
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly stereoselective spirocyclopropanation of various diazooxindoles with olefins catalyzed using Ru(II)-complex†
Masaya Tone a,
Yoko Nakagawaa,
Soda Chanthamatha,
Ikuhide Fujisawaa,
Naofumi Nakayamab,
Hitoshi Gotobc,
Kazutaka Shibatomia and
Seiji Iwasa*a
a,
Yoko Nakagawaa,
Soda Chanthamatha,
Ikuhide Fujisawaa,
Naofumi Nakayamab,
Hitoshi Gotobc,
Kazutaka Shibatomia and
Seiji Iwasa*a
aDepartment of Environmental and Life Sciences, Toyohashi University of Technology, Tempaku-cho, Toyohashi 441-8580, Japan. E-mail: iwasa@ens.tut.ac.jp
bCONFLEX Corporation, Shinagawa Center Building, 6F, 3-23-17 Takanawa Minato-ku, Tokyo 108-0074, Japan
cDepartment of Computer Science and Engineering, Toyohashi University of Technology, Tempaku-cho, Toyohashi 441-8580, Japan
First published on 28th November 2018
Abstract
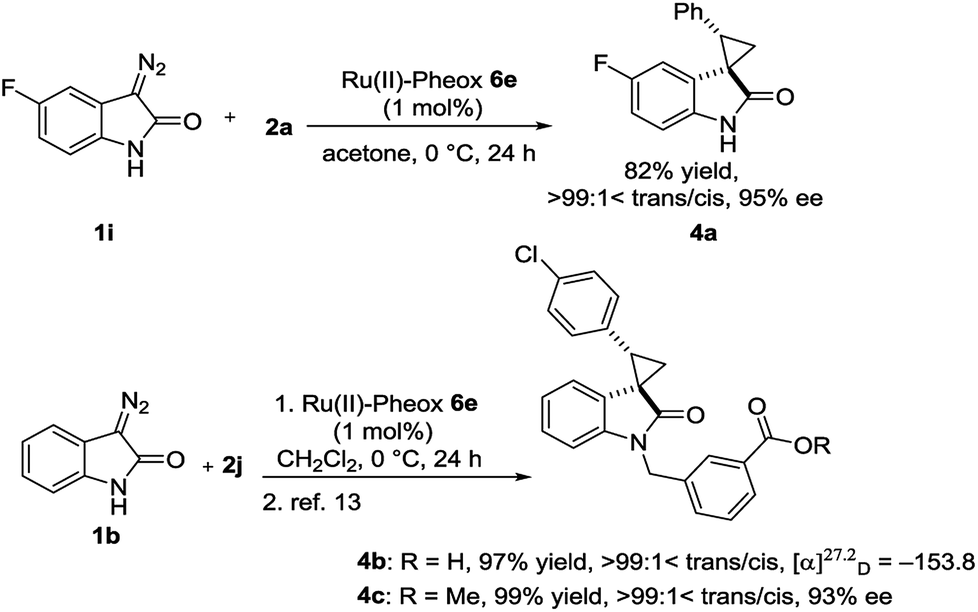
Optically active spirocyclopropyloxindole derivatives were efficiently synthesized from diazooxindoles and olefins in the presence of a Ru(II)-Pheox catalyst. Among a series of Ru(II)-Pheox catalysts, Ru(II)-Pheox 6e was determined to be the best catalyst for spirocyclopropanation reactions of diazooxindoles with various olefins in high yields (up to 98%) with high diastereoselectivities (up to trans:cis = >99:1<) and enantioselectivities (up to 99% ee). Furthermore, as the first catalytic asymmetric synthesis, anti-HIV active candidate 4a and a bioactive compound of AMPK modulator 4c were easily synthesized from the corresponding diazooxindoles 1i and 1b, respectively, in high yields with high enantioselectivities (4a: 82% yield, 95% ee, 4b: 99% yield, 93% ee).
The discovery of a new class of optically active cyclopropyl oxindoles, as shown below (Fig. 1), has stimulated intensive research interest in the development of biologically active new organic molecules in both academia and industry.1,2 These compounds were observed to be pharmacologically important substrates with potentially interesting biological activities—e.g., potent HIV inhibitors,3 and anti-cancer4 and inotrope agents.5 Furthermore, chiral spirocyclopropyl oxindoles are useful key intermediates for the ring-expansion reaction of spirocyclopropane rings via a concerted mechanism in the presence of a Lewis acid catalyst.6 As for the synthetic methodology, the transition-metal-catalyzed asymmetric cyclopropanation of diazooxindoles with olefins has been reported, which is one of the most direct and efficient pathways for the synthesis of chiral spirocyclopropyl oxindoles. After the first report by Arai7 and Zhou8 independently with Rh and Hg catalysts, Ding and co-workers reported the asymmetric cyclopropanation of diazooxindoles with alkenes using a C2-symmetric spiroketal bisphosphine/Au(I) complex with good enantioselectivity.9 More recently, Xu and co-workers reported efficient dirhodium catalysts, which exhibited good to excellent enantioselectivity control with alkyl alkenes.10 Although effective catalysts and catalytic systems have been reported for the asymmetric cyclopropanation of diazooxindoles with olefins, the development of effective and powerful methodologies to prepare the unique structural motif of a spirocyclopropyloxindole remains an active research area.
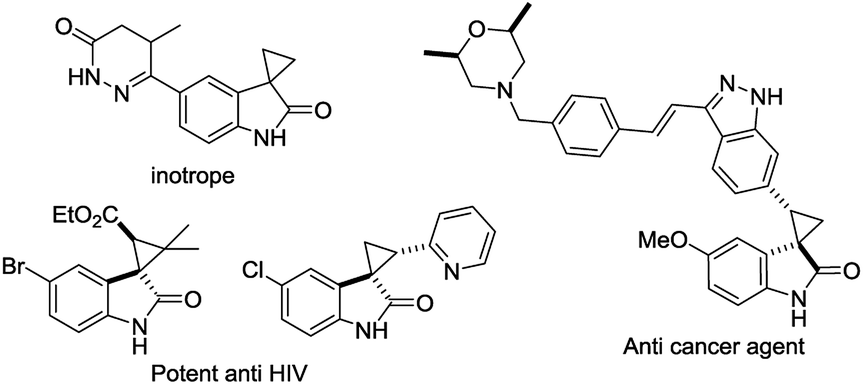 | ||
| Fig. 1 Bioactive spirocyclopropyl oxindoles. | ||
We reported a series of Ru(II) phenyloxazoline catalysts (Ru(II)-Pheox) in 2010, which effectively promoted the asymmetric intra- and intermolecular carbene transfer reactions to olefins, C–H, N–H, and Si–H bonds in high yields with high enantioselectivities.11 Based on this study, we employed Ru(II)-Pheox catalyst to synthesize a spirocyclopropanated oxindole from 5-bromo-3-methylene-indolin-2-one and diazoacetate.12 However, in this study, the reactivities and stereoselectivities were reported with moderate, or even high, diastereoselectivities. To improve the yield and enantioselectivity, we initially used Ru(II)-Pheox for the reaction of a diazooxindole of an olefin (Fig. 2).
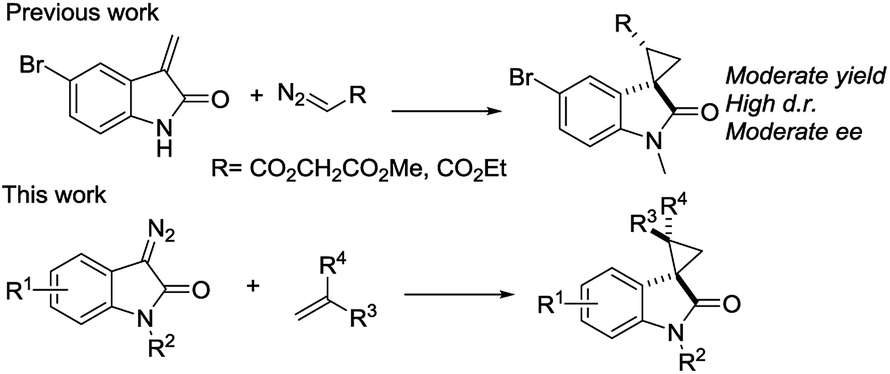 | ||
| Fig. 2 Synthetic strategies of optically active spirocyclopropyl oxindole. | ||
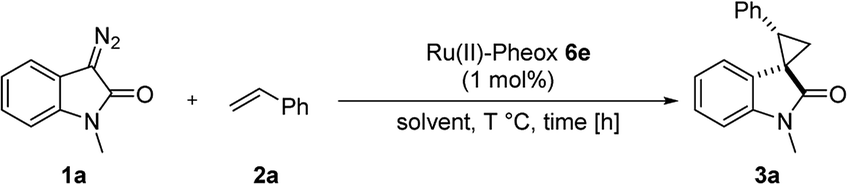
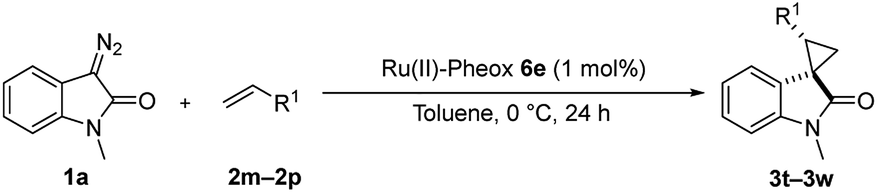
First, we attempted a catalytic asymmetric cyclopropanation of diazooxindole 1a with styrene 2a catalyzed by authorized complexes such as 413 and 514 and a series of Ru(II)-Pheox catalysts. The results are summarized in Table 1. The cyclopropanation with the commonly used catalysts 4 and 5 proceeded with low stereoselectivities (Table 1, entries 1 and 2). A series of Ru(II)-Pheox catalysts was also tested for the spirocyclopropanation reaction. The reaction of diazooxindole 1a proceeded smoothly at room temperature to afford the desired cyclopropane product 3a in high yield with moderate enantioselectivity (Table 1, entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time [h] | Yieldb [%] | trans/cisc | eed [%] |
| a Reaction condition: catalyst (1 mol%) and styrene 2a (5.0 equiv., 1 mmol) were dissolved in CH2Cl2 (2.0 mL), and diazooxindole 1a (0.2 mmol) in CH2Cl2 (2 mL) was added.b Isolated yield.c Determined by NMR.d Determined by chiral HPLC analysis. | |||||
| 1 | 4 | 10 | 90 | 69:31 | −92 |
| 2 | 5 | 4 days | 75 | 81:19 | 48 |
| 3 | 6a | 6 | 95 | 95:5 | 50 |
| 4 | 6b | 3 day | 95 | 97:3 | 46 |
| 5 | 6c | 24 | 93 | 95:5 | 49 |
| 6 | 6d | 5 | 91 | 95:5 | 61 |
| 7 | 6e | 6 | 91 | 94:6 | 92 |
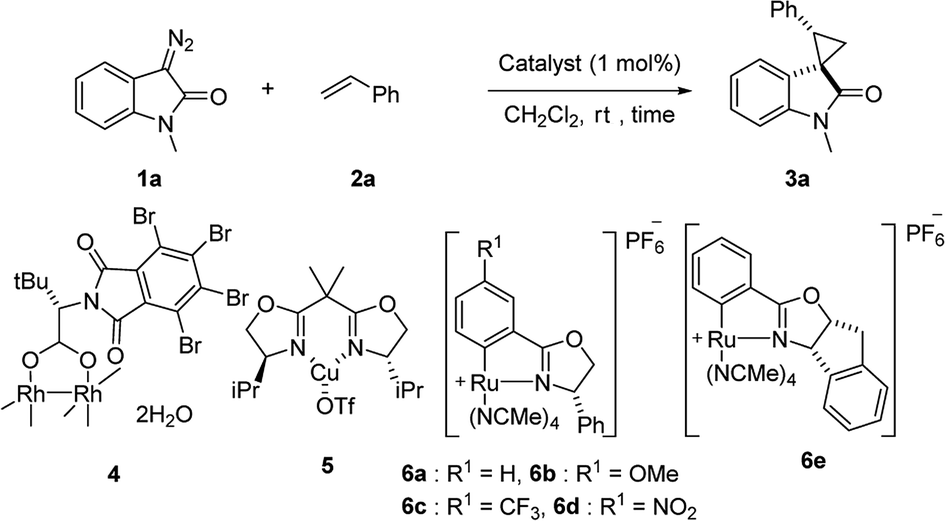
To improve the enantioselectivity, the Ru(II)-Pheox 6a catalyst was modified in terms of electron density on the aromatic ring connecting with Ru(II) and a chiral environment (6b–e). Catalyst screening of various Ru(II)-Pheox 6 catalysts is shown in Table 1 (entries 3–7). Consequently, we determined that the reactivity- and enantioselectivity-related electronic effect on the aromatic ring was slightly effective and increased the enantioselectivity to 61% ee with the use of Ru(II)-Pheox 6d having an electron-withdrawing group such as NO2 (Table 1, entry 6). However, a much more effective catalyst, Ru(II)-Pheox 6e15 having an indane-derived chiral environment, was observed to have higher activity and enantioselectivity than the other Ru(II)-Pheox 6a catalysts, resulting in the corresponding product in high yield (98%) with excellent diastereoselectivity (94:6) and high enantioselectivity (92% ee) (Table 1, entry 7).
To improve the diastereoselectivity and enantioselectivity of this catalytic system further, we examined the cyclopropanation in various solvents (Table 2, entries 1–5). For all tested solvents, spirocyclopropanation proceeded smoothly in high yield but with differing diastereoselectivities and enantioselectivities. Toluene led to an improvement of trans-selectivity (trans/cis = 97:3) and enantioselectivity (94% ee). Toluene and dichloromethane were observed to be suitable solvents for the spirocyclopropanation of diazooxindole and olefins catalyzed by Ru(II)-Pheox 6e. The effect of temperature on the reaction was also investigated (Table 2, entries 6–9), showing that enantioselectivity could be improved to 96% ee at a temperature of 0 °C (Table 2, entry 7). As such, 0 °C was determined to be the optimal temperature for this catalytic system.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | T [°C] | Time [h] | Yieldb [%] | trans/cisc | eed [%] |
a Reaction condition: catalyst (1 mol%) and styrene 2a (5.0 equiv., 1 mmol) were dissolved in CH2Cl2 (2.0 mL), and diazooxindole 1a (0.2 mmol) in CH2Cl2 (2 mL) was added.b Isolated yield.c Determined by NMR.d Determined by chiral HPLC analysis.e Toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 1:1. :CH2Cl2 = 1:1. |
||||||
| 1 | THF | rt | 7 | 77 | 96:4 | 94 |
| 2 | CH2Cl2 | rt | 6 | 91 | 94:6 | 92 |
| 3 | Acetone | rt | 6 | 87 | 94:6 | 91 |
| 4 | CH3CN | rt | 42 | 53 | 77:23 | 76 |
| 5e | Toluene:CH2Cl2 |
rt | 6 | 97 | 91:9 | 95 |
| 6 | Toluene | rt | 24 | 95 | 97:3 | 94 |
| 7 | Toluene | 0 | 24 | 94 | 94:6 | 96 |
| 8 | Toluene | −10 | 24 | 92 | 98:2 | 92 |
| 9 | Toluene | −20 | 24 | 92 | 92:8 | 91 |
Subsequently, we briefly examined the effect of N-substituted group under these optimized conditions. The result is summarized in Table 3. N-Alkyl diazooxindoles were subjected to these conditions with 2a. N-Benzyl group had some effects on the yield and resulted in the highest stereoselectivity in terms of both diastereoselectivity and enantioselectivity even far from the reactive site (Table 3, entry 4). When i-Pr group was used to substitute nitrogen, the yield was slightly decreased (Table 3, entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Product | Yieldb [%] | trans/cisc | eed [%] |
| a Reaction condition: catalyst (1 mol%) and styrene 2 (5.0 equiv., 1 mmol) were dissolved in CH2Cl2 (2.0 mL), and diazooxindole 1 (0.2 mmol) in CH2Cl2 (2 mL) was added.b Isolated yield.c Determined by NMR.d Determined by chiral HPLC analysis.e CH2Cl2 was used as the solvent. | |||||
| 1e | H | 3b | 98 | 93:7 | 92 |
| 2 | Et | 3c | 94 | 97:3 | 97 |
| 3 | iPr | 3d | 86 | 96:4 | 95 |
| 4 | Bn | 3e | 97 | >99:1< | 98 |
We subsequently explored the scope and generality of the catalytic system (Table 4). Various vinyl arenes were reacted with diazooxindole 1a in the presence of Ru(II)-Pheox 6e. Most of the vinyl arenes provided the corresponding spirocyclopropanation products in high yields (up to 98% yields) with high diastereoselectivities and enantioselectivities 93–99% ee (Table 4, entries 1–11). Germinal disubstituted olefin, α-methylstyrene 2g, also provided the product 3k in good yield with excellent diastereoselectivity and enantioselectivity (97% ee) (Table 4, entry 7). However, low enantioselectivity of 24% ee was observed in the case of cyclopropanation of 4-Me2N-substituted-styrene, which might be unstable during the purification following cyclopropane ring-opening reaction assisted by an electron-donating group (Table 4, entry 12).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Product | Yieldb [%] | trans/cisc | eed [%] |
| a Reaction condition: catalyst (1 mol%) and styrene 2 (5.0 equiv., 1 mmol) were dissolved in CH2Cl2 (2.0 mL), and diazooxindole 1 (0.2 mmol) in CH2Cl2 (2 mL) was added.b Isolated yield.c Determined by NMR.d Determined by chiral HPLC analysis.e Toluene:CH2Cl2 was used as the solvent.f Slow addition for 4 h, and stirring for 20 h. |
|||||||
| 1 | Ph | H | H | 3a | 94 | 94:6 | 96 |
| 2 | o-MeC6H4 | H | H | 3f | 92 | 92:8 | 95 |
| 3 | m-MeC6H4 | H | H | 3g | 80 | >99:1< | 99 |
| 4 | p-MeC6H4 | H | H | 3h | 96 | >99:1< | 96 |
| 5 | p-tBuC6H4 | H | H | 3i | 97 | >99:1< | 95 |
| 6e | Np | H | H | 3j | 83 | >99:1< | 96 |
| 7f | Ph | Me | H | 3k | 93 | 98:2 | 97 |
| 8e | p-NO2C6H4 | H | H | 3l | 85 | 96:4 | 94 |
| 9 | p-BrC6H4 | H | H | 3m | 98 | 96:4 | 94 |
| 10 | p-ClC6H4 | H | H | 3n | 98 | 96:4 | 93 |
| 11 | p-OMeC6H4 | H | H | 3o | 79 | >99:1< | 97 |
| 12 | p-NMe2C6H4 | H | H | 3p | 74 | >99:1< | 24 |
| 13 | Ph | H | 5-Br | 3q | 93 | 89:11 | 87 |
| 14 | Ph | H | 6-Cl | 3r | 98 | 96:4 | 99 |
| 15e | Ph | H | 6-OMe | 3s | 93 | 98:2 | 95 |
Furthermore, various oxindoles examined under similar conditions resulted in high yields and stereoselectivities, except 5-Br group (Table 4, entries 13–15). The cyclopropanation of aliphatic alkenes such as 1-hexane, multi-substituted olefins, inner alkene derivatives and α,β-unsaturated carbonyl compounds was also examined; however, no cyclopropane product was observed owing to rapid dimerization from the diazo compound.
Furthermore, hetero-atom-substituted olefins were examined for spirocyclopropanation reactions. The results are summarized in Table 5. Various N-alkyl diazooxindoles were subjected to these conditions with 2a, and all the tested diazooxindoles provided the cyclopropane product in good yields with high diastereoselectivity and enantioselectivity.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Product | Yieldb [%] | trans/cisc | eed [%] |
| a Reaction condition: catalyst (1 mol%) and styrene 2 (5.0 equiv., 1 mmol) were dissolved in CH2Cl2 (2.0 mL), and diazooxindole 1 (0.2 mmol) in CH2Cl2 (2 mL) was added.b Isolated yield.c Determined by NMR.d Determined by chiral HPLC analysis.e Toluene:CH2Cl2 was used as the solvent.f Slow addition for 4 h, and stirring for 20 h.g 98% conversion. |
|||||
| 1e,f | 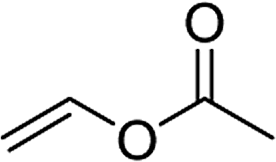 |
3t | 84 | 98:2 | 90 |
| 2e,f | 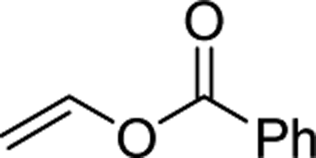 |
3u | 85 | >99:1< | 92 |
| 3 |  |
3v | 92 | 92:8 | 78 |
| 4e,f,g |  |
3w | 37 | 86:14 | 93 |
When the substrate has low solubility in toluene, a mixed solvent system such as CH2Cl2:toluene = 1:1 is efficient to improve the reactivity without decreasing the stereoselectivity (Table 5, entries 1, 2, and 4). Proton nuclear magnetic resonance (H-NMR) analysis suggested the quantitative yield for the spirocyclopropanation reaction of vinyl phthalimide 2o although the isolated yield was 37%. The product 3w from vinyl phthalimide was unstable during the purification through column chromatography on both silica gel and alumina (Table 5, entry 4).
Furthermore, we investigated the estimation of the transition state related to the reaction mechanism using computational chemical analysis for the metal–carbene complex derived from diazooxindole and Ru(II)-Pheox catalyst (Table S1, Fig. 3 and S1–S3†).17–19 The computational chemical analysis shows the Ru-oxindole carbene complex, which suggests that π–π interaction between oxindole and the indane aromatic ring controls the chiral environment to induce high enantioselectivity. Thus, an olefin may approach from only one side to provide high stereoselectivity as shown in Fig. 3. The absolute stereochemistry was determined using X-ray analysis (Scheme 1).
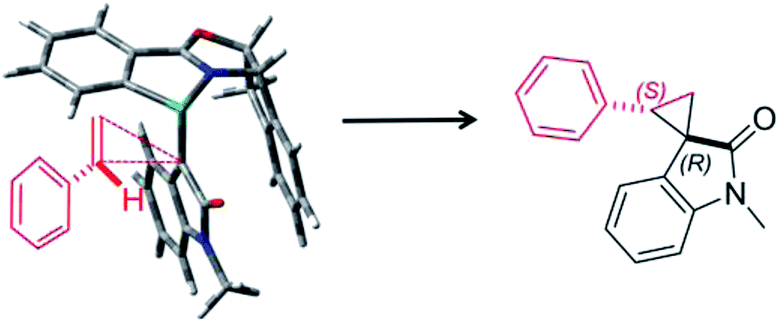 | ||
| Fig. 3 Plausible transition state of spirocyclopropanation. | ||
 | ||
| Scheme 1 Synthesis of bioactive compounds. | ||
To demonstrate the utility of our direct enantioselective cyclopropyloxindole synthesis, we demonstrated the first catalytic asymmetric synthesis of bioactive compounds such as anti-HIV activity candidate 4a and AMPK modulator 4c from diazooxindoles 1i and 1b, respectively. Both spirocyclopropanations proceeded smoothly, resulting in high yield with high enantioselectivity (4a: 82% yield, 95% ee, 4b: 99% yield, 93% ee as it is methyl ester).2e,16
In summary, the catalytic asymmetric cyclopropanation reaction of diazooxindoles and olefins by a series of Ru(II)-Pheox catalysts is very effective for the synthesis of optically active spirocyclopropyloxindole derivatives. The stereoselectivity is good to excellent in most cases and optically active spirocyclopropyloxindole derivatives can be synthesized under a mild reaction condition in a short step. Furthermore, this strategy could be applied for the first synthesis of optically active bioactive compounds such as anti-HIV candidates and an AMPK modulator.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. I. thank Ikeda Bussan Co. Ltd. for partial financial support for this study.Notes and references
- (a) C. Marit and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 11505 CrossRef PubMed; (b) N. Ye, H. Chen, E. A. Wold, P.-Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382 CrossRef CAS PubMed.
- (a) Z.-Y. Cao, Y.-H. Wang, X.-P. Zeng and J. Zhou, Tetrahedron Lett., 2014, 55, 2571 CrossRef CAS; (b) S. Muthusamy and R. Ramkumar, Tetrahedron Lett., 2014, 55, 6389 CrossRef CAS; (c) F. Pesciaioli, P. Righi, A. Mazzanti, G. Bartoli and G. Bencivenni, Chem.–Eur. J., 2011, 17, 2842 CrossRef CAS PubMed; (d) A. Noole, M. Ošeka, T. Pehk, M. Öeren, I. Järving, M. R. J. Elsegood, A. V. Malkov, M. Lopp and T. Kanger, Adv. Synth. Catal., 2013, 355, 829 CrossRef CAS; (e) M. Palomba, L. Rossi, L. Sancineto, E. Tramontano, A. Corona, L. Bagnoli, C. Santi, C. Pannecouque, O. Tabarrini and F. Marini, Org. Biomol. Chem., 2016, 14, 2015 RSC; (f) C. L. Ladd, D. S. Roman and A. B. Charette, Org. Lett., 2013, 15, 1350 CrossRef CAS PubMed; (g) S. Muthusamy, D. Azhagan, B. Gnanaprakasam and E. Suresh, Tetrahedron Lett., 2010, 51, 5662 CrossRef CAS; (h) G.-J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607 RSC; (i) Z.-Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443 CrossRef CAS PubMed; (j) N. Huang, L. Zou and Y. Peng, Org. Lett., 2017, 19, 5806 CrossRef CAS PubMed.
- (a) T. Jiang, K. L. Kuhen, K. Wolff, H. Yin, K. Bieza, J. Caldwell, B. Bursulaya, T. Y.-H. Wu and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 2105 CrossRef CAS PubMed; (b) T. Jiang, K. L. Kuhen, K. Wolff, H. Yin, K. Bieza, J. Caldwell, B. Bursulaya, T. Tuntland, K. Zhang, D. Karanewsky and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 2109 CrossRef CAS PubMed.
- (a) S.-W. Li, Y. Liu, P. B. Sampson, N. K. Patel, B. T. Forrest, L. Edwards, R. Laufer, M. Feher, F. Ban, D. E. Awrey, R. Hodgson, I. Beletskaya, G. Mao, J. M. Mason, X. Wei, X. Luo, R. Kiarash, E. Green, T. W. Mak, G. Pan and H. W. Pauls, Bioorg. Med. Chem. Lett., 2016, 26, 4625 CrossRef CAS PubMed; (b) J. M. Mason, D. C.-C. Lin, X. Wei, Y. Che, Y. Yao, R. Kiarash, D. W. Cescon, G. C. Fletcher, D. E. Awrey, M. R. Bray, G. Pan and T. W. Mak, Cancer Cell, 2014, 26, 163 CrossRef CAS PubMed.
- D. W. Robertson, J. H. Krushinski, G. D. Pollock, H. Wilson, R. F. Kauffman and J. S. Hayes, J. Med. Chem., 1987, 30, 824 CrossRef CAS PubMed.
- P.-W. Xu, J.-K. Liu, L. Shen, Z.-Y. Cao, X.-L. Zhao, J. Yan and J. Zhou, Nat. Commun., 2017, 8, 1619 CrossRef PubMed.
- (a) A. Awata and T. Arai, Synlett, 2013, 24, 29 CAS; (b) Y.-S. Xue, Y.-P. Cai and Z.-X. Chen, RCS Adv, 2015, 5, 57781 CAS.
- Z.-Y. Cao, F. Zhou, Y.-H. Yu and J. Zhou, Org. Lett., 2013, 15, 42 CrossRef CAS PubMed.
- Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197 CrossRef CAS PubMed.
- Y. Chi, L. Qiu and X. Xu, Org. Biomol. Chem., 2016, 14, 10357 RSC.
- (a) A. M. Abu-Elfotoh, K. Phemkeona, K. Shibatomi and S. Iwasa, Angew. Chem., Int. Ed., 2010, 49, 8439 CrossRef CAS PubMed; (b) S. Chanthamath, K. Phomkeona, K. Shibatomi and S. Iwasa, Chem. Commun., 2012, 48, 7750 RSC; (c) A. M. Abu-Elfotoh, D. P. T. Nguyen, S. Chanthamath, K. Phomkeona, K. Shibatomi and S. Iwasa, Adv. Synth. Catal., 2012, 354, 3435 CrossRef CAS; (d) S. Chanthamath, D. T. Nguyen, K. Shibatomi and S. Iwasa, Org. Lett., 2013, 15, 772 CrossRef CAS PubMed; (e) S. Chanthamath, S. Ozaki, K. Shibatomi and S. Iwasa, Org. Lett., 2014, 16, 3012 CrossRef CAS PubMed; (f) S. Chanthamath, H. W. Chua, S. Kimura, S. Shibatomi and S. Iwasa, Org. Lett., 2014, 16, 3408 CrossRef CAS PubMed; (g) Y. Nakagawa, S. Chanthamath, K. Shibatomi and S. Iwasa, Org. Lett., 2015, 17, 2792 CrossRef CAS PubMed; (h) S. Chanthamath, H. S. A. Mandour, T. T. M. Thu, K. Shibatomi and S. Iwasa, Chem. Commun., 2016, 52, 7814 RSC; (i) S. Chanthamath and S. Iwasa, Acc. Chem. Res., 2016, 49, 2080 CrossRef CAS PubMed; (j) H. S. A. Mandour, S. Chanthamath, K. Shibatomi and S. Iwasa, Adv. Synth. Catal., 2017, 359, 1742 CrossRef CAS.
- S. Chanthamath, S. Takaki, K. Shibatomi and S. Iwasa, Angew. Chem., Int. Ed., 2013, 52, 5818 CrossRef CAS PubMed.
- T. Goto, K. Takeda, M. Anada, K. Ando and S. Hashimoto, Tetrahedron Lett., 2011, 52, 4200 CrossRef CAS.
- (a) W. Kirmse, Angew. Chem., Int. Ed., 2003, 42, 1088 CrossRef CAS PubMed; (b) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151 CrossRef CAS PubMed; (c) G. Desimono, G. Fuita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef PubMed.
- K.-X. Huang, M.-S. Xie, G.-F. Zhao, G.-R. Qu and H.-M. Guo, Adv. Synth. Catal., 2016, 358, 3627 CrossRef CAS.
- L. Chen, M. Huang, L. Feng, Y. He and H. Yun, WO2011/069298 A1, 2011.
- (a) H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950 CrossRef CAS; (b) H. Goto and E. Osawa, J. Chem. Soc. Perkin Trans. 2, 1993, 187 RSC.
- H. Goto, S. Obata, N. Nakayama and K. Ohta, CONFLEX 8, CONFLEX Corporation, Tokyo, Japan, 2017 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1874398. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09212e |
| This journal is © The Royal Society of Chemistry 2018 |