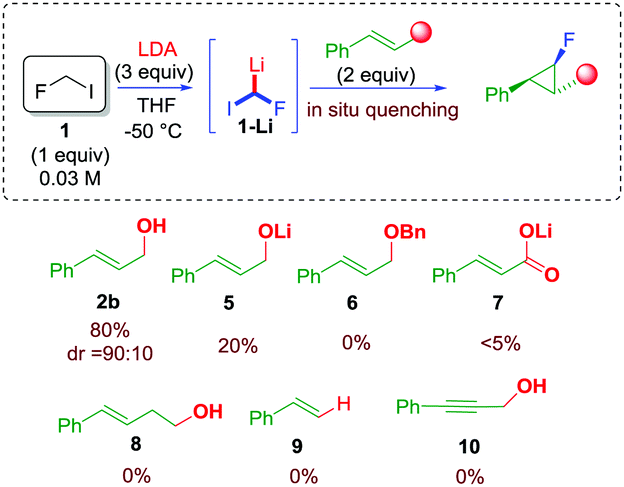 | ||
| Scheme 3 Testing unsaturated systems in fluorocyclopropanation. | ||
With the aim to apply this unprecedented and simple method to the stereoselective synthesis of fluorocyclopropanes, the scope of the reaction was investigated (Scheme 4). First, the use of trans-aryl substituted allylic alcohols (2b–2j) was considered, and the expected fluorocyclopropanes 4b–4j were obtained in good yields and with high stereoselectivity (dr >90:10). Electron-donating groups and halogens were tolerated as substituents of the aromatic ring. Interestingly, no interference was observed with the dimethylamino functionality (4g) or the bromine (4j) as aryl substituents. The presence of electron-withdrawing groups, as the substituents of the aromatic ring, affected the cyclopropanation reaction. In fact, no reaction was observed in the presence of the NO2-substituent (4ac, in Scheme 4), while the presence of a CF3-group was tolerated but the product 4ad (Scheme 4) was obtained in 15% yield although as a single diastereoisomer. The presence of an additional substituent at the carbinolic carbon, as for allylic alcohols 2k–n, resulted in moderate yields of the corresponding fluorocyclopropanes 4k–n and an almost unexpected high stereocontrol (dr >95:5), leading to a single stereoisomer.
 | ||
| Scheme 4 Scope of direct fluorocyclopropanation. | ||
It is worth noting that while the presence of the substituent at the carbinolic carbon affected the yield of the reaction, the additional stereogenic centre does not affect the stereoselectivity. This suggests that a very well organized transition state must likely be involved (see infra). The use of cyclic allylic alcohols such as 2a and 2o also successful led to 4a and 4o in reasonable yields although with a different stereocontrol with reference to the stereochemistry of the C–F carbon of the cyclopropane (see ESI†). Later, alkyl substituted allylic alcohols were also considered.
The use of conjugated allylic alcohol 2p resulted in a high chemo- and stereoselective reaction leading to 4p (Scheme 4). The use of allylic alcohols 2q–x furnished alkyl substituted derivatives 4q–x in good yields, and stereoselectivity depending on the configuration of the double bond. Very high stereoselectivity was observed using trans-configured alkenes (dr >95:5), while a slightly lower stereoselectivity was observed with the use of cis-configured allylic alcohols (i.e.2v, 2x). In derivatives 4v (dr 80:20) and 4x (dr 70:30) the double bond configuration was preserved, while the C–F configuration was likely affected by the alkene geometry. Almost unexpectedly, the use of terminal allylic alcohol 2ab didn’t provide the corresponding cyclopropane 4ab. The stereochemistry at the C–F carbon was also affected by the degree of substitution of the double bond of the allylic alcohol. In fact, the use of either cis- or trans-geraniol (Scheme 4) led to fluorocyclopropanes 4y and 4z, respectively, with good yields but low stereoselectivity. In addition, the reaction was found to be highly chemoselective, with cyclopropanation occurring only at the allylic position. Similarly, the use of peryllil alcohol produced a 70:30 mixture of fluorocyclopropane 4aa (Scheme 4).
Once the synthetic utility of this strategy was established, we were keen to find an explanation for the high level of chemo- and stereocontrol of the reaction, as well as to rationalize the electrophilic behaviour of such a highly reactive carbenoid. To this end, DFT calculations were used to get insights into the structure of 1-Li in the gaseous and the solvent (THF) phases (Fig. 2A, see the ESI† for further details). DFT studies for the monomeric species of 1-Li in THF disclosed a single structure for the carbenoid. In particular, a remarkable change in the geometry of the lithiated carbon was noticed from 1 to 1-Li. Furthermore, a pronounced effect was observed, when the calculations were carried out using THF as a solvent. The optimized geometry of 1-Li (in THF) clearly reveals that the Li, H and F atoms lie on the same plane, with the iodine sticking out from this plane. Pleasingly, analysis of the computed electronic density distribution also confirms the electrophilic behavior of fluoroiodomethyllithium carbenoid 1-Li in THF as a solvent (Fig. 2B). These results are perfectly in line with our experimental results, where the role of THF is believed to enhance the electrophilic nature of the involved carbenoid species. However, a detailed investigation is still underway to explore the possibilities of alkyllithium–lithium alkoxide aggregates.18
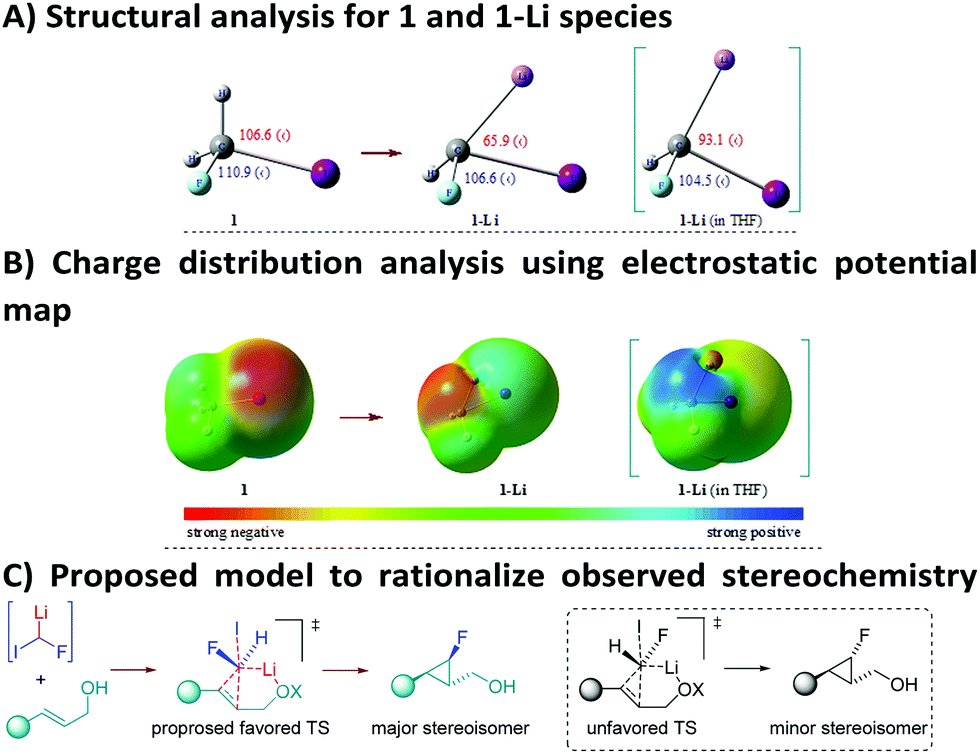 | ||
| Fig. 2 DFT studies [B3LYP/6-311++G(d,p) – LanL2DZ basis set for the I-atom] and proposed model for the observed stereoselectivity. | ||
On the basis of the computed structure, assuming a planar arrangement of Li, H and F atoms, it is reasonable to propose that the carbenoid would approach the double bond from the less hindered face (Fig. 2C). It is likely that the oxygen atom of the hydroxyl group would be playing a role in coordinating the lithium atom, thus introducing constraints that could justify the observed stereoselectivity.19 The lower stereocontrol, observed in some cases, could be due to the impossibility to realize such an ordered transition state (favored TS, Fig. 2C), hence promoting an attack of 1-Li with inverted stereochemistry at the C–F carbon (unfavored TS, Fig. 2C). The hypothesis of a well-organized transition state, involved in this process, was further confirmed by using the optically active allylic alcohol (R)-2m (er = 92:8) which provided the corresponding cyclopropane (R,S,R,R′)-4m with absolute preservation (er = 92:8) of the enantiopurity (Scheme 5). This particular result is quite remarkable, because the chiral allylic alcohol was able to induce stereoselectivity in the reaction of the fluorocarbenoid that formally consisted of a racemic mixture.20
 | ||
| Scheme 5 Validation of the observed stereoselectivity. | ||
In conclusion, an unprecedented and efficient chemo- and stereoselective synthesis of fluorocyclopropanes has been reported. The process was simple, fast, and highly stereoselective. Calculations helped to propose a model to explain the observed stereochemistry. Interestingly, the model for this direct fluorocyclopropanation was proved to be operating in a chiral synthesis of a cyclopropane occurring with absolute preservation of the enantiopurity of the starting allylic alcohol. Further work, aimed at expanding the methodology to other hetero-substituted allylic systems and at controlling the enantioselectivity, is underway and will be reported in due course.
This research was supported by the project Laboratorio Sistema code PONa300369 financed by MIUR, MISE, Horizon 2020 – PON 2014/2020 FARMIDIAB “code 338”; the University of Bari. N. S. S. thanks the Deanship of Scientific Research, King Faisal University for the research support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef PubMed; F. Menaa, B. Menaa and O. N. Sharts, J. Mol. Pharm. Org. Process Res., 2013, 1, 104 Search PubMed; M. Colella, P. Musci, C. Carlucci, S. Lillini, M. Tomassetti, A. Aramini, L. Degennaro and R. Luisi, ACS Omega, 2018, 3, 14841 CrossRef.
- V. Gouverneur, R. Szpera, D. F. J. Moseley, L. B. Smith and A. J. Sterling, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201814457; S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef PubMed.
- A. Pons, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Synthesis, 2016, 4060 Search PubMed; E. David, G. Milanole, P. Ivashkin, S. Couve-Bonnaire, P. Jubault and X. Pannecoucke, Chem. – Eur. J., 2012, 18, 14904 CrossRef CAS PubMed.
- X. Shen, W. Zhang, L. Zhang, T. Luo, X. Wan, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 6966 CrossRef CAS PubMed; P. Ivashkin, S. Couve-Bonnaire, P. Jubault and X. Pannecoucke, Org. Lett., 2012, 14, 2270 CrossRef PubMed; K. Hirotaki, Y. Takehiro, R. Kamaishi, Y. Yamada and T. Hanamoto, Chem. Commun., 2013, 49, 7965 RSC.
- M. Zhang, Y. Gong and W. Wang, Eur. J. Org. Chem., 2013, 7372 CrossRef CAS.
- V. N. G. Lindsay, C. Nicolas and A. B. Charette, J. Am. Chem. Soc., 2011, 133, 8972 CrossRef CAS PubMed; T. Shibue and Y. Fukuda, J. Org. Chem., 2014, 79, 7226 CrossRef PubMed; B. Zhang and A. Studerol, Org. Lett., 2014, 16, 1790 CrossRef PubMed; A. Pons, P. Ivashkin, T. Poisson, A. B. Charette, X. Pannecoucke and P. Jubault, Chem. – Eur. J., 2016, 22, 6239 CrossRef PubMed.
- V. Capriati, Modern Lithium Carbenoid Chemistry, in Contemporary Carbene Chemistry, ed. R. A. Moss and M. P. Doyle, John Wiley & Sons, Inc., Hoboken, NJ, 2013, ch. 11, pp. 327–362 RSC; V. H. Gessner, Chem. Commun., 2016, 52, 12011 RSC; L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC.
- M. Schlosser and G. Heinz, Angew. Chem., Int. Ed. Engl., 1968, 7, 820 CrossRef CAS; M. Schlosser, L. Van Chau and B. Spahić, Helv. Chim. Acta, 1975, 58, 2575 CrossRef; M. Schlosser and G. Heinz, Chem. Ber., 1971, 104, 1934 CrossRef.
- D. J. Burton and J. L. Hahnfeld, J. Org. Chem., 1977, 42, 828 CrossRef CAS.
- O. Tamura, M. Hashimoto, Y. Kobayashi, T. Katoh, K. Nakatani, M. Kamada, I. Hayakawa, T. Akiba and S. Terashima, Tetrahedron, 1994, 50, 3889 CrossRef CAS.
- C. Navuluri and B. Charette, Org. Lett., 2015, 17, 4288 CrossRef CAS PubMed; L.-P. B. Beaulieu, J. F. Schneider and A. B. Charette, J. Am. Chem. Soc., 2013, 135, 7819 CrossRef PubMed.
- G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef CAS PubMed.
- S. Monticelli, M. Colella, V. Pillari, A. Tota, T. Langer, W. Holzer, L. Degennaro, R. Luisi and V. Pace, Org. Lett., 2019, 21, 584 CrossRef CAS PubMed.
- D. J. Burton, Y. Zhen-Yu and Q. Weiming, Chem. Rev., 1996, 96, 1641 CrossRef CAS PubMed; W. B. Farnham, Chem. Rev., 1996, 96, 1633 CrossRef PubMed; D. L. S. Brahms and W. P. Dailey, Chem. Rev., 1996, 96, 1585 CrossRef PubMed; R. A. Moss, G. Kmiecik-Lawrynowicz and K. Krogh-Jespersen, J. Org. Chem., 1986, 51, 2168 CrossRef.
- L. Degennaro, A. Giovine, L. Carroccia and R. Luisi, Practical Aspects of Organolithium Chemistry, in Lithium Compounds in Organic Synthesis: From Fundamentals to Application, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 1st edn, 2014, ch. 18, pp. 513–538 Search PubMed.
- See ESI† during the optimization study, allylamine was also tested as acceptor. Nevertheless, the reaction returned a complex mixture.
- C. Bolm and D. Pupowicz, Tetrahedron Lett., 1997, 38, 7349 CrossRef CAS; M. J. Durán-Peña, M. E. Flores-Giubi, J. M. Botubol-Ares, L. M. Harwood, I. G. Collado, A. J. Macías-Sánchez and R. Hernández-Galán, Org. Biomol. Chem., 2016, 14, 2731 RSC.
- L. M. Pratt, O. Kwon, T. C. Ho and N. V. Nguyen, Tetrahedron, 2008, 64, 5314 CrossRef CAS.
- R. A. Moss, J. Org. Chem., 2017, 82, 2307 CrossRef CAS PubMed.
- The low yield, could likely be resulting from a sort of kinetic resolution of racemic fluorocarbenoid 1-Li. This intriguing aspect of the reactivity is still under investigation in our laboratory.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc03394g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |