
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective and catalytic carbon dioxide and heteroallene activation mediated by cerium N-heterocyclic carbene complexes†
Polly L.
Arnold
 *a,
Ryan W. F.
Kerr
ab,
Catherine
Weetman
a,
Scott R.
Docherty
a,
Julia
Rieb
ac,
Faye L.
Cruickshank
a,
Kai
Wang
a,
Christian
Jandl
ac,
Max W.
McMullon
ac,
Alexander
Pöthig
c,
Fritz E.
Kühn
*c and
Andrew D.
Smith
*b
*a,
Ryan W. F.
Kerr
ab,
Catherine
Weetman
a,
Scott R.
Docherty
a,
Julia
Rieb
ac,
Faye L.
Cruickshank
a,
Kai
Wang
a,
Christian
Jandl
ac,
Max W.
McMullon
ac,
Alexander
Pöthig
c,
Fritz E.
Kühn
*c and
Andrew D.
Smith
*b
aEaStCHEM School of Chemistry, University of Edinburgh, The King's Buildings, Edinburgh, EH9 3FJ, UK. E-mail: Polly.Arnold@ed.ac.uk
bEaStCHEM School of Chemistry, University of St. Andrews, North Haugh, St. Andrews, KY16 9ST, UK, E-mail: ads10@st-andrews.ac.uk
cMolecular Catalysis, Faculty of Chemistry and Catalysis Research Center, Technical University Munich, Lichtenbergstr. 4, 85748 Garching bei München, Germany
First published on 10th September 2018
Abstract
A series of rare earth complexes of the form Ln(LR)3 supported by bidentate ortho-aryloxide–NHC ligands are reported (LR = 2-O-3,5-tBu2-C6H2(1-C{N(CH)2N(R)})); R = iPr, tBu, Mes; Ln = Ce, Sm, Eu). The cerium complexes cleanly and quantitatively insert carbon dioxide exclusively into all three cerium carbene bonds, forming Ce(LR·CO2)3. The insertion is reversible only for the mesityl-substituted complex Ce(LMes)3. Analysis of the capacity of Ce(LR)3 to insert a range of heteroallenes that are isoelectronic with CO2 reveals the solvent and ligand size dependence of the selectivity. This is important because only the complexes capable of reversible CO2-insertion are competent catalysts for catalytic conversions of CO2. Preliminary studies show that only Ce(LMes·CO2)3 catalyses the formation of propylene carbonate from propylene oxide under 1 atm of CO2 pressure. The mono-ligand complexes can be isolated from reactions using LiCe(NiPr2)4 as a starting material; LiBr adducts [Ce(LR)(NiPr2)Br·LiBr(THF)]2 (R = Me, iPr) are reported, along with a hexanuclear N-heterocyclic dicarbene [Li2Ce3(OArCMe–H)3(NiPr2)5(THF)2]2 by-product. The analogous para-aryloxide–NHC proligand (p-LMes = 4-O-2,6-tBu2-C6H2(1-C{N(CH)2NMes}))) has been made for comparison, but the rare earth tris-ligand complexes Ln(p-LMes)3(THF)2 (Ln = Y, Ce) are too reactive for straightforward Lewis pair separated chemistry to be usefully carried out.
Carbon dioxide can be a useful and renewable C1 building block in the fine and bulk chemical industries due to its natural abundance and reactivity,1,2 and can provide carboxylic acids, esters and (cyclic) carbonates.3 Isoelectronic isocyanates and isothiocyanates are also valuable electrophilic elementary reagents used in polymerization and cyclisations,4–6 and thus chemistry which utilizes heteroallenes is of great interest.
Lewis basic N-heterocyclic carbenes (NHCs) are known to react with carbon dioxide, isocyanates and isothiocyanates as nucleophiles to form imidazolium carboxylates,7,8 imidazolium amidates9 and imidazolium carbimidothioates10 respectively A (Chart 1). While imidazolium carboxylates can successfully catalyse carbamate formation,11 NHCs react as organocatalysts with isocyanates to form cyclic ureas B through an azolium amidate intermediate.9
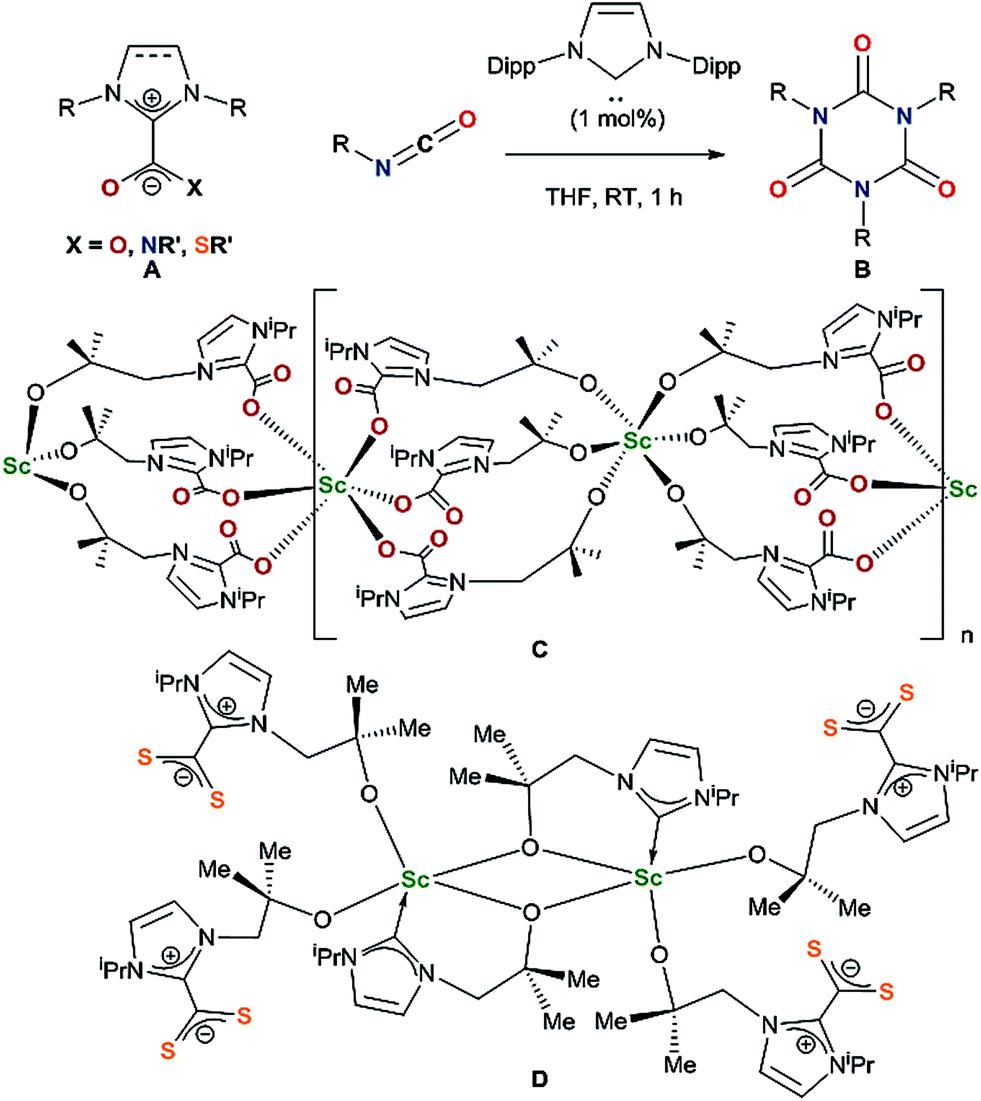 | ||
| Chart 1 | ||
Since the first reported isolation of lanthanide–NHC complexes in 1994,12,13 it has been shown that Lewis acidic rare-earth cations form hemilabile bonds with soft σ-donating NHCs.14,15 Between 2006 and 2010, Shen and co-workers published syntheses of aryloxide–NHC lanthanide complexes, however no subsequent reactivity was reported.16–19 In 2014, we reported the activation of carbon dioxide C and carbon disulfide D using a scandium alkoxide-NHC complex, achieving frustrated Lewis pair (FLP) like reactivity which resulted in metal–ligand scrambling to form a polymeric –(Sc–NHC–CO2)–n containing network owing to the flexible alkoxide tether.20
Cerium, the most abundant lanthanide has a relatively low toxicity; its trichloride is six times less toxic by ingestion than that of iron,21,22 and it has many applications in heterogeneous catalysis.23 Previously we showed that cerium-silylamido NHC complexes ([Ce(L)(N{SiMe3}2)2] L = bidentate alkoxy-tethered NHC ligand) react with CO2 to form an insoluble mixture while the uranium analogue [U(L)(N{SiMe3}2)2] yields an equivalent of isocyanate.24 In the latter instance it was not possible to isolate any intermediate that confirmed whether the NHC group was definitively involved in the CO2 activation.25 Recently, Suresh reported the first mononuclear N-carboxylate imidazolium lanthanide compounds, suggesting their potential use as single-molecule magnets.26 Here, we demonstrate that cerium (and other rare earth) complexes with aryloxide-tethered NHC ligands can successfully form homoleptic cerium imidazolium carboxylate complexes from CO2 insertion into the Ce–C carbene bonds. We show how to control the reversibility for the first time, and use this, and the extent of insertion of CO2 or isoelectronic heteroallenes (isocyanates and isothiocyanates) by changing the ligand steric and electronic properties, and by solvent effects. This is important as we show that only the complexes capable of reversible CO2-insertion are competent catalysts for the synthesis of cyclic carbonates from CO2 and epoxides.
Results and discussion
ortho-Aryloxide Ln–NHC complex synthesis
One objective for synthesizing lanthanide aryloxide tethered–NHC complexes is to combine valuable hemilability within a rigid framework for selective reactivity and we envisioned that varying coordination environments arising from respective alkyl and aryl substituents could give distinctive chemistry. A suspension of an ortho-aryloxide NHC proligand,27,28[o-H2LR][Br] where LR = 2-O-3,5-tBu2-C6H2(1-C{N(CH)2N(R)}) and R = iPr, tBu and Mes were treated with 6 equivalents of KN(SiMe3)2 and LnCl3(THF)n (Ln = Ce, Sm, Eu) in DME to afford bright yellow solutions with colourless precipitates of KCl and KBr (Scheme 1). After work-up, 1LnR (Ln(LR)3) can be afforded in moderate to good yields (15–76%), while over 8 g of 1CeiPr can be isolated in a single reaction.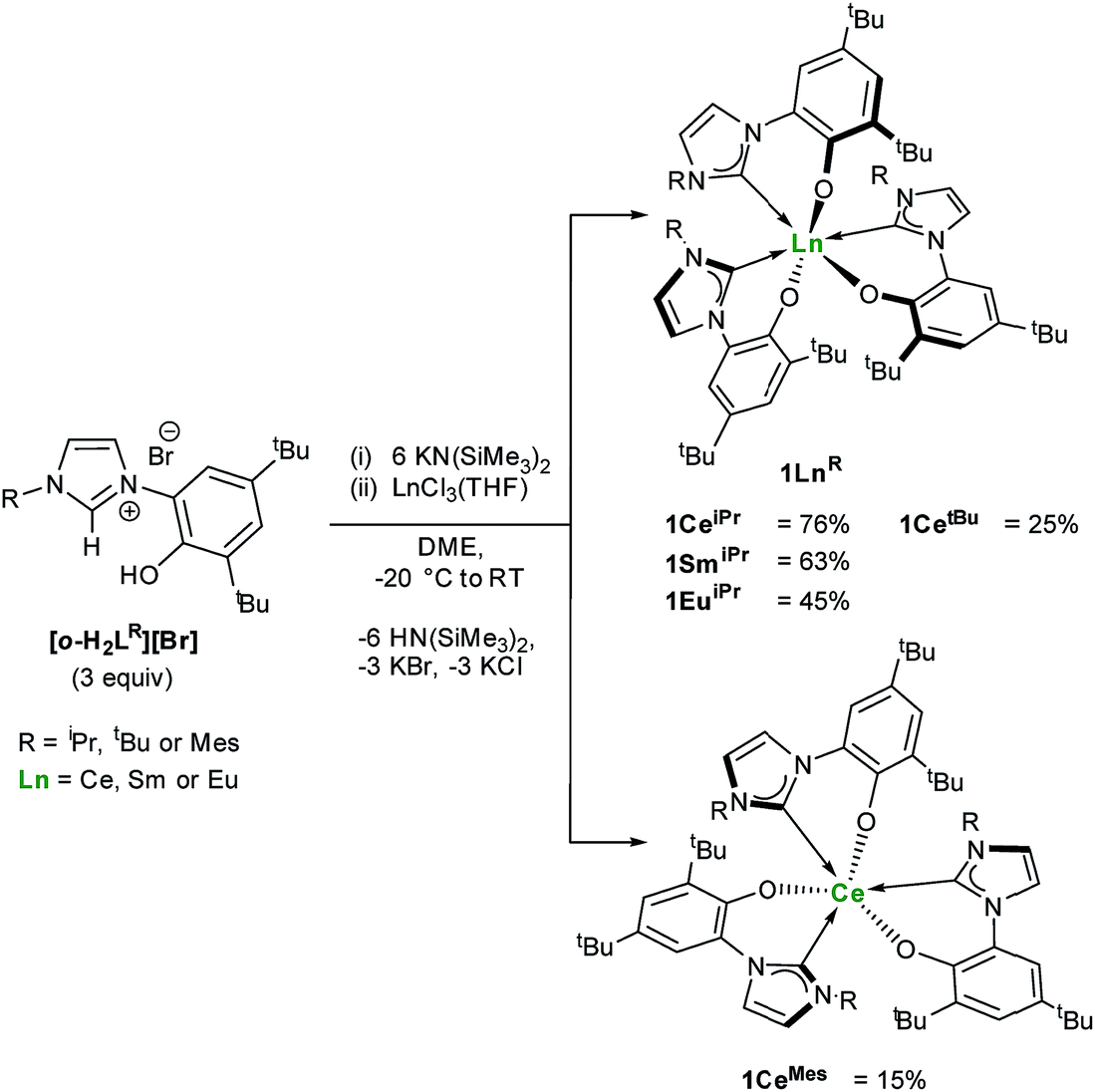 | ||
| Scheme 1 Synthesis of homoleptic lanthanide(III) complexes 1LnR. | ||
The 1H NMR spectra of all four lanthanide complexes bearing alkyl R groups (iPr or tBu) contain a complex set of paramagnetic resonances indicating C1 symmetry and a unique environment for each ligand. In agreement with the 1H spectrum of 1CeiPr the 13C{1H} NMR spectrum is also complicated, containing three carbene chemical shifts (δ = 174.8 ppm, 187.8 ppm, 192.3 ppm), slightly broadened compared to the rest of the spectrum (average fwhm 12 Hz), and shifted compared to similar diamagnetic lanthanide NHC complexes (normal region ≈ 200–238 ppm for YIII and CeIV).14,17,19,29,30 However in contrast, spectra of 1CeMes contain a single set of paramagnetically shifted resonances indicating C3 symmetry in solution on the 1H NMR spectroscopic timescale and 13C NMR spectroscopy of 1CeMes displays a single carbene resonance at 184.2 ppm.
These ligand orientation differences are rationalized by consideration that three planar mesityl groups pack more easily than the aryloxide/tert-butyl groups would, and that the tert-butyl/iso-propyl steric repulsions are less prescriptive. The C3-symmetric complex would also be favoured if π-stacking between the mesityl substituent and an adjacent imidazolin-2-ylidine ring is possible. This high degree of steric crowding is used to rationalise the failed synthesis of related but bulkier diisopropylphenyl containing aryloxide-carbene ligands. Reactions aimed at targeting the mono- and bis-alkoxy–NHC analogues using this synthetic method yielded only the tris-ligand complex and unreacted LnCl3 while the targeted synthesis of a Sm(II) analogue results in spontaneous oxidation and isolation of Sm(III) compound, 1SmiPr (see ESI†).
Single crystal X-ray analyses show that the fac- and mer-isomers are retained in the solid state for 1CeMes and 1CeiPr respectively (see Fig. 1). The coordination geometry of cerium in each is a pseudo-octahedral geometry defined by average C–Ce–C bond angles (172.93(18)°/88.41(18)° and 102.33(9)°) and OArCeOAr bond angles (154.05(15)°/102.44(16)° and 94.82(9)°). The average Ce–C bond distances of 1CeiPr and 1CeMes are 2.742(6) Å and 2.814(3) Å with the former within the regular range of a lanthanide–carbene bond. To the best of our knowledge the latter is the longest aryloxide tethered metal–carbene bond and amongst the longest lanthanide–carbene bonds known, consistent with the proposed high degree of hemilability. For 1CeMes there is a conceivable offset aromatic donor–acceptor interaction between the electron deficient imidazolin-2-ylidine and the electron rich mesityl with an average centroid distance of 4.36 Å, within the upper limits of face-centred π-stacking.31,32
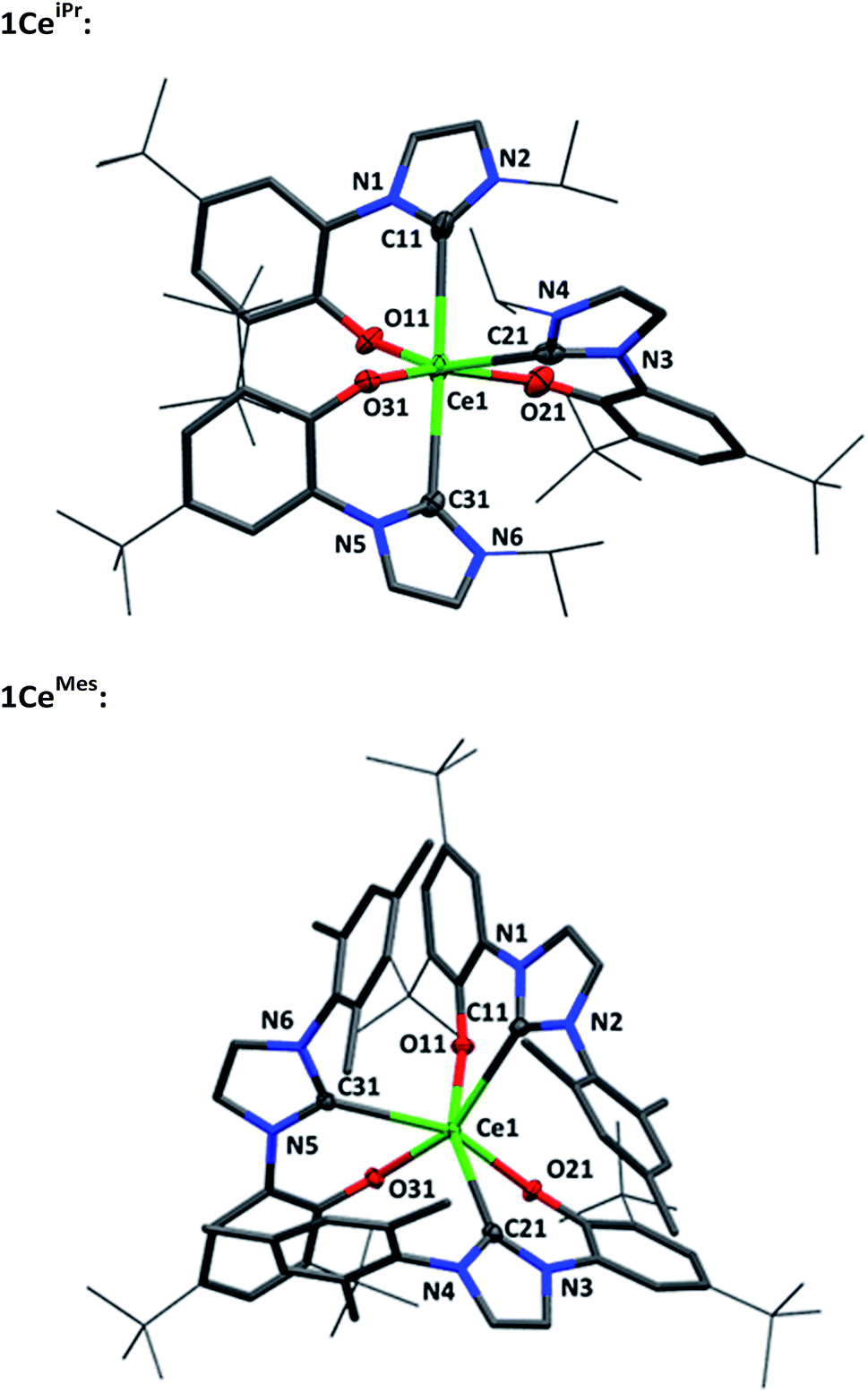 | ||
| Fig. 1 Molecular structures of 1CeiPr (upper) and 1CeMes (lower) with Ce, O and Ccarbene shown at 50% ellipsoid probability, framework and peripheral carbon atoms drawn capped stick and wireframe respectively, and H and lattice solvent omitted for clarity. Selected distances (Å) and angles (°) for 1CeiPr: Ce1–C11 2.747(6), Ce1–C21 2.694(6), Ce1–C31 2.785(7), Ce1–O11 2.349(4), Ce1–O21 2.277(4), Ce1–O31 2.283(5), C11–Ce1–C21 88.28(18), C11–Ce1–C31 172.93(18), C21–Ce1–C31 88.53(18), O11–Ce–O21 154.05(15), O11–Ce–O31 107.42(15), O21–Ce–O31 97.45(16), C11–Ce1–O11 68.68(16), C21–Ce1–O21 69.09(18), C31–Ce1–O31 69.66(17); for 1CeMes: Ce1–C11 2.823(3), Ce1–C21 2.814(3), Ce1–C31 2.806(3), Ce1–O11 2.266(2), Ce1–O21 2.264(2), Ce1–O31 2.251(2), C11–Ce1–C21 105.49(9), C11–Ce1–C31 100.01(9), C21–Ce1–C31 101.48(9), O11–Ce1–O21 93.66(8), O11–Ce1–O31 96.58(8), O21–Ce1–O31 94.23(8), C11–Ce1–O11 65.85(9), O21–Ce1–O21 66.82(8), O31–Ce1–O31 66.25(9). | ||
Reactivity of 1CeR complexes
Exposure of a solution of 1CeR to an atmosphere of carbon dioxide results in the instant and quantitative formation of 2CeR (Ln(LR·CO2)3) as observed by the precipitation of a beige solid (hexanes reaction solvent) or monitoring by 1H NMR spectroscopy (benzene reaction solvent), Scheme 2. As anticipated for a complex with a hemilabile metal–NHC bond, the CO2 exclusively inserts into the three Ce–C bonds, and pleasingly, and in contrast to the complexes with more flexible, bidentate alkoxide–NHCs, the rest of the molecule remains relatively unperturbed, with no evidence of ligand redistribution between metal centres. Samples of 2CeiPr and 2CetBu held at elevated temperatures under dynamic vacuum (100 °C, 10−3 mbar) show no loss of CO2. However, a sample of 2CeMes shows some loss of CO2 under dynamic vacuum (25 to 100 °C, 10−3 mbar), that is fully reversible. Solution phase analysis of the material formed shows it to be a complicated mixture that could be oligomeric, but the material is quantitatively converted back to 2CeMes upon re-exposure to an atmosphere of CO2.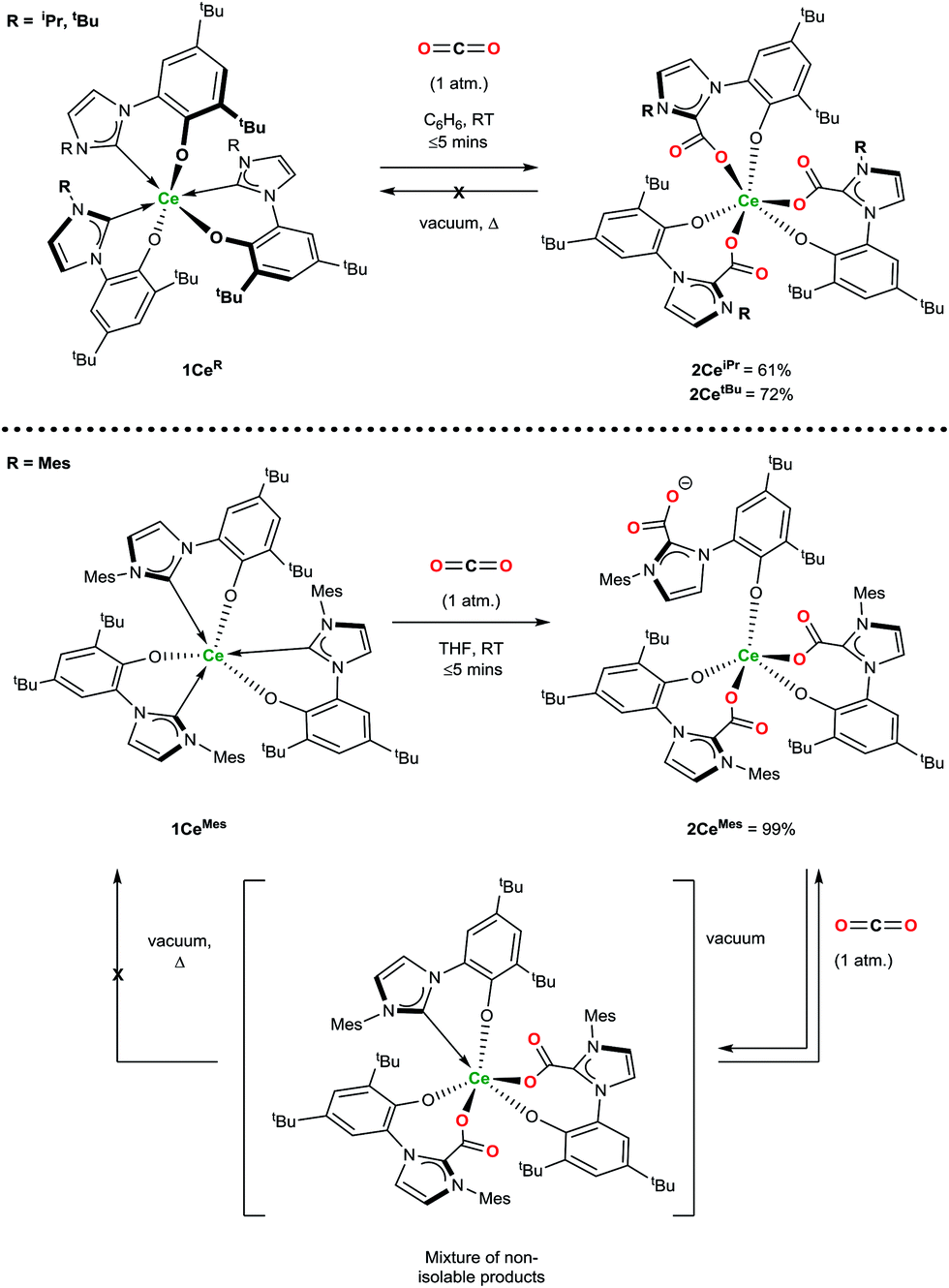 | ||
| Scheme 2 Reactivity of 1CeR with CO2 that forms the triply CO2–NHC inserted adducts irreversibly (2CeiPr and 2CetBu) and reversibly (2CeMes). | ||
1H NMR spectroscopic analysis reveals that the N-alkyl functionalised 2CeR complexes have C3 symmetry, i.e. a fac-conformation of the three bidentate ligands. The 13C NMR spectra contain diagnostic CO2 carbon resonances for 2CeiPr and 2CetBu (δ = 173.1 ppm and 173.5 ppm respectively) at significantly higher frequency than known organic NHC·CO2 compounds (≈20 ppm)7,8 as might be anticipated from proximity to the paramagnetic metal center. The FTIR spectrum of 2CeiPr shows a characteristic absorption at 1666 cm−1 (typical range ∼ 1630–1690).8,33,34 The conversion of 1CeMes to 2CeMes results in a lowering of symmetry from C3 to C1 according to room temperature solution spectroscopies. The 1H NMR spectrum shows three broadened sets of paramagnetic ligand resonances, and two C–O stretches observable in the FTIR spectrum (1678 and 1715 cm−1). We suggest that due to steric hindrance of three mesityl groups that one of the imidazolium carboxylate units is non-coordinating in solution.
Single crystal X-ray analysis confirms that CO2 insertion products 2CeiPr and 2CetBu have a pseudo-trigonal prismatic geometry with C3-symmetric fac-arrangement described by the average OAr–Ce–OAr bond angles (97.03(10)° and 95.94(8)° respectively) and OCO–Ce–OCO bond angles (77.77(10)° and 80.48(8)°) (Fig. 2). The average Ce–OCO bond length is within the regular bond length range at (2.472(6) Å and 2.473(2) Å respectively) suggesting a strong degree of stabilisation despite an increase of metal chelate ring size from 6 to 8.
The substrate scope was further explored with carbon disulfide and other isoelectronic (hetero)allenes shown in Scheme 3. Interestingly, treatment of a benzene solution of 1CeiPr with excess carbon disulfide at temperatures up to 80 °C shows no reaction. This differs from the alkoxide-tethered carbene complex D for which the product arising from the insertion of CS2 into two (of the three) M–C bonds was characterized.20 The higher reactivity of CO2 compared to CS2 in this system is reasonable considering the stronger affinity of Ce for oxygen, and the lower dipole moment in the latter reagent.
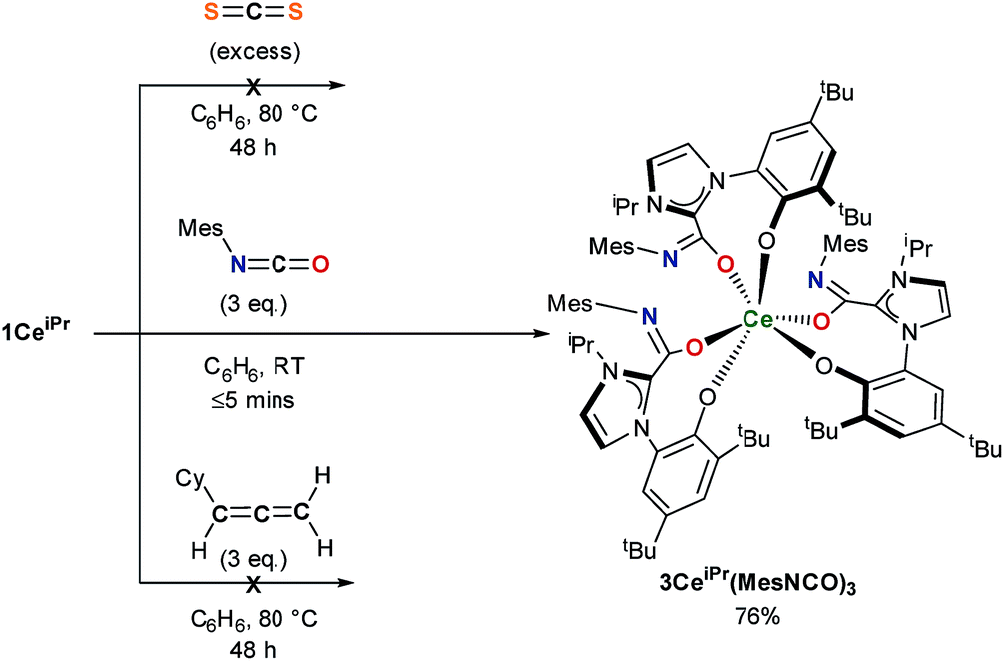 | ||
| Scheme 3 Treatment of 1CeiPr with reagents isoelectronic to CO2. | ||
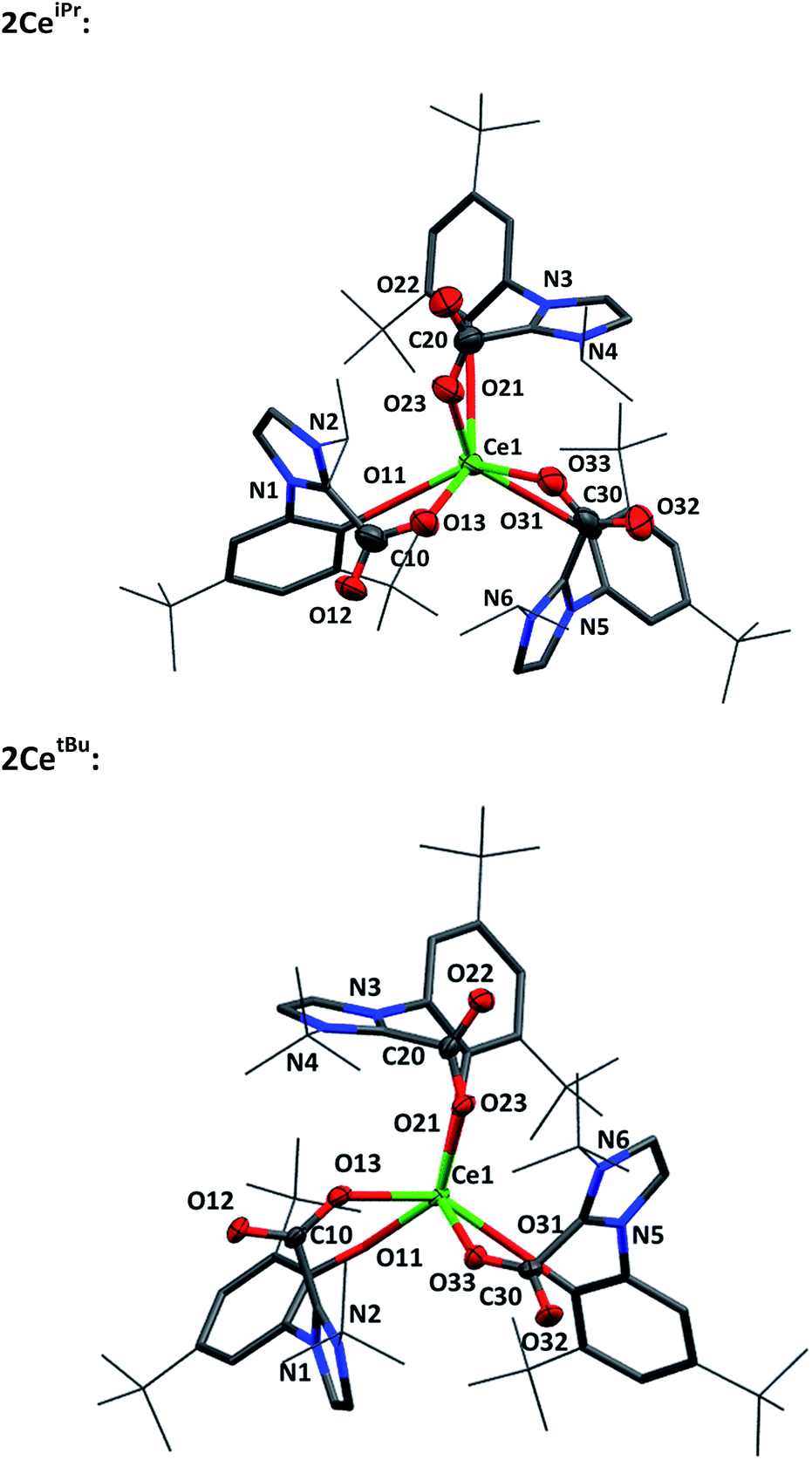 | ||
| Fig. 2 Molecular structures of 2CeiPr (upper) and 2CetBu (lower) with carboxylate and Ce atoms shown at 50% ellipsoid probability, framework and peripheral carbon atoms drawn capped stick and wireframe respectively, and H and lattice solvent omitted for clarity. Selected distances (Å) and angles (°) for 2CeiPr: Ce1–O11 2.790(5), Ce1–O21 2.274(5), Ce1–O31 2.256(6), Ce1–O13 2.468(6), Ce1–O23 2.482(6), Ce1–O33 2.466(6), O11–Ce1–O21 97.18(19), O11–Ce1–O31 99.3(2), O21–Ce1–O31 94.8(2), O13–Ce1–O23 77.02(19), O13–Ce1–O33 76.68(19), O23–Ce1–O33 79.6(2), O11–Ce1–O13 75.72(19), O21–Ce1–O23 75.7(2), O31–Ce1–O33 75.9(2). For 2CetBu: Ce1–O11 2.261(2), Ce1–O21 2.268(2), Ce1–O31 2.274(2), Ce1–O13 2.473(2), Ce1–O23 2.477(2), Ce1–O33 2.470(2), O11–Ce1–O21 95.38(8), O11–Ce1–O31 95.25(8), O21–Ce1–O31 97.20(8), O13–Ce1–O23 76.76(8), O13–Ce1–O33 80.89(8), O23–Ce1–O33 80.80(8), O11–Ce1–O13 75.50(8), O21–Ce1–O23 74.77(2), O31–Ce1–O33 75.03(8). | ||
Treatment of a benzene or THF solution of 1CeiPr with three equivalents of mesityl isocyanate (MesNCO) immediately results in the insertion of isocyanate into all three Ce–NHC bonds, affording a pale-yellow solution from which the tris-azoliumamidate 3CeiPr(MesNCO)3 can be readily isolated as colourless microcrystalline powder in 76% yield, Scheme 3. No dimer or trimer isocyanate products were observed as a comparison to “free” NHC isocyanate chemistry.9 As could be expected, the non-polar and more sterically hindered cyclohexylallene shows no reactivity with 1CeiPr.
In the reaction of 1CeiPr with three equivalents of tert-butyl isocyanate (tBuNCO) in benzene or THF, two molecules of isocyanate insert to form 3CeiPr(tBuNCO)2, however in DME solution, three molecules insert to form 3CeiPr(tBuNCO)3 as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the fac- and mer-isomers observable by 1H NMR spectroscopy, Scheme 4. We suggest that in the former two solvents, the steric bulk of the tert-butyl groups restricts access to the third equivalent, but the stronger, bidentate donor solvent DME increases the lability of the NHC groups, enabling three insertions to occur. If 1CeiPr is treated with 3 equivs of tert-butyl isothiocyanate (tBuNCS) at 80 °C in benzene or THF, a single equivalent of isothiocyanate inserts to form 3CeiPr(tBuNCS) while in DME two equivalents of isothiocyanate insert to form 3CeiPr(tBuNCS)2.
:1 mixture of the fac- and mer-isomers observable by 1H NMR spectroscopy, Scheme 4. We suggest that in the former two solvents, the steric bulk of the tert-butyl groups restricts access to the third equivalent, but the stronger, bidentate donor solvent DME increases the lability of the NHC groups, enabling three insertions to occur. If 1CeiPr is treated with 3 equivs of tert-butyl isothiocyanate (tBuNCS) at 80 °C in benzene or THF, a single equivalent of isothiocyanate inserts to form 3CeiPr(tBuNCS) while in DME two equivalents of isothiocyanate insert to form 3CeiPr(tBuNCS)2.
 | ||
| Scheme 4 Differences in reactivity of 1CeiPr with sterically hindered isocyanates and isothiocyanates depending on solvent, and ligand substituents to afford 3CeiPr(tBuNCO)2, 3CeiPr(tBuNCO)3, 3CeiPr(tBuNCS), and 3CeiPr(tBuNCS). | ||
Single crystals of 3CeiPr(tBuNCS)2 were grown by slow diffusion of heptane into a toluene solution. An X-ray diffraction study, Fig. 3, reveals a pseudo-trigonal prismatic molecular geometry at the metal center in the solid state. The Ce–S bond lengths average at 3.022 Å, and the Ce–C bond (2.716 Å) is only a little shorter than the average Ce–C bond length in the parent compound 1CeiPr (2.742 Å). The obtuse S–Ce–S bond angle (143.91°) and chelate angle of each bidentate ligand is within the expected range; S–Ce–OAr (78.75° avg.) and C–Ce–OAr (69.93°).
 | ||
| Fig. 3 Molecular structure of 3CeiPr(tBuNCS)2 with selected C and non-C/H atoms shown at 50% ellipsoid probability, framework and peripheral carbon atoms drawn capped stick and wireframe respectively, and H and lattice solvent omitted for clarity. Selected average distances (Å) and angles (°) for 3CeiPr(tBuNCS)2: Ce1–S10 2.996(12), Ce1–S20 3.048(12), Ce1–C31 2.716(4), Ce1–O11 2.280(3), Ce1–O21 2.273(3) Ce1–O31 2.277(3), S10–Ce1–S20 143.91(3), S10–Ce1–C31 126.96(9), S20–Ce1–C31 80.70(7), S10–Ce1–O11 78.71(8), S20–Ce1–O21 78.79(7), C31–Ce1–O31 69.93(11). | ||
Catalytic applications of 2CeR complexes
The formation of cyclic carbonates from epoxides and carbon dioxide was chosen for a preliminary study of the catalytic activity of the tris(ligand) CO2 adducts 2CeiPr and 2CeMes. Both free base NHCs and imidazolium carboxylates can be used as catalysts for the formation of cyclic carbonates from epoxides and carbon dioxide under high temperatures and pressures (up to 120 °C and 20 atm), while rare earth initiators are known to function at lower temperatures and/or pressures, a co-catalyst is usually required.11,35Scheme 5 shows how under an atmosphere of carbon dioxide, 1 mol% of 2CeMes catalyses the conversion of propylene oxide to propylene carbonate with 22% conversion at 80 °C in THF over 7 days, a much higher activity than the imidazolium carboxylates alone. On the other hand, the more compact 2CeiPr shows no reactivity. The solid-state structures show a higher steric congestion in the LMes adduct 1CeMes, and IR and NMR spectroscopies confirm different ligand solution environments for 2CeMes, suggesting both the Ce–Ccarbene and Ce–CCO2 interactions are weaker and more labile for the Mes system. We propose that the catalysis requires a combination of Lewis base type NHC–CO2 activation, and Lewis acid type Ce-epoxide activation.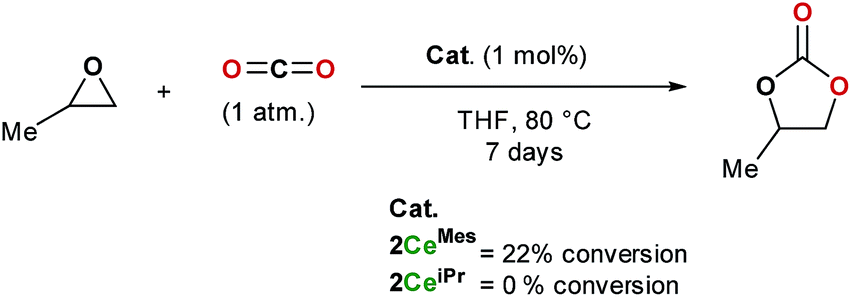 | ||
| Scheme 5 Catalytic formation of propylene carbonate from propylene oxide in an atmosphere of carbon dioxide using 2CeMes and 2CeiPr. | ||
Synthesis of the heteroleptic substituted NHC analogues
To target reactions with single equivalents of CO2, reactions designed to make complexes containing a single NHC ligand were carried out. The reactions of the ligands [o-H2LR][Br], R = Me, iPr and equimolar amounts of Li(THF)[CeN(iPr2)4] only afford clean material in low yields and significant decomposition can be observed. Adding an additional bromide source improves the yield of the mono-NHC–Ce complexes 4CeMe and 4CeiPr (Ce2Br4LR(iPr2N)2Li2(THF)2) to a moderate level (20% and 38% respectively, see Scheme 6). | ||
| Scheme 6 Reactions to target mono–NHC Ln complexes that afford 4CeMe, 4CeiPr and the hexanuclear 5CeMe that is the by-product isolated as single X-ray quality crystals for R = Me. | ||
Crystallographic analysis reveals a dimeric structure still containing unreacted base and lithium ions (see ESI†). A complicated bis(ligand) Li4Ce6 cluster 5CeMe, in which each ligand has been deprotonated at the NHC backbone (in the 4-position) yielding a dianionic OC ligand that bridges two cerium cations, is isolated in low yield as orange crystals that are suitable for single crystal diffraction studies (Fig. 4 and ESI†). Syntheses to target 4 or 5 in the absence of an additional bromide source, or from cerium bromide starting materials, yield only complicated mixtures of compounds in our hands.
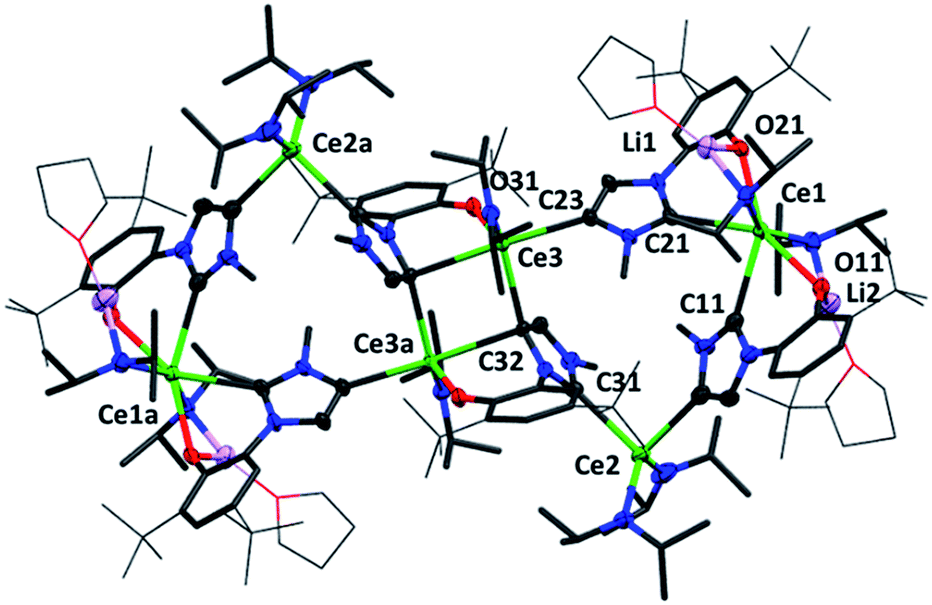 | ||
| Fig. 4 Molecular structure of 5CeMe with imidazolium, Ce, O and Li atoms shown at 50% ellipsoid probability, framework atoms drawn capped stick and coordinated solvents, peripheral carbon atoms wireframe, and H and lattice solvent omitted for clarity. Selected distances (Å) and angles (°) for 5CeMe: Ce1–C11 2.743(5), Ce1–C21 2.728(5), Ce2–C13 2.667(7), Ce2–Ce31 2.710(5), Ce3–C23 2.651(6), Ce3–C32 2.854(4), Ce3–C32a 2.685(6), Ce1–O11 2.517(4), Ce1–C21 2.462(4), Ce3–O31 2.232(3), C11–Ce1–O11 65.1(2), C11–Ce1–C21 82.1(2), C21–Ce1–O21 65.9(1), C13–Ce2–C31 94.4(2), Ce3–C32–Ce3a 98.5(1), C32–Ce3–C32a 81.5(1), C32–Ce3–O31 71.1(1), C32–Ce3–C23a 163.8(2). | ||
Synthesis of the para-aryloxide substituted NHC analogues
The analogous complexes of the para-substituted aryloxide ligand p-LR separate the Lewis acid and Lewis base centers, and thus offer a potential insight into the importance of the adjacent Ln centre and the nucleophilic NHC in the combined activation of CO2 and the other unsaturated substrates. A modification of Wang's proligand synthesis using saturated-backbone imidazoline analogues allows access to the para-functionalized proligand in 15% yield.27 Treatment of this N-mesityl functionalized proligand [p-H2LMes][X], (p-LMes = 4-O-2,6-tBu2-C6H2(1-C{N(CH)2NMes}), X = PF6, Br) with either MN(SiMe3)2 (M = Na or K) in THF at room temperature affords the group 1 NHC salts 6MMes [(M(p-LMes)]n, (M = Na, K) in quantitative yield, Scheme 7. The solid-state structures of both are polymeric, according to single crystal X-ray data, with 6NaMes displaying repeating C–[Na–(μ-ArO)2–Na]–C diamond units, while 6KMes displays a perpendicular ArO–K–C arrangement, see ESI.† | ||
| Scheme 7 Reaction to target the para-ligand adducts 7LnMes (Ln = Y, Ce). | ||
Salt 6MMes can be treated with YCl3 or CeCl3 at −20 °C to synthesise 7LnMes (Ln(p-LMes)3(THF)2 where Ln = Y, Ce) in 56% and 30% yield as yellow powders. Due to their high reactivity all the compounds start to degrade rapidly making further analysis difficult, and the complexes are best stored in their protonated form, i.e. [Ln(p-HLMes)3(THF)2]X3.
Analysis of 7YMes by 13C NMR spectroscopy reveals a characteristic carbene signal (δ 238.2 ppm) is observed as a singlet indicating that the carbene does not bind to yttrium in solution. These complexes were found to be extremely air sensitive, were only ever isolated as KCl and HCl salts and became highly insoluble in a range of solvents so were not pursued further (see ESI†).
Conclusions
The tris(ortho-aryloxide–NHC) rare earth complexes Ln(LR)3 are readily isolated and are the thermodynamic sink in this system. Insertion of CO2 or a range of isoelectronic (hetero)allenes into the labile cerium carbene bond in Ce(LR)3 shows a dependence on solvent and N-R group on LR that enables control of the degree of insertion. The CO2-insertion products form cleanly at ambient pressure, but only reversibly for the bulky mesityl substituted Ce(LMes)3. The reversibility of the CO2 insertion appears to be crucial for further reactivity as only Ce(LMes·CO2)3 is an active catalyst for the conversion of propylene oxide to propylene carbonate. Although yields in these preliminary tests using low temperatures and one atmosphere of CO2 are low, the catalyst is more active than a monodentate NHC, and when the ligands are better fit to the metal in Ce(LiPr·CO2)3, the complexes are inactive. We propose that the catalysis requires a combination of Lewis base type NHC–CO2 activation, and Lewis acid type Ce-epoxide activation.Although the tris-ligand complexes are the thermodynamic sink in the system, the mono-ligand complexes can be isolated from reactions using LiCe(NiPr2)4 as a starting material; LiBr adducts [Ce(LR)(NiPr2)Br·LiBr(THF)]2 (R = Me, iPr) are reported, along with a hexanuclear N-heterocyclic dicarbene complex [Li2Ce3(OArCMe–H)3(NiPr2)5(THF)2]2 which is formed as a by-product. The analogous para-aryloxide–NHC proligand p-LMes has been made for comparison. The group 1 salts [Na(p-LMes)]n and [K(p-LMes)]n form two different types of infinite coordination polymers through metal carbene-bonds. Synthesis of the analogous lanthanide para-aryloxide NHC complexes Ln(p-LMes)3(THF)2 (Ln = Y, Ce) is possible but they are all highly reactive leading to rapid degradation. Therefore straightforward Lewis pair separated chemistry cannot usefully be carried out.
Further work is underway to use the C3-symmetric tris(ortho-aryloxide–NHC)–CO2 adducts in asymmetric catalysis and to expand the scope of the CO2 functionalisation.
Experimental
General details
All manipulations were carried out under a dry, oxygen-free atmosphere of nitrogen using standard Schlenk and glovebox technique. All gases were supplied by BOC gases UK. All glassware items, cannulae and Fisherbrand 1.2 μm retention glass microfibre filters were dried in a 170 °C oven overnight before use. Benzene and DME were distilled from potassium and stored over 4 Å molecular sieves. Hexane, heptane, THF, and toluene were degassed and purified by passage through activated 4 Å molecular sieves or activated alumina towers and stored over 4 Å molecular sieves. Deuterated solvents, benzene-d6, THF-d8 and pyridine-d5 were dried over potassium, vacuum-transferred, and freeze–pump–thaw degassed prior to use. 1H and 13C NMR spectra were recorded on Bruker AVA400, AVA500, or PRO500 spectrometers at 300 K. Chemical shifts are reported in parts per million, δ, referenced to residual proton resonances, and calibrated against external TMS. Infrared spectra were recorded on a Perkin Elmer Spectrum 65 FT-IR spectrometer as nujol mulls between KBr disks. Mass spectra were acquired using a SolariX FT-ICR (12 T) (Bruker UK Ltd) equipped with a Bruker APPI source. Samples were prepared as ca. 1 mM toluene solutions of the substrate. Elemental analyses were carried out at London Metropolitan University, London, UK.NaN(SiMe3)2, KN(SiMe3)2,36 and the [o-H2LR][Br]27,28 pro-ligands were prepared according to the literature procedures. YCl3(H2O)n and LnCl3(H2O)n were purchased and stirred overnight with TMSCl (40 equiv.) in THF before vacuum drying for several hours.
General procedure 1 – synthesis of 1LnR
To a suspension of [o-H2LR][Br] (3 equiv.) in DME (0.1 M) KN(SiMe3)2 (6 equiv.) was added and the resulting mixture was stirred for 5 min at −20 °C while and warmed to room temperature. LnCl3(THF)n (1 eq.) was added, and the resulting mixture was stirred at room temperature for 2 h. Volatiles were removed under reduced pressure, the crude product was extracted three times with hexane and the combined filtrates were concentrated to saturation and cooled to −20 °C overnight. The resulting suspension was filtered and the solid collected and dried under vacuum to give the title compound which was stored at −20 °C under a nitrogen atmosphere.General procedure 2 – synthesis of 2CeR
A solution of 1CeR (3 equiv.) in benzene, toluene, hexane or THF (0.5 M) was freeze–pump–thaw degassed 3 times and exposed to an atmosphere of dry CO2 in a Teflon-valved ampoule. The solvent was removed under reduced pressure, and the crude product was extracted with toluene and concentrated to saturation and cooled to −30 °C overnight. The resulting suspension was filtered and dried under vacuum to yield the title compound which was stored at −20 °C under a nitrogen atmosphere.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding through the Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT, EP/L016419/1, R. W. F. K.), EP/J018139/1 and the UK Catalysis Hub (EP/K014714/1, P. L. A., C. W.), EP/M010554/1 (P. L. A.). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 740311, P. L. A.). K. W. thanks the China Scholarship Council (CSC) for a postgraduate fellowship. P. L. A., M. W. M., J. R. and F. E. K. thank the Technische Universität München – Institute for Advanced Study, funded by the German Excellence Initiative. A. D. S. thanks the Royal Society for a Wolfson Research Merit Award. C. J. thanks the DAAD for a scholarship, and C. J. and J. R. thank the TUM Graduate School for financial support. We would also like to thank Dr Colin Logan Mackay of the University of Edinburgh for mass spectrometry and the University of Edinburgh SIRCAMS facility for use of the FTICRMS.Notes and references
- D. H. Gibson, Chem. Rev., 1996, 96, 2063–2096 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933–5948 CrossRef PubMed.
- S. Ozaki, Chem. Rev., 1972, 72, 457–496 CrossRef.
- B. Bantu, G. M. Pawar, U. Decker, K. Wurst, M. Schmidt Axel and R. Buchmeiser Michael, Chem.–Eur. J., 2009, 15, 3103–3109 CrossRef CAS PubMed.
- B. C. Norris and C. W. Bielawski, Macromolecules, 2010, 43, 3591–3593 CrossRef CAS.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- H. A. Duong, M. J. Cross and J. Louie, Org. Lett., 2004, 6, 4679–4681 CrossRef CAS PubMed.
- B. C. Norris, D. G. Sheppard, G. Henkelman and C. W. Bielawski, J. Org. Chem., 2011, 76, 301–304 CrossRef CAS PubMed.
- H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS PubMed.
- A. J. Arduengo, M. Tamm, S. J. McLain, J. C. Calabrese, F. Davidson and W. J. Marshall, J. Am. Chem. Soc., 1994, 116, 7927–7928 CrossRef CAS.
- R. D. Fischer, Angew. Chem., Int. Ed. Engl., 1994, 33, 2165–2168 CrossRef.
- (a) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed; (b) H. Schumann, M. Glanz, J. Winterfeld, H. Hemling, N. Kuhn and T. Kratz, Angew. Chem., Int. Ed., 1994, 33, 1733–1734 CrossRef.
- T. Mehdoui, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Chem. Commun., 2005, 2860–2862 RSC.
- Y. Haisheng, Z. Yong, S. Hongmei and S. Qi, Eur. J. Inorg. Chem., 2009, 2009, 1920–1925 CrossRef.
- H. Yao, J. Zhang, Y. Zhang, H. Sun and Q. Shen, Organometallics, 2010, 29, 5841–5846 CrossRef CAS.
- J. Zhang, H. Yao, Y. Zhang, H. Sun and Q. Shen, Organometallics, 2008, 27, 2672–2675 CrossRef CAS.
- Z.-G. Wang, H.-M. Sun, H.-S. Yao, Q. Shen and Y. Zhang, Organometallics, 2006, 25, 4436–4438 CrossRef CAS.
- P. L. Arnold, I. A. Marr, S. Zlatogorsky, R. Bellabarba and R. P. Tooze, Dalton Trans., 2014, 43, 34–37 RSC.
- ThermoFisher Scientific, Safety Data Sheet: FeCl3, https://www.fishersci.com/msds?productName=AC217091000%26produc, accessed 9 July, 2018.
- ThermoFisher Scientific, Safety Data Sheet: CeCl3,https://www.fishersci.com/shop/msdsproxy?productName=AC369750050%26productDescription=CERIUM, accessed 9 Jul, 2018.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- P. L. Arnold, Z. R. Turner, A. I. Germeroth, I. J. Casely, G. S. Nichol, R. Bellabarba and R. P. Tooze, Dalton Trans., 2013, 42, 1333–1337 RSC.
- W. Ding and D. Wang, Organometallics, 2014, 33, 7007–7010 CrossRef CAS.
- G. Prabusankar and P. Suresh, ChemistrySelect, 2017, 2, 9920–9923 CrossRef CAS.
- H. Ren, P. Yao, S. Xu, H. Song and B. Wang, J. Organomet. Chem., 2007, 692, 2092–2098 CrossRef CAS.
- M. Nirmala, G. Prakash, P. Viswanathamurthi and J. G. Malecki, J. Mol. Catal. A: Chem., 2015, 403, 15–26 CrossRef CAS.
- L. Arnold Polly, R. Turner Zoë, N. Kaltsoyannis, P. Pelekanaki, M. Bellabarba Ronan and P. Tooze Robert, Chem.–Eur. J., 2010, 16, 9623–9629 CrossRef CAS PubMed.
- I. J. Casely, S. T. Liddle, A. J. Blake, C. Wilson and P. L. Arnold, Chem. Commun., 2007, 5037–5039 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS PubMed.
- A. Tudose, A. Demonceau and L. Delaude, J. Organomet. Chem., 2006, 691, 5356–5365 CrossRef CAS.
- E. Brule, V. Guerineau, P. Vermaut, F. Prima, J. Balogh, L. Maron, A. M. Z. Slawin, S. P. Nolan and C. M. Thomas, Polym. Chem., 2013, 4, 2414–2423 RSC.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- J. Eppinger, E. Herdtweck and R. Anwander, Polyhedron, 1998, 17, 1195–1201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1856101–1856106, 1868204–1868209. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03312a |
| This journal is © The Royal Society of Chemistry 2018 |