
Organocatalytic asymmetric synthesis of cornolactones A and B, and formal synthesis of brasoside and littoralisone†
Hui
Hou
ab,
Qi
Gu
a,
Xuan
Wang
a,
De-Qun
Sun
b and
Bing-Feng
Sun
 *a
*a
aCAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai 200032, China. E-mail: bfsun@sioc.ac.cn
bMarine College, Shandong University at Weihai, 180 Wenhua West Road, Weihai 264209, China
First published on 11th October 2017
Abstract
The asymmetric total synthesis of cornolactones A and B as well as the formal asymmetric synthesis of brasoside and littoralisone were accomplished in short steps, from simple starting materials. The key features of the synthesis include an efficient organocatalytic access to cyclopentenal 15, an intramolecular Michael addition reaction and an intramolecular oxa-Diels–Alder reaction.
The chemical synthesis of iridoids has attracted much attention from the scientific community owing to the biological and ecological significance of iridoids.1 Cornolactone A (1) and cornolactone B (2) were isolated in 2014 by West and co-workers from Cornus florida, a tree native to eastern North America that has been traditionally used for the treatment of malaria.2 The synthesis of cornolactone A was documented as an synthetic intermediate in the total synthesis of semperoside A,3 but to date no total synthesis of cornolactone B has been reported. Littoralisone (3), an iridoid isolated from the traditional medicinal plant Verbena littoralis in 2001 by Ohizumi, exhibited enhanced activity of NGF-mediated neurite outgrowth from PC12D cells.4 Littoralisone possesses an unprecedented heptacyclic molecular architecture and is biogenetically related to brasoside (4).5 The first total synthesis of littoralisone and brasoside was achieved elegantly from a common synthetic precursor by MacMillan's group in 2005.6a In this paper, we report the asymmetric total synthesis of cornolactones A and B and the formal synthesis of littoralisone and brasoside by employing a novel synthetic strategy.
A divergent synthetic plan for target molecules 1–4 was envisaged, as illustrated in Scheme 1. Cornolactone A (1) and cornolactone B (2) were anticipated to be derived from compound 5 which in turn was to be constructed via the intramolecular Michael addition reaction of 6. By employing a ring-contraction strategy, 6 was traced back to the known chiral compound 7. On the other hand, in MacMillan's pioneering work, compound 8 served as the key common synthetic precursor to littoralisone (3) and brasoside (4).6a The tricyclic framework of 8 was envisioned to stem from the critical intramolecular oxa-Diels–Alder reaction of the key precursor 9. Compound 9 may similarly stem from 7.
 | ||
| Scheme 1 Cornolactones A (1) and B (2), littoralisone (3), and brasoside (4), and the collective retrosynthetic analysis. | ||
The synthetic journey commenced with the preparation of 7 (Scheme 2). According to Jørgensen's one-pot procedure, acetoacetate 10 reacted with crotonaldehyde (11) in the presence of the chiral amine 12 (10 mol%) at −78 °C before exposure to TsOH in toluene at 80 °C to furnish 7 in 52% yield with 83% ee.7 Compound 7 was subjected to a diastereoselective reduction with Dibal-H at −78 °C before acetylation to give 13 in 86% yield over two steps. Notably, use of Ac as the protecting group for the hydroxyl group was found beneficial for success in the subsequent synthetic sequence. Our initial endeavors with Bn as the masking group met with difficulties. Alkene 13 was then exposed to ozonolysis to give the crude dial 14 after reductive workup (Scheme 2).
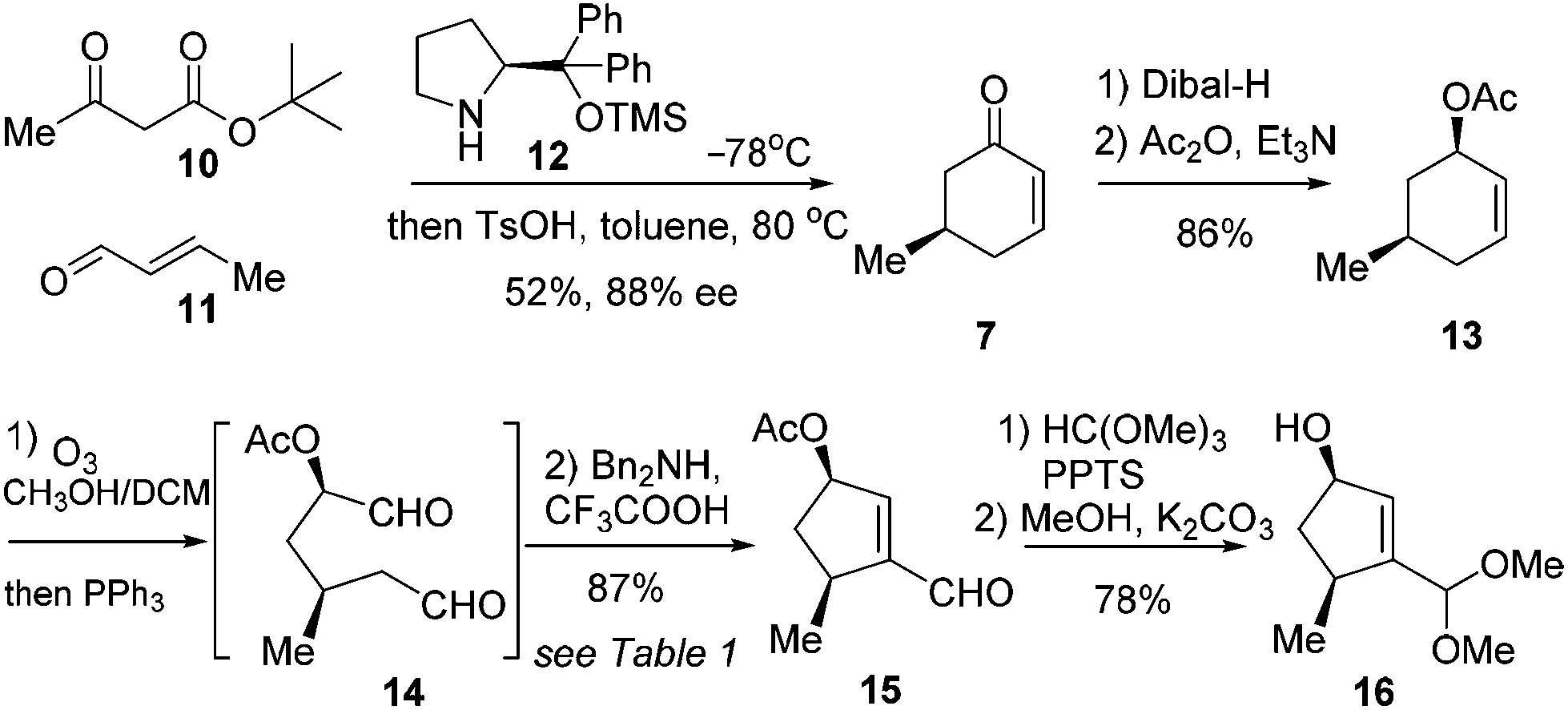 | ||
| Scheme 2 Synthesis of 16. | ||
This crucial intramolecular aldol condensation of 14 to cyclopentenal 15 was investigated by subjecting the crude mixture containing 14 to various conditions. As summarised in Table 1, the reaction with TsOH in heated toluene gave 15 in 15% yield (entry 1).8 Tertiary amines in the presence of AcOH provided the product in slightly higher yields (entries 2 and 3). The catalytic activities of secondary amines were next examined. While piperidine/AcOH in ether only generated 15 in a meager yield,9L-proline in DMSO gave a significantly higher yield of 45% (entries 4 and 5). To our delight, the combination of Bn2NH and CF3COOH in DCM could deliver the desired product in a yield of 87% (entry 6).10 Considering that this is the overall yield covering two steps from 13, this cleavage/condensation protocol is highly efficient. Compound 15 was converted to 16via sequential acetalization and deacylation in a good yield of 78% (Scheme 2).
With the key intermediate 16 in hand, we first set out to explore the total synthesis of 1 and 2 (Scheme 3). To set up the C6 stereocenter, the Mitsunobu reaction of 16 with monomethyl malonate gave 17 which, after hydrolysis of the acetal group, provided the key precursor enal 6. The critical intramolecular Michael addition reaction was studied (Table 2). When DBU or piperidine was employed as the base, 5 was isolated as an inseparable 1.5/1 diastereomeric mixture in trace amount (entries 1 and 2). Using Et3N, t-BuOK, or NaH as the base, modest yields were obtained (entries 3–5). The optimal result was achieved with TBAF, offering 5 as an inconsequential diastereomeric mixture in a satisfactory yield of 78% (entry 6). Three stereogenic centres were created in this single step, including C4, C5 and C9. Among these, the stereochemistries at C5 and C9 are critical for the subsequent success of the total synthesis. To our delight, although the diastereoselectivity at C4 was found to be variably poor (ca. 1/1), the selectivity was favorably good at C9 (ca. 10/1) and excellent at C5 (>20/1). The selectivity at C5 could be ascribed to the high propensity to form the cis-[3.3.0] bicyclic ring system. The selectivity at C9 was deemed a kinetically controlled result and probably arose from the preferred protonation from the more accessible convex face of the bicyclic system.
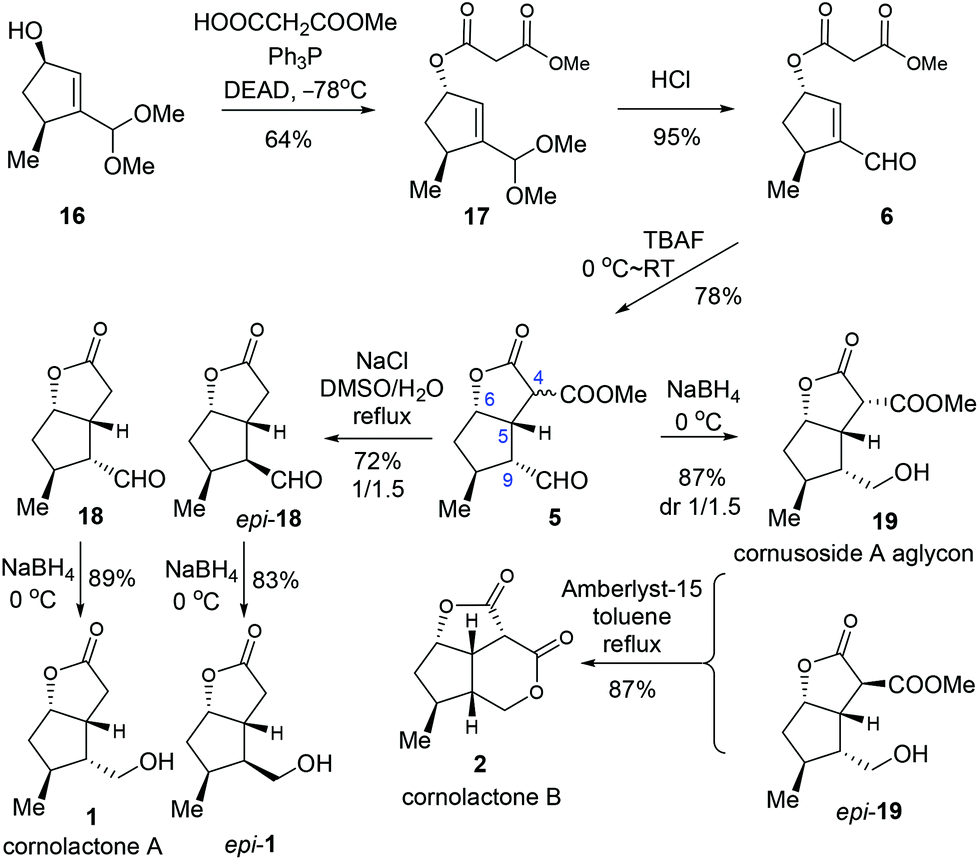 | ||
| Scheme 3 Total synthesis of cornolactone A (3) and cornolactone B (4). | ||
| Entry | Conditions | Yield (%) |
|---|---|---|
| 1 | DBU, THF, r.t. | 5 |
| 2 | Piperidine, THF, r.t. | 10 |
| 3 | Et3N, THF, r.t. | 25 |
| 4 | t-BuOK, THF, r.t. | 20 |
| 5 | NaH, THF, 0 °C–r.t. | 38 |
| 6 | TBAF, THF, 0 °C–r.t. | 78 |
With the key intermediate 5 in hand, we moved on to finish the total synthesis of 1 and 2 (Scheme 4). First, a decarboxylation reaction was realized by exposing 5 to NaCl in refluxing DMSO/H2O, furnishing 18 (29%) and epi-18 (43%). Reduction of 18 with NaBH4 gave cornolactone A (1) in 89% yield. And epi-1 was obtained in 83% yield with the same procedure. On the other hand, compound 5 was reduced to give a 1/1.5 mixture of 19 and epi-19 in a combined yield of 87%. Treatment of the mixture with Amberlyst-15 in refluxing toluene furnished cornolactone B (2) in a yield of 87%.
 | ||
| Scheme 4 Formal synthesis of littoralisone (3) and brasoside (4). | ||
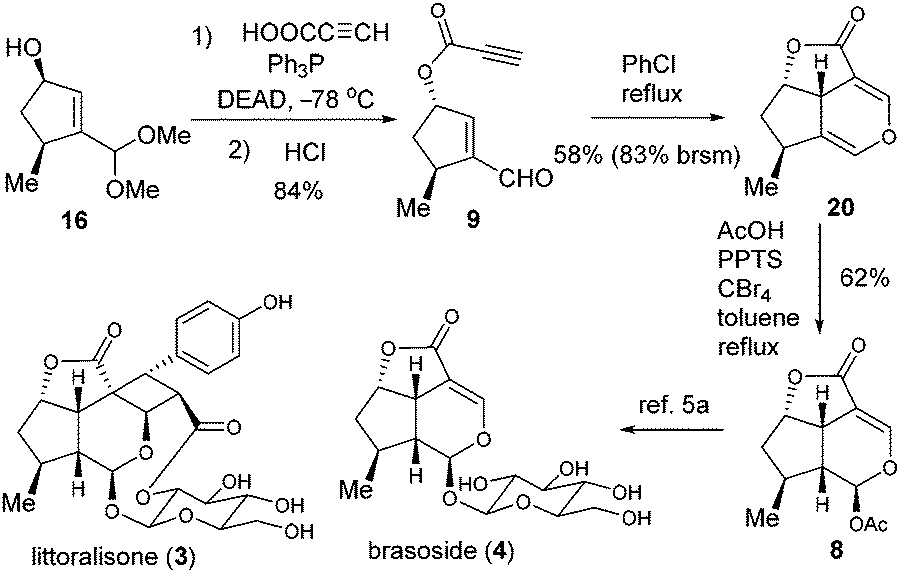
Encouraged by the success of the total synthesis of cornolactones, we moved on to the formal synthesis of 3 and 4 (Scheme 4). The Mitsunobu reaction of 16 was effected with propiolic acid before being further converted to 9 in 84% yield over two steps. The key intramolecular oxa-Diels–Alder reaction of 9 was then explored.11 Fortunately, when the solution of 9 in toluene was refluxed for 8 hours, the desired cycloaddition product 20 was isolated. Although the yield of 20 based on recovered 9 was as high as 91%, the conversion was only 18%. To our delight, the conversion could be dramatically enhanced to 70% when refluxing chlorobenzene was employed, with 20 being isolated in 58% yield (83% brsm). To the best of our knowledge, this represents the first example of an oxa-Diels–Alder reaction between enal and propiolate. The last challenge was to convert 20 to MacMillan's intermediate 8 by selective acetoxylation. After examination of various conditions, 20 was treated with AcOH in the presence of PPTS/CBr4 in refluxing toluene to successfully engender 8 in 62% yield, thus forming the formal synthesis of littoralisone (1) and brasoside (2).
Conclusions
In summary, the asymmetric total synthesis of cornolactone A and cornolactone B as well as the formal asymmetric synthesis of brasoside and littoralisone were accomplished in a collective and concise fashion from simple starting materials. Salient transformations include the efficient organocatalytic access to the key cyclopentenal 15, the intramolecular Michael addition of 6 that set up the critical C5 and C9 stereocenters, and the unprecedented intramolecular oxa-Diels–Alder reaction of 9. Some analogues, including epi-1, cornusoside A aglycone (19) and epi-19, were achieved as well. The newly demonstrated synthetic strategy may readily lend itself to the total synthesis of a broad scope of iridoid natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21290180, 21472210, and 21672243), and the Youth Innovation Promotion Association CAS (2012202).Notes and references
- For reviews on iridoid chemistry, see: (a) L. J. El-Naggar and J. L. Beal, J. Nat. Prod., 1980, 43, 649–707 CrossRef CAS PubMed; (b) C. A. Boros and F. R. Stermitz, J. Nat. Prod., 1990, 53, 1055–1147 CrossRef CAS; (c) H. Franzyk, Progress in the Chemistry of Organic Natural Products, Springer-Verlag, Wien, 2000, pp. 1–114 Search PubMed.
- Y. He, J. Peng, M. T. Hamann and L. M. West, J. Nat. Prod., 2014, 77, 2138–2143 CrossRef CAS PubMed.
- P. Piccinini, G. Vidari and G. Zononi, J. Am. Chem. Soc., 2004, 126, 5088–5089 CrossRef CAS PubMed.
- Y.-S. Li, K. Matsunaga, M. Ishibashi and Y. Ohizumi, J. Org. Chem., 2001, 66, 2165–2167 CrossRef CAS PubMed.
- A. Franke and H. Rimpler, Phytochemistry, 1987, 26, 3015–3020 CrossRef CAS.
- (a) I. K. Mangion and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3696–3697 CrossRef CAS PubMed. For an earlier synthetic approach to brasoside, see: (b) J. Robertson, M. Ménard and R. Ford, Synlett, 2004, 2788–2790 CrossRef CAS.
- A. Carlone, M. Marigo, C. North, A. Landa and K. A. Jørgensen, Chem. Commun., 2006, 4928–4930 RSC.
- A. Fukuzawa, H. Sato and T. Masamune, Tetrahedron Lett., 1987, 28, 4303–4306 CrossRef CAS.
- (a) R. B. Woodward, F. Sondheimer, D. Taub, K. Heusler and W. M. McLamore, J. Am. Chem. Soc., 1952, 74, 4223–4251 CrossRef CAS; (b) B. B. Snider and K. Yang, Tetrahedron Lett., 1989, 30, 2465–2468 CrossRef CAS.
- S. A. Snyder and E. J. Corey, Tetrahedron Lett., 2006, 47, 2083–2086 CrossRef CAS.
- For examples of intermolecular oxa-Diels–Alder reaction, see: (a) M. A. Terzidis, E. Dimitriadou, C. A. Tsoleridis and J. Stephanidou-Stephanatou, Tetrahedron Lett., 2009, 50, 2174–2176 CrossRef CAS; (b) H. Dückert, V. Khedkar, H. Waldmann and K. Kumar, Chem. – Eur. J., 2011, 17, 5130–5137 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00843k |
| This journal is © the Partner Organisations 2018 |