
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed asymmetric hydrogenation of 2-substituted ketones via dynamic kinetic resolution†
Olga V.
Zatolochnaya
 a,
Sonia
Rodríguez
*a,
Yongda
Zhang
*a,
Kendricks S.
Lao
a,
Sergei
Tcyrulnikov
b,
Guisheng
Li
a,
Xiao-Jun
Wang
a,
Bo
Qu
a,
Soumik
Biswas
a,
Hari P. R.
Mangunuru
a,
Daniel
Rivalti
a,
Joshua D.
Sieber
a,
Jean-Nicolas
Desrosiers
a,
Joyce C.
Leung
a,
Nelu
Grinberg
a,
Heewon
Lee
a,
Nizar
Haddad
a,
Nathan K.
Yee
a,
Jinhua J.
Song
a,
Marisa C.
Kozlowski
*b and
Chris H.
Senanayake
a
a,
Sonia
Rodríguez
*a,
Yongda
Zhang
*a,
Kendricks S.
Lao
a,
Sergei
Tcyrulnikov
b,
Guisheng
Li
a,
Xiao-Jun
Wang
a,
Bo
Qu
a,
Soumik
Biswas
a,
Hari P. R.
Mangunuru
a,
Daniel
Rivalti
a,
Joshua D.
Sieber
a,
Jean-Nicolas
Desrosiers
a,
Joyce C.
Leung
a,
Nelu
Grinberg
a,
Heewon
Lee
a,
Nizar
Haddad
a,
Nathan K.
Yee
a,
Jinhua J.
Song
a,
Marisa C.
Kozlowski
*b and
Chris H.
Senanayake
a
aChemical Development, Boehringer Ingelheim Pharmaceuticals, Inc., 900 Old Ridgebury Road, Ridgefield, CT 06877, USA. E-mail: yongda.zhang@boehringer-ingelheim.com; sonrodrod@hotmail.com
bDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA. E-mail: marisa@sas.upenn.edu
First published on 23rd April 2018
Abstract
A new class of tunable heterophosphole dimeric ligands have been designed and synthesized. These ligands have enabled the first examples of Cu-catalyzed hydrogenation of 2-substituted-1-tetralones and related heteroaryl ketones via dynamic kinetic resolution, simultaneously creating two contiguous stereogenic centers with up to >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 98:2 er. The ligand-Cu complexes were isolated and characterized by single crystal X-ray, and DFT calculations revealed a novel heteroligated dimeric copper hydride transition state.
:1 dr and 98:2 er. The ligand-Cu complexes were isolated and characterized by single crystal X-ray, and DFT calculations revealed a novel heteroligated dimeric copper hydride transition state.
Transition metals such as Pd, Pt, Rh, Ru and Ir have been widely utilized for a large variety of catalytic transformations.1 However, the long-term sustainability of such processes has become an increasing concern due to the high cost, limited abundance and observed toxicity of noble metals.2 To overcome these issues, in recent years, there has been a major endeavor in the field to explore new synthetic methods utilizing non-precious metal catalysis.3 To this end, we have recently reported several novel methodologies employing Fe, Ni, Co and Cu-centered catalysts.4 For example, our laboratories have developed the first Cu-catalyzed asymmetric propargylation reaction of aldehydes4a whereas the key to obtaining high enantioselectivities was the use of our bidentate MeO-BIBOP ligand to control the stereochemical course of the reaction. MeO-BIBOP belongs to a family of P-chiral benzooxaphosphole (BOP) ligands which have demonstrated superior abilities to catalyze a range of asymmetric transformations.5 The modular design of the ligand core allows for high tunability of steric and electronic properties of the ligand to afford the desired reactivities and stereoselectivities (Scheme 1). Encouraged by our success with the BIBOP-Cu system, we aspired to design new bidentate benzooxaphosphole ligands to promote other non-precious metal catalyzed fundamental reactions such as asymmetric hydrogenation of ketones. Herein we report the discovery of a new class of heterophosphole dimeric ligands (BABIPhos) and their application in the first copper-catalyzed asymmetric hydrogenation of 2-substituted-1-tetralones and related heteroaryl ketones via dynamic kinetic resolution.
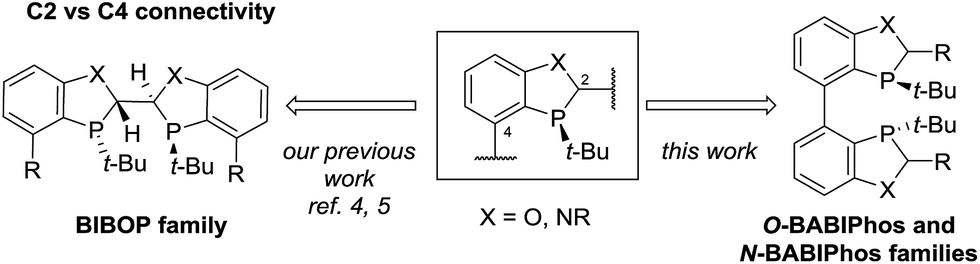 | ||
| Scheme 1 Dimeric benzoheterophosphole ligands. | ||
The Cu-catalyzed asymmetric reduction of ketones has been described in the literature using both silanes and hydrogen.6 The silane-based methods often require cryogenic temperatures and generate stoichiometric amounts of silane byproducts rendering them unattractive for scaleup.7 On the other hand, greener alternatives based on catalytic hydrogenation are more challenging and significantly less developed. Shimizu and co-workers reported the first asymmetric Cu-catalyzed ketone hydrogenation using BDPP as ligand obtaining good selectivities with ortho-substituted aryl and heteroaryl ketones.8 Beller's group described a more general system using monodentate binaphthophosphepine ligands that provided selectivities up to 77% ee.9 More recently, Hatcher and co-workers reported the use of modified BoPhoz ligands for ketone hydrogenation to afford optically enriched alcohol products with 75:25 to 98:2 er.10 As part of our long-standing interest in non-precious metal catalysis and engineering innovative catalytic systems, we sought to develop more efficient methods for Cu-catalyzed asymmetric ketone hydrogenation.
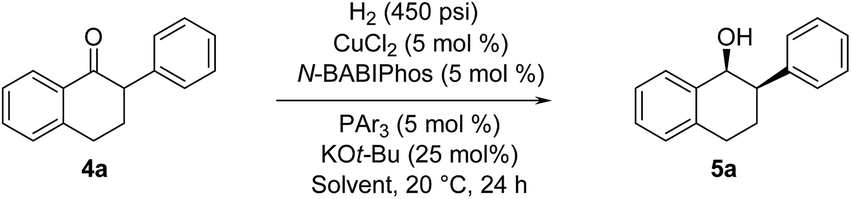
2-Aryl-1-tetralone derivatives were chosen as initial substrates for the study because the resulting chiral alcohols were required for one of our ongoing programs. Furthermore the relatively low pKa of the keto-benzylic position offers an opportunity for dynamic kinetic resolution (DKR), creating two adjacent stereocenters in one single operation. It should be noted that no examples of Cu-catalyzed asymmetric reduction of ketones via DKR have been previously reported.11
The asymmetric hydrogenation of racemic 2-phenyl-1-tetralone 4a was first evaluated using protocols reported by Shimizu,8 Beller9 and Hatcher10 (Table 1). Low conversions were observed under Shimizu's BDPP and Beller's CatASium KPh conditions. Hatcher's modified BoPhoz ligand provided 70% conversion after 24 h at 400 psi with moderate stereoselectivity of 88:12 dr and 83:17 er for the major diastereoisomer. Evaluation of chiral BIBOP ligand provided full conversion exclusively to the cis-diastereomer with 77:23 er, while the reaction with MeO-BIBOP gave less gratifying results. Phenyl and cyclohexyl BIBOP analogs exhibited no reactivity in this reaction. Despite the observed modest stereoselectivities in the initial screening, these results clearly showed that the anticipated DKR has occurred under the reaction conditions.
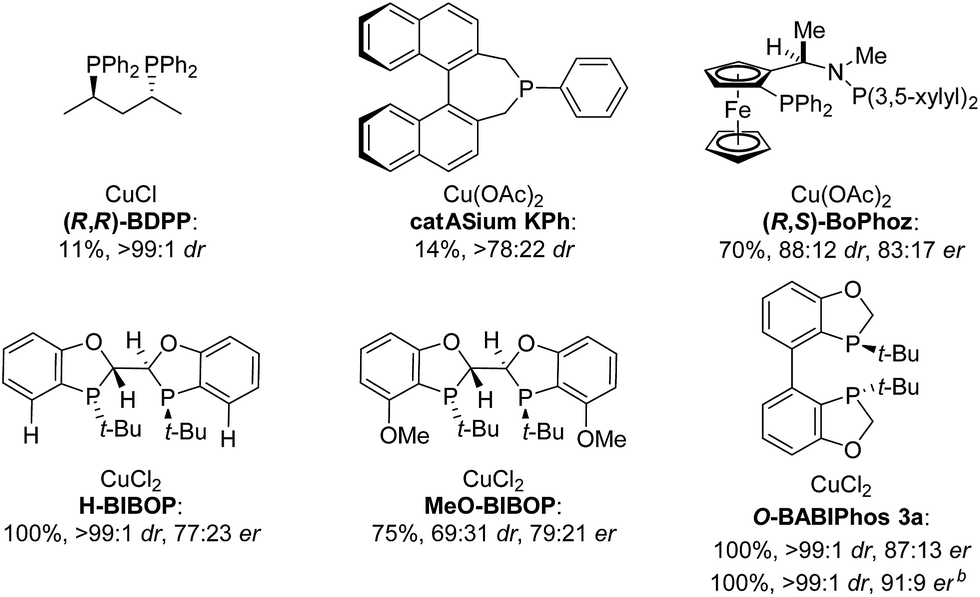
To further improve the reactivity and selectivity of the reaction, we decided to synthesize the alternative BOP dimer via the C4-coupling which might provide new ligands with different properties (Scheme 1).12 Our quest for this type of biaryl ligands began by exploring the reductive homocoupling of the corresponding benzooxaphosphole oxide triflate 1a (Scheme 2). After extensive investigation of the Pd-13 and Ni-catalyzed14 protocols for dimerizing aryl halides and triflates,15 it was found that under Pd-catalysis in the presence of BI-DIME, aryl triflate 1a was converted to the corresponding biaryl 2a in 84% yield. Additionally, the oxaphosphole ring of 2a was alkylated to form C2, C2′-substituted oxide analogs 2c–e. The phosphine oxides were subsequently reduced to the corresponding phosphines 3a,c–e (O-BABIPhos) giving access to a new class of bidentate ligands. This route was demonstrated on several hundred grams scale.
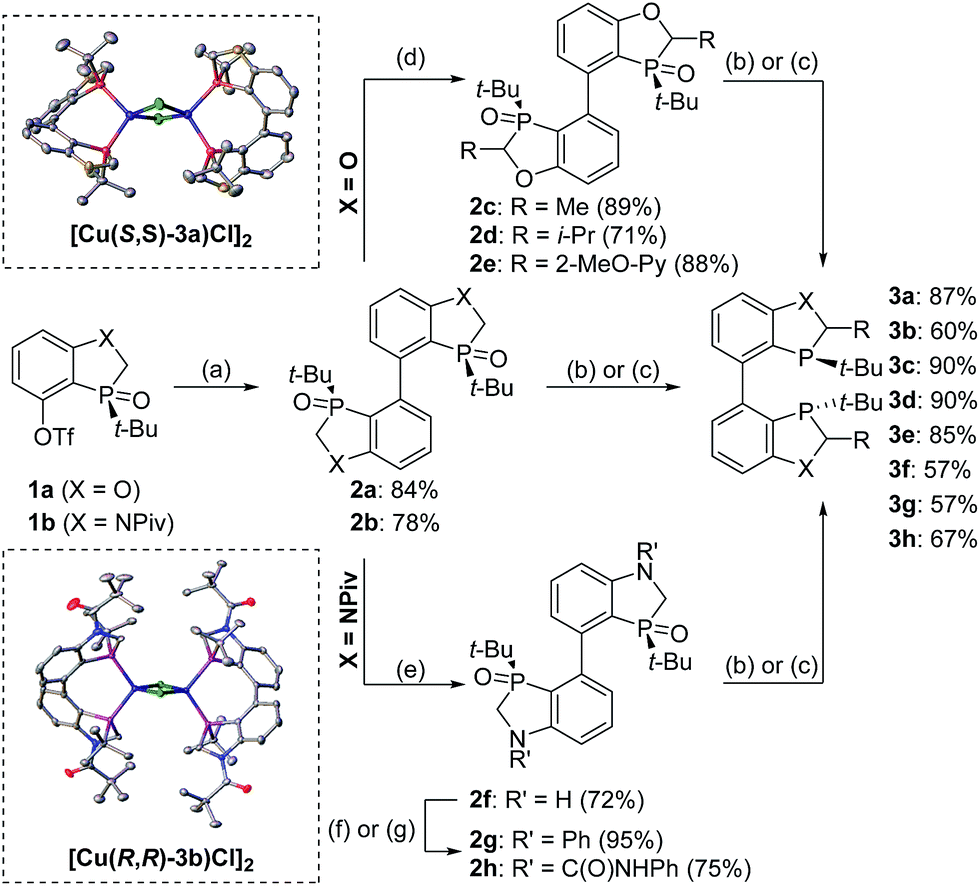 | ||
| Scheme 2 Synthesis of O-BABIPhos and N-BABIPhos. Conditions: (a) 2a (1 equiv.), Pd2(dba)3 (2.5 mol%), BI-DIME (5 mol%, 2a) or DTBPF (5 mol%, 2b), Et4NI (2 equiv.), Zn dust (3 equiv.), DMAc, 140 °C; (b) (Me2HSi)2O (3 equiv.), Ti(Oi-Pr)4 (2.3 equiv.), THF, 65 °C; (c) Et3SiH (3 equiv.), Et3N (3 equiv.), toluene, 110 °C; (d) LDA (2.5 equiv.); then RX (2.5 equiv.; 2c: MeI, 2d: i-PrI, 2e: 2-methoxy-6-(phenylsulfonyl)pyridine), THF, −78 °C to r.t.; (e) NaOH, MeOH/H2O, 50 °C; (f) 2g: PhBr, Pd2(dba)3 (5 mol%), RuPhos (20 mol%), NaOt-Bu (2.4 equiv.), THF, reflux; (g) 2h: PhNHCO (10 equiv.), i-Pr2NEt (3.6 equiv.), THF, reflux. BI-DIME = 3-(tert-butyl)-4-(2,6-dimethoxyphenyl)-2,3-dihydrobenzo[d] [1,3]oxaphosphole, DTBPF = 1,1′-bis(di-tert-butylphosphino)ferrocene, LDA = lithium diisopropylamide, RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl. | ||
With these ligands in hand, the ketone hydrogenation was carried out, and we were pleased to find that the reaction with O-BABIPhos ligand 3a provided high reactivities and selectivities up to 91:9 er of single diastereoisomer 5a (Table 1). However, modification of the R group in O-BABIPhos did not further improve the enantioselectivity. To obtain more selective ligands, a new class of dimeric azaphosphole ligands, N-BABIPhos were designed and prepared to further modulate the electronic properties of the ligand by introducing various functionalities on the nitrogen atom (Scheme 2). Dimerization of the nitrogen-containing triflate 1b proved to be much more challenging and a wide-scope ligand screen led to the identification of DTBPF as the best ligand that gave good yield of biaryl product 2b. Further modification at nitrogen gave quick access to N-BABIPhos analogs 3f–h.
We were delighted to find that the Cu-catalyzed hydrogenation of 2-phenyl-1-tetralone in t-AmOH using N-BABIPhos 3b as the ligand proceeded in complete diastereoselectivity and 97:3 er (Table 2, entry 1). Reactions with other azaphospholes bearing different N-substitutions gave overall less satisfactory results (entries 2–4). t-AmOH proved to be the solvent of choice for this transformation. Lower reactivity was observed when performing the reactions in t-BuOH as solvent while i-PrOH provided good reactivity but only 92:8 er (entries 5–6)16. The nature of ancillary phosphine played a key role in the reactivity of our new catalytic system. Thus, no reaction was observed in the absence of ancillary phosphine or using electron-deficient triarylphosphines (entries 7–8). The source of this phenomenon was further elucidated via DFT calculations (vide infra).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | PAr3 | %5ab | drc | erc |
| a Conditions: H2 (450 psi), CuCl2 (5 mol%), N-BABIPhos (5 mol%), PAr3 (5 mol%), KOt-Bu (25 mol%), t-AmOH, 20 °C. b Determined by comparison of relative HPLC integration of alcohol to ketone at 220 nm. c Determined by chiral HPLC. d t-BuOH as solvent. e i-PrOH as solvent. | |||||
| 1 | 3b | P(3,5-xylyl)3 | 100 | >99:1 |
97:3 |
| 2 | 3f | P(3,5-xylyl)3 | 50 | >99:1 |
96:4 |
| 3 | 3g | P(3,5-xylyl)3 | 100 | >99:1 |
86:14 |
| 4 | 3h | P(3,5-xylyl)3 | 50 | >99:1 |
94:6 |
| 5d | 3b | P(3,5-xylyl)3 | 66 | >99:1 |
96:8 |
| 6e | 3b | P(3,5-xylyl)3 | 100 | >99:1 |
92:8 |
| 7 | 3b | — | 0 | — | — |
| 8 | 3b | P(3,5-CF3-Ph)3 | 1 | — | — |
Both the Cu–O-BABIPhos and Cu–N-BABIPhos complexes were obtained in quantitative yields by treatment of CuCl with a stoichiometric amount of respective ligand in dichloromethane at reflux temperature for 2 h. The molecular structures of the complexes were unambiguously characterized by single crystal X-ray crystallography17 revealing a chloride-bridged dicopper complex such as [Cu(μ-Cl) (κ2-N-BABIPhos)]2 with a distorted tetrahedral geometry at both copper(I) centers, as observed for similar Cu complexes in the literature (Scheme 2).18
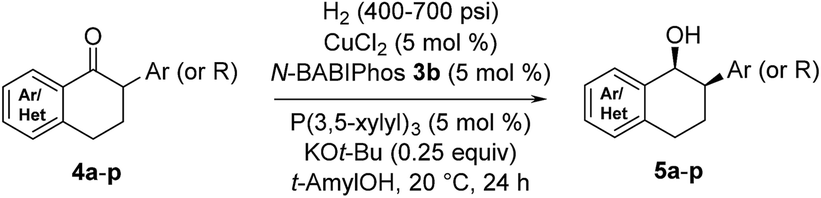
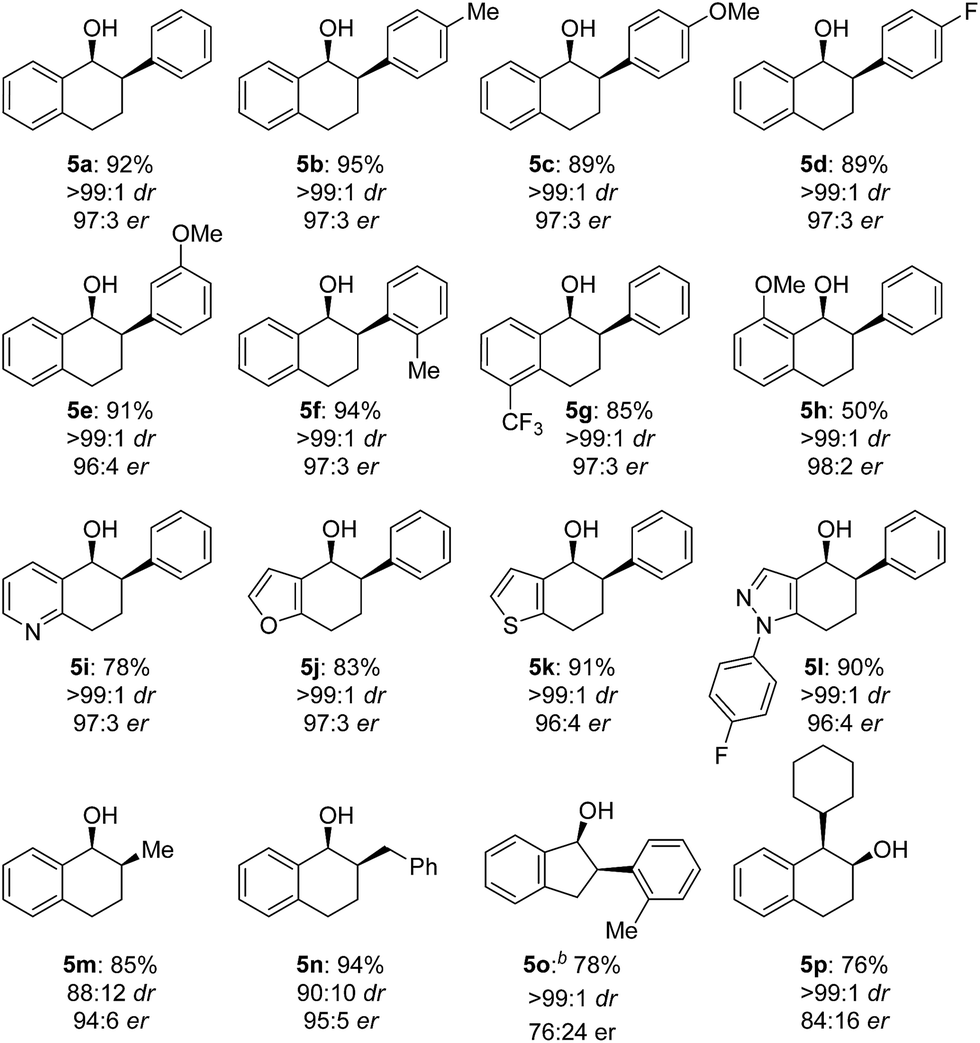
The scope of the reaction was next examined (Table 3). A variety of racemic 2-aryl-1-tetralones were smoothly reduced under our standard conditions with excellent enantioselectivities (96:4 to 98:2 er, products 5a–h). Reactions with heterocycle-containing substrates were equally effective. Pyridine, furan, thiophene and pyrazole-fused cyclohexanones underwent the Cu-catalyzed hydrogenation in high stereoselectivities (96:4 to 97:3 er, products 5i–l). Furthermore, the hydrogenative DKR of 2-alkyl-1-tetralones 4m and 4n proved to be highly enantioselective with good diastereoselectivities. Substituted indanone substrate 4o was reduced under the reaction conditions, albeit with a more modest er (76:24). Finally, hydrogenation of a 1-cyclohexyl-2-tetralone (4p) was also successful furnishing the desired product in synthetically useful enantioselectivity (84:16).
The resulting optically enriched tetralols are valuable building blocks for synthesis of bioactive molecules. For example, compound 5q (>99:1 dr, 97:3 er from asymmetric hydrogenation of 4q, Scheme 3) can be readily deoxygenated to 2-aryltetralin 6 (97:3 er) which is a key precursor to pharmaceutically important compounds for treatment of arrhythmias by inhibiting the Na+/Ca2+ exchange mechanism.19 The only known asymmetric route to such 2-aryltetralins required the use of Pd-catalyzed arylation of 1-tetralone employing an equimolar amount of highly toxic tributyltin methoxide.20
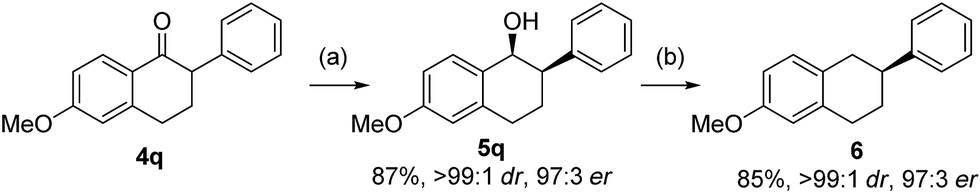 | ||
| Scheme 3 Synthesis of 7-methoxy-2-phenyl tetralin. Conditions: (a) H2 (700 psi), CuCl2 (5 mol%), 3b (5 mol%), P(3,5-xylyl)3 (5 mol%), KOt-Bu (0.25 equiv.), t-AmOH, 20 °C, 24 h; (b) Et3SiH (8 equiv.), TFA (4 equiv.), CH2Cl2, 0 °C. | ||
To understand the observed mixed ligand effect and to facilitate further development of the efficient catalysts, we initiated DFT study of the mechanism of this transformation. The reaction is believed to proceed via in situ formation of copper hydrides, where multiple species could be acting as the hydride source (Scheme 4).21
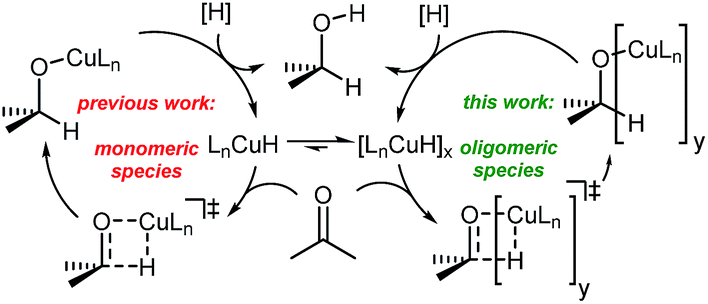 | ||
| Scheme 4 Generic catalytic cycle for the Cu-catalyzed ketone reduction. | ||
Copper hydrides are known to exist in oligomeric form, with the exact nature of the oligomer strongly depending on the identity of the ligand and the reaction conditions22. Thus, for the LCuH species, where L is a phosphine, there is evidence for existence of the corresponding dimers23, trimers24, pentamers25, hexamers26 and octamers27. For biaryl bidentate ligands, analogous to our ligand of interest, only a SEGPHOS-based system28 has been examined29; NMR and MS experiments did not provide any conclusive results about the exact nature of this LCuH species, but suggested the presence of oligomers. Kinetic studies also pointed to the oligomeric character of the copper hydride. Previously published computational studies support the relative instability of the monomer30. Given the well-known propensity of the LCuH species to form less reactive oligomers, the greater reactivity observed here when two different ligands are employed [bidentate L and monodentate P(3,5-xylyl)3] may arise from the formation of different aggregates. Specifically, we theorize that the homoaggregates [L alone or P(3,5-xylyl)3 alone] are less reactive than mixed aggregates incorporating the two different ligands.
DFT analysis of some model higher aggregates demonstrated that this is not the case, and the heteroligated aggregates average the stabilities of the homoligated systems (Scheme 5). This finding led us to propose the involvement of transition states incorporating two Cu atoms, where different ligands can be coordinated to each Cu, to explain the mixed ligand effect. To the best of our knowledge, such transition states incorporating dimer Cu species had not been previously calculated for this type of reaction.
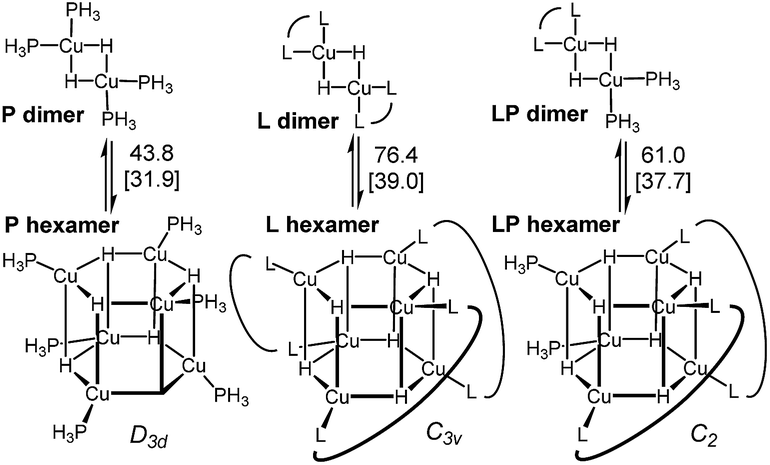 | ||
| Scheme 5 Energy of the model dimers relative to the corresponding hexameric CuH aggregates. Ligands are truncated (see ESI† for details).31 | ||
In analyzing hydride transfer transition states for the reduction of substituted tetralone, transition states incorporating two copper centers were lower than those incorporating one copper center for both monodentate and bidentate ligands (Scheme 6).
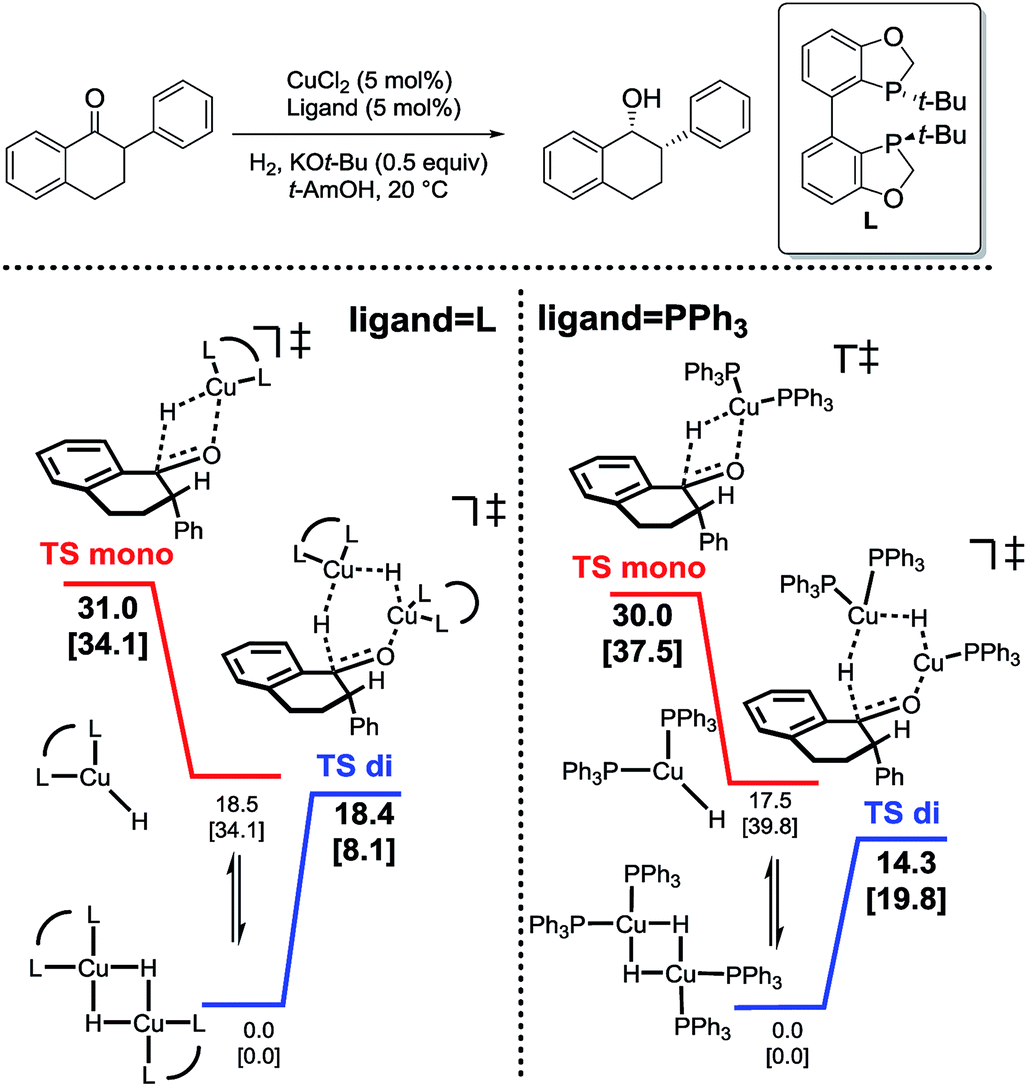 | ||
| Scheme 6 Relative energetics of monomeric (red) and dimeric (blue) hydride transfer pathways for bidentate (left) and monodentate (right) ligands.31 | ||
Following discovery of this lower energy reaction pathway, we analyzed the relative energetics of the most stable homo- and hetero-ligated dimeric transition states (Scheme 7).
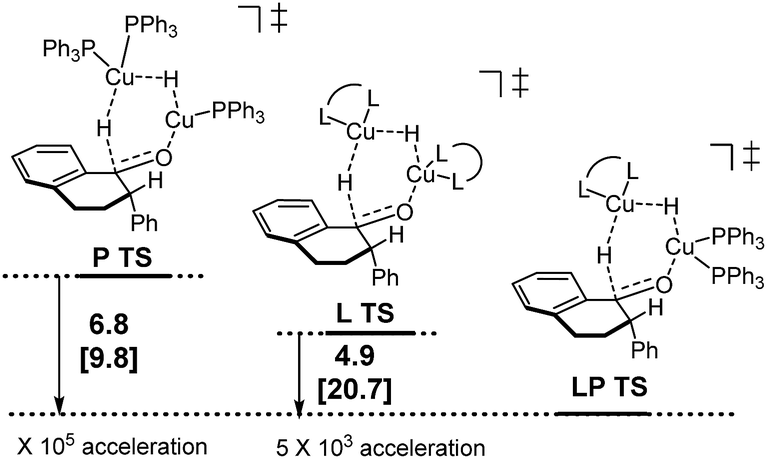 | ||
| Scheme 7 Relative energetics of the homoligated (leftmost and center) and mixed dimeric hydride transfer transition states. | ||
These results indicate that the heteroligated dimeric transition state is lower in energy than the monoligated dimeric transition states, providing more than 1000-fold acceleration of the hydride transfer. Interestingly, the stabilization of the heteroligated dimeric transition state is not a purely entropic phenomenon. That is, P TS and LP TS differ in energy, even though both have the same molecularity. We hypothesize that the mixed ligand environment better supports the charge distribution required for the effective hydride transfer (Scheme 8). Specifically, one Cu center engages in Lewis acid activation of the carbonyl, for which an electron-deficient L2 would be superior. And, the second copper center delivers nucleophilic hydride, which is assisted by an electron-rich L1.
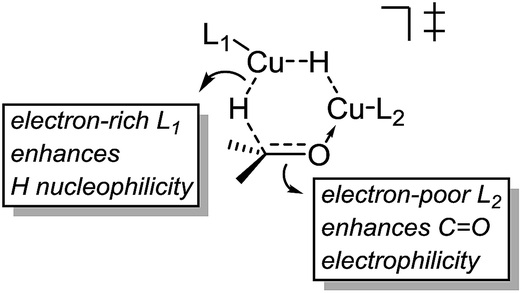 | ||
| Scheme 8 Favorable electronics for the dimeric hydride transfer transition state. | ||
In summary, a new class of heterophosphole dimeric ligands (O-BABIPhos and N-BABIPhos) have been designed and synthesized. These ligands are highly modular and tunable and have enabled the first example of Cu-catalyzed hydrogenation of 2-substituted-1-tetralones and related heteroaryl ketones via dynamic kinetic resolution, simultaneously establishing two contiguous stereogenic centers with up to >99:1 dr and 98:2 er. The ligand–Cu complexes were isolated and characterized by single crystal X-ray, and DFT calculations revealed a novel heteroligated dimeric copper hydride transition state. Application of this new series of oxa- and azaphosphole bisphosphine ligands to other important catalytic reactions is currently under investigation and will be disclosed in due course.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Scott Pennino and Keith McKellop of Boehringer Ingelheim Pharmaceuticals for HRMS analysis. M. C. K. thanks the NIH (GM087605 M. C. K.) and Boehringer Ingelheim Pharmaceuticals for financial support. Computational support was provided by XSEDE (TG-CHE120052).Notes and references
- Selected reviews on metal catalysis: (a) C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS; (b) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS PubMed; (c) Transition metals for Organic Synthesis, ed. Beller M. and Bolm C, Wiley-VCH, 2004 Search PubMed.
- (a) F. Roschangar, J. Colberg, P. J. Dunn, F. Gallou, J. D. Hayler, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Mergelsberg, J. L. Tucker, R. A. Sheldon and C. H. Senanayake, Green Chem., 2017, 19, 281–285 RSC; (b) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, University Press, Oxford, 1998 Search PubMed; (c) M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed; (d) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed; (e) P. T. Anastas, B. Han, W. Leitner and M. Poliakoff, Green Chem., 2016, 18, 12–13 RSC; (f) M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807–810 CrossRef CAS PubMed; (g) R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS PubMed.
- J. R. Dunetz, D. R. Fandrick and H.-J. Federsel, Org. Process Res. Dev., 2015, 19, 1325–1326 CrossRef CAS.
- (a) D. R. Fandrick, K. R. Fandrick, J. T. Reeves, Z. Tan, W. Tang, A. G. Capacci, S. Rodriguez, J. J. Song, H. Lee, N. K. Yee and C. H. Senanayake, J. Am. Chem. Soc., 2010, 132, 7600–7601 CrossRef CAS PubMed; (b) C. A. Malapit, M. D. Visco, J. T. Reeves, C. A. Busacca, A. R. Howell and C. H. Senanayake, Adv. Synth. Catal., 2015, 357, 2199–2204 CrossRef CAS; (c) J. N. Desrosiers, L. Hie, S. Biswas, O. V. Zatolochnaya, S. Rodriguez, H. Lee, N. Grinberg, N. Haddad, N. K. Yee, N. K. Garg and C. H. Senanayake, Angew. Chem., 2016, 128, 12100–12103 ( Angew. Chem. Int. Ed. , 2016 , 55 , 11921–11924 ) CrossRef; (d) F. G. Buono, Y. Zhang, Z. Tan, A. Brusoe, B.-S. Yang, J. C. Lorenz, R. Giovannini, J. J. Song, N. K. Yee and C. H. Senanayake, Eur. J. Org. Chem., 2016, 2599–2602 CrossRef CAS.
- (a) W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176–179 CrossRef CAS PubMed; (b) W. Li, S. Rodriguez, A. Duran, X. Sun, W. Tang, A. Premasiri, J. Wang, K. Sidhu, N. D. Patel, J. Savoie, B. Qu, H. Lee, N. Haddad, J. C. Lorenz, L. Nummy, A. Hossain, N. K. Yee, B. Lu and C. H. Senanayake, Org. Process Res. Dev., 2013, 17, 1061–1065 CrossRef CAS; (c) S. Rodriguez, B. Qu, K. R. Fandrick, F. Buono, N. Haddad, Y. Xu, M. A. Herbage, X. Zeng, S. Ma, N. Grinberg, H. Lee, Z. S. Han, N. K. Yee and C. H. Senanayake, Adv. Synth. Catal., 2014, 356, 301–307 CrossRef CAS; (d) R. Tan, X. Zheng, B. Qu, C. A. Sader, K. R. Fandrick, C. H. Senanayake and X. Zhang, Org. Lett., 2016, 18, 3346–3349 CrossRef CAS PubMed.
- A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 8318–8372 CrossRef CAS PubMed.
- (a) S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349–358 CrossRef PubMed; (b) B. H. Lipshutz, Synlett, 2009, 509–524 CrossRef CAS; (c) G. A. Carmela, Mini-Rev. Org. Chem., 2009, 6, 159–167 CrossRef.
- (a) H. Shimizu, D. Igarashi, W. Kuriyama, Y. Yusa, N. Sayo and T. Saito, Org. Lett., 2007, 9, 1655–1657 CrossRef CAS PubMed; (b) H. Shimizu, T. Nagano, N. Sayo, T. Saito, T. Ohshima and K. Mashima, Synlett, 2009, 3143–3146 CrossRef CAS.
- K. Junge, B. Wendt, D. Addis, S. Zhou, S. Das, S. Fleischer and M. Beller, Chem. - Eur. J., 2011, 17, 101–105 CrossRef CAS PubMed.
- S. W. Krabbe, M. A. Hatcher, R. K. Bowman, M. B. Mitchell, M. S. McClure and J. S. Johnson, Org. Lett., 2013, 15, 4560–4563 CrossRef CAS PubMed.
- For reviews on metal-catalyzed DKR, see: (a) H. Pellissier, Tetrahedron, 2008, 64, 1563–1601 CrossRef CAS; (b) W. Li, B. Lu and Z. Zhang, Chem. Rec., 2016, 16, 2506–2520 CrossRef CAS PubMed. For selected examples, see: (c) P. Peach, D. J. Cross, J. A. Kenny, I. Mann, I. Houson, L. Campbell, T. Walsgrove and M. Wills, Tetrahedron, 2006, 62, 1864–1867 CrossRef CAS; (d) C. Liu, J.-H. Xie, Y.-L. Li, J.-Q. Chen and Q.-L. Zhou, Angew. Chem., 2013, 125, 621–624 ( Angew. Chem. Int. Ed. , 2013 , 52 , 593–596 ) CrossRef; (e) S. Hashiguchi, A. Fujii, K.-J. Haack, K. Matsumura, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1997, 36, 288–290 CrossRef CAS.
- During preparation of this manuscript, some of the O-BABIPhos ligands were disclosed in palladium-catalyzed asymmetric hydrogenation: W. Jiang, Q. Zhao and W. Tang, Chin. J. Chem., 2018, 36, 153–156 CrossRef CAS.
- For selected examples, see: (a) A. Jutand and A. Mosleh, J. Org. Chem., 1997, 62, 261–274 CrossRef CAS PubMed; (b) V. Penalva, J. Hassan, L. Lavenot, C. Gozzi and M. Lemaire, Tetrahedron Lett., 1998, 39, 2559–2560 CrossRef CAS; (c) D. D. Hennings, T. Iwama and V. H. Rawal, Org. Lett., 1999, 1, 1205–1208 CrossRef CAS; (d) M. Kuroboshi, Y. Waki and H. Tanaka, J. Org. Chem., 2003, 68, 3938–3942 CrossRef CAS PubMed; (e) C. F. Nising, U. K. Schmid, M. Nieger and S. Bräse, J. Org. Chem., 2004, 69, 6830–6833 CrossRef CAS PubMed; (f) C. Qi, X. Sun, C. Lu, J. Yang, Y. Du, H. Wu and X.-M. Zhang, J. Organomet. Chem., 2009, 694, 2912–2916 CrossRef CASFor reviews, see: (g) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (h) T. D. Nelson and R. D. Crouch, Org. React., 2004, 63, 265–555 CAS.
- For selected examples, see: (a) V. Percec, J.-Y. Bae, M. Zhao and D. H. Hill, J. Org. Chem., 1995, 60, 176–185 CrossRef CAS; (b) G.-Q. Lin and R. Hong, J. Org. Chem., 2001, 66, 2877–2880 CrossRef CAS PubMed; (c) N. Iranpoor and F. Panahi, Org. Lett., 2015, 17, 214–217 CrossRef CAS PubMed; (d) J. Maddaluno and M. Durandetti, Synlett, 2015, 26, 2385–2388 CrossRef CAS ; see also ref. 13a, g and h.
- See ESI† for optimization of homocoupling reaction conditions..
- See ESI† for detailed optimization of the reaction conditions..
- CCDC 1816601 – [O-BABIPhos-CuCl]2 and CCDC 1816602 – [N-BABIPhos-CuCl]2 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.
- (a) B. H. Lipshutz, B. Frieman and H. Birkedal, Org. Lett., 2004, 6, 2305–2308 CrossRef CAS PubMed; (b) G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef CAS PubMed.
- T. Koskelainen, L. Otsomaa, A. Karjalainen, P. Kotovuori, J. Tenhunen, S. Rasku, P. Nore, E. Tiainen and O. Tormakangas, WO 2003/006452, 2003.
- Z. Huang, L. H. Lim, Z. Chen, Y. Li, F. Zhou, H. Su and J. Zhou, Angew. Chem., 2013, 125, 5006–5011 ( Angew. Chem. Int. Ed. , 2013 , 52 , 4906–4911 ) CrossRef.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- (a) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (b) R. S. Dhayal, W. E. van Zyl and C. W. Liu, Acc. Chem. Res., 2016, 49, 86–95 CrossRef CAS PubMed.
- G. V. Goeden, J. C. Huffman and K. G. Caulton, Inorg. Chem., 1986, 25, 2484–2485 CrossRef CAS.
- M. S. Eberhart, J. R. Norton, A. Zuzek, W. Sattler and S. Ruccolo, J. Am. Chem. Soc., 2013, 135, 17262–17263 CrossRef CAS PubMed.
- C. F. Albert, P. C. Healy, J. D. Kildea, C. L. Raston, B. W. Skelton and A. H. White, Inorg. Chem., 1989, 28, 1300–1306 CrossRef CAS.
- E. L. Bennett, P. J. Murphy, S. Imberti and S. F. Parker, Inorg. Chem., 2014, 53, 2963–2967 CrossRef CAS PubMed.
- T. H. Lemmen, K. Folting, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1985, 107, 7774–7775 CrossRef CAS.
- B. H. Lipshutz and B. A. Frieman, Angew. Chem., 2005, 117, 6503–6506 ( Angew. Chem. Int. Ed. , 2005 , 44 , 6345–6348 ) CrossRef.
- (a) S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 15913–15916 CrossRef CAS PubMed; (b) B. H Lipshutz, K. Noson, W. Chrisman and A. Lower, J. Am. Chem. Soc., 2003, 125, 8779–8789 CrossRef PubMed.
- J.-T. Issenhuth, F.-P. Notter, S. Dagorne, A. Dedieu and S. Bellemin-Laponnaz, Eur. J. Inorg. Chem., 2010, 4, 529–541 CrossRef.
- Gibbs free energies and enthalpies (in brackets) are computed with RM06/6-311+G(d,p), Cu:LANL2DZ, IEFPCM:tBuOH//RB3LYP/6-31G(d), Cu:SDD. Values are in kcal mol−1.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1816601 and 1816602. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00434j |
| This journal is © The Royal Society of Chemistry 2018 |