
Photoredox-catalyzed sulfonylation of alkyl iodides, sulfur dioxide, and electron-deficient alkenes†
Shengqing
Ye‡
a,
Danqing
Zheng‡
b,
Jie
Wu
 *ab and
Guanyinsheng
Qiu
*bc
*ab and
Guanyinsheng
Qiu
*bc
aInstitute for Advanced Studies, Taizhou University, 1139 Shifu Avenue, Taizhou 318000, China. E-mail: jie_wu@fudan.edu.cn
bDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, China
cCollege of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China. E-mail: qiuguanyinsheng@mail.zjxu.edu.cn
First published on 25th January 2019
Abstract
A photoredox-catalyzed sulfonylation of alkyl iodides, sulfur dioxide, and electron-deficient alkenes under mild conditions is achieved. This reaction proceeds through alkyl radicals formed in situ from alkyl iodides under visible light irradiation in the presence of a photoredox catalyst. The alkyl radical intermediates would react with sulfur dioxide leading to alkylsulfonyl radicals, which would be trapped by electron-deficient alkenes giving rise to alkyl sulfones. Various functional groups including nitro, halo, acetyl, sufonyl, and pyridinyl are all tolerated under the photoredox conditions.
Transition metal-catalyzed coupling reactions of aryl/alkyl halides have been utilized broadly, due to aryl/alkyl halides being easily available and cheap.1 Recently, reactions of aryl/alky halides under photoredox catalysis have attracted much attention. It is well recognized that the photoinduced C–X bond dissociation of aryl/alkyl halides would produce the corresponding carbon radicals via electron transfer.2 Therefore, the possibility of the β-H elimination of alkyl halides under transition metal catalysis may be avoided under photoinduced conditions. So far, progress of photoinduced C–X bond dissociation has been witnessed. For example, Peters and Fu reported photoinduced Ullmann C–N coupling by using a stoichiometric or catalytic amount of copper salt under visible light irradiation at room temperature.2a
In the past decade, synthesis of sulfonyl compounds through the insertion of sulfur dioxide has developed rapidly,3 which avoids the utilization of pre-installed sulfonyl precursors.4 Currently, the sulfur dioxide surrogates including DABCO·(SO2)2 (1,4-diazabicyclo[2.2.2]octane-sulfur dioxide)5–7 and inorganic sulfites8 have been used broadly in the sulfonylation process. As part of a program for the generation of sulfonyl compounds, we are interested in the radical process with the insertion of sulfur dioxide.7 So far, various radical precursors have been employed in the sulfonylation reaction including aryldiazonium tetrafluoroborates,7 aryl/alkyl halides,9 diaryliodonium salts,10 and potassium alkyltrifluoroborates.11 Among these precursors, aryl/alkyl halides are especially attractive, as they are easily available and cheap. For instance, aryl/alkyl halides could react with sulfur dioxide and hydrazines under ultraviolet irradiation.8a Although this transformation was efficient, the reaction could not be extended to other partners due to the ultraviolet irradiation. Thus, method development for the sulfonylation of aryl/alkyl halides with the insertion of sulfur dioxide under mild conditions, especially in the presence of visible light, would be highly desirable. Herein, we report a photoredox-catalyzed sulfonylation of alkyl iodides, sulfur dioxide, and electron-deficient alkenes under mild conditions. This reaction proceeds through alkyl radicals formed in situ from alkyl iodides under visible light irradiation in the presence of a photoredox catalyst, leading to diverse sulfones. Various functional groups including nitro, halo, acetyl, sufonyl, and pyridinyl are all tolerated under the photoredox conditions (Scheme 1).
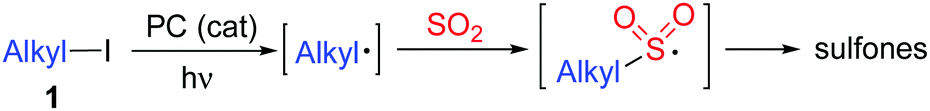 | ||
| Scheme 1 Generation of sulfones under photoredox catalysis with the insertion of sulfur dioxide. | ||
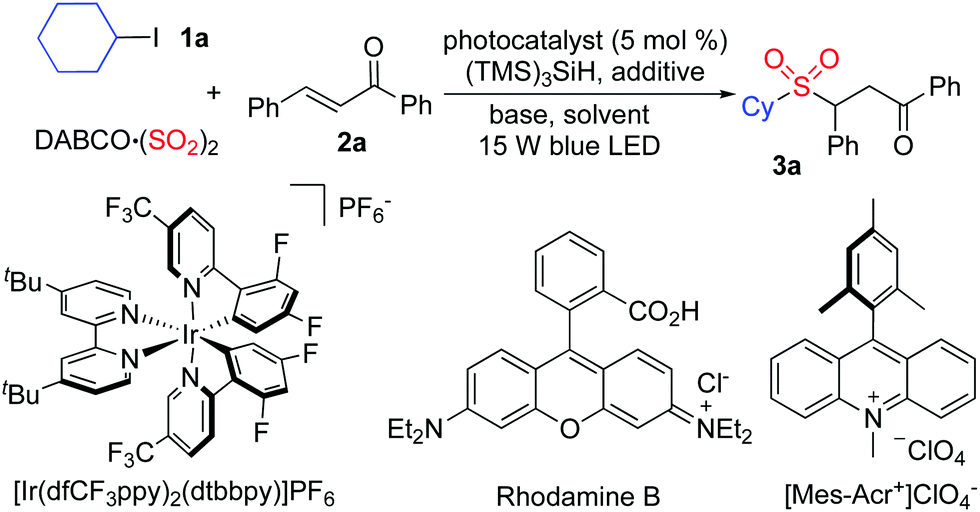
At the outset, a model reaction of cyclohexyl iodide 1a, DABCO·(SO2)2, and (E)-chalcone 2a in the presence of TTMS and photocatalyst (5 mol%) was explored. The results are summarized in Table 1. Initially, the reaction was catalyzed by [Ir(dfCF3ppy)2(dtbbpy)]PF6 in 1,2-dichloroethane irradiated by a 15 W blue LED (Table 1, entry 1). To our delight, the corresponding sulfone 3a was obtained in 31% yield. Inferior results were observed when the solvent was changed to 1,2-dioxane, MeCN, or MeOH (Table 1, entries 2–4). No reaction occurred when the photocatalyst was replaced by rhodamine B (Table 1, entry 5). A slightly higher yield was afforded when 9-Mes-10-methyl acridinium perchlorate was used as the photocatalyst (Table 1, entry 6). Several inorganic bases were then screened (Table 1, entries 7–11), and it was found that the transformation worked well when sodium acetate was used leading to the expected product 3a in 42% yield (Table 1, entry 11). Since the presence of halide would promote the conversion, the reaction was examined with the addition of sodium bromide or sodium iodide (Table 1, entries 12 and 13). Gratifyingly, the reaction with the addition of sodium iodide provided the desired product 3a in 66% isolated yield (Table 1, entry 13). Further exploration revealed that the yield was enhanced to 71% when iodine was employed instead (Table 1, entry 14). No change was observed when the amount of base was increased (Table 1, entry 15). The yield could not be improved by changing the amount of TTMS (Table 1, entry 16). No reaction occurred in the absence of the photocatalyst (Table 1, entry 17). Only a trace amount of the product was obtained when the reaction was performed in the dark (Table 1, entry 18).
|
|
|||||
|---|---|---|---|---|---|
| Entry | PC | Additive | Base | Solvent | Yieldb (%) |
| a Reaction conditions: chalcone 1a (0.2 mmol), iodocyclohexane 2a (2.0 equiv. 0.4 mmol), DABSO (0.8 equiv.), TTMS (1.0 equiv., 0.2 mmol), additives (10 mol%), base (1.5 equiv. 0.3 mmol), photocatalyst (5 mol%), solvent (3.0 mL), N2, rt, under blue LED irradiation (15 W) for 48 h. b Isolated yield based on 1a. c [Ir(dfCF3ppy)2(dtbbpy)]PF6. d In the presence of base (2.0 equiv. 0.4 mmol). e In the presence of TTMS (1.2 equiv. 0.24 mmol). f In the absence of photocatalyst. g In the dark. | |||||
| 1 | [Ir]c | — | — | DCE | 31 |
| 2 | [Ir]c | — | — | Dioxane | 23 |
| 3 | [Ir]c | — | — | MeOH | Trace |
| 4 | [Ir]c | — | — | MeCN | 18 |
| 5 | Rhodamine B | — | — | DCE | 0 |
| 6 | Mes-Acr+ | — | — | DCE | 35 |
| 7 | Mes-Acr+ | — | K2CO3 | DCE | 22 |
| 8 | Mes-Acr+ | — | Na2CO3 | DCE | 24 |
| 9 | Mes-Acr+ | — | NaHCO3 | DCE | 37 |
| 10 | Mes-Acr+ | — | NaOAc | DCE | 42 |
| 11 | Mes-Acr+ | — | Na2HPO4 | DCE | 35 |
| 12 | Mes-Acr+ | NaBr | NaOAc | DCE | 44 |
| 13 | Mes-Acr+ | NaI | NaOAc | DCE | 66 |
| 14 | Mes-Acr+ | I2 | NaOAc | DCE | 71 |
| 15d | Mes-Acr+ | I2 | NaOAc | DCE | 72 |
| 16e | Mes-Acr+ | I2 | NaOAc | DCE | 63 |
| 17f | — | I2 | NaOAc | DCE | 0 |
| 18g | Mes-Acr+ | I2 | NaOAc | DCE | Trace |
The generality of the reaction scope was then investigated under the above optimized conditions. The result is shown in Scheme 2. It was found that this transformation with a range of alkyl iodides was efficient, and proceeded smoothly giving rise to the corresponding sulfones 3 as expected. Not only 4-iodotetrahydro-2H-pyran but also 3-iodooxetane was a suitable substrate in the reaction of DABCO·(SO2)2 and (E)-chalcone 2a, leading to the desired product 3b and 3e, respectively. It was noteworthy that only alkyl iodide was effective in this transformation, and alkyl chloride was inert under these conditions. For example, compound 3g was produced in 66% yield, and the chloro group was retained. The reaction of an ester-containing substrate also worked well, affording the desired product 3h in 58% yield. Other substituted chalcones were examined subsequently, and various functional groups including methoxy, nitro, and chloro were all tolerated. Further exploration revealed that cyclohexyl iodide 1a reacted with DABCO·(SO2)2 and 4-vinylpyridine giving rise to the corresponding product 3t in 72% yield. Compound 3u was generated in 46% yield when (vinylsulfonyl)benzene was employed as the substrate. However, the reactions failed to produce the corresponding products when other acrylates, styrenes or Michael acceptor systems holding a β-alkyl substituent were used as substrates.
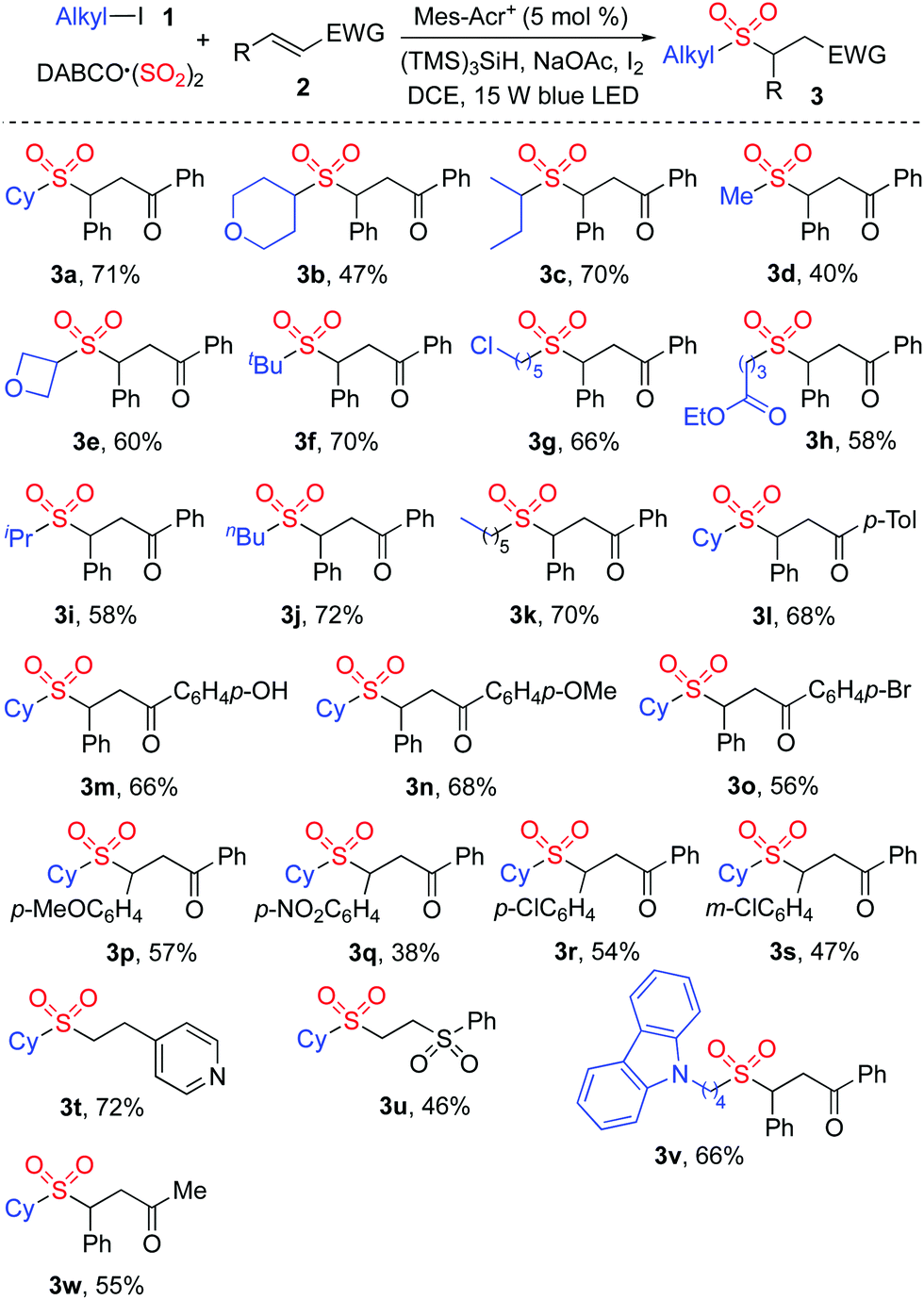 | ||
| Scheme 2 Scope investigation for the photoredox-catalyzed sulfonylation of alkyl iodides 1, sulfur dioxide, and electron-deficient alkenes 2. | ||
As mentioned above, we proposed that under photoredox catalysis, alkyl radical intermediates would be produced to initiate the reaction. Therefore, several control experiments were performed, as presented in Scheme 3. As expected, no reaction occurred with the addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in the mixture of cyclohexyl iodide 1a, DABCO·(SO2)2, and (E)-chalcone 2a under the standard conditions, and compound 4 was detected by HRMS (Scheme 3, eqn (a)). Additionally, (iodomethyl)cyclopropane was employed in the reaction of DABCO·(SO2)2 with (E)-chalcone 2a, resulting in the formation of compound 3x in 56% yield (Scheme 3, eqn (b)). This result demonstrated that a cyclopropylmethyl radical might be generated during the reaction process. These results indicated that a radical process might be involved, as proposed in Scheme 4.
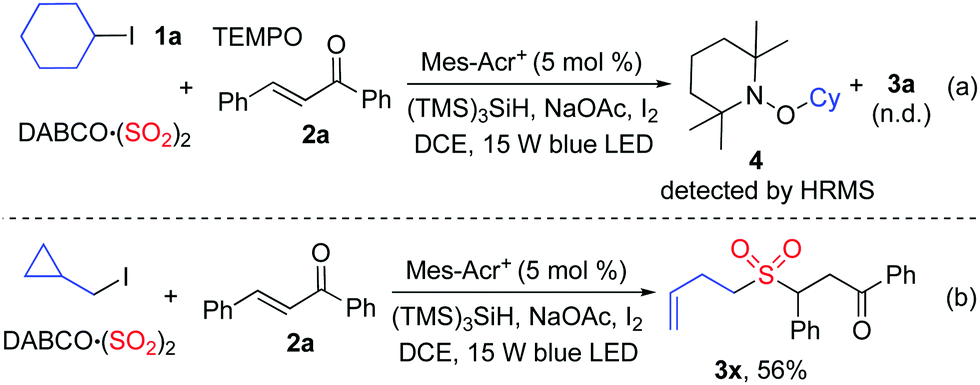 | ||
| Scheme 3 Control experiments. | ||
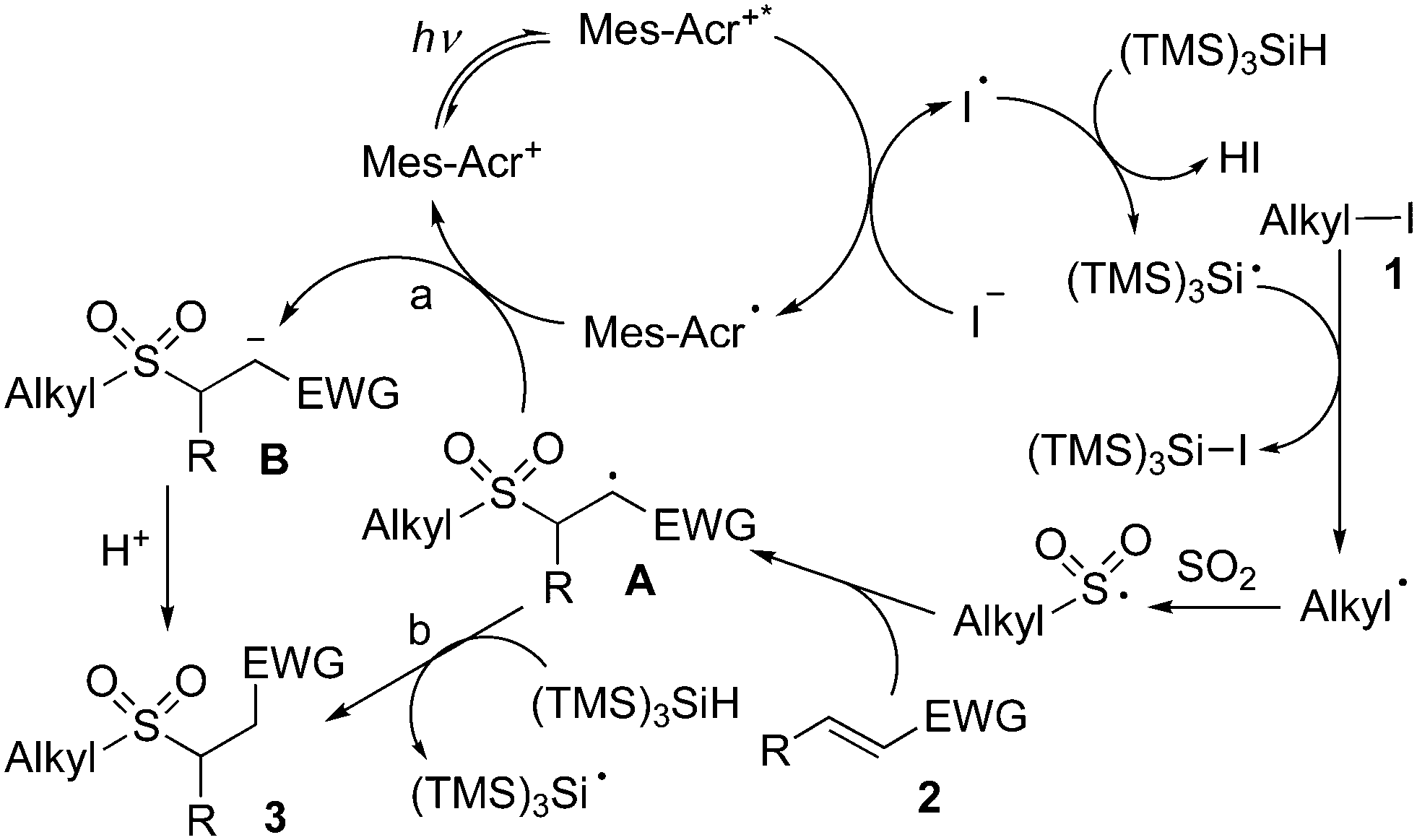 | ||
| Scheme 4 Plausible mechanism. | ||
On the basis of the above results and previous reports,11 we postulated that under visible light irradiation, the excited state of the photocatalyst would assist the formation of a trimethylsilylsilyl radical (Scheme 4). Thus, the trimethylsilylsilyl radical would react with alkyl iodide 1 leading to an alkyl radical intermediate, which would be captured by sulfur dioxide to provide an alkylsulfonyl radical. Then, the alkylsulfonyl radical would attack the double bond of electron-deficient alkene 2, giving rise to radical intermediate A. Subsequently, two possible pathways might occur. In path a, radical intermediate A would undergo a reductive single electron transfer (SET) to produce anion intermediate B. Further protonation would afford the corresponding sulfone 3. Alternatively, radical A would go through proton abstraction from TTMS, giving rise to the desired product 3 (path b).
In summary, we have described a photoredox-catalyzed sulfonylation of alkyl iodides, sulfur dioxide, and electron-deficient alkenes under mild conditions. This reaction proceeds through alkyl radicals formed in situ from alkyl iodides under visible light irradiation in the presence of a photoredox catalyst. The alkyl radical intermediates would react with sulfur dioxide leading to alkylsulfonyl radicals, which would be trapped by electron-deficient alkenes giving rise to alkyl sulfones. Various functional groups including nitro, halo, acetyl, sufonyl, and pyridinyl are all tolerated under the photoredox conditions.
Financial support from the National Natural Science Foundation of China (No. 21672037 and 21532001) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected examples, see: (a) Transition Metal Reagents and Catalysts, ed. J. Tsuji, John Wiley & Sons, Ltd, 2000 Search PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490 CrossRef CAS PubMed.
- For selected examples, see: (a) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS PubMed; (b) D. T. Ziegler, J. Choi, J. M. Muñoz-Molina, A. C. Bissember, J. C. Petersm and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 13107 CrossRef CAS PubMed; (c) L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328 CrossRef CAS PubMed; (d) Y. C. Tan, J. M. Munoz-Molina, G. C. Fu and J. C. Peters, Chem. Sci., 2014, 5, 2831 RSC; (e) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed; (f) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef PubMed; (g) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (h) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS PubMed; (i) L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929 CrossRef CAS PubMed.
- For reviews: (a) D. Zheng and J. Wu, Sulfur Dioxide Insertion Reactions for Organic Synthesis, Nature Springer, Berlin, 2017 CrossRef; (b) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691 RSC; (c) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592 RSC; (d) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393 RSC; (e) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 2701 Search PubMed; (f) E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602 CrossRef CAS; (g) K. Hofman, N.-W. Liu and G. Manolikakes, Chem. – Eur. J., 2018, 24, 11852 CrossRef CAS PubMed; (h) G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405 RSC; (i) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561 RSC; (j) S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013 RSC; (k) J. Zhu, W.-C. Yang, X.-D. Wang and L. Wu, Adv. Synth. Catal., 2018, 360, 386 CrossRef CAS.
- For selected examples, see: (a) L.-Y. Xie, S. Peng, F. Liu, G.-R. Chen, W. Xia, X. Yu, W.-F. Li, Z. Cao and W.-M. He, Org. Chem. Front., 2018, 5, 2604 RSC; (b) W. Li, G. Yin, L. Huang, Y. Xiao, Z. Fu, X. Xin, F. Liu, Z. Li and W.-M. He, Green Chem., 2016, 18, 4879 RSC; (c) L.-Y. Xie, S. Peng, L.-L. Jiang, X. Peng, W. Xia, X. Yu, X.-X. Wang, Z. Cao and W.-M. He, Org. Chem. Front., 2019, 2, 167 RSC; (d) L.-Y. Xie, S. Peng, J.-X. Tan, R.-X. Sun, X. Yu, N.-N. Dai, Z.-L. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 16976 CrossRef CAS; (e) Y. Su, X. Zhou, C. He, W. Zhang, X. Ling and X. Xiao, J. Org. Chem., 2016, 81, 4981 CrossRef CAS PubMed; (f) L.-Y. Xie, Y.-J. Li, J. Qu, Y. Duan, J. Hu, K.-J. Liu, Z. Cao and W.-M. He, Green Chem., 2017, 19, 5642 RSC.
- P. S. Santos and M. T. S. Mello, J. Mol. Struct., 1988, 178, 121 CrossRef CAS.
- For selected examples, see: (a) B. Nguyen, E. J. Emmet and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372 CrossRef CAS PubMed; (b) F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570 CrossRef CAS PubMed; (c) Y. Wang, L. Deng, Y. Deng and J. Han, J. Org. Chem., 2018, 83, 4674 CrossRef CAS; (d) J. Sheng, Y. Li and G. Qiu, Org. Chem. Front., 2017, 4, 95 RSC; (e) A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226 CrossRef CAS PubMed; (f) D. Yang, P. Sun, W. Wei, F. Liu, H. Zhang and H. Wang, Chem. – Eur. J., 2018, 24, 4423 CrossRef CAS PubMed; (g) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS PubMed; (h) Z.-J. Shen, Y.-N. Wu, C.-L. He, L. He, W.-J. Hao, A.-F. Wang, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 445 RSC; (i) D. Sun, K. Yin and R. Zhang, Chem. Commun., 2018, 54, 1335 RSC; (j) X. Wang, L. Xue and Z. Wang, Org. Lett., 2014, 16, 4056 CrossRef CAS PubMed; (k) V. Vedovato, E. P. A. Talbot and M. C. Willis, Org. Lett., 2018, 20, 5493 CrossRef CAS; (l) Y. Wang, L. Deng, J. Zhou, X. Wang, H. Mei, J. Han and Y. Pan, Adv. Synth. Catal., 2018, 360, 1060 CrossRef CAS; (m) H. Wang, B. Wang, S. Sun and J. Cheng, Org. Chem. Front., 2018, 5, 2547 RSC; (n) T. Wang, F. Wang, J. Shen, T. Pang, Y. Ren, B. Wu and X. Zhang, Tetrahedron Lett., 2018, 59, 1183 CrossRef CAS; (o) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2015, 54, 1168 CrossRef CAS PubMed; (p) N. Wolff, J. Char, X. Frogneux and T. Cantat, Angew. Chem., Int. Ed., 2017, 56, 5616 CrossRef PubMed; (q) C. C. Chen and J. Waser, Org. Lett., 2015, 17, 736 CrossRef CAS PubMed; (r) Y. Luo, X. Pan, C. Chen, L. Yao and J. Wu, Chem. Commun., 2015, 51, 180 RSC; (s) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747 CrossRef CAS PubMed; (t) A. S. Tsai, J. M. Curto, B. N. Rocke, A. R. Dechert-Schmitt, G. K. Ingle and V. Mascitti, Org. Lett., 2016, 18, 508 CrossRef CAS PubMed; (u) A. L. Tribby, I. Rodríguez, S. Shariffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294 CrossRef CAS; (v) J. Zhang, Y. An and J. Wu, Chem. – Eur. J., 2017, 23, 9477 CrossRef CAS PubMed; (w) Y. Chen, P. R. D. Murray, A. T. Davies and M. C. Willis, J. Am. Chem. Soc., 2018, 140, 8781 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451 CrossRef CAS PubMed; (b) D. Zheng, J. Yu and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 11925 CrossRef CAS PubMed; (c) R. Mao, Z. Yuan, R. Zhang, Y. Ding, X. Fan and J. Wu, Org. Chem. Front., 2016, 3, 1498 RSC; (d) Y. An and J. Wu, Org. Lett., 2017, 19, 6028 CrossRef CAS PubMed; (e) T. Liu, W. Zhou and J. Wu, Org. Lett., 2017, 19, 6638 CrossRef CAS PubMed; (f) H. Xia, Y. An, X. Zeng and J. Wu, Chem. Commun., 2017, 53, 12548 RSC; (g) Y. Xiang, Y. Kuang and J. Wu, Chem. – Eur. J., 2017, 23, 699 Search PubMed; (h) T. Liu, D. Zheng and J. Wu, Org. Chem. Front., 2017, 4, 1079 RSC; (i) J. Zhang, F. Zhang, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 3891 RSC; (j) K. Zhou, J. Zhang, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 7459 RSC; (k) H. Xia, Y. An, X. Zeng and J. Wu, Org. Chem. Front., 2018, 5, 366 RSC; (l) K. Zhou, M. Chen, L. Yao and J. Wu, Org. Chem. Front., 2018, 5, 371 RSC; (m) X. Wang, Y. Li, G. Qiu and J. Wu, Org. Chem. Front., 2018, 5, 2555 RSC; (n) J. Zhang, K. Zhou and J. Wu, Org. Chem. Front., 2018, 5, 813 RSC; (o) F.-S. He, X. Cen, S. Yang, J. Zhang, H. Xia and J. Wu, Org. Chem. Front., 2018, 5, 2437 RSC; (p) F.-S. He, Y. Wu, J. Zhang, H. Xia and J. Wu, Org. Chem. Front., 2018, 5, 2940 RSC.
- For selected examples by using inorganic sulfites as the source of sulfur dioxide, see: (a) S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037 RSC; (b) M. Wang, S. Chen and X. Jiang, Org. Lett., 2017, 19, 4916 CrossRef CAS PubMed; (c) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS PubMed; (d) Y. Wang, B. Du, W. Sha, J. Han and Y. Pan, Org. Chem. Front., 2017, 4, 1313 RSC; (e) J. Zhang, Y. An and J. Wu, Chem. – Eur. J., 2017, 23, 9477 CrossRef CAS PubMed; (f) A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226 CrossRef CAS PubMed; (g) A. Shavnya, K. D. Hesp, V. Mascitti and A. C. Smith, Angew. Chem., Int. Ed., 2015, 54, 13571 CrossRef CAS PubMed; (h) H. Konishi, H. Tanaka and K. Manabe, Org. Lett., 2017, 19, 1578 CrossRef CAS PubMed; (i) X. Gong, J. Chen, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 11172 RSC; (j) N.-W. Liu, S. Liang and G. Manolikakes, Adv. Synth. Catal., 2017, 359, 1308 CrossRef CAS; (k) H. Zhu, Y. Shen, Q. Deng, J. Chen and T. Tu, Chem. Commun., 2017, 53, 12473 RSC; (l) K. Zhou, J. Zhang, G. Qiu and J. Wu, Org. Lett., 2019, 21, 278 Search PubMed; (m) J. Zhang, K. Zhou, G. Qiu and J. Wu, Org. Chem. Front., 2019, 6, 36 RSC; (n) H. Liao, A. Chatupheeraphat, C. C. Hsiao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 15540 CrossRef CAS PubMed; (o) M. Wang, Q. Fan and X. Jiang, Green Chem., 2018, 20, 5469 RSC.
- (a) Y. Li, D. Zheng, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 574 RSC; (b) K. Zhou, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 865 RSC; (c) X. Gong, Y. Ding, X. Fan and J. Wu, Adv. Synth. Catal., 2017, 359, 2999 CrossRef CAS; (d) J. Zhang, K. Zhou, G. Qiu and J. Wu, Org. Chem. Front., 2019, 6, 36 RSC.
- (a) X. Gong, J. Chen, J. Liu and J. Wu, Org. Chem. Front., 2017, 4, 2221 RSC; (b) N.-W. Liu, S. Liang and G. Manolikakes, Adv. Synth. Catal., 2017, 359, 1308 CrossRef CAS; (c) M. Wang, S. Chen and X. Jiang, Org. Lett., 2017, 19, 4916 CrossRef CAS PubMed; (d) Z. Chen, N.-W. Liu, M. Bolte, H. Ren and G. Manolikakes, Green Chem., 2018, 20, 3059 RSC.
- (a) T. Liu, Y. Li, L. Lai, J. Cheng, J. Sun and J. Wu, Org. Lett., 2018, 20, 3605 CrossRef CAS PubMed; (b) T. Liu, Y. Ding, X. Fan and J. Wu, Org. Chem. Front., 2018, 5, 3153 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data, and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c9cc00347a |
| ‡ S. Ye and D. Zheng contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |