DOI:
10.1039/C7QM00590C
(Research Article)
Mater. Chem. Front., 2018,
2, 929-934
Synthesis and properties of spiro-type heterasumanenes containing group 14 elements as bridging atoms†
Received
15th December 2017
, Accepted 15th February 2018
First published on 15th February 2018
Abstract
Spiro-type heterasumanenes composed of two triphenylenodithiophene units and group 14 bridging atoms were synthesized by stepwise lithiations of hexaalkoxytriphenylene derivatives and treatment with heteroatom reagents. The molecular structures of the spirobiheterasumanenes, which contain orthogonal two π-frameworks, were determined by single-crystal X-ray diffraction analyses. The stepwise and reversible two-electron oxidation process revealed that the orthogonal π-frameworks interact with each other through spiro-conjugation in their oxidized state.
Introduction
Spiro-type π-conjugated molecules have been widely studied in terms of their unique electronic communication between the orthogonal π-frameworks,1 and morphological stability in the solid state, which is highly advantageous for the application of these materials to organic electronic devices.2 On the other hand, heteroatom-containing π-conjugated molecules have attracted considerable attention because their electronic structures can be tuned by changing the heteroatoms introduced into the π-frameworks.3 Heterasumanenes,4 heteroatom analogues of sumanene,5 are known as two-dimensionally π-extended molecules, and their chalcogen,6 silicon,6e,7 germanium,7c,8 nitrogen,9 phosphorus,10 and other analogues11 have been synthesized (Fig. 1). A synthetic challenge in the chemistry of π-extended heteroaromatics is to utilize the reactivity of the main group elements to extend the molecular geometry as well as their electronic perturbation. Here, we report the synthesis and properties of spiro-type heterasumanenes 1–3 containing two triphenylenodithiophene units with group 14 elements (Si, Ge, and Sn) as bridging atoms.
 |
| | Fig. 1 Examples of sumanene and heterasumanenes, and the new compounds studied in this work. | |
Results and discussion
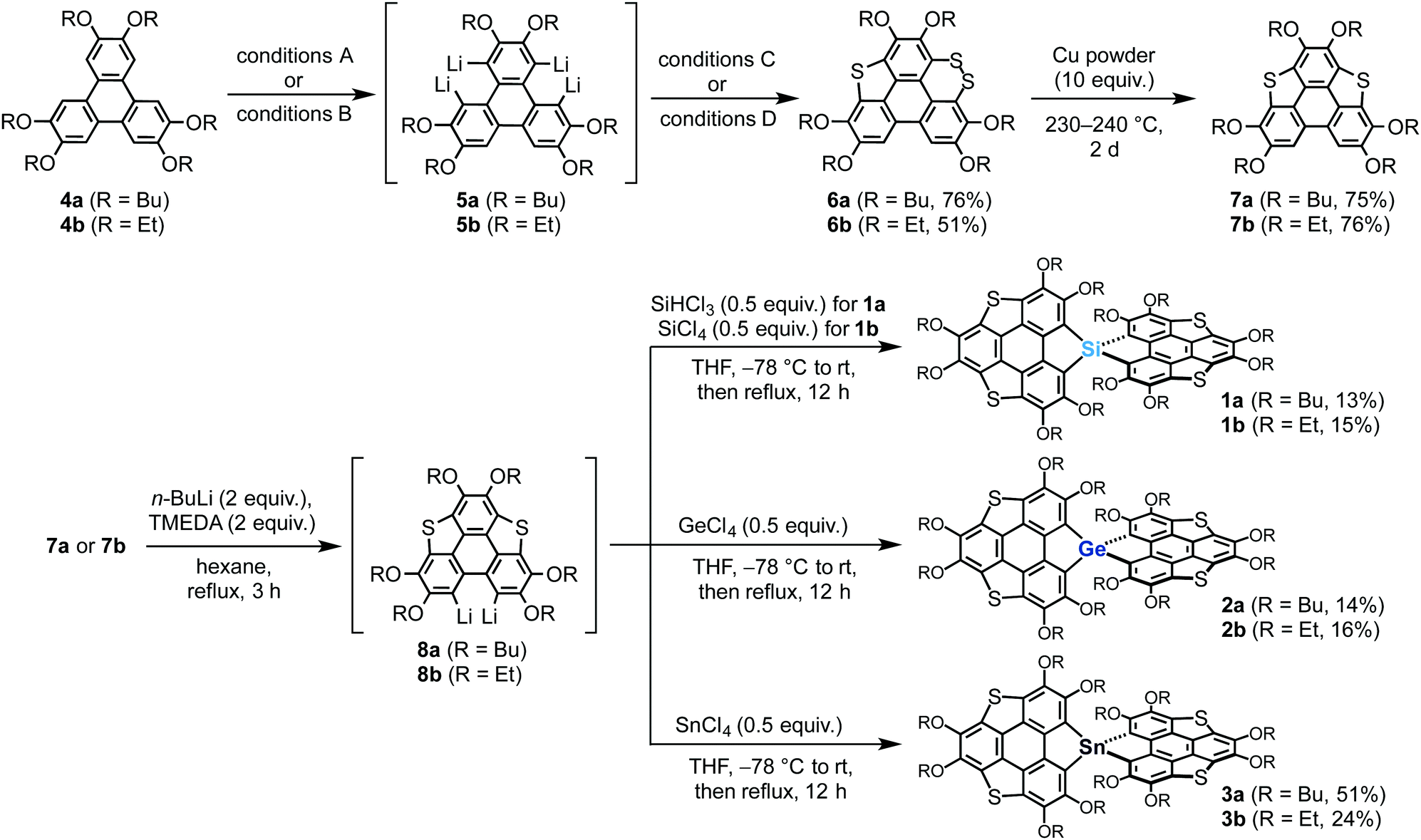
Our previously reported synthetic approach based on the hexalithiation of hexaalkoxytriphenylenes (HATs)10 triggered the synthesis of the target spirobiheterasumanenes 1–3. We succeeded in synthesizing 1–3 by applying stepwise lithiations and introduction of heteroatom substituents to the HAT derivatives (Scheme 1). Treatment of 2,3,6,7,10,11-hexabutoxytriphenylene (4a) with 4 equivalents of n-BuLi and N,N,N′,N′-tetramethylenediamine (TMEDA) in hexane under reflux conditions resulted in the in situ formation of the corresponding tetralithiated species (5a) (conditions A). A subsequent treatment of 5a with elemental sulfur, followed by the addition of iodine (conditions C), afforded dithiin derivative 6a in 76% yield. We also synthesized ethoxy derivatives by a modified method for the synthesis of the butoxy derivatives. Treatment of 2,3,6,7,10,11-hexaethoxytriphenylene (4b) with a small excess (6.5 equiv.) of n-BuLi in hexane under reflux conditions afforded the corresponding tetralithiated species 5b (conditions B). The addition of elemental sulfur into the intermediate 5b gave dithiin 6b in 51% yield (conditions D). Triphenylenodithiophenes 7 were synthesized by desulfurization of 6 using copper powder in good yields (7a (75%), 7b (76%)).
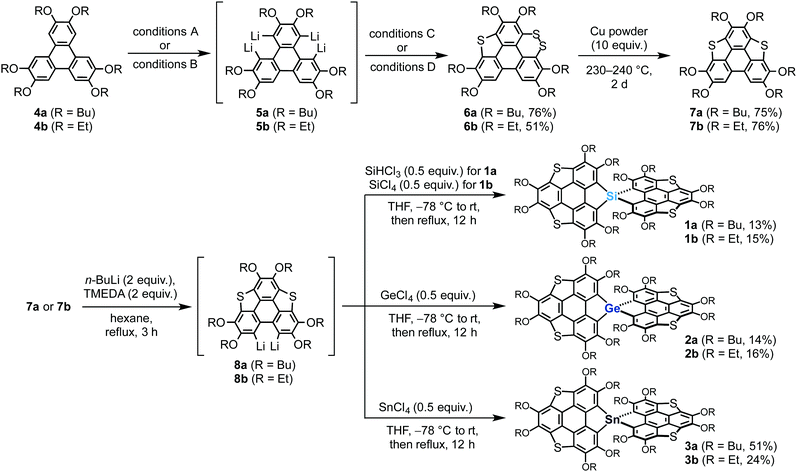 |
| | Scheme 1 Synthesis of spiro-type heterasumanenes 1–3. Conditions A (from 4a to 5a): n-BuLi (4 equiv.), TMEDA (4 equiv.), hexane, rt, 3 h. Conditions B (from 4b to 5b): n-BuLi (6.5 equiv.), hexane, reflux, 3 h. Conditions C (from 5a to 6a): (1) S8 (2 equiv.), THF, −78 °C to rt, (2) I2 (8 equiv.), CCl4, rt, 30 min, then reflux 12 h. Conditions D (from 5b to 6b): S8 (2 equiv.), THF, −78 °C to rt. | |
Transformation of the precursors 7 to the target spiro compounds 1–3 was achieved by the treatment of 7 with n-BuLi (2 equiv.) and TMEDA, followed by the addition of the corresponding heteroatom reagents (0.5 equiv.) into the in situ generated dilithiated species 8. After purification by column chromatography on silica, we obtained the isolated silicon (1a (13%), 1b (15%)), germanium (2a (14%), 2b (16%)), and tin (3a (51%), 3b (24%)) derivatives.
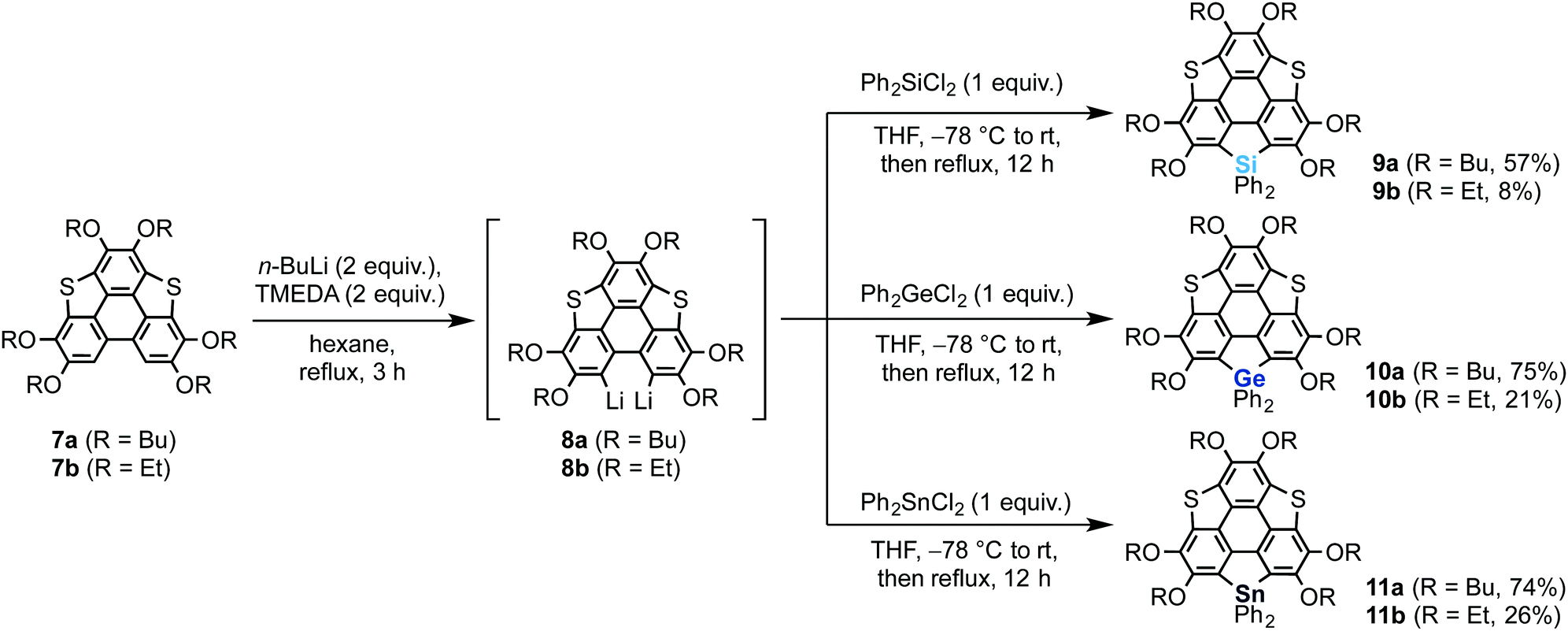
We also synthesized non-spiro-type heterasumanenes 9–11 as reference compounds by a similar method to that described for the synthesis of the spiro compounds (Scheme 2). The dilithiated species 8 were prepared from 7, then the corresponding heteroatom reagents (Ph2ECl2, E = Si or Ge or Sn) (1 equiv.) were added to the intermediate to obtain each product: silicon (9a (57%), 9b (8%)), germanium (10a (75%), 10b (21%)), and tin (11a (74%), 11b (26%)) derivatives.
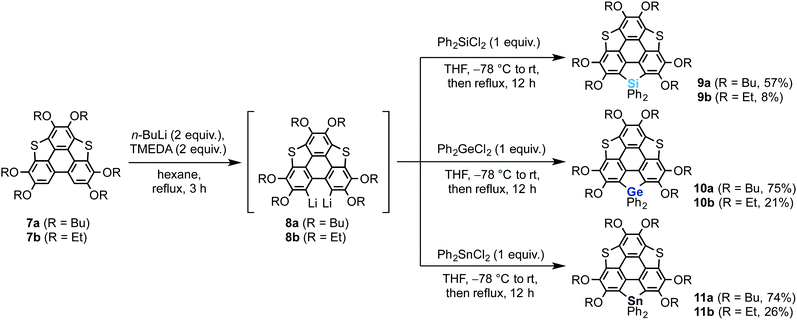 |
| | Scheme 2 Synthesis of heterasumanenes 9–11. | |
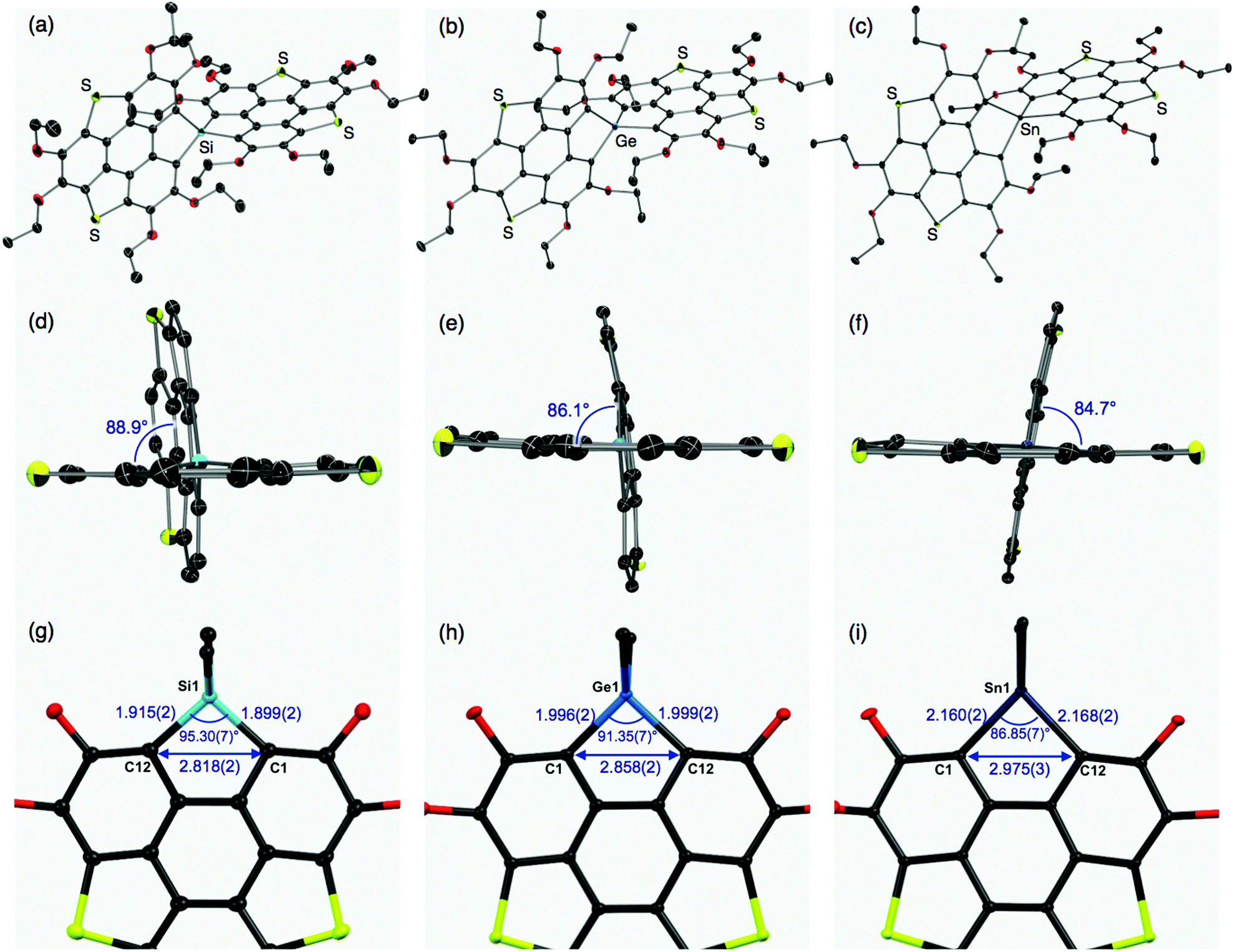
The molecular structures of the thus-obtained spiro compounds 1b, 2b, and 3b were determined by single-crystal X-ray diffraction analyses and are shown in Fig. 2 and Table S1 (ESI†). Structural refinement of butoxy derivatives 1a–c failed because of severe disorders found in the peripheral butoxy groups. The triphenylenodithiophene frameworks connected with group 14 atoms were almost perpendicular with dihedral angles of 88.9° (1b), 86.1° (2b), and 84.7° (3b) (Fig. 2d–f). The slight difference in the dihedral angles would originate from intermolecular interactions in the crystals and the degree of hybridization between the bridging heavy atoms and carbon atoms. The tendency of heavy atoms (Ge, Sn) to avoid unfavorable participation in hybridization and their intrinsic large radii result in flexible, less strained structures found in 2b and 3b, compared with the silicon analogue 1b. In the silicon analogue 1b, one of the π-frameworks is slightly distorted from planarity to afford its bowl shape (depth = 0.24 Å),12 and the other is almost planar. The structure of the germanium analogue 2b is similar to that of 1b: one of the π-frameworks has a shallow bowl shape with a depth of 0.20 Å, and the other has a planar structure. In tin analogue 3b, both π-frameworks are planar. The difference in the π-frameworks (bowl/planar) should be mainly due to the atomic radii of the group 14 atoms (Fig. 2g–i). The silicon analogue 1b has shorter C–E (E = Si) bond lengths (C1–Si1: 1.899(2) Å, C12–Si1: 1.915(2) Å) and a larger bond angle (∠C1–Si1–C12: 95.30(7)°) than those of the germanium (C1–Ge1: 1.996(2) Å, C12–Ge1: 1.999(2) Å, ∠C1–Ge1–C12: 91.35(7)°) and tin (C1–Sn1: 2.160(2) Å, C12–Sn1: 2.168(2) Å, ∠C1–Sn1–C12: 86.85(7)°) analogues. These structural changes subsequently affect the atomic distances between C1 and C12 (1b: 2.818(2) Å, 2b: 2.858 (2) Å, and 3b: 2.975(3) Å). The corresponding C–C distance of a reported triphenylenodithiophene is 3.063 Å,13 which is larger than that of 1b–3b. Bridging the two carbon atoms with the smaller group 14 atoms should increase the strain of the triphenylenodithiophene skeletons, resulting in the formation of bowl shape structures.
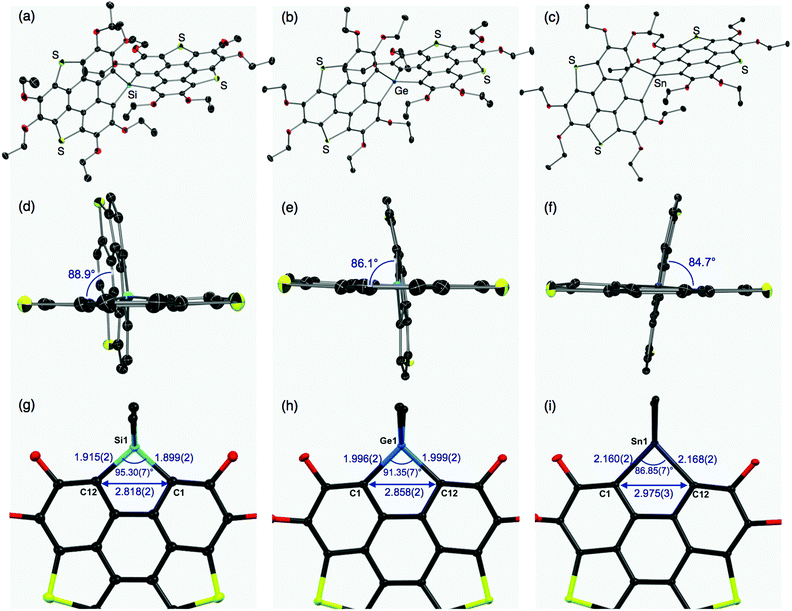 |
| | Fig. 2 Whole and side views of the molecular structures of spirobiheterasumanenes 1b (a and d), 2b (b and e), and 3b (c and f) determined by single-crystal X-ray diffraction analyses. Thermal ellipsoids are set at 50% probability, and all hydrogen atoms and solvent molecules (2b, 3b) are omitted for clarity. In (d–f), ethoxy groups are omitted for clarity. Partial structures, and selected distances and angles of 1b (g), 2b (h), and 3b (i). | |
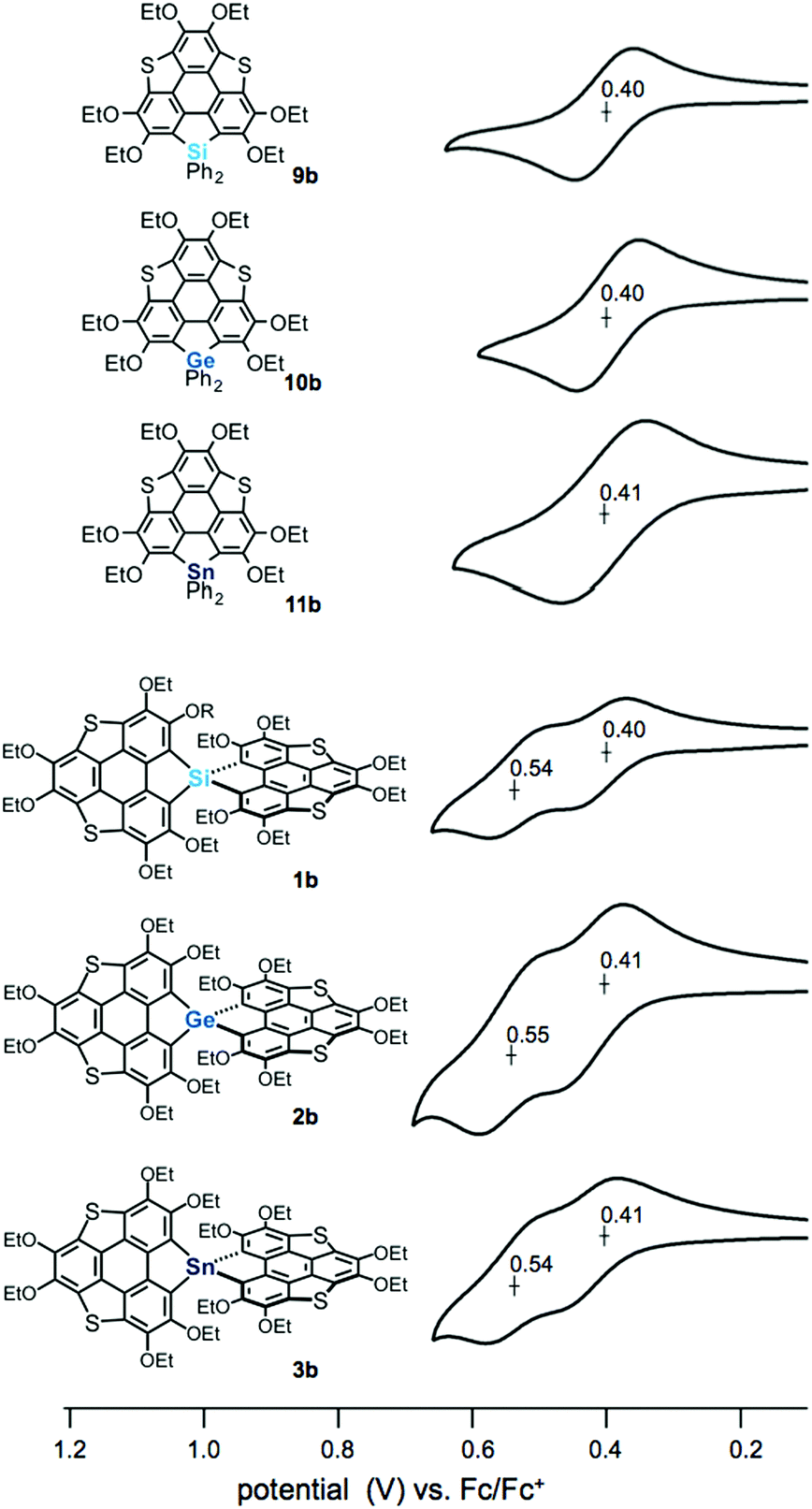
The electrochemical properties of the spiro compounds 1b–3b and the reference compounds 9b–11b were studied by cyclic voltammetry (CV) (Fig. 3, Table 1 and Fig. S2, ESI†). The non-spiro-type 9b–11b showed quasi-reversible one electron oxidation waves at E1/2 = 0.40 V (9b), 0.40 V (10b) and 0.41 V (11b), suggesting the generation of stable radical cations. The germanium and tin analogues also showed irreversible oxidation waves at Epa = 0.74 V (10b) and 0.72 V (11b), respectively (Fig. S1, ESI†). In contrast, the spiro compounds showed two reversible oxidation waves (E1/2 = 0.40 V, 0.54 V (1b); 0.41 V, 0.55 V (2b); 0.41 V, 0.54 V (3b)). Generally, spiro-type π-conjugated molecules containing heavy group 14 elements as bridging atoms show irreversible oxidation waves. Therefore, the reversible oxidation waves of 1b–3b indicate that these compounds provide the corresponding oxidized species with greater stablity than those of the conventional spiro compounds upon oxidation, resulting from the extended π-conjugation of the triphenylenothiophene units through the spiro conjugation.14 To elucidate the oxidation process, we carried out chemical oxidation of spiro-compound 3b using 1 equiv. of silver hexafluoroantimonate(V) (Scheme S1, ESI†). We measured an electron spin resonance (ESR) spectrum of the product, and observed a signal at a g value of 2.0061 (Fig. S2, ESI†). This g value is close to a value obtained in the oxidation of dibenzothiophene (g = 2.0052),15 suggesting that the radical cation of 3b was generated in the oxidation reaction. Therefore, the two reversible waves observed in the spiro compounds should correspond to stepwise one-electron oxidation processes. The peak separation of ΔE1/2 = ca. 0.14 V indicates that the two π-frameworks in the spiro compounds interact with each other through spiro-conjugation.
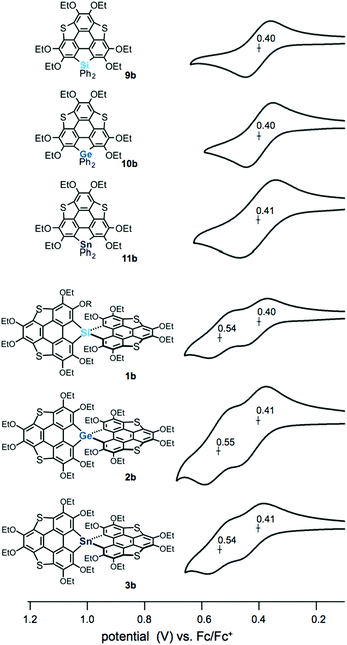 |
| | Fig. 3 Cyclic voltammograms of heterasumanenes 9a, 10b, and 11b and spirobiheterasumanenes 1b, 2b, and 3b using 0.4–0.7 mM samples with 0.1 M n-Bu4NClO4 in CH2Cl2 at a scan rate of 100 mV s−1. | |
Table 1 Electrochemical properties and UV-vis absorption maxima of 9–11 and 1–3
|
|
9
|
10
|
11
|
1
|
2
|
3
|
|
Data obtained from ethoxy derivatives 9b–11b and 1b–3b.
Data obtained from butoxy derivatives 9a–11a and 1a–3a.
|
|
E
1/2
[V] |
0.40 |
0.40 |
0.41 |
0.40 |
0.41 |
0.41 |
| — |
— |
— |
0.54 |
0.55 |
0.54 |
|
λ
max
[nm] |
397 |
396 |
396 |
403 |
400 |
400 |
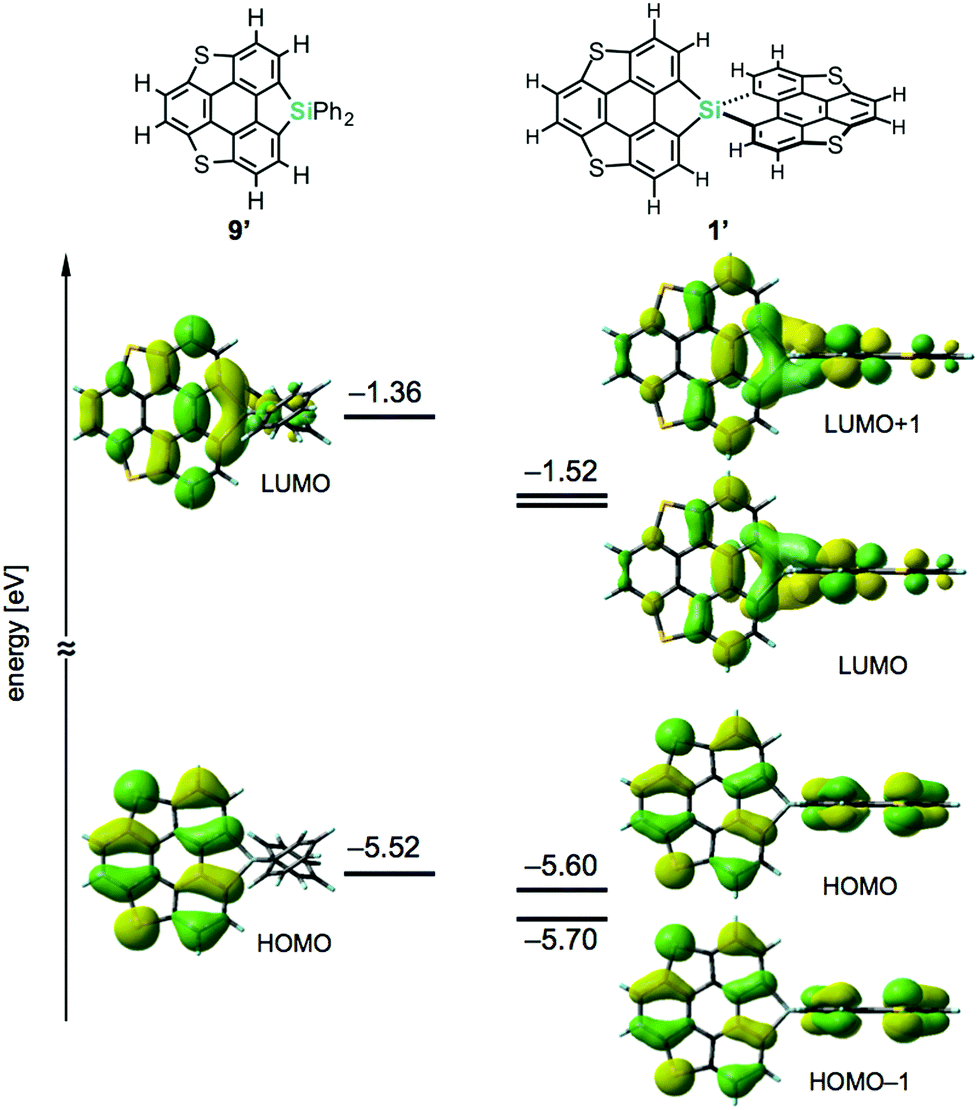
Density functional theory (DFT) calculations on model compounds 1′ and 9′ (Fig. 4), in which all alkoxy groups were replaced by hydrogen atoms, and UV-vis absorption spectra of the synthesized compounds 1a–3a and 9a–11a (Table 1 and Fig. 5) confirmed the notion of the interaction between the π-frameworks. The HOMO and HOMO−1 of 1′ are delocalized through the orthogonal π-frameworks by the spiro-conjugations, which would be a reason for the reversible oxidation of the spiro compounds. Interestingly, the bridging group 14 atoms do not contribute to the occupied frontier orbitals. Accordingly, the group 14 atoms in the spiro heterasumanenes function only as bridges without effective electronic effects. A small difference was found in the HOMO energy levels of 1′ (−5.60 eV) and 9′ (−5.52 eV), and the calculated results are consistent with the small difference in the oxidation potentials of 1b and 9b. In contrast to the small change of the HOMOs, the LUMO of 1′ (−1.52 eV) was lower in energy than that of 9′ (−1.36 eV), and delocalized through a σ*–π* conjugation of the σ*-orbitals of the C–Si bonds and π*-orbitals of the triphenylene units. UV-vis absorption spectra of the spiro-type compounds (Table 1) showed longer absorption maxima (λmax) at 403 nm (1a), 400 nm (2a), and 400 nm (3a) than those of the non-spiro compounds (397 nm (9a), 396 nm (10a), and 396 nm (11a)), which would originate from the lowering of the energy levels of the LUMOs.
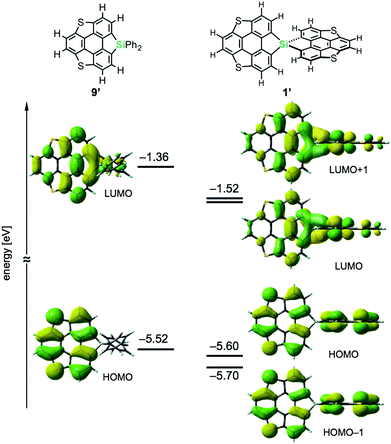 |
| | Fig. 4 Frontier orbitals and energy diagrams of the model compounds 1′ and 9′. The DFT calculations were performed at the B3LYP/6-31G(d) level of theory. | |
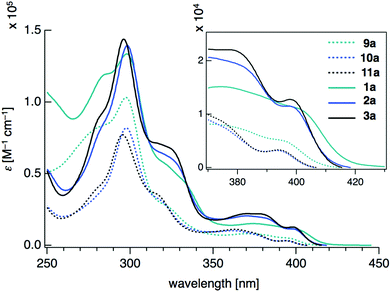 |
| | Fig. 5 UV-vis absorption spectra of 9a–11a and 1a–3a measured at a concentration of 10−5 mol L−1 in CH2Cl2. | |
Conclusions
In summary, we synthesized geometrically extended spiro-type heterasumanenes composed of two triphenylenodithiophene units and group 14 bridging atoms by stepwise lithiations of hexaalkoxytriphenylene derivatives followed by treatment with heteroatom reagents. The dihedral angles of the spiro compounds 1b–3b are almost right angles, and are slightly affected by the choice of group 14 element. The spiro-conjugation between the orthogonal π-frameworks, extended by group 14 atoms, brings about effective stabilization of their oxidized states.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Grants-in-Aid for Scientific Research on Innovative Areas “π-System Figuration: Control of Electron and Structural Dynamism for Innovative Functions” (26102006 for M. S.) and “Stimuli-responsive Chemical Species for the Creation of Functional Molecules”, as well as Grants-in-Aid for Exploratory Research (15H00918 and 16K13945 for S. F.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Notes and references
-
(a) Y. Hirao, M. Urabe, A. Ito and K. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 3300–3303 CrossRef CAS PubMed;
(b) A. Ito, K. Hata, K. Kawamoto, Y. Hirao, K. Tanaka, M. Shiro, K. Furukawa and T. Kato, Chem. – Eur. J., 2010, 16, 10866–10878 CrossRef CAS PubMed;
(c) A. Ito, M. Urabe and K. Tanaka, Angew. Chem., Int. Ed., 2009, 48, 5785 CrossRef CAS;
(d) T. Agou, M. D. Hossain, T. Kawashima, K. Kamada and K. Ohta, Chem. Commun., 2009, 6762–6763 RSC.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
-
(a) S. Yamaguchi and K. Tamao, Chem. Lett., 2005, 34, 2–7 CrossRef CAS;
(b) S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC.
-
(a) M. Saito, S. Furukawa, J. Kobayashi and T. Kawashima, Chem. Rec., 2016, 16, 64–72 CrossRef CAS PubMed;
(b) X. Li and X. Shao, Synlett, 2014, 1795–1798 CAS;
(c) X.-Q. Hou, Y.-T. Sun, L. Liu, S.-T. Wang, R.-L. Geng and X.-F. Shao, Chin. Chem. Lett., 2016, 27, 1166–1174 CrossRef CAS.
-
(a) H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed;
(b) T. Amaya and T. Hirao, Chem. Commun., 2011, 47, 10524 RSC;
(c) T. Amaya and T. Hirao, Chem. Rec., 2014, 15, 310–321 CrossRef PubMed.
-
(a) K. Imamura, K. Takimiya, T. Otsubo and Y. Aso, Chem. Commun., 1999, 1859–1860 RSC;
(b) X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535–538 CrossRef CAS PubMed;
(c) Y.-M. Liu, D. Xia, B.-W. Li, Q.-Y. Zhang, T. Sakurai, Y.-Z. Tan, S. Seki, S.-Y. Xie and L.-S. Zheng, Angew. Chem., Int. Ed., 2016, 55, 13047–13051 CrossRef CAS PubMed;
(d) S. Wang, X. Li, X. Hou, Y. Sun and X. Shao, Chem. Commun., 2016, 52, 14486–14489 RSC;
(e) Q. Tan, D. Zhou, T. Zhang, B. Liu and B. Xu, Chem. Commun., 2017, 53, 10279–10282 RSC.
-
(a) S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192–14193 CrossRef CAS PubMed;
(b) T. Tanikawa, M. Saito, J. D. Guo and S. Nagase, Org. Biomol. Chem., 2011, 9, 1731–1735 RSC;
(c) D. Zhou, Y. Gao, B. Liu, Q. Tan and B. Xu, Org. Lett., 2017, 19, 4628–4631 CrossRef CAS PubMed.
- T. Tanikawa, M. Saito, J. D. Guo, S. Nagase and M. Minoura, Eur. J. Org. Chem., 2012, 7135–7142 CrossRef CAS.
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed.
- S. Furukawa, Y. Suda, J. Kobayashi, T. Kawashima, T. Tada, S. Fujii, M. Kiguchi and M. Saito, J. Am. Chem. Soc., 2017, 139, 5787–5792 CrossRef CAS PubMed.
- M. Saito, T. Tanikawa, T. Tajima, J. D. Guo and S. Nagase, Tetrahedron Lett., 2010, 51, 672–675 CrossRef CAS.
- The bowl depth is defined as the average distance between the plane formed by the central benzene ring of the triphenylene core and that of the three heteroatoms.
- M. Saito, T. Tanikawa, T. Tajima, J. D. Guo and S. Nagase, J. Organomet. Chem., 2010, 695, 1035–1041 CrossRef CAS.
- K. Murakami, Y. Ooyama, H. Higashimura and J. Ohshita, Organometallics, 2016, 35, 20–26 CrossRef CAS.
- L. Petrakis, P. L. Meyer and G. L. Jones, J. Phys. Chem., 1980, 84, 1029–1038 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, detailed experimental procedures and computational data. CCDC 1590628 (1b), 1590627 (2b), and 1590629 (3b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00590c |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Keisuke
Hayashi
,
Ken
Yamagishi
and
Masaichi
Saito
*,
Keisuke
Hayashi
,
Ken
Yamagishi
and
Masaichi
Saito