
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of Cu(I) with the Met-X3-Met motif of alpha-synuclein: binding ligands, affinity and structural features†
Iñaki
Gentile‡
a,
Hugo A.
Garro‡
a,
Susana
Delgado Ocaña
a,
Nazareno
Gonzalez
a,
Timo
Strohäker
b,
Daniela
Schibich
a,
Liliana
Quintanar
 c,
Luis
Sambrotta
a,
Markus
Zweckstetter
bd,
Christian
Griesinger
b,
Mauricio
Menacho Márquez
a and
Claudio O.
Fernández
*ab
c,
Luis
Sambrotta
a,
Markus
Zweckstetter
bd,
Christian
Griesinger
b,
Mauricio
Menacho Márquez
a and
Claudio O.
Fernández
*ab
aMax Planck Laboratory for Structural Biology, Chemistry and Molecular Biophysics of Rosario (MPLbioR, UNR-MPIbpC) and Instituto de Investigaciones para el Descubrimiento de Fármacos de Rosario (IIDEFAR, UNR-CONICET), Universidad Nacional de Rosario, Ocampo y Esmeralda, S2002LRK Rosario, Argentina. E-mail: fernandez@iidefar-conicet.gob.ar; cfernan@gwdg.de; Tel: +54 341 4237868 ext. 752
bDepartment of NMR-based Structural Biology, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, D-37077 Göttingen, Germany
cDepartment of Chemistry, Centro de Investigación y de Estudios Avanzados (Cinvestav), Av. Instituto Politécnico Nacional 2508, 07360, D.F., Mexico
dDeutches Zentrum für Neurodegenerative Erkrankungen (DZNE), von-Siebold-Str. 3a, 37075, Göttingen, Germany
First published on 24th September 2018
Abstract
The identity of the Cu(I) binding ligands at Met-X3-Met site of AcαS and its role into the affinity and structural properties of the interaction were elucidated by NMR spectroscopy. We provide evidence that the source of ligands for Cu(I) binding to the Met-X3-Met site comes from the N-terminal acetyl group and the Met-1, Asp-2 and Met-5 residues. From the study of site-directed mutants and synthetic peptide models of αS we demonstrated the critical role played by Met-1 and Met-5 residues on the binding affinity of the Cu(I) complex, acting as the main metal anchoring residues. While having a more modest impact in the affinity features of Cu(I) binding, as compared to the Met residues, the N-terminal acetyl group and Asp-2 are important in promoting local helical conformations, contributing to the stabilization of these structures by favoring Cu(I) binding.
Significance to metallomicsProtein–metal interactions play an important role in αS aggregation and might link the pathological processes of protein aggregation, oxidative damage and neuronal cell toxicity. Advances in the bioinorganic chemistry of Parkinson's disease require that details of the binding specificity of Cu(I) to the protein α-synuclein and its conformational consequences to be better understood. In this work we have elucidated identity of the Cu(I) binding ligands at the Met-X3-Met Motif of AcαS and its role into the affinity and structural properties of the interaction. Our findings might have both physiological and pathological implications. |
Misfolding and aberrant self-assembly of proteins are considered key molecular events in several neurodegenerative disorders such as Creutzfeldt–Jakob's disease, Alzheimer's (AD) and Parkinson's disease (PD).1,2 Although these structural transformations have been observed for a range of proteins, the mechanisms behind the self-assembly of proteins into fibrillar deposits remain often unknown.
Parkinson's disease, the second most prevalent neurodegenerative disease after AD,3 is characterized by the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta affecting motor and non-motor functions.4 A hallmark of PD is the intraneuronal aggregation of the protein α-synuclein (αS) into amyloid fibrillar formations.5,6 α-Synuclein is an intrinsically disordered protein (IDP) making up 1% of total brain-soluble proteins in humans and may play roles in uptake, storage, recycling of neurotransmitter vesicles and maintenance of dopamine.7–9 The protein comprises 140 amino acid residues (Fig. 1), which constitute the amphipathic region at the N-terminal region (resides 1–60), the hydrophobic non-amyloid-β component (NAC) region (residues 61–95), and the acidic region at the C-terminal region (96–140). In solution, the NAC region tends to be partially shielded from the solvent, induced by transient intramolecular interactions that delay intermolecular aggregation.10,11 However, in disorders such as PD or dementia αS may complex with Lewy bodies and adopt conformations that trigger toxicity and neuronal cell death, reflected by the presence of amyloid fibrillar aggregates.12,13
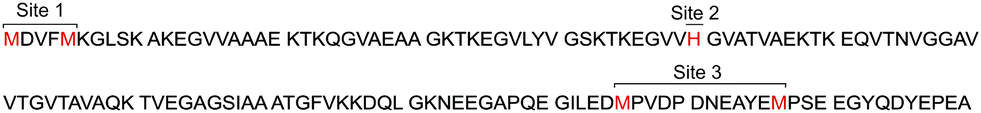 | ||
| Fig. 1 Primary sequence of full-length αS. Met and His residues acting as main anchoring residues in Cu(I) binding sites are highlighted. | ||
Transition metal ion homeostasis (copper, iron, zinc) plays an important role in neurodegenerative disorders, because these ions are considered as one of the possible factors leading to protein aggregation.14–16 Indeed, metal–protein interactions often impact the kinetics of amyloid aggregation and the neurotoxicity of protein aggregates; particularly, this has been demonstrated for the case of copper and zinc interactions with the amyloid beta peptide,17–19 associated to AD.
Protein–metal interactions play an important role in αS aggregation15,20 and might link the pathological processes of protein aggregation, oxidative damage and neuronal cell toxicity.14,16,21 A recent study demonstrated that under aerobic conditions, iron(II)-mediated O2 chemistry locks αS into an oligomeric conformation that prevents a parallel β-sheet fold that would normally progress into fibrils, suggesting that the elevated toxicity of the iron-αS coupling could be linked to the antiparallel right-twisted conformation of αS that occurs following iron(II) coordination in the presence of O2.22 It was also shown recently that calcium mediates distinct pathways of αS aggregation, involving interfibrillar aggregation and formation of large, toxic aggregates as the final products.23,24 Notably, copper was shown to be the most effective metal ion promoting αS fibril formation,15,25 subsequently causing cytotoxicity26,27 or seeding αS aggregation.28 Adding biological relevance to these findings, brain concentrations of copper appears dysregulated in patients with PD, suggesting that a positive correlation might be established between an increase of copper levels and disease severity.29–34
The ability of copper to accept or donate electrons implicates it in the production of reactive oxygen species, an established pathway of PD.35 This mechanism is a highly selective, site-specific process that involves interactions of the protein with both oxidation states of the copper ion.15,36–43 Added to the abundant evidence revealing that αS undergoes N-terminal acetylation in vivo (AcαS),44,45 it was recently reported that this modification of αS abolishes high-affinity Cu(II) binding.46 Since copper ions are predominantly found in their Cu(I) state in the reducing environment of living cells, characterization of the physiologically relevant AcαS–Cu(I) complexes is important.
NMR-based studies revealed that the main anchoring groups for Cu(I) binding – Met1/Met5 (site 1), His50 (site 2) and Met116/Met127 (site 3)- were preserved in the acetylated form of the protein42,47–49 (Fig. 1). In addition, formation of the AcαS–Cu(I) complex at site 1 stabilizes local conformations that contain substantial α-helical secondary structure and have restricted mobility.42 Linked to the fact that the Met-X3-Met motif (site 1) at the N-terminus of AcαS resembles motifs found in helical copper transport proteins, the formation of AcαS–Cu(I) complex at site 1 might have physiologically relevant implications in processes related to metal-transport, membrane binding or protein aggregation, which are enhanced by increased α-helical content at the N-terminus of the protein.
With regard to the coordination environment of Cu(I) in site 1 of the αS protein, EXAFS investigations revealed a 2S2O/N coordination sphere for the metal ion, thus indicating the presence of a four coordinated Cu(I) ion, probably with a tetrahedral geometry.37 While the role of the two thioether groups of Met-1 and Met-5 as metal anchoring groups for Cu(I) binding to site 1 was demonstrated,41,44,46,50,51 the identity of the residues providing the oxygen/nitrogen ligands is less well known. From NMR structural calculations obtained for the complex between Ag(I) and the 1–15 peptide of β-synuclein (βS), an homologue protein of αS that coordinates Cu(I) through Met residues at position 1, 5 and 10, it was proposed that Asp residue at position 2 of βS might act as a potential source for the establishment of a Cu–O bond.37 Based on these data, a similar role was proposed for Asp-2 in Cu(I) coordination to the protein αS.37 Regarding the additional oxygen/nitrogen atom bound to Cu(I) in site 1, it was attributed to a water or acetonitrile molecule.
In this work we sought to delineate the coordination environment and binding specificity of Cu(I) to the Met-X3-Met motif of the protein αS. The identity of the Cu(I) binding ligands and its role into the affinity and structural properties of the interaction was investigated by the combined application of NMR spectroscopy and the design of site-directed mutants and synthetic peptide models of the protein. Previously, we demonstrated the role of Met-1 and Met-5 as anchoring residues for the binding of Cu(I) to the protein αS.49 In order to assess the role of N-terminal acetylation and Asp-2 on metal coordination to the Met-X3-Met site, we investigated the details of Cu(I) binding to the synthetic peptide Ac-1MDVFMK6 (P1AS) and its non-acetylated variants P1AS and D2A P1AS, and compared their Cu(I)-binding features. The backbone amide regions in the 1D 1H NMR spectra of the different peptide species in the absence and presence of Cu(I) are shown in Fig. 2A–F. This spectral region contains well-resolved resonances and thus constitutes an excellent probe to analyze the metal–peptide interaction process. Whereas the amide resonances of Met-1 and Asp-2 cannot be detected in the experiments performed with the non N-acetylated peptide forms because of solvent-exchange effects, the signals from these residues become detectable upon acetylation of the N-terminus (Fig. 2E, F and Fig. S1, ESI†).
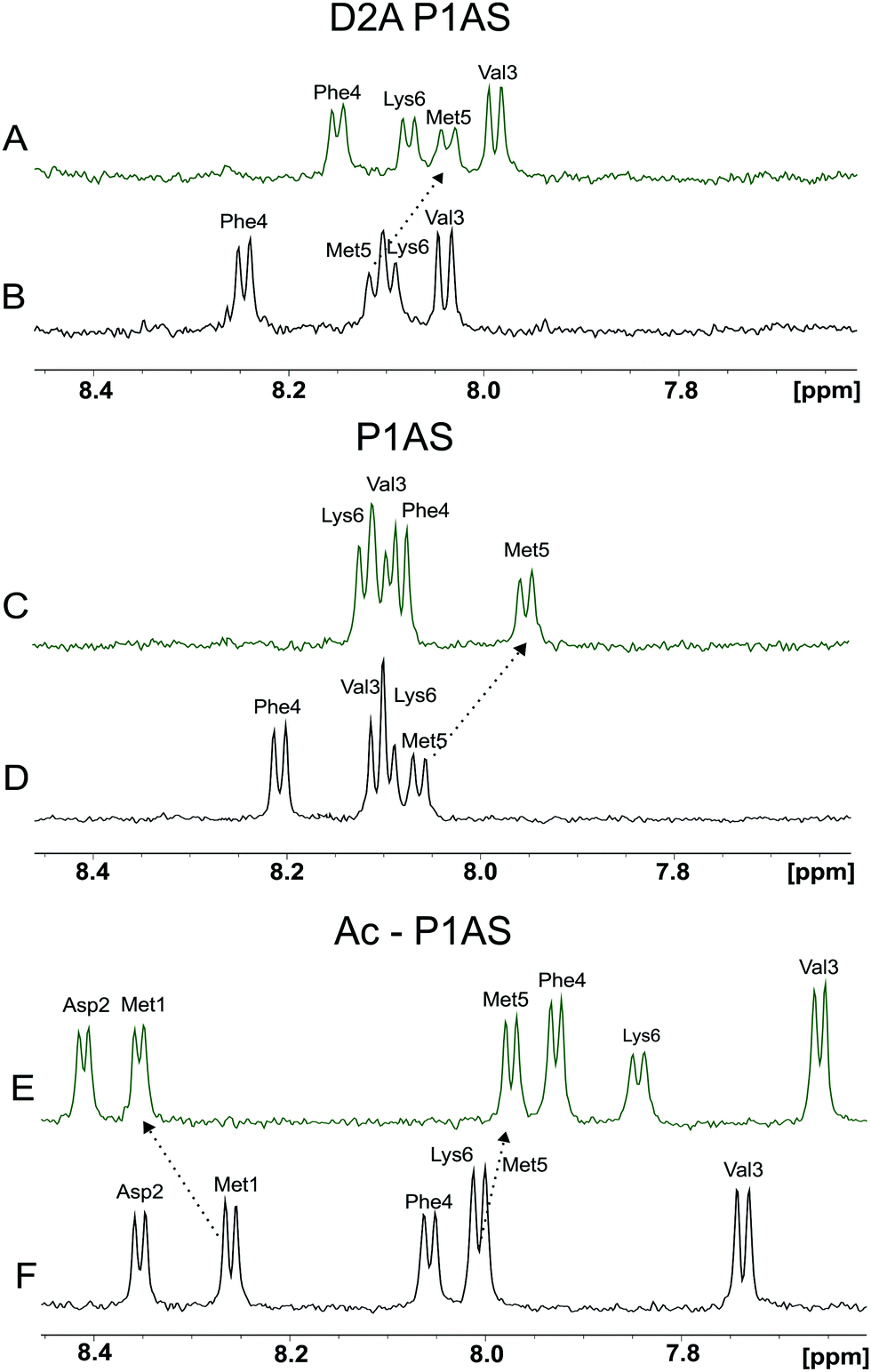 | ||
| Fig. 2 NMR analysis of Cu(I) binding to αS synthetic model peptides. 1D 1H NMR spectra (7.5–8.5 ppm) of D2A P1AS (A and B), P1AS (C and D) and Ac-P1AS (E and F) peptides in the absence (black) and presence (green) of 1 equiv. of Cu(I). In all cases, the addition of EDTA abolished the changes induced by the metal ion, confirming the reversibility of the interaction (data not shown). All experiments were recorded on 50 μM peptide samples dissolved in buffer A at 15 °C. | ||
As reflected by Fig. 2E and F resonances of amide protons corresponding to Met-1 in the Ac-P1AS peptide were clearly most affected by the interaction with the metal ion than those corresponding to Met-5, indicating that the interaction of the metal ion with Met-1 is favored. Notably, the effects of Cu(I) binding on the amide resonances of Met-5 were more pronounced in the variants P1AS (Fig. 2C and D) and D2A (Fig. 2A and B) compared to the Ac-P1AS peptide, suggesting that acetylation at the N-terminus and/or the presence of an Asp residue at position 2 influences Cu(I) binding at site 1.
We further characterized the binding interaction of Cu(I) with the P1AS variants of Fig. 2 by inspecting the chemical shifts changes induced by Cu(I) binding on the S-CH3 methyl resonances belonging to Met residues. To detect the characteristic Hε proton resonances corresponding to the S-CH3 groups of Met-1 and Met-5 we performed 1D 1H NMR experiments (data not shown). Consistent with the results described above, the degree of perturbation induced by one equivalent of Cu(I) on the S-CH3 resonances of Met-1 decreases in the order: Ac-P1AS (Δδ = 0.14 ppm) > P1AS (Δδ = 0.11 ppm) > D2A P1AS (Δδ = 0.08 ppm), giving further support to the role of N-terminal acetylation and Asp-2 as structural factors promoting Cu(I) coordination at site 1. Overall, these results confirm that the effects observed on the amide groups of the peptide variants in the presence of Cu(I) reflect the direct interaction of the metal ion with the sulfur atoms of the Met-1 and Met-5 residues, consistent with the binding preference of Cu(I) to coordinate sulfur atoms of Met residues in metalloproteins.
Next, to quantify the impact of these changes on the affinity features of Cu(I) binding to the Met-X3-Met site, we determined the dissociation constants of Cu(I) complexes with the P1AS variants studied. The resonances corresponding to the S-CH3 groups of Met-1 and Met-5 were well-resolved over the entire Cu(I) titration experiments and thus well-suited for calculation of the dissociation constant. Fig. S2 (ESI†) shows the binding curves of Cu(I) to Ac-P1AS and the P1AS and D2A P1AS variants. The derived conditional dissociation constants (cKd1) for the complexes of Cu(I) with Ac-P1AS, P1AS and D2A P1AS variants were 4.8 ± 0.7 nM, 8.5 ± 0.5 nM and 13.4 ± 1.0 nM, respectively. From the estimation of the conditional affinity for the complexes of Cu(I) with the M5I and M1I P1AS peptide variants the values 65 ± 5 nM and 163 ± 10 nM were obtained, respectively.49 These results allow us to conclude that: (i) the binding affinity for Cu(I) decrease in the order Ac-P1AS > P1AS > D2A P1AS > M5I P1AS > M1I P1AS; (ii) Met-1 and Met-5 residues act as the main anchoring moieties for Cu(I) binding to site 1, providing S–Cu binding modes; (iii) N-terminus acetyl group and Asp residue in position 2 sequence play a more modest role in terms of Cu(I) binding affinity, acting as potential sources for the establishment of Cu–O binding modes.
To confirm the findings derived from our analysis of Cu(I) binding to peptide models, we then analyzed the structural and affinity features of the Cu(I) complex with the Met-X-Met motif in the N-terminal region of the protein αS. To this purpose, we used 15N isotopically enriched AcαS and the M5I/H50A and D2A mutants. The D2A mutant of αS is not a substrate for the NatB acetylase and thus lacks that post-translational modification, being referred as D2A αS further on.
The Cu(I) complexed states of the AcαS protein and its M5I/H50 AcαS and D2A αS mutant species were analyzed by 2D NMR spectroscopy (Fig. 3 and 4). As reported previously, upon titration of 15N-enriched AcαS with increasing concentrations of Cu(I), the 1H–15N heteronuclear single quantum correlation (HSQC) spectra retained the excellent resolution of the uncomplexed protein but demonstrated large chemical shift changes in a discrete number of amide resonances belonging to residues involved in Cu(I) binding at site 1 (Fig. 3A). As shown in Fig. 4, the resonances corresponding to amide groups of residues involved in Cu(I) binding to site 1 of the D2A αS protein were affected to a lesser extent by the presence of the metal ion, whereas almost no changes were observed in that set of signals for the M5I/H50 AcαS mutant. From these experiments, the conditional affinity for Cu(I) binding at site 1 in AcαS was cKd1 = 3.9 ± 1.0 nM (Fig. 3B), consistent with previous studies.42,50 Interestingly, the value reported for the Cu(I)-complex in the non-acetylated protein was cKd1 = 7.8 ± 1.0 nM,48 in line with the affinity differences found for Cu(I) binding to the N-acetylated and free amine P1AS peptides. The value of cKd1 for complexation of Cu(I) to the Met-X3-Met site in M5I/H50A AcαS and the D2A αS were 63 ± 5 nM and 14 ± 2 nM, respectively. Overall, these data demonstrate that the affinity features observed for Cu(I) binding in the synthetic peptide models are preserved in the proteins.
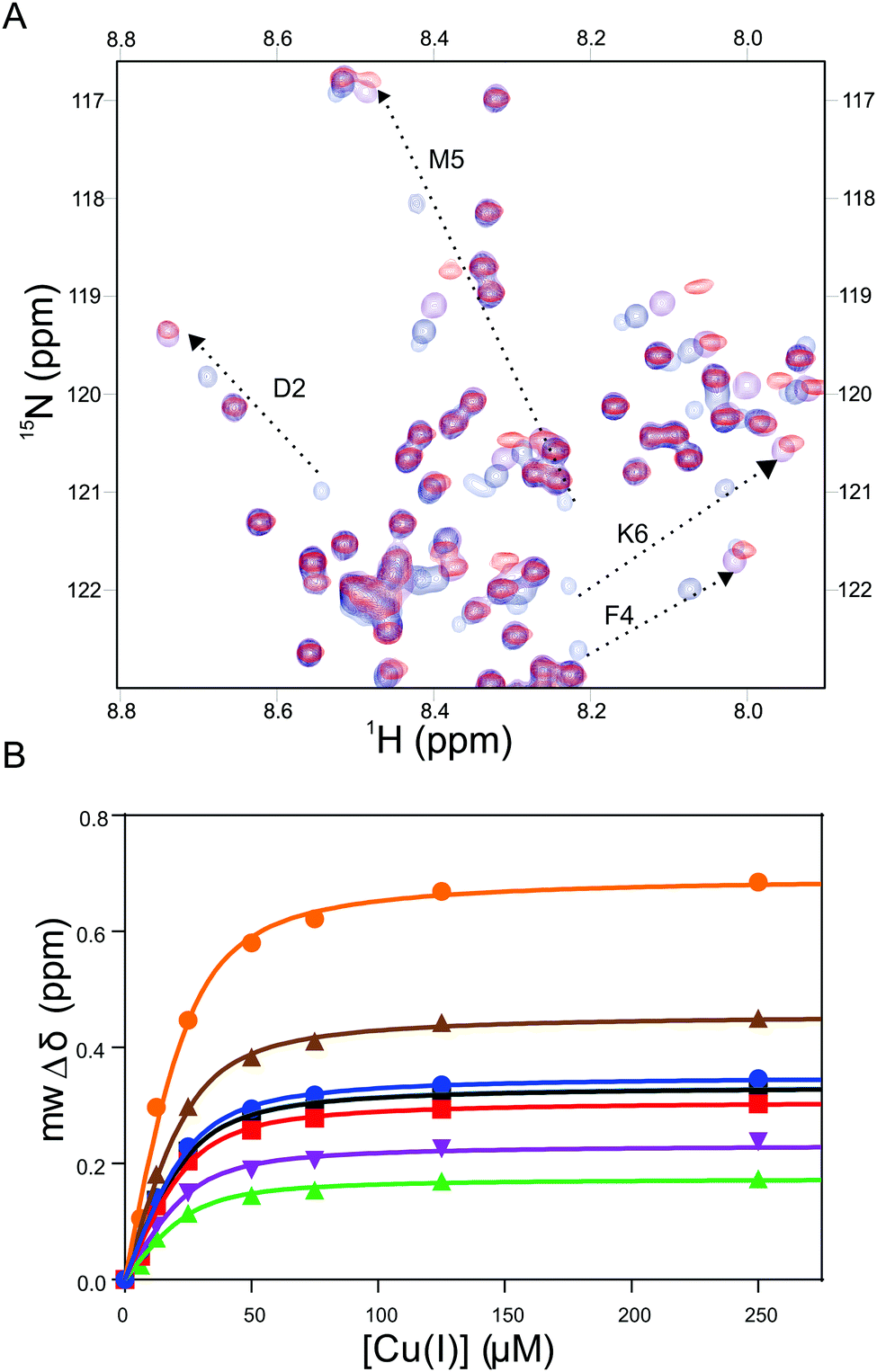 | ||
Fig. 3 NMR analysis of Cu(I) binding to AcαS. (A) Overlaid 1H–15N HSQC spectra of AcαS in the absence and presence of increasing Cu(I) concentrations. From blue to red: 0, 1.0, 3.0 and 5.0 equiv. of Cu(I). Most-affected residues involved in Cu(I) binding to the Met-X3-Met site are labeled. (B) Binding curves of Cu(I) to the Met-X3-Met site of AcαS as monitored by the average change in the mwΔδ for 1H and 15N of most affected amide resonances: Met-1 ( ), Asp-2 ( ), Asp-2 ( ), Val-3 ( ), Val-3 ( ), Phe-4 ( ), Phe-4 ( ), Met-5 ( ), Met-5 ( ), Lys-6 (■) and Gly-7 ( ), Lys-6 (■) and Gly-7 ( ). Curves represent the fit to a model incorporating complexes of Cu(I) into three classes of independent, non-interactive binding sites, using the program DynaFit. Experiments were recorded at 15 °C using 15N isotopically enriched AcαS (50 μM) samples dissolved in buffer A. ). Curves represent the fit to a model incorporating complexes of Cu(I) into three classes of independent, non-interactive binding sites, using the program DynaFit. Experiments were recorded at 15 °C using 15N isotopically enriched AcαS (50 μM) samples dissolved in buffer A. | ||
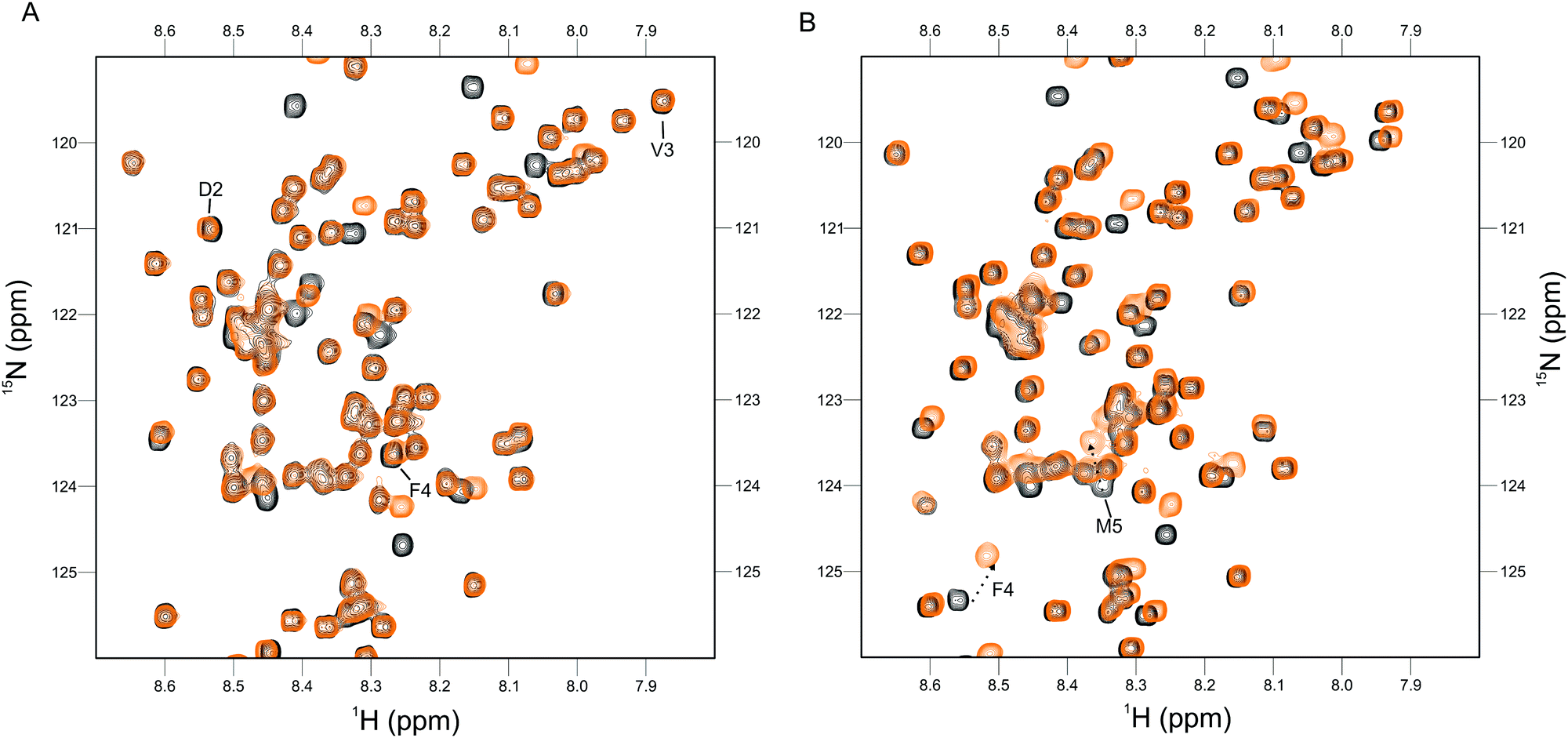 | ||
| Fig. 4 NMR analysis of Cu(I) binding to the M5I/H50A and D2A protein variants. (A) Overlaid 1H–15N HSQC spectra of M5I/H50A AcαS in the absence (black) and presence (orange) of 1 equiv. of Cu(I). (B) Overlaid 1H–15N HSQC spectra of D2A αS in the absence (black) and presence (orange) of 1 equiv. of Cu(I). For comparative purposes with AcαS-Cu(I), residues well-isolated and involved in Cu(I) binding to site 1 are labeled. Experiments were recorded at 15 °C using 15N isotopically enriched protein (50 μM) samples dissolved in Buffer A. | ||
The structural implications of Cu(I) binding to these proteins were also evaluated in terms of the dynamic properties of their Cu(I) complexed forms. To this purpose, we measured 15N R1 and R2 relaxation rates. This set of experiments was first measured on the free state of the AcαS protein and the M5I/H50A AcαS and D2A αS mutants (Fig. 5). In all cases, the relaxation parameters showed a similar sequence dependence, with lower values at the termini of the protein and a plateau at the center of the relaxation profile, showing R1 values between 1.2 and 1.7 s−1 and R2 values between 2.0 and 4.0 s−1 (Fig. 5). Complexation with Cu(I) resulted in a slight increase in the R1 values for the 1–10 segment of the AcαS sequence (mean R1 values of 1.5 s−1 and 1.9 s−1 in the free and complexed protein, respectively); however, more pronounced deviations were found in the R2 values (mean R2 values of 2.2 s−1 and 5.8 s−1 for the free and complexed protein, respectively). These data indicate restricted local sampling in the pico- to nanosecond time scale for the 1–10 segment of AcαS–Cu(I) relative to the free protein, reflecting the loss of flexibility due to the stabilization of local conformations with α-helical secondary structure, as previously published.42,50 On the other hand, the slight increase of R2 values around His 50 and Met-116/127 residues in AcαS–Cu(I) reflects the low affinity interaction and fast exchange of the metal ion at these secondary sites.42,50 Interestingly, the backbone dynamic profiles of the Cu(I) complexed states of M5I/H50 AcαS and D2A αS measured under the same experimental conditions do not show changes in their R1 values relative to their metal-free states. As shown in Fig. 5, the differences observed between the R2 profiles of AcαS–Cu(I), M5I/H50 AcαS–Cu(I) and D2A αS–Cu(I) states are centered at the 1–10 segment. The R2 values in the 1–10 segment of the D2A αS–Cu(I) are increased to a lower extent compared to the AcαS–Cu(I) state, whereas only a slight deviation is observed for the R2 values in the M5I/H50 AcαS–Cu(I) form. These differences correlate well with the different affinities determined for the binding of the metal ion at these sites; however, the lower R2 values for the D2A αS–Cu(I) form might be also reflecting a small increase of α-helical propensity near the N-terminus, induced by the interaction with Cu(I) at the Met-X3-Met site. These results motivated us to evaluate the Cu(I) protein complexes in terms of their conformational properties. 3JHN-Hα couplings are reliable quantitative reporters of the time-averaged distribution of the backbone torsion angles, φ, and are frequently used to probe the propensity of intrinsically disordered proteins to sample different regions of conformational space. Therefore, we measured residue-specific 3JHN-Hα couplings in both free and copper-bound states of these proteins (Fig. S3, ESI†). With the exception of the decrease in 3JHN-Hα for the first 10 residues of AcαS upon Cu(I) binding, the values measured for the two forms of the M5I/H50A AcαS and D2A αS proteins were essentially indistinguishable. With an averaged 3JHN-Hα of 4.5 Hz expected for an ideal α-helix and a 3JHN-Hα of ∼7 Hz for random coil, our results indicate that a conformational transition toward increased α-helix structures is clearly observed only for the AcαS–Cu(I) form.
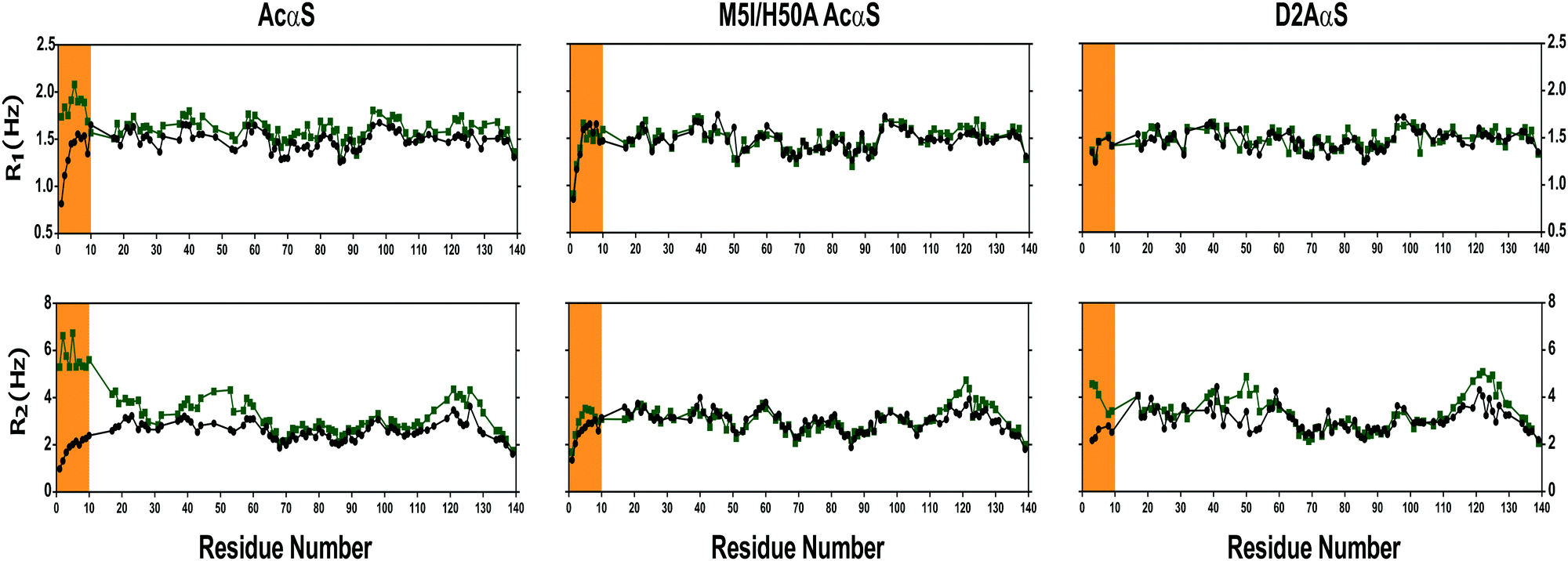 | ||
| Fig. 5 15N relaxation parameters of αS variants and its Cu(I) complexes. R1 and R2, relaxation rates of the proteins AcαS, M5I/H50A AcαS and D2A αS in the absence (black) and presence (green) of Cu(I). Experiments were recorded at 15 °C using 15N isotopically enriched protein (200 μM) samples dissolved in Buffer A in the absence and presence of 2 equivalents of Cu(I). The increase of R2 values around His-50 and Met-116/127 residues in AcαS-Cu(I) reflects the fast exchange of Cu(I) at these secondary sites, as previously reported.42,50 | ||
These results allow us to conclude that the N-terminal acetyl group and the Met-1, Asp-2 and Met-5 residues provide the binding ligands for the coordination environment of Cu(I) at the Met-X3-Met site of AcαS. Met-1 and Met-5 residues are critical for the binding affinity of the Cu(I) complex, acting as the main anchoring residues for metal binding. While having a more modest impact in the affinity features of Cu(I) binding at this site, as compared to the Met residues, the N-terminal acetyl group and Asp-2 are important in promoting local helical conformations, contributing to the stabilization of these structures by favoring Cu(I) binding. Thus, the increased helicity in AcαS–Cu(I) can be rationalized by stabilization of the helix macrodipole and formation of energetically more favorable hydrogen bond interactions triggered by the removal of the α-amino positive charge upon acetylation, by the role of Asp-2 diminishing the dipole moment to its N-terminus and by the Cu(I)-induced structural rearrangement of Met-1 and Met-5 side chains, respectively.51,52
Our study demonstrates that perturbing the coordinating residues involved in Cu(I) binding at site 1 of αS has an effect also in the redox properties of the complex. Specifically, for the series of peptide and protein variants studied here, the trend in Cu(I) binding affinity follows: Ac-P1AS > P1AS > D2A-P1AS > M5I-P1AS > M1I-P1AS. Consistent with our results and with previous findings,49 the Met residues play a key role in Cu(I) coordination and affinity features, having Met-1 a more important role in stabilizing Cu(I) binding. In terms of the redox properties of the site, the decrease in Cu(I) binding affinity observed for the M1I modification translates into an 76 mV decrease in the reduction potential (53 mV for the M5I mutant), consistent with previous reports.53 A new finding from this study is that Asp-2 also plays a role in Cu(I) binding; the D2A modification causes a ∼1.5 fold decrease in binding affinity for Cu(I), which would translate into a decrease of ∼15 mV in the reduction potential, if this mutation were not to impact Cu(II) binding affinity as it does (Table S1, ESI†). While having a more modest impact in the redox properties of site 1, Asp-2 is certainly also playing a role in stabilizing Cu(I) coordination to αS. Finally, the acetylation of the N-terminal group causes a ∼1.7 fold increase in Cu(I) binding affinity, which would translate into a ∼17 mV increase in the reduction potential of the complex, if it were not to impact Cu(II) binding affinity. However, it is important to note that acetylation also has a drastic impact in Cu(II) coordination, since it abolishes metal binding at this site.46 Thus, the combined effect of acetylation of the N-terminal group is expected to stabilize Cu(I) while significantly destabilizing Cu(II), contributing to a significant increase of ∼258 mV of the reduction potential of the site, as compared to the non-acetylated form (Table S1, ESI†). Overall, these results underscore the important role that acetylation and the Asp-2 residue play together to stabilize the reduced form of the αS–Cu complex.
Another important impact of the acetylation of αS is the promotion of local helical conformation upon Cu(I) binding.42,50 It has been reported that chemical induction of alpha-helical conformation in αS by using fluorinated solvents increases the reduction potential of the αS–Cu complex by ∼90 mV, possibly stabilizing the Cu(I) form.54 From this shift, an increase in Cu(I) binding affinity of an order of magnitude was estimated for the alpha-helical αS form, as compared to the unstructured state. In line with this, a more recent work reported that Cu(I) binding to the 1–15 fragment of αS in the presence of SDS micelles resulted in a dissociation constant two orders of magnitude smaller than that found for the complex with the full protein in aqueous buffer.47 On the other hand, our data does not show such a dramatic change in Cu(I) affinity upon induction of α-helical conformations by complexation of the metal ion to the N-acetylated form of the protein. Although this could be attributed to a shorter length and/or the more dynamic (transient) properties of helical structures induced by αS–Cu(I) complexation at the Met-X3-Met site, as compared to that induced by the fluorinated solvent or SDS micelles, studies performed under more physiologically relevant conditions are clearly needed to evaluate the role of local conformations over the affinity and redox potential properties of αS–Cu(I) complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. O. F. thanks Universidad Nacional de Rosario (CUA-DAHZ 007/11), ANPCyT-FONCyT (PICT 2014-3704) and the Alexander von Humboldt Foundation (P4507) for financial support. C. O. F. and C. G. thank the Max Planck Society (P10390) for support. L. Q. thanks the National Council for Science and Technology in Mexico (CONACYT) grants 221134 and 193318. Open Access funding provided by the Max Planck Society.References
- C. M. Dobson, Semin. Cell Dev. Biol., 2004, 15, 3–16 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef PubMed.
- S. Gandhi and N. W. Wood, Nat. Neurosci., 2010, 13(7), 789–794 CrossRef PubMed.
- C. Henchcliffe and F. M. Beal, Nat. Clin. Pract. Neurol., 2008, 4(11), 600–609 CrossRef PubMed.
- S. Papapetropoulos, N. Adi, J. Ellul, A. A. Argyriou and E. Chroni, Neurodegener. Dis., 2007, 4(6), 424–427 CrossRef PubMed.
- M. G. Spillantini, R. A. Crowther, R. Jakes, N. J. Cairns, P. L. Lantos and M. Goedert, Neurosci. Lett., 1998, 251(3), 205–208 CrossRef PubMed.
- L. Stefanis, Cold Spring Harbor Perspect. Med., 2012, 2(2), a009399 Search PubMed.
- A. Sidhu., C. Wersinger and P. Vernier, FASEB J., 2004, 18, 637–647 CrossRef PubMed.
- L. Yavich, H. Tanila, S. Vepsäläinen and P. Jäkälä, J. Neurosci., 2004, 24(49), 11165–11170 CrossRef PubMed.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2005, 127, 476–477 CrossRef PubMed.
- C. W. Bertoncini, Y. S. Yung, C. O. Fernández, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef PubMed.
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef PubMed.
- M. J. Volles and P. T. Lansbury, Biochemistry, 2003, 42, 7871–7878 CrossRef PubMed.
- D. R. Brown, Metallomics, 2011, 3, 226–228 RSC.
- A. Binolfi, L. Quintanar, C. W. Bertoncini, C. Griesinger and C. O. Fernández, Coord. Chem. Rev., 2012, 256, 2188–2201 CrossRef.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef PubMed.
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47(8), 2252–2259 CrossRef PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52(21), 12193–12206 CrossRef PubMed.
- A. Binolfi, E. E. Rodriguez, D. Valensin, N. D’Amelio, E. Ippoliti, G. Obal, R. Duran, A. Magistrato, O. Pritsch, M. Zweckstetter, G. Valensin, P. Carloni, L. Quintanar, C. Griesinger and C. O. Fernández, Inorg. Chem., 2010, 49, 10668–10679 CrossRef PubMed.
- S. Bolognin, L. Messori and P. Zatta, NeuroMol. Med., 2009, 11, 223–238 CrossRef PubMed.
- D. L. Abeyawardhane, C. O. Fernández, C. J. Murgas, D. R. Heitger, A. K. Froney, M. K. Crozier and H. R. Lucas, J. Am. Chem. Soc., 2018, 140, 5028–5032 CrossRef PubMed.
- J. Y. Han, T. S. Choi and H. I. Kim, Sci. Rep., 2018, 8, 1895 CrossRef PubMed.
- J. Lautenschläger, A. D. Stephens, G. Fusco, F. Ströhl, N. Curry, M. Zacharopoulou, C. H. Michel, R. Laine, N. Nespovitaya, M. Fantham, D. Pinotsi, W. Zago, P. Fraser, A. Tandon, P. St George-Hyslop, E. Rees, J. J. Phillips, A. De Simone, C. F. Kaminski and G. S. K. Schierle, Nat. Commun., 2018, 9(1), 712 CrossRef PubMed.
- R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4294–4299 CrossRef PubMed.
- X. Wang, D. Moualla, J. A. Wright and D. R. Brown, J. Neurochem., 2010, 113, 704–714 CrossRef PubMed.
- J. A. Wright, X. Wang and D. R. Brown, FASEB J., 2009, 23, 2384–2393 CrossRef PubMed.
- A. Villar-Piqué, T. Lopes da Fonseca, R. Sant’Anna, P. M. Szegö, L. Fonseca-Ornelas, R. Pinho, A. Carija, E. Gerhardt, C. Masaracchia, E. Abad Gonzalez, G. Rossetti, P. Carloni, C. O. Fernández, D. Foguel, I. Milosevic, M. Zweckstetter, S. Ventura and T. F. Outeiro, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6506–E6515 CrossRef PubMed.
- E. Carboni and P. Lingor, Metallomics, 2015, 7, 395–404 RSC.
- C. J. Brewer, Chem. Res. Toxicol., 2010, 23(2), 319–326 Search PubMed.
- Y. Pushkar, G. Robinson, B. Sullivan, S. X. Fu, M. Khone, W. Jiang, S. Rohr, B. Lai, M. A. Marcus, T. Zakharova and W. Zheng, Aging Cell, 2013, 12(5), 823–832 CrossRef PubMed.
- F. Rose, M. Hodak and J. Bernholc, Sci. Rep., 2011, 1, 11 CrossRef PubMed.
- N. Arnal and M. J. de Alaniz, Brain Res., 2010, 1319, 118–130 CrossRef PubMed.
- K. M. Davies, D. J. Hare, V. Cottam, N. Chen, L. Hilgers, G. Halliday, J. F. B. Mercer and K. L. Double, Metallomics, 2013, 5, 43–51 RSC.
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef PubMed.
- A. Binolfi, G. R. Lamberto, R. Duran, L. Quintanar, C. W. Bertoncini, J. M. Souza, C. Cerveñansky, M. Zweckstetter, C. Griesinger and C. O. Fernández, J. Am. Chem. Soc., 2008, 130, 11801–11812 CrossRef PubMed.
- R. De Ricco, D. Valensin, S. Dell'Acqua, L. Casella, P. Dorlet, P. Faller and C. Hureau, Inorg. Chem., 2015, 16(16), 2319–2328 Search PubMed.
- A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernández, J. Am. Chem. Soc., 2006, 128, 9893–9901 CrossRef PubMed.
- S. C. Drew, S. Ling Leong, C. L. L. Pham, D. J. Tew, C. L. Masters, L. A. Miles, R. Cappai and K. J. Barnham, J. Am. Chem. Soc., 2008, 130, 7766–7773 CrossRef PubMed.
- J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2008, 130, 6898–6899 CrossRef PubMed.
- A. Binolfi, A. A. Valiente-Gabioud, R. Duran, M. Zweckstetter, C. Griesinger and C. O. Fernández, J. Am. Chem. Soc., 2011, 133, 194–196 CrossRef PubMed.
- M. C. Miotto, A. A. Valiente-Gabioud, G. Rossetti, M. Zweckstetter, P. Carloni, P. Selenko, C. Griesinger, A. Binolfi and C. O. Fernández, J. Am. Chem. Soc., 2015, 137, 6444–6447 CrossRef PubMed.
- F. Camponeschi, D. Valensin, I. Tessari, L. Bubacco, S. Dell’Acqua, L. Casella, E. Monzani, E. Gaggelli and G. Valensin, Inorg. Chem., 2013, 52, 1358–1367 CrossRef PubMed.
- T. Bartels, J. G. Choi and D. J. Selkoe, Nature, 2011, 477, 107–110 CrossRef PubMed.
- B. Fauvet, M. B. Fares, F. Samuel, I. Dikiy, A. Tandon, D. Eliezer and H. A. Lashuel, J. Biol. Chem., 2012, 287, 28243–28262 CrossRef PubMed.
- G. M. Moriarty, C. A. Minetti, D. P. Remeta and J. Baum, Biochemistry, 2014, 53, 2815–2817 CrossRef PubMed.
- S. Dell’Acqua, V. Pirota, E. Monzani, F. Camponeschi, R. De Ricco, D. Valensin and L. Casella, Inorg. Chem., 2016, 55, 6100–6106 CrossRef PubMed.
- M. C. Miotto, A. Binolfi, M. Zweckstetter, C. Griesinger and C. O. Fernández, J. Inorg. Biochem., 2014, 141, 208–211 CrossRef PubMed.
- M. C. Miotto, E. E. Rodriguez, A. A. Valiente-Gabioud, V. Torres-Monserrat, A. Binolfi, L. Quintanar, M. Zweckstetter, C. Griesinger and C. O. Fernández, Inorg. Chem., 2014, 53, 4350–4358 CrossRef PubMed.
- M. C. Miotto, M. D. Pavese, L. Quintanar, M. Zweckstetter, C. Griesinger and C. O. Fernández, Inorg. Chem., 2017, 56, 10387–10395 CrossRef PubMed.
- W. G. Hol, Adv. Biophys., 1985, 19, 133–165 CrossRef PubMed.
- S. Tyanova, J. Cox, J. Olsen, M. Mann and D. Frishman, PLoS Comput. Biol., 2013, 9(1), e1002842 CrossRef PubMed.
- E. E. Rodríguez, T. Arcos-López, L. G. Trujano-Ortiz, C. O. Fernández, F. J. González, A. Vela and L. Quintanar, J. Biol. Inorg. Chem., 2016, 21(5-6), 691–702 CrossRef PubMed.
- B. Zhou, Y. Hao, C. Wang, D. Li, Y. N. Liu and F. B. Zhou, J. Inorg. Biochem., 2013, 118, 68–73 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mt00232k |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2018 |