DOI:
10.1039/C8QM00141C
(Research Article)
Mater. Chem. Front., 2018,
2, 1475-1480
Pillararene-based host–guest recognition facilitated magnetic separation and enrichment of cell membrane proteins†
Received
1st April 2018
, Accepted 24th May 2018
First published on 24th May 2018
Abstract
Here we report a new strategy to separate and enrich cell membrane proteins by means of pillararene-based host–guest recognition and magnetic solid-phase extraction. Due to the high affinity between a water-soluble per-phosphate pillar[5]arene (WP5-P) and its cationic guest, after coating WP5-P on the surface of Fe3O4 nanoparticles, the modified nanoparticles exhibited both magnetic responsiveness and host–guest recognition ability. Furthermore, the WP5-P modified magnetic nanoparticles were successfully applied in separating and enriching guest-tagged cell membrane proteins by means of an external magnetic field from a cell lysate.
Introduction
Cell membrane proteins, which interact with or are part of biological membranes, play indispensable roles in cell life, including transportation,1 intercellular communication,2 molecular recognition3 and signaling cascades.4 Therefore, it is necessary to figure out the structures of these proteins and how they regulate or respond to cellular signals. These studies will further benefit drug design and even help us to understand the therapies of diseases from the molecular level.5 Up to now, several feasible methods have been developed to analyze cell membrane proteins,6,7 typically mass spectroscopy based protein analysis.8 Although powerful in their abilities to analyze protein compositions or protein–protein interactions, the efficiency and effectiveness of these methods are limited by the low purity and concentration of proteins. To improve the isolating efficiency of membrane proteins, numerous approaches have been established, such as aqueous two-phase extraction and gradient centrifugation.9 These two methods exhibit non-denaturing and mild conditions for protein separation, but the purity requires amelioration. Another well-developed strategy is the biotin–streptavidin (BT–SA) system-based solid-phase extraction (SPE), in which living cells are incubated with water-soluble and membrane-impermeable reactants (biotinylation reagents), followed by capturing the biotinylated membrane proteins with streptavidin-coated insoluble beads.10 The BT–SA system is frequently used for cell membrane protein separation due to its high affinity and specificity. Nevertheless, there are still some inevitable drawbacks. For example, endogenous biotin or naturally biotinylated intracellular proteins tremendously interfere with the separation process.11 Moreover, owing to the protein nature of streptavidin, enzymes, high temperature or other harsh conditions significantly impair the effectiveness of the BT–SA system because of the degradation of streptavidin or detachment from the beads.12 In view of these flaws, finding new methods for fishing cell membrane proteins with specificity and convenience is still a challenge.
Here we show that the challenge of finding new enrichment methods for cell membrane proteins can be tackled via the emerging supramolecular chemistry by combining pillar[5]arene-based host–guest interactions and magnetic solid-phase extraction (MSPE). Pillararenes, a new class of supramolecular hosts after crown ethers,13 cyclodextrins (CD),14 calixararenes15 and cucurbiturils (CB),16 have attracted a lot of attention due to their unique host–guest properties.17 Since the host–guest interactions between water-soluble anionic or neutral pillararenes and cationic guests (such as ammonium, imidazolium and pyridinium salts) are highly selective and reliable,18 it's important to explore the application of pillararene-based host–guest chemistry in the field of protein separation. As shown in Scheme 1, since the phosphate group is much more stable on the surfaces of magnetic nanoparticles (Fe3O4, MNP) than the carboxylic acid group,19 we synthesized a water-soluble per-phosphate pillar[5]arene (WP5-P) with an easy procedure and coated it on the surfaces of MNP. These coated nanoparticles, MNP-P5, were further applied in MSPE. The host–guest complexation between WP5-P and the imidazolium cation guest G endowed MNP-P5 with the ability to associate with guest-labeled molecules including guest-labeled proteins. While a lysate of the guest-labeled cells was extracted by MNP-P5, the guest-labeled membrane proteins were successfully separated and enriched, whereas the intracellular proteins were nearly eliminated because of the high selectivity in pillararene-based host–guest recognitions. Moreover, the overall procedure within the MSPE strategy, which can be rapidly and conveniently carried out by magnets, is more simple and efficient than the traditional SPE method.20
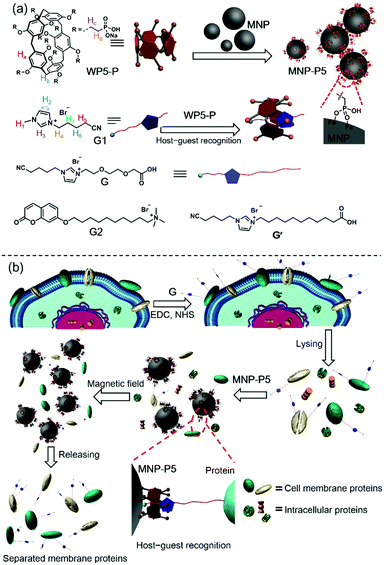 |
| | Scheme 1 (a) Structures and cartoon representations of WP5-P, MNP-P5 and guest molecules. (b) Cartoon representations of the host–guest recognition-involving MSPE strategy for membrane protein separation. The membrane proteins are labelled with imidazolium guests by EDC coupling, and the labelled proteins are selectively recognized by MNP-P5via host–guest interactions in the cell lysate. Upon exposure to a magnetic field, the captured proteins can be separated from the cell lysate and released by heating. | |
Results and discussion
Studies of host–guest interactions
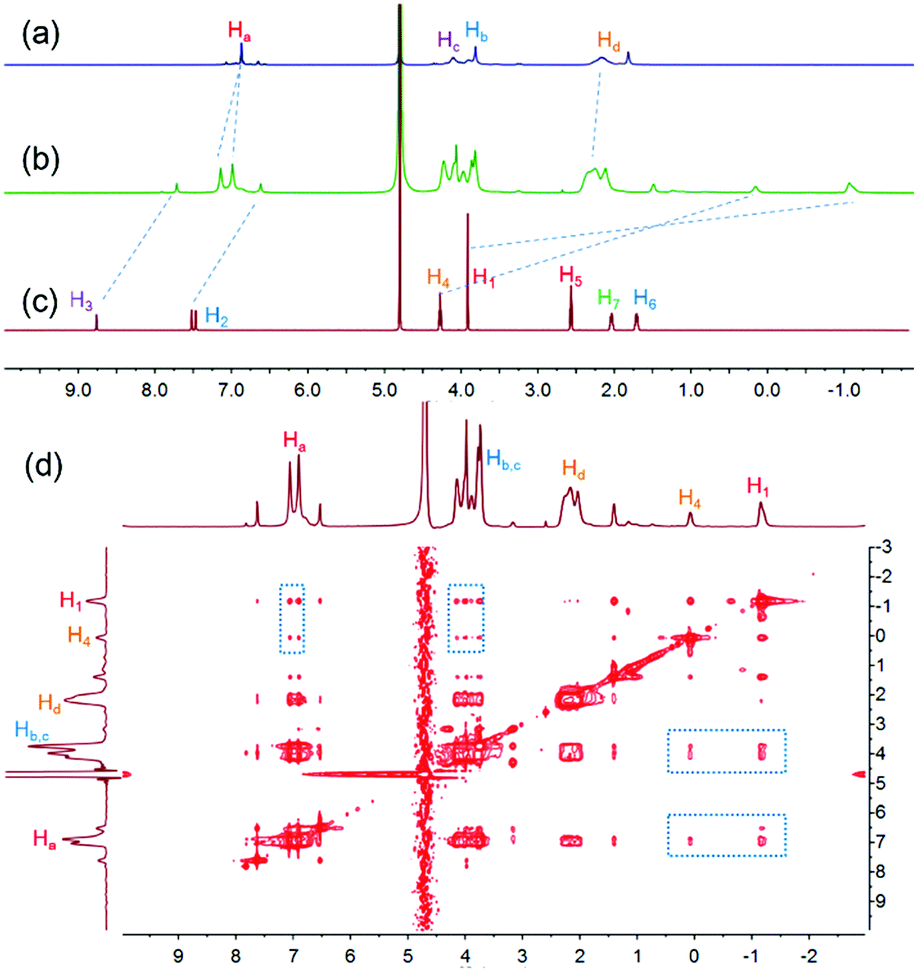
To study the host–guest interactions between WP5-P21a and cationic guests, the imidazolium guest G1 (Scheme 1) was used as a model guest because of its simple structure. From the proton nuclear magnetic resonance (1H NMR) spectra of WP5-P, G1 and their mixture, the peaks related to H1, H2, H3 and H4 of G1 shifted upfield, and the peaks related to Ha and Hd of WP5-P shifted downfield after the complexation, indicating that G1 could be associated with WP5-P (Fig. 1a–c). Meanwhile, the 2D nuclear overhauser effect spectroscopy (NOESY) spectrum of a mixture of WP5-P and G1 displayed NOE correlation signals between protons Ha, Hb, and Hc of WP5-P and protons H1 and H4 of G1, providing convincing evidence of the host–guest recognition between WP5-P and G1 (Fig. 1d). The association constant (Ka) was measured to be 1.10 × 106 M−1 by isothermal titration calorimetry (ITC, ESI,† Fig. S23) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation mode.
:1 complexation mode.
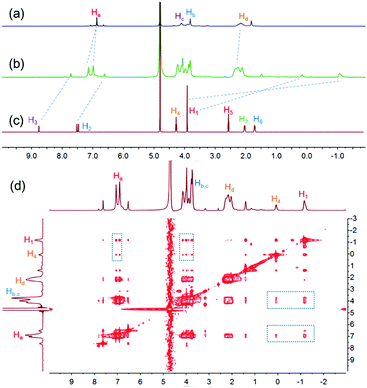 |
| | Fig. 1
1H NMR spectra (400 MHz, D2O, 298 K): (a) WP5-P (10.0 mM); (b) WP5-P (10.0 mM) and G1 (10.0 mM); (c) G1 (10.0 mM). (d) 2D NOESY spectrum of a mixture of WP5-P (10.0 mM) and G1 (10.0 mM). The NOE correlation signals that confirm the host–guest interactions are marked on the spectrum. | |
Synthesis and characterization of MNP-P5
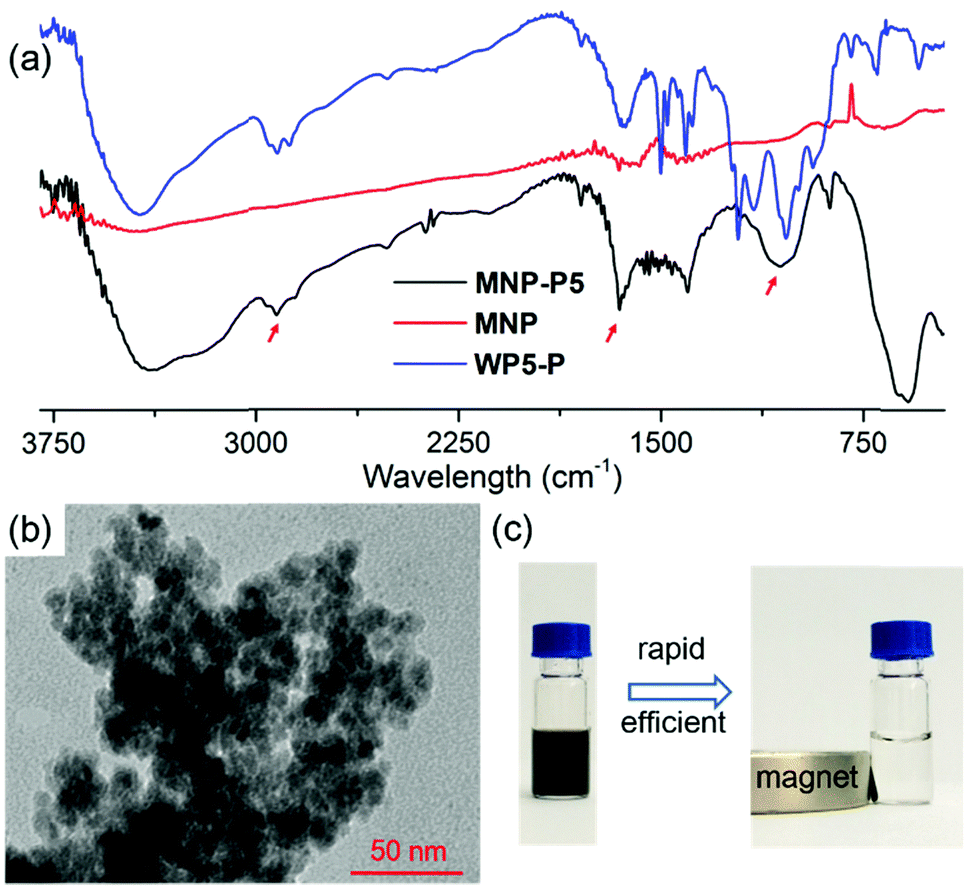
After the establishment of the WP5-P⊃G1 host–guest recognition motif, we explored the synthesis of MNP-P5. Briefly, MNP was synthesized by a co-precipitation method of ferrous and ferric salts in alkaline medium.22 Then, WP5-P was added into the freshly prepared MNP. The resulting MNP-P5 was washed and separated by means of external magnetic force (NdFeB magnet). Fourier transform infrared spectroscopy (FT-IR) was used to investigate the binding between WP5-P and MNP. As shown in Fig. 2a, the IR spectrum of MNP-P5 displayed peaks at 2922 cm−1 and 1647 cm−1, which demonstrated the existence of alkyl groups and aryl groups, respectively, and no obvious peaks were shown in the same regions in the spectrum of uncoated MNP. The broad band centered at 1050 cm−1 probably indicated the formation of a mono- or binuclear complex composed of a phosphate ligand and iron atom.23 The successful chemical modification of MNP could be concluded from these results. The existence of WP5-P was also confirmed by energy-dispersive X-ray spectroscopy (EDS, ESI,† Fig. S24), and the spectrum showed the content of phosphorus and carbon in MNP-P5. Moreover, the thermal stabilities of MNP and MNP-P5 were analyzed by thermogravimetric analysis (TGA, ESI,† Fig. S25). From the TGA curves, MNP was thermally stable in an inert atmosphere, but the sample of MNP-P5 lost 10.9% of weight at 600 °C, which was assigned to the organic coating on the nanoparticles. The morphology of MNP-P5 was studied by scanning electronic microscopy (SEM, ESI,† Fig. S26) and transmission electron microscopy (TEM, Fig. 2b). Due to the ten phosphate groups on its two rims, WP5-P acted as a cross-linker to aggregate MNP to form irregular aggregates with an average size of nearly 200 nm. However, when these irregular aggregates were magnified, nearly spherical nanoparticles with diameters of 5–10 nm were observed. The size of MNP-P5 was also verified by dynamic light scattering (DLS, ESI,† Fig. S27), and this was about 239 nm. Besides, due to the negatively charged WP5-P, the zeta potential of MNP-P5 decreased to −35.2 mV. Therefore, the nanoparticles showed good stability in water and could be well re-dispersed after magnetic separation. Furthermore, we also characterized the magnetic properties of MNP and MNP-P5via a vibrating sample magnetometer (VSM, ESI,† Fig. S28). Both of them were superparamagnetic at 300 K,24 and the saturated magnetization (Ms) value, 54.1 emu g−1, of MNP was larger than the Ms value, 44.6 emu g−1, of MNP-P5. This phenomenon was ascribed to the nonmagnetic shell coating on MNP.25 When a suspension of MNP-P5 was exposed to an external magnetic field, MNP-P5 could be fairly separated in 20 seconds, indicating the convenience of the MSPE strategy (Fig. 2c).
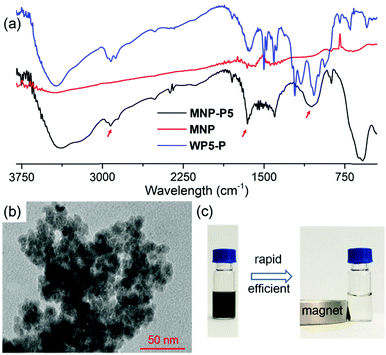 |
| | Fig. 2 (a) FT-IR spectra of MNP-P5 (black line), MNP (red line) and WP5-P (blue line). The peaks of the alkyl groups, aryl groups and phosphate–iron complexation were marked separately with arrows in the spectra. (b) TEM images of MNP-P5. (c) Photographs of MNP-P5 dispersions before (left) and after (right) exposure to an external magnetic field (NdFeB magnet). | |
Evaluation of the guest capture ability of MNP-P5
In order to evaluate the capture and separation ability of MNP-P5 by the designed MSPE, the coumarin derivative G2 (Scheme 1), as a cationic guest-containing molecule,18a,21b was chosen to firstly examine the association ability and removal efficiency of MNP-P5 by measuring the fluorescence of G2 at 400 nm in water. (ESI,† Fig. S29). After incubation with MNP-P5, the fluorescence of the solution dropped intensively. Moreover, by heating the nanoparticles to 80 °C, the fluorescence of the supernatant was nearly recovered. These results indicated that WP5-P hosts on the surfaces of MNP-P5 could associate with cationic guests.
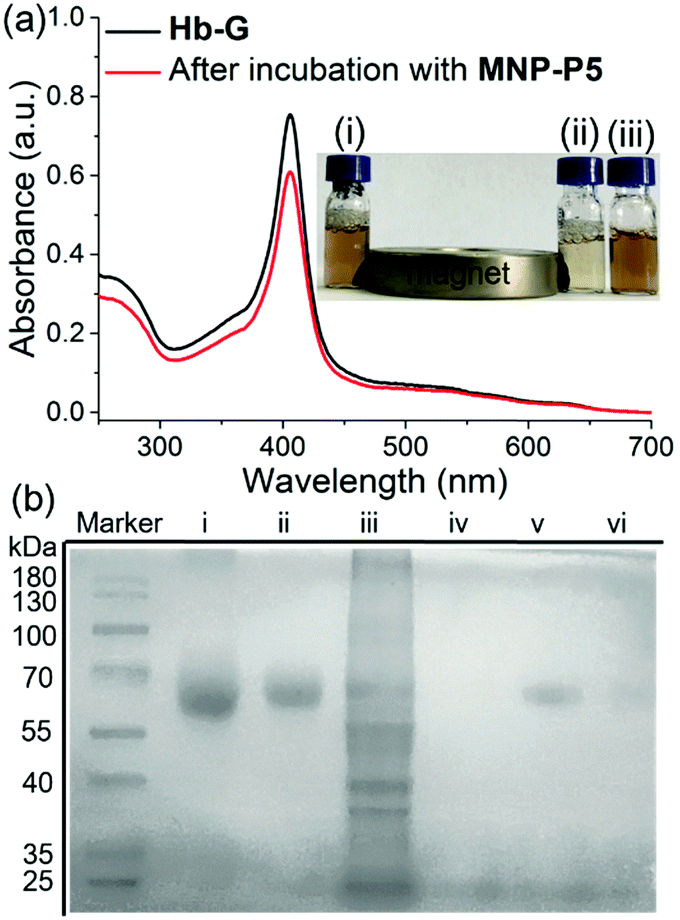
Furthermore, the protein capture ability of MNP-P5 was evaluated by using imidazolium labeled hemoglobin Hb-G as a model protein owing to its distinct color and UV absorbance at 405 nm. After incubation of MNP-P5 (1.00 mg) with a solution of Hb-G (ca. 1.00 mg mL−1, 1.00 mL) in phosphate buffered saline (PBS), MNP-P5 and absorbed protein were removed by a magnet, and the supernatant was analyzed by UV-vis spectroscopy. By measuring the absorbance at 405 nm, the absorption intensities before and after separation were 0.752 and 0.607, respectively. Therefore, approximately 19.3% of Hb-G was captured (Fig. 3a). For visualizing the capture process, excess MNP-P5 was added into an orange solution of Hb-G and then separated by a magnet. The color of the solution changed to pale yellow, indicating that the concentration of Hb-G obviously decreased (Fig. 3a, insert photograph).
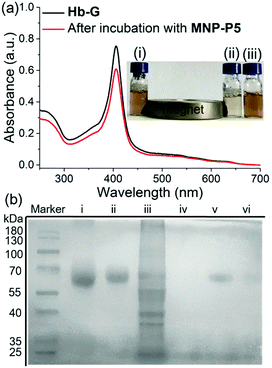 |
| | Fig. 3 (a) UV-vis spectra of Hb-G before (black line) and after (red line) being incubated with MNP-P5. Insert photograph: Hb-G with (i) excess MNP, (ii) excess MNP-P5, and (iii) no additive. (b) SDS-PAGE analysis of capturing the target protein from protein mixtures: native BSA (lane i), BSA-G (lane ii), BSA-G + IPEC-J2 cell lysate (lane iii); captured proteins recovered from MNP-P5 incubated in native BSA + IPEC-J2 cell lysate (lane iv), or BSA-G + IPEC-J2 cell lysate (lane v); captured proteins recovered from MNP incubated in BSA-G + IPEC-J2 cell lysate (lane vi). | |
With these preliminary separation results in hand, we further investigated whether MNP-P5 could separate a target protein from a mixture of the target protein and cell lysate (Fig. 3b, lane iii). Bovine serum albumin (BSA) was selected as a model protein in this step because it didn’t contain any subunit. The target protein, imidazolium labeled BSA (BSA-G), was prepared and confirmed by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectroscopy (ESI,† Fig. S30). After incubation of MNP-P5 with a mixture of BSA-G and cell lysate (intestinal porcine epithelial IPEC-J2 cells were used), the nanoparticles and captured proteins were isolated by a magnet. The solid was washed with PBS buffer and then heated to 95 °C for 10 min to recover and denature the proteins in a protein loading buffer. After the magnetic separation, the supernatant was subsequently analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, Fig. 3b). As expected, only a single band corresponding to BSA-G was observed (Fig. 3b, lane v), illustrating that BSA-G could be selectively captured by MNP-P5. Compared with native BSA (Fig. 3b, lane i), the synthesized BSA-G (Fig. 3b, lane ii) displayed a single band with a larger molecular weight, indicating that the derivatization of BSA was successful. As a control, other samples were also loaded on SDS-PAGE. When the same procedure was performed on a mixture of native BSA and IPEC-J2 cell lysate, no proteins were captured by MNP-P5 (Fig. 3b, lane iv). Nevertheless, when MNP was used in the procedure of capturing BSA-G from IPEC-J2 cell lysate, a small amount of BSA-G was observed in the recovered solution (Fig. 3b, lane vi). A possible reason was that the negatively charged MNP (containing hydroxyl groups on the surface) slightly attracted the positively charged BSA-G (containing the imidazolium cation) via electrostatic interactions, and thus a small amount of BSA-G could be captured.26 The results of SDS-PAGE clearly verified that MNP-P5 had the ability to capture the target protein from protein mixtures by means of host–guest recognition.
Separation and enrichment of cell membrane proteins by MNP-P5
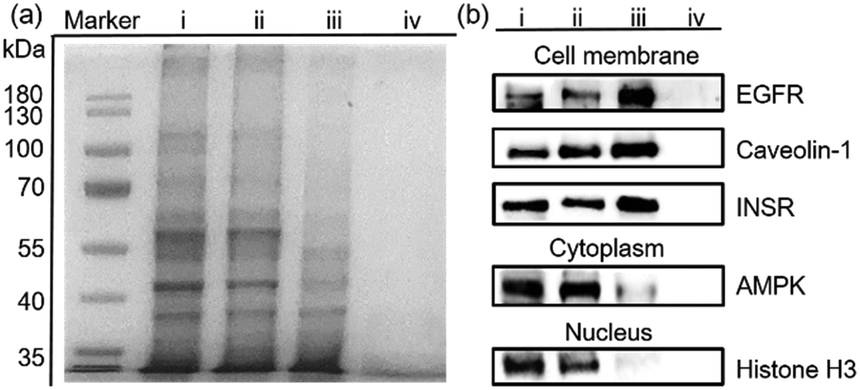
Encouraged by the separation of BSA-G from cell lysate, we further used our MSPE strategy to enrich the total plasma membrane proteins. To label the membrane proteins, we treated adherent monolayers of IPEC-J2 cells with a labeling agent (containing G, NHS and EDC). As a control, we used G′ to replace G in order to confirm that G didn’t destroy cell structures because its polar polyglycol group rendered the membrane impermeant.6 As expected, after treatment with G for 30 minutes, the morphologies of the IPEC-J2 cells didn’t change (ESI,† Fig. S32c). However, treatment with G′ rapidly caused an obvious morphological change of the cells, suggesting potential cell death as a result of the surfactant capability of G′ (ESI,† Fig. S32b). Therefore, the labeling of G on the extracellular proteins didn’t jeopardize the survival of the cells in such a short time. After the labeled cells were lysed in RIPA buffer, the lysate was incubated with MNP-P5 followed by magnetic separation. After the captured proteins were recovered and denatured at 95 °C, we used SDS-PAGE to analyze the proteins. As shown in Fig. 4a, the composition and the molecular weights of these proteins of the lysate of imidazolium labeled cells (Fig. 4a, lane ii) were similar to those of the native cell lysate (without tags, Fig. 4a, lane i). The bands of proteins captured by MNP-P5 (Fig. 4a, lane iii) were shown at the same positions as those of the lysate of imidazolium labeled IPEC-J2 cells (Fig. 4a, lane ii). Compared with lane ii, several bands were observed at lower intensities in lane iii. Moreover, nearly no protein bands (Fig. 4a, lane iv) were observed when we used MNP instead of MNP-P5 to extract the lysate of the imidazolium labeled cells. Therefore, WP5-P on the surfaces of MNP was the hinge for selective protein separation. To investigate the captured proteins, western blotting experiments were carried out. Antibodies against several crucial membrane proteins including the epidermal growth factor receptor (EGFR, relevant to diseases and tumors), caveolin-1 (involving in receptor-independent endocytosis) and the insulin receptor (INSR, regulating glucose homeostasis) were used in western blotting. Moreover, to estimate the efficiency and selectivity of our MSPE strategy, for comparison, antibodies against nuclear protein (histone H3) and cytoplasmic protein (adenosine 5′-monophosphate-activated protein kinase, AMPK) were used to examine whether the intracellular proteins were captured by MNP-P5. As shown in Fig. 4b, after being extracted by MNP-P5 (Fig. 4b, lane iii), all of the membrane proteins (EGFR, caveolin-1 and INSR) remained in the mixture whereas the intracellular proteins (histone H3 and AMPK) were nearly eliminated. Notably, EGFR was remarkably enriched in the MSPE procedure compared with caveolin-1 and INSR in the final solution, and yet AMPK couldn’t be fairly removed because it could be localized to cell membranes via a lipid anchored modification.27 As a control, bare MNP was used in the same procedure, and neither membrane proteins nor intracellular proteins were found (Fig. 4b, lane iv), indicating that the host–guest chemistry was indispensable in the MSPE strategy.
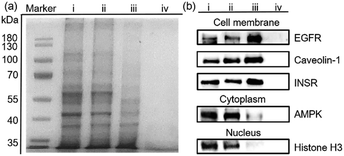 |
| | Fig. 4 (a) SDS-PAGE analysis of capturing cell membrane proteins from the lysate of imidazolium labeled IPEC-J2 cells: native IPEC-J2 cell lysate (lane i), the lysate of the imidazolium labeled IPEC-J2 cells (lane ii), captured proteins recovered from MNP-P5 (lane iii) or MNP (lane iv) incubated in the lysate of imidazolium labeled IPEC-J2 cells. (b) Western blotting analysis of subcellular marker proteins: total proteins of the IPEC-J2 cells (lane i), the imidazolium labeled IPEC-J2 cells (lane ii), captured proteins recovered from MNP-P5 (lane iii) or MNP (lane iv) incubated in the lysate of the imidazolium labeled IPEC-J2 cells. | |
Conclusion
In this work, we have successfully applied pillararene-based host–guest chemistry to develop a new MSPE strategy to separate and enrich cell membrane proteins. Comparisons between this study and previously reported methods are listed in Section S13 in the ESI.† We initially studied the strong interactions between WP5-P and the imidazolium guest G1. By coating WP5-P on the surfaces of MNP, MNP-P5 exhibited both magnetic responsiveness and guest affinity and they could be used to remove a fluorescent coumarin derivative and guest-labeled hemoglobin from aqueous media by means of an external magnetic field. Additionally, the imidazolium guest labeled BSA-G was efficiently separated from a protein mixture. Moreover, this MSPE strategy was successfully applied in the enrichment of membrane proteins. The pillararene-based MSPE strategy established here has advantages of simple synthesis, fast separation and good performance, and offers us a new perspective to design magnetic field-responsive materials, benefiting the development of both supramolecular chemistry and biochemistry.
Experimental
Materials preparation
WP5-P,21aG221b and per-(2-bromoethyl)pillar[5]arene (DBEP5)21c were synthesized according to the published methods. The detailed synthetic routes of G and G′ are described in the ESI.† The water used in this manuscript was Milli Q water. All reagents including native model proteins were commercially available and used as supplied without further purification.
Preparation of MNP-P5
Bare MNP was prepared by a co-precipitation method of ferrous and ferric salts in alkaline medium.22 0.200 g of ferric sulfate heptahydrate and 0.349 g of ferric chloride hexahydrate were dissolved in 10.0 mL of water, and 2.50 mL of 25% NH3 (aq.) was added under vigorous stirring. After the black precipitate formed, MNP was separated by a NdFeB magnet and washed with water several times. The MNP was re-dispersed in water by sonication, and 0.200 g of WP5-P was added into the suspension. The mixture was stirred overnight. MNP-P5 was separated by a magnet and washed with water. Finally MNP-P5 was suspended in 3.00 mL of water and stored in liquid nitrogen. The concentration of the MNP-P5 suspension was estimated to be 7.88 mg mL−1 by drying 1.00 mL of the suspension under vacuum.
Preparation of imidazolium labeled proteins (BSA-G and Hb-G)
The preparation of BSA-G is provided as an example. A solution of G (2.50 mg, 6.67 μmol), EDC·HCl (1.50 mg, 7.92 μmol) and NHS (1.00 mg, 8.77 μmol) was stirred in 20.0 μL of dry DMF for 2 hours in an ice bath. Then 5.00 mg of BSA in 2.00 mL of phosphate buffered saline (PBS, pH = 7.40) was added. The mixture was stirred for 2 hours in an ice bath. By centrifugal filtration (with a molecular weight cut-off of 10.0 kDa), excess reagents were removed. The residue (BSA-G) was dissolved in 5.00 mL of PBS buffer for further analysis and the concentration of total proteins was determined to be 3.66 μg mL−1 (see the ESI,† Section S11). The molecular weight of BSA-G was determined to be 70.7 kDa, which was larger than that of BSA (66.4 kDa).
Separation of cell membrane proteins from a cell lysate
A solution containing G (3.75 mg, 10.0 μmol), EDC·HCl (0.328 mg, 2.00 μmol) and NHS (0.230 mg, 2.00 μmol) was stirred in 50.0 μL of water for 2 hours in an ice bath in order to activate G. The resulting solution was added into 4.50 mL of PBS buffer and then rapidly added into adherent monolayers of PBS washed IPEC-J2 cells. The cells were incubated for 30 minutes. After being washed with PBS buffer three times, the cells were harvested by scraping and then lysed in 200 μL of RIPA buffer (10.0 μL of the lysate was used in SDS-PAGE, Fig. 4a, lane ii; 40.0 μL of the lysate was used in western blotting analysis, Fig. 4b, lane ii). The insoluble precipitate was discarded by centrifuging. The lysate containing intracellular proteins and G-labeled membrane proteins was incubated with 800 μL of a MNP-P5 suspension (7.88 mg mL−1) and 200 μL of 2× PBS buffer for 1 hour in an ice bath. After washing, the captured proteins were recovered by heating MNP-P5 at 95 °C in 40.0 μL of protein loading buffer. After magnetic separation, the supernatants were used in SDS-PAGE (10 μL for each, Fig. 4a, lane iii) and western blotting analysis (40 μL for each time, Fig. 4b, lane iii), respectively.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (91527301).
Notes and references
-
(a) Y. Ye, Y. Shibata, C. Yun, D. Ron and T. A. Rapoport, Nature, 2004, 429, 841 CrossRef PubMed;
(b) C. Paulino, V. Kalienkova, A. K. M. Lam, Y. Neldner and R. Dutzler, Nature, 2017, 552, 421 CrossRef PubMed.
-
(a) D. A. Goodenough, J. A. Goliger and D. L. Paul, Annu. Rev. Biochem., 1996, 65, 475 CrossRef PubMed;
(b) C. Soekmadji, P. J. Russell and C. C. Nelson, Cancers, 2013, 5, 1522 CrossRef PubMed.
- J. Abramson, I. Smirnova, V. Kasho, G. Verner, R. H. Kaback and S. Iwata, Science, 2003, 301, 610 CrossRef PubMed.
-
(a) M. J. Langton, F. Keymeulen, M. Ciaccia, C. A. Hunter and N. H. Williams, Nat. Chem., 2017, 9, 426 CrossRef PubMed;
(b) H. Kouhara, Y. R. Hadari, T. Spivk-Kroizman, J. Schilling, D. Bar-Sagi, I. Lax and J. Schlessinger, Cell, 1997, 89, 693 CrossRef PubMed.
-
(a) J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993 CrossRef PubMed;
(b) A. Krogh, B. R. Larsson, G. Von Heijne and E. L. L. Sonnhammer, J. Mol. Biol., 2001, 305, 567 CrossRef PubMed;
(c) C. Mamot, D. C. Drummond, U. Greiser, K. Hong, D. B. Kirpotin, J. D. Marks and J. W. Park, Cancer Res., 2003, 12, 3154 Search PubMed.
- K. H. Loh, P. S. Stawski, A. S. Draycott, N. D. Udeshi, E. K. Lehrman, D. K. Wilton, T. Svinkina, T. J. Deerinck, M. H. Ellisman, B. Stevens, S. A. Carr and A. Y. Ting, Cell, 2016, 166, 1295 CrossRef PubMed.
-
(a) T. Gopinath, S. E. D. Nelson and G. J. Veglia, Magn. Reson., 2017, 285, 101 CrossRef PubMed;
(b) A. J. Miles and B. A. Wallace, Chem. Soc. Rev., 2016, 45, 4859 RSC;
(c) I. N. Dahmke, A. Verch, J. Hermannsdoerfer, D. B. Peckys, R. S. Weatherup, S. Hofmann and N. de Jonge, ACS Nano, 2017, 11, 11108 CrossRef PubMed;
(d) M. Pfreundschuh, D. Harder, Z. Ucurum, D. Fotiadis and D. J. Muller, Nano Lett., 2017, 17, 3261 CrossRef PubMed.
-
(a) M. P. Washburn, D. Wolters and J. R. Yates, Nat. Biotechnol., 2001, 19, 242 CrossRef PubMed;
(b) G. Shen, S. Li, W. Cui, S. Liu, Y. Yang, M. Gross and W. Li, Biochemistry, 2018, 57, 286 CrossRef PubMed.
-
(a) D. Josic, M. K. Brown, F. Huang, H. Callanan, M. Ručevic, A. Nicoletti, J. Clifton and D. C. Hixson, Electrophoresis, 2005, 26, 2809 CrossRef PubMed;
(b) R. T. Leonard and W. J. Vanderwoude, Plant Physiol., 1976, 57, 105 CrossRef PubMed.
-
(a) Y. Zhao, W. Zhang, Y. Kho and Y. Zhao, Anal. Chem., 2004, 76, 1817 CrossRef PubMed;
(b) M. Wilchek and E. A. Bayer, Biomol. Eng., 1999, 16, 1 CrossRef PubMed;
(c) D. J. Josic and G. Clifton, Proteomics, 2007, 7, 3010 CrossRef PubMed.
- R. Takechi, A. Taniguchi, S. Ebara, T. Fukui and T. Watanabe, J. Nutr., 2008, 138, 680 CrossRef PubMed.
- C. E. Chivers, E. Crozat, C. Chu, V. T. Moy, D. J. Sherratt and M. Howarth, Nat. Methods, 2010, 7, 3913 CrossRef PubMed.
-
(a) Z.-J. Zhang, H.-Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834 CrossRef PubMed;
(b) J. W. Jones, L. N. Zakharov, A. L. Rheingold and H. W. Gibson, J. Am. Chem. Soc., 2002, 124, 3381 Search PubMed.
-
(a) A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128 CrossRef PubMed;
(b) H. Yan, C. Teh, S. Sreejith, L. Zhu, A. Kwok, W. Fang, X. Ma, K. T. Nguyen, V. Korzh and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 8373 CrossRef PubMed;
(c) S. Sreejith, N. V. Menon, Y. Wang, H. Joshi, S. Liu, K. C. Chong, Y. Kang, H. Sun and M. C. Stuparu, Mater. Chem. Front., 2017, 1, 831 RSC.
-
(a) D.-S. Guo, K. Wang, Y.-X. Wang and Y. Liu, J. Am. Chem. Soc., 2012, 134, 10244 CrossRef PubMed;
(b) Y. Yeon, S. Leem, C. Wagen, V. M. Lynch, S. K. Kim and J. L. Sessler, Org. Lett., 2016, 18, 4396 CrossRef PubMed.
-
(a) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef PubMed;
(b) X. Zhou, X. Su, P. Pathak, R. Vik, B. Vinciguerra, L. Isaacs and J. Jayawickramarajah, J. Am. Chem. Soc., 2017, 139, 13916 CrossRef PubMed;
(c) S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222 CrossRef PubMed.
-
(a) M. Xue, Y. Yao, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294 CrossRef PubMed;
(b) L.-L. Tan, H. Li, Y. Tao, S. X.-A. Zhang, B. Wang and Y.-W. Yang, Adv. Mater., 2014, 26, 7027 CrossRef PubMed;
(c) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937 CrossRef PubMed;
(d) X.-F. Ji, D.-Y. Xia, X. Yan, H. Wang and F.-H. Huang, Acta Polym. Sin., 2017, 9 Search PubMed.
-
(a) B. Shi, K. Jie, Y. Zhou, J. Zhou, D. Xia and F. Huang, J. Am. Chem. Soc., 2016, 138, 80 CrossRef PubMed;
(b) B. Li, Z. Meng, Q. Li, X. Huang, Z. Kang, H. Dong, J. Chen, J. Sun, Y. Dong, J. Li, X. Jia, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2017, 8, 4458 RSC;
(c) L. Jiang, X. Huang, D. Chen, H. Yan, X. Li and X. Du, Angew. Chem., Int. Ed., 2017, 56, 2655 CrossRef PubMed.
- S. Zuluaga, P. Manchanda, Y.-Y. Zhang and S. T. Pentelides, ACS Omega, 2017, 2, 4480 CrossRef.
-
(a) T. Nguyen, N. S. Joshi and M. B. Francis, Bioconjugate Chem., 2006, 17, 869 CrossRef PubMed;
(b) D.-W. Lee, K. M. Park, M. Banerjee, S. H. Ha, T. Lee, K. Suh, S. Paul, H. Jung, J. Kim, N. Selvapalam, S. H. Ryu and K. Kim, Nat. Chem., 2011, 3, 154 CrossRef PubMed.
-
(a) X.-Y. Hu, X. Liu, W. Zhang, S. Qin, C. Yao, Y. Li, D. Cao, L. Peng and L. Wang, Chem. Mater., 2016, 28, 3778 CrossRef;
(b) C. Yang, H. Shi, S. Li and Q. Li, RSC Adv., 2017, 7, 1593 RSC;
(c) Y. Ma, X. Ji, F. Xiang, X. Chi, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Chem. Commun., 2011, 47, 12340 RSC.
- Y. Sun, X. Ding, Z. Zheng, X. Cheng, X. Hu and Y. Peng, Eur. Polym. J., 2007, 43, 762 CrossRef.
-
(a) T. J. Daou, S. Begin-Colin, J. M. Grenèche, F. Thomas, A. Derory, P. Bernhardt, P. Legaré and G. Pourroy, Chem. Mater., 2007, 19, 4494 CrossRef;
(b) E. Tronc, A. Ezzir, R. Cherkaoui, C. Chanéac, M. Noguès, H. Kachkachi, D. Fiorani, A. M. Testa, J. M. Grenéche and J. P. Jolivet, J. Magn. Magn. Mater., 2000, 221, 63 CrossRef.
- H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782 CrossRef PubMed.
- S. Zhang, H. Niu, Y. Zhang, J. Liu, Y. Shi, Z. Zhang and Y. Cai, J. Chromatogr. A, 2012, 1238, 38 CrossRef PubMed.
- Y. Sun, X. Ding, Z. Zheng, C. Cheng, X. Hu and Y. Peng, Macromol. Rapid Commun., 2007, 28, 346 CrossRef.
- K. A. Wong and H. F. Lodish, J. Biol. Chem., 2006, 281, 36434 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterizations and other materials. See DOI: 10.1039/c8qm00141c |
| ‡ H. Z. and J. L. contributed equally. |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Huanhuan
Wang
b,
Zhengwei
Mao
a,
Huanhuan
Wang
b,
Zhengwei
Mao