
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How should multicomponent supramolecular gels be characterised?
Emily R.
Draper
 * and
Dave J.
Adams
*
* and
Dave J.
Adams
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Emily.Draper@Glasgow.ac.uk; Dave.Adams@Glasgow.ac.uk
First published on 8th February 2018
Abstract
Low molecular weight gels, or supramolecular gels, are formed when small molecules self-assemble into fibrous structures. Above a critical concentration, the entanglement and cross-linking of these structures leads to the formation of a self-supporting gel. There are many examples where a single component is used to form such gels. There is however an ever-increasing interest in using multiple components. Here, if each component is able to form a gel by itself, a range of fibre types are possible, formed by either random or specific associations between the low molecular weight gelators (LMWG). The properties of the networks will depend on how the LMWG assemble into the primary fibrous structures and then how these primary structures entangle. As such, to understand these gels, it is necessary to understand the networks across multiple length scales. Here, we discuss the current state of the art, the effectiveness of the different techniques that have been used, and hopefully provide the impetus for the field to move away from the cartoon-level discussion of assembly.
 Dave J. Adams (left) and Emily R. Draper (right) | Emily Draper (right) completed a Masters in Chemistry at the University of Liverpool where she then carried out her PhD in Chemistry working with photoresponsive gelators. She was awarded a PhD in 2015 and continued as a PDRA working on multi-component hydrogels. In 2016, Emily moved to the University of Glasgow as a PDRA working on multi-component hydrogels for solar cell applications. In September 2017, Emily then was awarded an Early Career Fellowship from the Leverhulme Trust and a Lord Kelvin Adam Smith Leadership award from the University of Glasgow, where she is currently working on self-assembled small molecules for organic electronic devices. |
Dave Adams (left) received his DPhil from the University of York in 1999. After postdoctoral research at York, Leeds, and Leicester, he joined Unilever R&D for four years. In 2008, he joined the University of Liverpool before joining the University of Glasgow in 2016. He is currently an EPSRC Fellow. His current research interests encompass many areas of materials chemistry including soft matter (supramolecular polymers and gels, self-assembled polymer nanostructures), as well as materials for strategically important gas storage and carbon capture (high surface area polymers, clathrates and porous solids). |
Key learning points1. Multicomponent supramolecular gels can be formed by the self-assembly of two or more independent gelators into fibrous structures that entangle.2. On self-assembly, different possibilities exist: the gelators can interact specifically to lead to ordered fibres containing both gelators or randomly to form fibres that contain random amounts of both gelator. Self-sorted fibres may also be formed, where each fibre contains only one of the gelators. Whilst often not stressed, mixtures of these possibilities are conceptually possible. 3. Once these primary structures are formed, entanglement happens over longer length scales and the fibres can interact in different ways. 4. The properties of the gels will not only be a result of this primary assembly, but also to how the fibres entangle, cross-link and interact. 5. To understand these systems fully therefore, it is necessary to characterise the gels across multiple length-scales, from molecular interactions to bulk properties. |
Introduction
Gels can be formed by the self-assembly of small molecules called low molecular weight gelators (LMWGs).1–3 To form the gels, the LMWG assemble into long fibres, which cross-link in some manner, either by entanglement or branching for example, to form a network. The networks are held together only by the non-covalent forces that induce the self-assembly of the LMWG and so the gels tend to be reversible. These LMWG have been investigated for a wide range of applications, for example for controlled release, cell culturing, optoelectronics and directing crystallisation.1,4–6For the majority of cases, a single LMWG is used to form the gels. There are also a number of examples where more than one component is needed to form a gel; essentially here different components come together in situ to form a LMWG.7 Recently, there has been more and more interest in the idea of gels formed from multiple LMWG. In these cases, each of the components is a LMWG in its own right.7,8 There are a number of reasons why mixing different LMWG is interesting. For example, one could imagine a case where one LMWG provides the structural elements of the gel, whilst another drives specific interactions with a desired target.9 There are examples where the different components have different electronic properties, allowing the formation of an analogy of a bulk heterojunction.10 It may also be possible to access different properties by mixing components than can be achieved with a single component, such as a temporal trigger for gelation,11 or this approach can allow gels that can be photo-patterned to be formed.12,13
Whilst there is an ever-increasing number of papers on multicomponent gels, there are very few detailed discussions as to how best to characterise these systems. Characterising single component systems fully across all length-scales can be challenging. Moving to multicomponent systems increases the complexity dramatically. Conceptually, when two LMWG are mixed, there are a range of possibilities. Assuming that the overall self-assembly process still leads to the formation of fibres, the LMWG can interact specifically to lead to ordered fibres containing both gelators (e.g.Fig. 1a), or randomly to form fibres that contain random amounts of both LMWG (Fig. 1b).
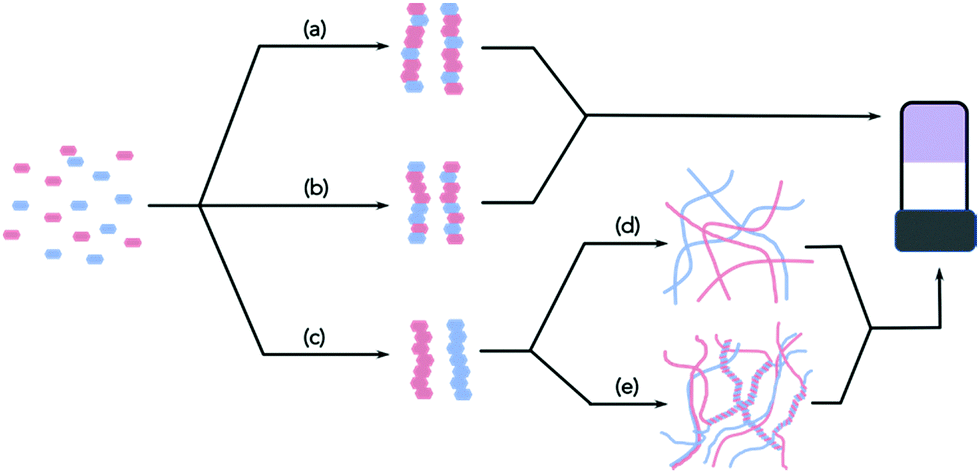 | ||
| Fig. 1 In a mixture of two LMWG, the primary assembled fibres can be formed by (a) specific interactions leading to ordered fibres; (b) random incorporation of the two gelators; (c) self-sorted fibres. Self-sorted fibres can go on to form (d) homo- or (e) hetero-aggregates. | ||
Self-sorted fibres may also be formed, where each fibre contains only one of the LMWG (Fig. 1c). Of course, it is possible that the assembly leads to a mixture of these possibilities, or mainly but not exclusively one type of assembly. If there are more than two LMWG, the number of scenarios becomes increasingly complex, but all of the examples reported so far are (to the best of our knowledge) only formed by two components.
Importantly, the properties of the gels will not only be a result of this primary assembly, but also to how the fibres entangle, cross-link and interact. Hence, in general, it is important to understand not just the primary assembled structures, but also the structure on a longer length-scale. This is often the most difficult aspect to understand.
Here, we will discuss how these multicomponent gels can be characterised and understood. Currently, the literature has a number of examples, but there are cases where the assembly is assumed, or very little data used to prove the assembly type. In some cases, of course, full characterisation may not be needed, but our contention is that the understanding is necessary such that the systems can be designed. As a single example, if we assume that self-sorted fibres are formed and one network is formed before the second, the properties of the gel will be a result of the two networks, how those networks are interacting (if at all), whether the first network was modified by the presence of the second (at the time un-gelled) component, and whether the second network was affected by growing in the presence of the first. This is important; it has been shown that gel networks can be significantly affected by growing in the presence of a non-gelling additive.14 To understand how to carry this out reproducibly, the gel would need to be characterised at many length-scales. Similarly, if we wish one component to interact specifically with something (e.g. with a cell), we would need this component to be placed in a position where this interaction can occur; again, characterisation using a range of techniques will be necessary to understand if this is the case.
In this Tutorial Review, we discuss the current state of the art. We do not attempt to cover how multicomponent systems can be designed to co-assemble or self-sort, but rather we focus on how we can tell the difference post-gelation. For the interested reader, recent reviews cover the design elements of multicomponent systems.7,8 As this is a Tutorial Review, we are restricted in the overall number of references. We have therefore chosen a range of papers to discuss that cover the concepts. We are in no way suggesting that these are the only, nor necessarily the most important multicomponent gel papers, only that these allow us to cover the widest concepts within the restricted reference list. We have also chosen to focus our discussion around gels formed primarily by assembly of the LMWG into fibrous structures; assembly into other structures such as vesicles and lamellar structures is also known to be possible to form gels.1,2 As far as we know, there are no examples where mixed systems have been prepared from such LMWG however. Finally, many of the examples presented here are for where at least one of the components is a LMWG based around an amino acid or oligopeptide. However, this is not always the case as we have chosen to show the widest range of techniques.
Multicomponent assembly
There are now a number of papers that describe the use of multicomponent LMWG systems.7,8 A number of these are based on functionalised amino acids or peptides. As noted, whilst there is conceptually no limit to the number of components that could be mixed, the examples reported so far focus on a mixture of two LMWGs. It is also most common to focus on a single ratio and absolute concentration of each LMWG, but of course this does not need to be the case. As such, whilst the potential complexity is very high, most reports limit this where possible.It is unsurprisingly most common to mix LMWG whose self-assembly as a single component has also been examined. As such, in many cases, the two-component system is compared to that of the single component and differences are used to infer information about the two-component system. However, it is clear that using this approach, using only a single technique is sometimes perhaps not as informative as might be assumed. For example, both self-sorted and co-assembled gels (as proven by a range of techniques) were found to have rheology data higher for the two-component gel than for gels formed from either of the two components alone;11 only by using a number of complementary techniques could the absolute assembly type be assigned. It is also surprising that data are often presented without any comments on reproducibility. It is also very common for a single type of assembly to be assigned, as in 100% self-sorted. It is however, not clear that it would be possible to tell if a mixture of assembly types had occurred and to what degree.
With these caveats in mind, we believe that it is necessary to characterise the gels across multiple length-scales. Here, we mean that it is necessary to understand how the system has self-assembled from the molecular level to the multi-micron length-scale. The interactions at the molecular scale essentially will inform us as to the assembly in the primary structures that are shown in cartoon format in Fig. 1. Once the primary fibres have formed, these can interact in different ways, but this will occur on the nanometre scale (for example, lateral association of fibres will mean that the diameters could be on the order of 10–100 nm, Fig. 2a and b), with the distribution of fibres in space being perhaps homogeneous or heterogeneous on the multi-micron length-scale with potentially different underlying microstructures (Fig. 2c and d). As such, it is necessary to use a range of techniques to access information across these dimensions. We discuss the techniques that have been used below. These techniques have also of course been used for single component systems,15 but we focus on how these can be used to understand multicomponent systems.
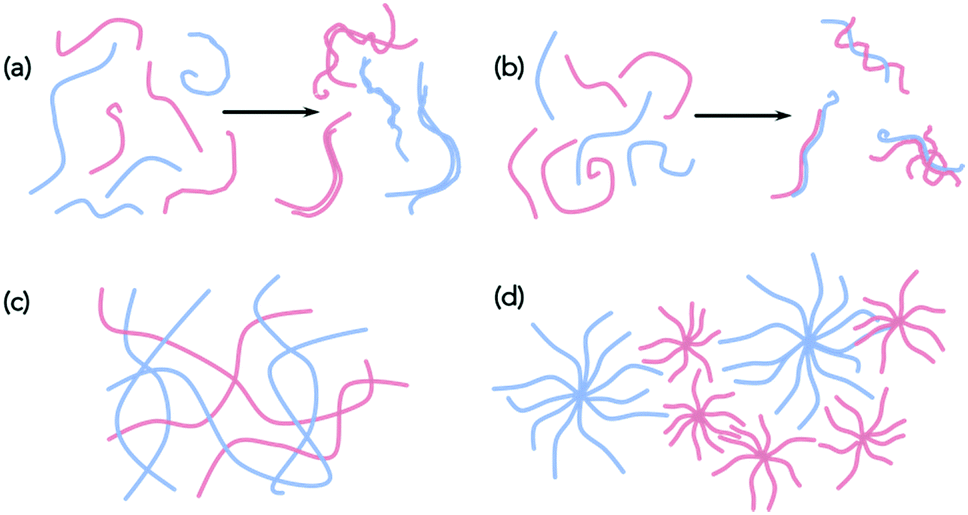 | ||
| Fig. 2 Assembly of self-sorted fibres could lead to homo-(a) aggregates or (b) hetero-aggregates by (a) lateral association. Different microstructures are conceptually possible, for example (c) and (d), which would both be consistent with a self-sorted system. | ||
Molecular-level assembly
In terms of our discussion here, we will order the techniques in terms of length-scale. We begin by discussing techniques that can probe the assembly at the molecular level, before moving to the fibre length-scale, and then finally discussing the techniques that can be used on bulk samples.Spectroscopy
The initial assembly is at the molecular level. In a mixture of gelators, the assembly can be such that the molecules may wish to co-assemble or self-sort (Fig. 1). At this length scale, tools such as infra-red spectroscopy, UV-vis absorption spectroscopy, fluorescence spectroscopy and circular dichroism (CD) spectroscopy can be powerful tools to probe molecular packing, hydrogen bonding etc.As with most of the techniques discussed here these techniques also rely on the individual components being significantly different to one another or the resulting network being different than the two starting materials. UV-vis absorption spectroscopy in particular can be used to look at aggregation states of molecules and is often used to determine whether aggregates are H-aggregated or J-aggregated, forming extended stacks etc. Conceptually, these spectra can also be used to investigate whether the packing in a multicomponent system is co-assembly or self-sorting. If a system were self-sorted (or at least the majority is self-sorted), then we would expect no change in the absorption of the individual components and the combined spectrum should look like an overlay of the two spectra for the individual components. For example, Sugiyasu et al. have shown that the absorption spectrum for the mixture was simply an addition of the spectra for the two components.16 The absence of any new peaks attributable to charge transfer implied that self-sorting had occurred. Monitoring the UV-vis absorption spectra on heating and cooling was used to show that changes occurred at the same temperatures as expected from the data for the single components. A similar overlay of the expected UV-vis absorption spectra was found for a mixture of a gelators.17
Alternatively, if there were some co-assembly (either social or random) then we would expect that there could be a change the absorption of the two molecules as the energy levels would be changed by the aggregation. It can be difficult to distinguish whether these systems are socially self-sorted or randomly assembled, or anywhere in between with this technique, or indeed quantify how much of each there is. It may be possible to model the aggregation and resulting spectrum with computational methods, but the number of molecules involved in the stack makes these calculations difficult. However, in some cases, distinct changes in the spectrum can occur on mixing due to charge transfer and this can be used to determine co-assembly. For example, when applied to multicomponent systems, specific co-assembly has been shown for mixtures of electron-rich and electron-poor gelators. For example, Das and Ghosh reported that a mixture of a naphthalene-based gelator could be mixed with a naphthalene diimide based gelator (Fig. 3).18 The gels formed from the single components were transparent and pale yellow respectively. On mixing, specific co-assembly could be shown by the formation of a charge transfer complex, resulting in a deep red gel. A different mixture was designed such that self-sorting was expected. In this case, the gel was pale yellow, showing the absence of charge transfer.
 | ||
| Fig. 3 (left) Structure of the gelators used by Das and Ghosh.18 DAN-1 and NDI-1 form a self-sorted mixture, whilst DAN-1 and NDI-2 specifically co-assemble. (right) Photos of gels in methylcyclohexane formed by DAN-1 (a), NDI-1 (b), DAN-1 + NDI-1 (c), DAN-1 + NDI-2 (d) and NDI-2 (e). Reproduced from ref. 18 with permission from The Royal Society of Chemistry. | ||
Dramatic changes in the UV-vis absorption spectra will only be expected if suitably functionalised gelators are used.
Fluorescence spectroscopy can be used in other cases, where energy transfer effects can be used to show that the molecules must be close to one another. This can be used to imply co-assembly. For example, Chen et al. showed that energy transfer could occur in a mixture of a naphthalene-functionalised dipeptide and an anthracene-functionalised dipeptide.19 Later work by Felip-León et al. however assigned the observation of energy transfer to a self-sorted system.20 George and co-workers showed both a self-sorted and a co-assembled system which were assigned on the basis on Förster resonance energy transfer and using donor and acceptor type molecules. They showed a chirality driven self-assembly process and used the observation of energy transfer and the CD data to show that the system was co-assembled. They could also visualize their fibres using fluorescence microscopy as a result. Effective energy transfer requires that the donor and acceptor are sufficiently close to one another and this can be achieved either by co-assembly or intimate mixing of fibres. Hence, distinguishing between these two possibilities on the basis of spectroscopy alone is difficult.
CD can provide information on chiral assemblies. For self-sorted systems, the CD spectrum has been shown to be a simple overlap of the sum of those obtained from the individual components.16 In a case where co-assembly occurred, the CD spectrum was not the same as the expected sum of those of the components.11
Fourier-Transform Infrared (FTIR) spectroscopy FTIR works similarly to the above techniques where a difference in the spectrum for the mixed system compared to those for the components indicates co-assembly, whilst an overlay of the two separate components can suggest that co-assembly is occurring.21 FTIR can also give us an insight as to how interactions are occurring, for example between COOH groups or H-bonding. This technique could therefore be used to understand how the molecules are stacking and even be used to quantify how much co-assembly is occurring. For example, Hao and co-workers used a host–guest system of two gelling components where they could trigger gelation in two different ways giving different types of interactions.22 This could be seen directly in the IR spectra, with new peaks appearing and other peaks shifting. The spectra also revealed that incomplete self-sorting had occurred in the self-sorted system as a small amount of interaction with the second gelator could be detected. This a rare example of where this has been shown.
Nuclear Magnetic Resonance (NMR) spectroscopy can be used to follow the gelation process. Most commonly, before assembly the molecules can be detected by NMR. As they assemble into fibres, they become NMR-invisible. Hence, this technique can be used to follow the process if the kinetics of gelation are slow, and be used to show that sequential disappearance (indicative of self-sorting)23 or simultaneous disappearance of the peaks11 from the different gelators occurs.
For all of these techniques, caution should be used, as they are very concentration dependent. LMWGs can have concentration dependent assembly and so it is important to compare data at the same concentration across all the different techniques. With CD spectroscopy, the HT data needs to below a threshold value in order to be reliable and so should always be shown. Since it is important to use the same concentration for different techniques, dilution is not optimum as a means of (for example) reduce scattering artefacts in CD or the intensity of absorbance in UV-vis absorption spectroscopy. Instead, it is preferable to lower the path-length of cuvettes used.
These techniques can be extremely powerful, but it is important to realise that these can often only be used to show assembly at the molecular level; at best, they imply that the primary structures are formed by co-assembled or self-sorted gelators. To return to Fig. 1, this means that these techniques show whether step (a), (b), or (c) has occurred, but provide little information as to the next length-scale.
Fibre-level assembly
To probe the next length-scale, most commonly microscopy is used to image the self-assembled fibres, and small angle scattering can also be used to probe these structures.Microscopy
For most LMWGs, optical microscopy does not have sufficient resolution to allow the network to be imaged. As such, it is most common to probe the fibrous structures using scanning electron microscopy (SEM) or transmission electron microscopy (TEM). Here, the samples are typically dried, so the images are collected for xerogels, not the gels themselves. For hydrogels, cryo-TEM can be used, which has the advantage that the structures are still in a hydrated environment, as opposed to dried under high vacuum, which is the case for TEM. However, the film thickness that is used for cryo-TEM means that it is difficult to truly capture the gel structure; rather it is the structural components that are assumed to be imaged as opposed to the 3D gel network.For LMWGs, it is not always clear whether the structures that are imaged using SEM or TEM represent those in the gel phase, as drying artefacts can occur, leading to aggregation of structures, and potentially even structural changes. For example, we have shown that drying a dipeptide-based LMWG leads to the fibres becoming much thicker than the primary structures which are observed using cryo-TEM.24 Further issues can arise from the use of a stain for TEM, which can potentially lead to structural changes. It should also be stated that both SEM and TEM have such high magnifications that only a tiny fraction of a sample can be imaged in a realistic timeframe, meaning that there is always the question as to whether the images are representative. It is also worth highlighting that some specialised gels can be difficult to image, for example those where the solvent is an ionic liquid and therefore difficult to dry.
Saying all of this, microscopy can be an invaluable tool for characterising gels, and in some cases highly informative for multicomponent systems. A wide range of structural types have been imaged for LWMGs, including fibres, tubes, helical structures, and plates, as well as hierarchical structures where primary structures can be imaged forming larger objects. Unfortunately, from the perspective of differentiating between gelators in a multicomponent system, when most LMWGs are imaged, fibrous structures with similar dimensions are found. In our cartoons, it is easy to distinguish between red and blue fibres (for example), but, in reality, often no simple means of differentiation exists. In multicomponent systems, this of course means that it is very difficult to tell if the structures are a mixture of fibres from the individual components or whether new structures are formed. This is often exacerbated by the fact that many gels show the presence of a distribution of fibre widths when imaged. For example, for a mixture of dipeptide-based LMWGs that we were able to show formed a self-sorted system by other techniques, it was extremely difficult to ascertain this by microscopy.23 Whilst the fibres formed from the two LMWGs formed apparently different fibres when alone, the microscopy for the multicomponent system showed a range of fibres which could not be categorically assigned. Elsewhere, Chen et al. reported simply that the distribution of fibre widths was greater in a mixed system of two peptide-based LMWG than in either of the individual components.25 From this alone, it is clearly impossible to determine whether there are two networks or whether the distribution of fibres of one (or both) component has been modified by growing in the presence of the other LMWG.
However, in some cases, the microscopy is very clear. Moffat and Smith showed both that images could be collected for a self-sorted system where two clearly different fibre types were visible (Fig. 4a).26 The two fibre types had very different diameters, and could be seen to be co-existing. In a second case, where co-assembly was expected from gel melting data, SEM showed that a more homogeneous network was formed where the fibre diameters were greater than for either of the two single component systems. Similarly, Escuder's group showed TEM images again of different diameter self-sorted fibres, but with interaction between the two with the thinner fibres being wrapped around the larger one.27 For two amino acid-based LMWGs, we were able to show by image analysis that the distribution of fibre diameters in a self-sorted mixture was bimodal, mirroring the expected two distributions from the individual components.28 Similarly, atomic force microscopy (AFM) has been used to image co-existing fibres with different widths.29
 | ||
| Fig. 4 (a) SEM image showing two different fibre networks in a two-component xerogel. The scale bar represents 200 nm. Adapted from Moffat and Smith.26 Adapted from ref. 26 with permission from The Royal Society of Chemsitry. (b) A three-dimensional confocal laser scanning microscope image showing self-sorted network in the gel state. The fibres are stained specifically such that one type of fibre has a different colour from the other.31 Adapted with permission from Onogi et al.ref. 31, Copyright 2016, Nature Publishing Group. | ||
In other cases, SEM was used to show that helical structures were formed in a mixture of two gelators, whilst flat ribbons were imaged in the gels formed from either of the single components.30 This strongly implies co-assembly has led to the formation of a new type of structure.
The identity of the components forming the fibres that are imaged by TEM, SEM, and AFM has to be inferred on the basis of structure or diameter as it is not trivial to differentiate between the fibres chemically. Conceptually, if one of the constituent molecules contained an element that was not present in the second component, it might be possible to show the location of this component using energy-dispersive X-ray spectroscopy, although to the best of our knowledge this has never been carried out. An alternative approach to localising different components is to label the gelators with a fluorescent dye and use confocal microscopy. Selectively staining only one gelator is difficult as the dye is simply incorporated into all structures in a multicomponent system.
A more effective approach here is to covalently attach the dye to the gelator itself. Here, it is of course very likely that the covalent modification of a gelator will to some degree affect the gelation ability. Nonetheless, if a fluorescently-labelled gelator can be co-assembled with the non-labelled analogue, conceptually this effect should be minimised. Using this approach, Onogi et al. were able to selectively label two different gelators in a two-component mixture. Using confocal laser scanning microscopy (CLSM) and stimulated emission depletion (STED) microscopy, the authors showed the first example where directly imaged self-sorted fibres could be proved in the gel state, and a three-dimensional image constructed (Fig. 4b). This could impressively be extended to imaging the real-time evolution of the self-sorted gel networks.
A similar approach has recently been used to differentiate between co-assembled and self-sorted systems formed from low-complexity domains of a protein.32 Block structures could also be formed and imaged, and this approach was also used to show that the fibres were dynamic, with mixing of the building blocks occurring even when pre-formed fibres were mixed. A very recent report used stochastic reconstruction microscopy (STORM) to likewise show that mixing two separately labelled samples (albeit of the same peptide) could be used to provide evidence of fibre bundling (which as noted above is very hard to resolve using TEM or AFM) to show little monomer exchange.33 These high-resolution microscopy techniques are becoming more available, and we anticipate that there will soon be a number of papers showing their power for multicomponent systems.
Small angle scattering
Small angle scattering can be used to probe both primary fibres and some features of the network.34 The advantages of scattering over most microscopy techniques is that it is non-destructive and can be carried out on the solvated, bulk samples. The disadvantage is that access to large-scale facilities is often needed. Small angle neutron scattering (SANS) also requires contrast, which is generally achieved by using either a deuterated solvent, or a deuterated LMWG. This normally therefore requires that the system is not absolutely the same as for the other techniques used, and also adds cost. SANS and small angle X-ray scattering (SAXS) can be used to look at primary fibres structures34 and ultra-small angle neutron scattering (USANS) can be used to look at longer length scales, and hence probe some elements of the network35 (although there seem to currently be no examples of using USANS for multicomponent systems). Generally, the scattering data is fitted to a model, with the choice of model used being based on the best fit to the data. For LMWGs, the best fits usually are best to a cylinder, a flexible cylinder, a hollow cylinder, or some other long, anisotropic structure. The fit provides information as to the radius of the structure, the length, persistence length (if appropriate) etc. It has also recently been shown that SANS can be used to follow the drying process if suitable contrast is achieved using deuterated dipeptide-based gelators.24 The data showed that the scattering primarily probed the primary fibres, not aggregated fibres that were imaged using SEM.Such scattering techniques can be used to determine whether self-sorting or co-assembly has occurred if the two networks scatter significantly differently from each other,36 or the resulting new network is sufficiently different from than those of the two individual components. As an example, a mixture of two dipeptide-based LMWGs was prepared (Fig. 5a). The SANS data from the gels formed from the individual components were significantly different (Fig. 5b).23
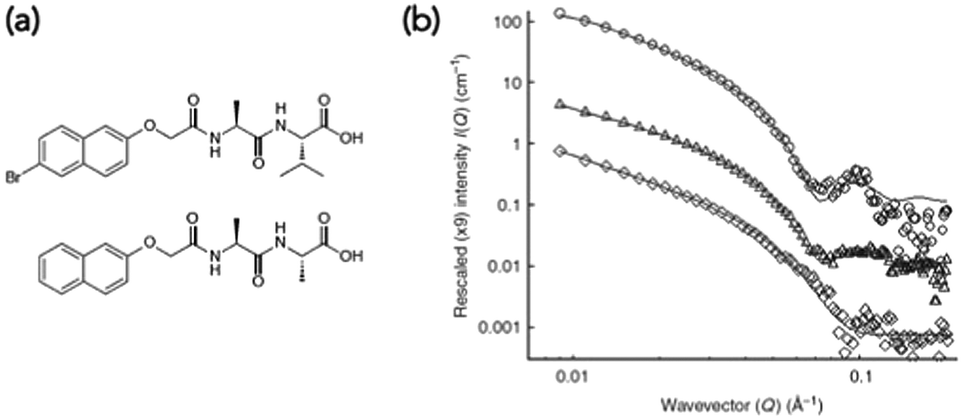 | ||
| Fig. 5 (a) Structures of functionalised dipeptide LMWGs 1 (top) and 2 (bottom). (b) Small angle neutron scattering (SANS) data. SANS data for (○) 1 alone, (◊) 2 alone and (△) 1 and 2. Adapted with permission from Morris et al.ref. 23, Copyright 2013, Nature Publishing Group.23 | ||
The data for LMWG 1 could be fitted to a hollow cylinder model, whilst that for LMWG 2 could be fitted to a flexible cylinder. Again, as for the discussion above, these diameters were lower than those imaged by microscopy, implying that SANS probes the primary assemblies. For the gel formed by mixing the two LMWGs, the scattering had key features from the scattering data for both individual gelators, where the core–shell structure could still be seen for LMWG 1 for example by the peak at a Q of 0.1 Å−1. A partially-deuterated analogue of LMWG 2 was prepared, which significantly reduced the contribution of this LMWG to the overall scattering. In this mixture, the scattering was very similar to that of LMWG 1 alone, strongly implying that self-sorted fibres had been formed.
This latter aspect shows the power of contrast matching in SANS and conceptually it should be possible to modify the ratio of H2O to D2O in the solvent mixture to specifically contrast match a single component of a multicomponent system, although this has yet to be shown. The ability to selectively deuterate (and so contrast match) part of a molecule also should allow for a greater understanding of the molecular packing of a LMWG within a fibre, although again this has yet to be fully exploited.
Elsewhere, a co-assembled system was shown to scatter very similarly to one of the individual components, implying that the structures formed by the co-assembly are driven by this gelator.11 Elsewhere, the SAXS data for a co-assembled system was shown to be different from that expected from either of the individual components, showing that a new type of structure was formed in the co-assembled system.37
Computational modelling
Many groups have used computational methods to investigate the molecular packing of these self-assembled or co-assembled materials.38 Some have even been able to look at stacks of molecules, simulating what a fibre could look like.9,39 The problems facing computational methods is that the more molecules are involved in the calculations, the longer and more difficult the calculations are to do. It is also difficult to take into account the solvent environment and changing conditions that occur as the molecules are assembling (gels are triggered from a dissolved or dispersed state to a gelled state, so the self-assembly occurs generally in a changing environment). Presently, most people using computational methods are basing their models on data already collected on the samples. In essence, this is post-rationalising the results, rather than predicting whether the sample is self-sorted or co-assembled.There are various calculations that can be used depending on what data has been collected, the number of molecules included and environment. Caneschi and co-workers used Merck Molecular Force Field (MMFF) in vacuo combined with X-Ray Diffraction (XRD) data to model the anti and gauche rotation of the fibres.40 They could then use the CD spectra within the coupled oscillator framework (DeVoe method) to computationally predict the spectra and compare with the experimental data. The data matched up and they could show the self-sorted fibres had a twisted conformation, although could not predict the handedness of the twist.
For all of these examples, the data are generally useful to describe whether the fibres that have been formed are a result of co-assembly or self-sorting. However, this still does not allow us to fully describe the system. The network forms as a result of the entanglement of the fibres, and the primary fibre assembly does not necessarily allow us to discuss this.
Network-level assembly
It is extremely difficult to access direct information on the gel networks. Generally, electron microscopy and scattering can only inform on the primary fibres, and so understanding the type and relative degrees of crosslinking and or lateral association of fibres is difficult. Generally, therefore the networks have to be differentiated by inference.Physical properties
First, it can be possible to infer a network type by eye, or by a change in behaviour. The change in properties is not necessary able to explain the exact assembled nature of the samples nor the degree to which assembly has occurred, but it can provide a quick suggestion of the assembly type. For example, when mixing two samples which independently give stable transparent gels, if the mixture then gives a turbid gel this could be an indication that some co-assembly could be happening. However, this change in appearance could however be also due to a change in concentration or the samples not mixing effectively and so therefore other techniques would need to be used.The kinetics at which turbidity changes occur on gelation can be indicative of different processes occurring (Fig. 6).41,42 Likewise, a change in colour of the gel could also be an indication that different LMWGs are interacting with each, perhaps suggesting co-assembly, as described above for gels which undergo charge transfer.18 Self-sorting of a system can be seen by eye in rare cases. For example, if a multicomponent gel network is formed where one component undergoes a gel-to-crystal transition in the single component gel, then the observation that crystallisation is occurring in the mixture, with a gel being maintained overall is highly indicative that a self-sorted interpenetrated network has been formed. If the system were co-assembled, crystallisation from the gel phase would be expected to destroy the overall gel network as a result. We have observed this crystallisation from a multicomponent system,11 but do stress that this is a highly unusual situation.
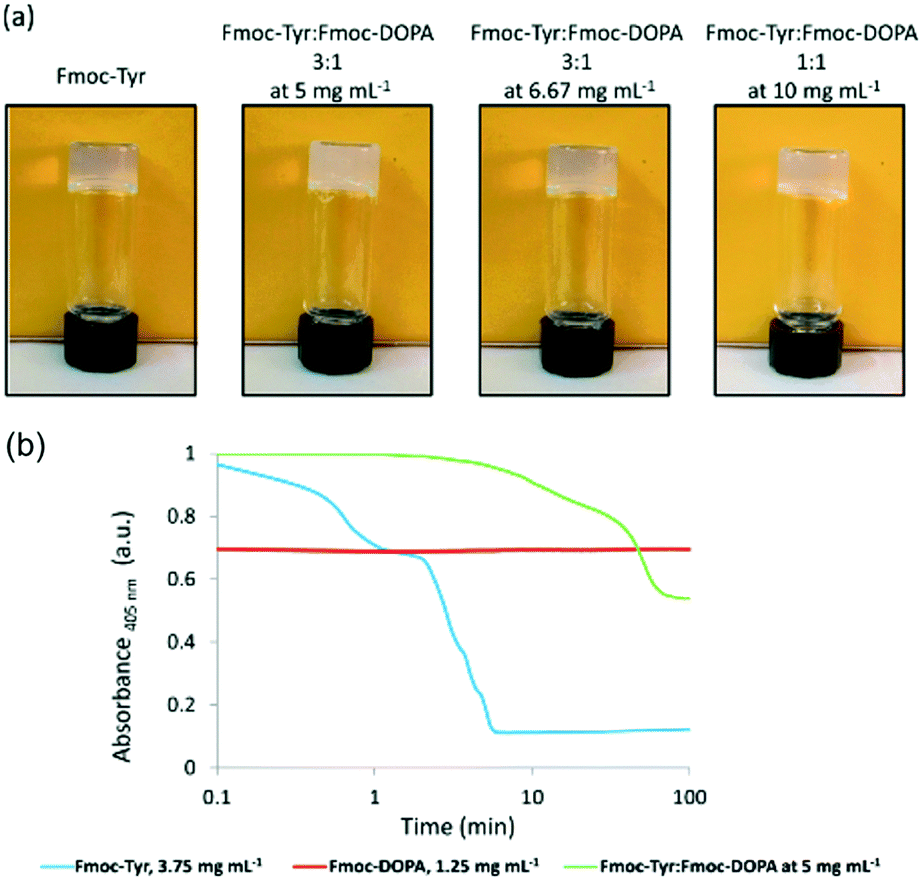 | ||
| Fig. 6 (a) Photographs showing the change in turbidity upon mixing of two gelators at different concentrations, compared to that of the single component. (b) Graph showing the change in turbidity during gelation of the single components (blue and red) and then the mixed gel (green), showing very different gelation kinetics are occurring.42 Adapted from ref. 42 with permission from The Royal Society of Chemsitry. | ||
Another example of a change in properties that can arise in multicomponent systems is when using LMWG for electronic applications. The use of a n-type system mixed with p-type gelling system could result in a p–n heterojunction being formed in the gel.16 This would result in a change in resistance in the material formed. The extent of the change in resistance could indicate whether the sample was most likely self-sorted or co-assembled or even whether the sample was hetero- or homo-aggregated. For example, Draper et al. used a perylene bisimide based n-type LMWG mixed with a stilbene-based p-type LMWG to make a p–n heterojunction xerogel.28 The xerogel formed from the n-type component alone only showed a photocurrent when UV light was used, and the single component p-type xerogel did not produce any current. When mixed together, the xerogel was now responsive to blue light. This shows that there was donation of an electron from the p-type LMWG into the n-type LMWG, reducing the amount of energy of light needed to give a response. This indicates that the samples must be self-sorted and not significantly hetero-aggregated. If co-assembly or too intimate a mixing of the two fibres had occurred, one would expect effective recombination of charges, leading to a reduction in the current from the xerogel.
Melting points
The melting temperature of a gel is a common parameter to be measured and quoted. Here, the assumption is that the gel's melting temperature is indicative of the underlying network. Typically, the melting temperature is determined by vial inversion; when the network has sufficiently melted, the sample flows when the vial is turned over. This might happen uniformly throughout the sample, or perhaps sufficient quantities of the network melts to lubricate the material.Moffat and Smith showed that gel melting temperatures could be used to suggest whether self-sorted or mixed systems are formed in multicomponent systems.26 In one example, two amino-acid based bolaamphiphile based gelators were used which had a significant difference in melting temperatures. In the mixture, the melting temperature was found to be close to that of the higher melting single component. At the very least, this implies that the network formed by this gelator is similar to that in the single component and hence implies self-sorting has occurred. For another mixture, the melting temperature was higher than the lowest melting component, but lower than that of the highest melting component. This implies that a different network type has formed, and hence that co-assembly has occurred. These data agreed with the microscopy (see above). These data were obtained from bulk melting of the gel. If there were two networks with one melting at a significantly lower temperature, but the second network were able to maintain a self-supporting network, it would not be possible to show this using this method.
Elsewhere, a mixture of two bis(amino acid) gelators was shown to be synergistic, with the mixtures being able to immobilise significantly more solvent than the individual components.43 The melting point of the gel was around 20–30 °C higher for the mixture as compared to that for the gels formed from the single components.
As alternative methods, differential scanning calorimetry (DSC) or NMR spectroscopy can be used. DSC measures the endo- or exotherms that arise from sol–gel and gel–sol transitions. Using this method, Smith and Smith were able to show that there were two distinct transitions in a two-component mixture at significantly different temperatures, providing clear evidence of two co-existing networks (Fig. 7).44 The endotherm for the lower melting gelator was around 6 °C below that for the pure component and the authors speculated that this showed that the network was compromised in the mixture by the presence of the other component. For the same mixture, NMR was also used. On heating, the integrals for the peaks from the gelator increase as the network melts and the gelator becomes more mobile in solution. Using variable temperature NMR, it was shown that the integrals increased on heating with a very similar profile for the lower melting gelator in either the single or two-component network. This implies that the networks are independent. An interesting caveat here is that the DSC, the NMR, and the bulk melting data are not perfectly in agreement; the NMR data implies that the lower melting component melts between 30 and 60 °C, the DSC implies that this component melts at 48 °C, and the tube inversion implies that the gel melts at 32 °C. The authors rationalise this on the basis of the size of sample environment and geometry. It could also be the case that different absolute properties are being measured; tube inversion is only possible when a sample tube spanning network is complete, so a slight change in the network might mean that tube inversion is not possible even though the network has not formally melted.
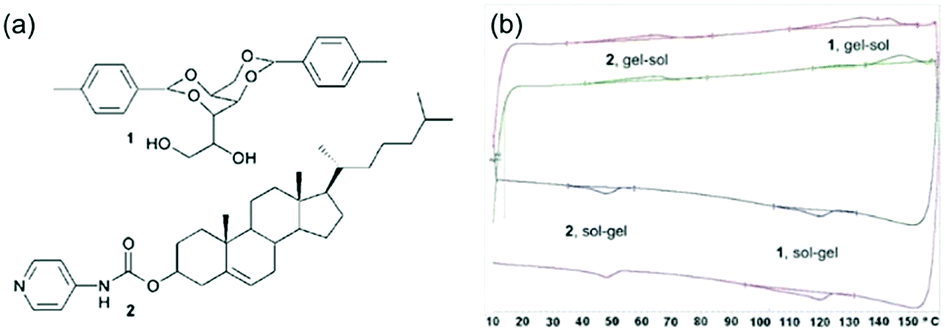 | ||
| Fig. 7 (a) Structure of gelator 1 (top) and gelator 2 (bottom) (b) DSC plots showing distinct melting points and gelling points in the mixed system for the separate components.44 Adapted from ref. 44, with permission from The Royal Society of Chemsitry. | ||
Rheology
Rheology can be used to analyse the mechanical properties of bulk gel samples.45 The rheological properties are determined by a number of factors, including the morphology of the fibres, the fibre persistence lengths and strengths, the number and type of crosslinks and the distribution of the fibres in space. This is simple to state, but it is often extremely difficult to determine why a specific gel is stiffer than another as any or all of these parameters might have changed.When two gelators are mixed, a difficult decision is what data should be compared with what. Most examples compare the rheological properties of each single component gel with that of the multi-component gel, with the concentrations of the single component gels being carried over to the multi-component system. Hence, if gelators 1 and 2 are at a concentration of 5 mg mL−1 in the single component gel, then this is the concentration also used in the multicomponent systems. However, this means that the multicomponent system is now at a total gelator concentration of 10 mg mL−1; the rheological properties of such gels are generally very concentration dependent (most basically, the more gelator, the higher the number of fibres), so we would expect that it would be very likely that the multicomponent system would have higher values of the storage modulus (G′) and loss modulus (G′′) no matter what. However, this is not always observed, although it is rare for this to be commented upon.
As such, the absolute values of the G′ and G′′ alone cannot be used to determine whether a system is self-sorted or co-assembled. There are examples where upon mixing, the moduli increase for the system as compared to the single components. For example, Li et al. showed that a mixture of two gelators formed a gel with a storage modulus an order of magnitude above that of either of the individual components.46 As mentioned above, this could simply be because there is more gelator in the system. It could also be due to new fibres being formed upon co-assembly making the new system stronger than the individual ones, for example by the incorporation of molecule that provides cross-links making the system stronger.47 Alternatively, it could be the result of a self-sorted system having fibres with favourable interactions with one another (hence leading to entangling, increased hydrogen bonding etc.) which increases the gel strength. This was shown by Schneider and co-workers where they used different enantiomers to enhance mechanical properties of a self-sorted gel.48 Kinetics are also important; in some cases for low molecular weight gels, the quicker the network forms, the higher the number of crosslinks and the lower the homogeneity; both of these factors can affect the rheological properties. Hence, sometimes the data are very difficult to deconvolute. For example, Halperin-Sternfield et al. mixed a Fmoc-dipeptide and a Fmoc-amino acid.41 The rheological data was shown to vary with the ratio of the two LMWG, with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture showing significantly higher moduli than either of the two components. However, the kinetics of the assembly process was also shown to vary as the ratio was varied. Hence, it is difficult to link an increase in modulus to one specific effect on the basis of the rheology alone.
:1 mixture showing significantly higher moduli than either of the two components. However, the kinetics of the assembly process was also shown to vary as the ratio was varied. Hence, it is difficult to link an increase in modulus to one specific effect on the basis of the rheology alone.
There are also examples were the mixing of the two gelators creates a gel that is of a similar stiffness or perhaps weaker than that formed from either of the two individual components. For example, Chen et al. have reported a gel where the values of G′ and G′′ are similar to those of the strongest single component gel.25 Similar data was reported by Horgan et al., where the value of G′ in the mixed gel was very similar to that of the stiffest single component network.37 However, here the value of G′′ was different to that of the stiffest single component gel, meaning that the value of tanδ (G′′/G′) was more similar to that of the weaker single component.
The explanation for the mixed gels being no stronger than the individual gels could be simply that the second network is essentially so weak that the first dominates. Alternatively, it could be that co-assembled new fibres are formed that are weaker or less entangled than the network in the two single component gels. In a self-sorted system, the two individual networks could have unfavourable interactions with other or not want to interact or mix with each other, resulting in a weaker overall network. An alternative explanation could be that there are steric crowding effects in the system. As an example of the complexity, Liyanage and Nilsson produced a range of Fmoc-amino acid based LMWG and found the increase or decrease in gel strength did not correlate to whether systems containing two of these LMWGs were co-assembled or self-sorted.49
Rheological frequency tests, nanoindentation, and cavitational rheology that give us only G′ and G′′ values are not therefore always that useful in providing information on the assembly type. However, if one of the networks can be changed with an external trigger, it can be possible to infer information about the system. For example, Smith and co-workers showed a mixed system where a UV light can be used to trigger the gelation of one of the components in the presence of another gel formed by a change in pH.13 Hence, here one network is grown first, followed by the second. This shows that the networks are self-sorted, but does not necessarily show whether the two networks are interpenetrating or whether the second network grows on the first for example.
Draper et al. showed a self-sorted system where the assembly was triggered sequentially by a slow pH change.12 It was possible to selectively remove one of the networks post-gelation using UV light; one of the gelators underwent a isomerisation under UV light, rendering it ineffective as a LMWG. As a result, the network formed by this gelator fell apart on irradiation. The network remaining after irradiation was found to have the same rheological property as the single component gel. This is a rare example of where is has been shown that one networks is not affected by the other network that already formed and there are apparently two completely independent networks, consistent with Fig. 2a. From other data, they concluded that their system was self-sorted and the two fibres were strongly interacting with one another causing the change in rheological properties. Since this method used UV-light to erase one network, it was possible to pattern a gel, such that one region contained a self-sorted network, and another the single component (Fig. 8).
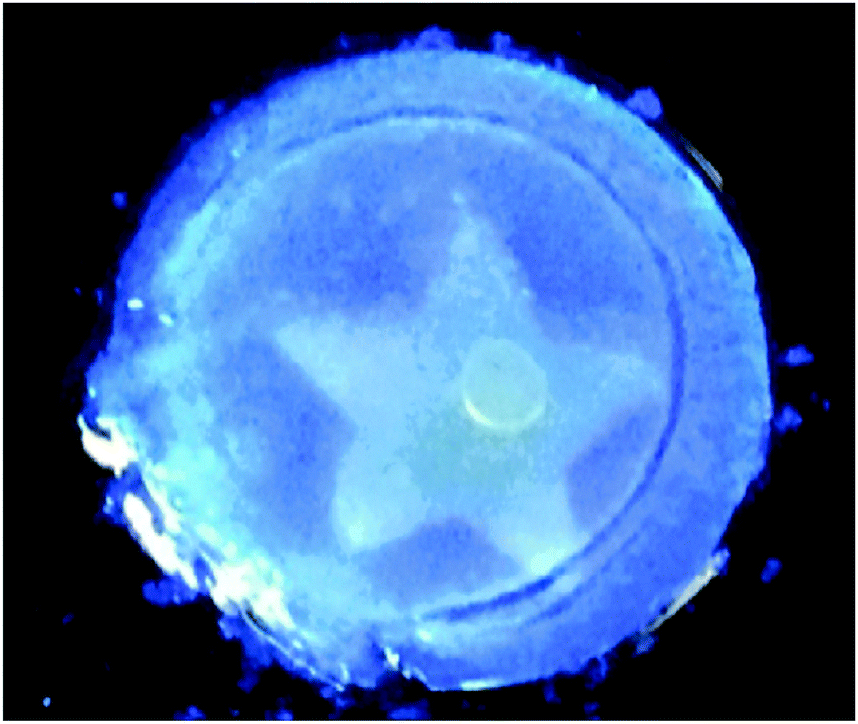 | ||
| Fig. 8 Photograph of a photopatterned gel under 365 nm UV light. The star has been irradiated with to photo-erase one network, whilst outside the star contains a two-component self-sorted network. Adapted with permission from Draper et al., ref. 12, Copyright 2015, Nature Publishing Group. | ||
Rheological techniques that can be used to show a change in network include using a strain sweep and looking at how the network breaks down. This relies on the two individual components having a different breakdown behaviour (maybe in multiple steps, having a sharp break at a specific strain, creaming behaviour, or perhaps a large difference in the strain required to break the network). If co-assembly has occurred, a different breakdown behaviour may be seen as the fibres in the network will have changed. If self-sorting has occurred, two different breakdown profiles may still be seen even if the G′ and G′′ are different compared to the single component systems. At the moment, it is difficult to determine the type of network from these data. Instead, we can only infer that there is a difference. As an example, Che et al. showed that their mixed gel had significantly longer linear viscoelastic region than the single components.50 As a result, it is clear that the gel network is not simply a combination of the two networks formed from the single components. Self-sorting was proved by other means, and the rheological data were interpreted on the basis of interactions between the different types of fibre. Hence, this correlates with the situation in Fig. 2b. Differences in the strain sweeps were also observed by Xing et al.22
Finally, if the gelation method is suitable, it is possible to show the assembly type using a rheological time sweep. We have described a number of systems where a slow pH trigger is used to induce gelation. In many cases, the two LMWG are chosen to have different pH at which assembly and gelation occurs.12,23 Hence, the slow pH change allows sequential assembly of the networks and this can be seen in a multi-step increase in G′ and G′′ during gelation. This can be combined with techniques such as NMR to show that one component becomes NMR-invisible as it gels, and there is a concomitant increase in the rheology at this point. In cases where co-assembly occurs, both LMWG simultaneously become NMR-invisible and there is a single increase in the rheological data.11
Conclusions
In many cases, preparing gels from mixtures of two LMWG is relatively straightforward if the triggering method is the same for both. However, the complexity that can be achieved from even a two-component system is high. This is a fascinating, intellectually stimulating problem, which requires significant input of time and effort to pull apart. For the primary assembled structures, co-assembly, self-sorting, or a combination of these can occur. At the next length scale, self-sorted primary structures can conceptually also co-assemble, self-sort or form a combination. Hence, the simple statement that two LMWG form a specific type of assembly is often extremely difficult to prove across all length scales. This however does not stop such statements being routinely made.Here, we hope that we have shown that a wide range of techniques can be used to probe mixed systems across the length scales. Not all will be appropriate in all cases, and it may be that only a small number of techniques will be able to suggest the assembly type. It is also important to consider the hierarchy level at which the technique is able to provide information. It is also not clear that techniques that show co-assembly has occurred can always pull apart whether the co-assembly is random or specific. Nonetheless, by applying many of these techniques in concert, we believe that it is generally possible to explain the assembly type. This requires using a number of techniques in unison, and often using several pieces of information.
As a single example, we showed that sequential assembly could be proved by the rate at which the peaks from two different gelators disappeared out of the NMR spectrum when a slow gelation trigger was used.23 This shows only sequential assembly, but does not prove self-sorted fibres are formed. Microscopy was unclear, as this was not a case where two distinct fibre populations could be imaged. SANS was informative, with the scattering being consistent with the system containing a mixture of self-sorted fibres from the two individual components, but was not conclusive. Using fibre X-ray diffraction however, we were able to absolutely demonstrate that the primary fibres were self-sorted. Whilst this was a clear demonstration that sequential assembly could be used to form self-sorted systems, this still does not inform as to how the fibres are distributed in space and whether the self-sorted fibres form homo- or hetero-aggregates. There are limited methods which are able to do this. Hence, there is still a way to go in understanding how to characterise these systems.
In some cases, it could be argued that it is unimportant to known definitively. However, we would respond by stating that generally mixed systems are used to add information or signals to a system. As such, design of these is predicated on the understanding of the assembly type. As a single example, if one component has a signalling unit on it, the density of these groups will be very different if the components form co-assembled structures (where the signal will be ‘diluted’ in a fibre by the other component) or self-sorted fibres.
There are ever increasing numbers of papers discussing mixed systems. In many cases, it seems to us that the assignment of the structure as co-assembled or self-sorted can be at the suggestion of the authors, with little data to categorically demonstrate this. We hope that this review will provide the impetus for the field to move away from the cartoon-level discussion of assembly to the general requirement that proof is provided for each assignment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. R. D. thanks the Leverhulme Trust for funding (ECF-2017-223) and the University of Glasgow for an LKAS Leadership Fellowship. D. J. A. thanks the EPSRC for a Fellowship (EP/L021978/1).Notes and references
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, Royal Society of Chemistry, Cambridge, 2014 Search PubMed.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jäckel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC.
- C. Colquhoun, E. R. Draper, E. G. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC.
- E. R. Draper, E. G. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848 CrossRef CAS PubMed.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS PubMed.
- Y. J. Adhia, T. H. Schloemer, M. T. Perez and A. J. McNeil, Soft Matter, 2012, 8, 430–434 RSC.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- K. Sugiyasu, S.-I. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed., 2015, 54, 946–950 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Chem. Commun., 2011, 47, 8922–8924 RSC.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC.
- C. Felip-León, S. Díaz-Oltra, F. Galindo and J. F. Miravet, Chem. Mater., 2016, 28, 7964–7972 CrossRef.
- Y. M. Abul-Haija, S. Roy, P. W. J. M. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973–979 CrossRef CAS PubMed.
- P. Xing, X. Chu, S. Li, F. Xin, M. Ma and A. Hao, New J. Chem., 2013, 37, 3949–3955 RSC.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O’Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- G. Chen, J. Li, M. Song, Z. Wu, W. Zhang, Z. Wang, J. Gao, Z. Yang and C. Ou, Adv. Funct. Mater., 2017, 27, 1701798 CrossRef.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC.
- N. Singh, K. Zhang, C. A. Angulo-Pachon, E. Mendes, J. H. van Esch and B. Escuder, Chem. Sci., 2016, 7, 5568–5572 RSC.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jackel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC.
- N. Singh, C. Maity, K. Zhang, C. A. Angulo-Pachón, J. H. van Esch, R. Eelkema and B. Escuder, Chem. – Eur. J., 2017, 23, 2018–2021 CrossRef CAS PubMed.
- J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem. – Eur. J., 2014, 20, 279–291 CrossRef CAS PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
- B. An, X. Wang, M. Cui, X. Gui, X. Mao, Y. Liu, K. Li, C. Chu, J. Pu, S. Ren, Y. Wang, G. Zhong, T. K. Lu, C. Liu and C. Zhong, ACS Nano, 2017, 11, 6985–6995 CrossRef CAS PubMed.
- H. Cox, P. Georgiades, H. Xu, T. A. Waigh and J. R. Lu, Biomacromolecules, 2017, 18, 3481–3491 CrossRef CAS PubMed.
- J.-B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- R. A. Hule, R. P. Nagarkar, A. Altunbas, H. R. Ramay, M. C. Branco, J. P. Schneider and D. J. Pochan, Faraday Discuss., 2008, 139, 251–264 RSC.
- A. M. Castilla, E. R. Draper, M. C. Nolan, C. Brasnett, A. Seddon, L. L. Mears, N. Cowieson and D. J. Adams, Sci. Rep., 2017, 7, 8380 CrossRef PubMed.
- C. C. Horgan, A. L. Rodriguez, R. Li, K. F. Bruggeman, N. Stupka, J. K. Raynes, L. Day, J. W. White, R. J. Williams and D. R. Nisbet, Acta Biomater., 2016, 38, 11–22 CrossRef CAS PubMed.
- R. K. Das, R. Kandanelli, J. Linnanto, K. Bose and U. Maitra, Langmuir, 2010, 26, 16141–16149 CrossRef CAS PubMed.
- Y. M. Abul-Haija, G. G. Scott, J. K. Sahoo, T. Tuttle and R. V. Ulijn, Chem. Commun., 2017, 53, 9562–9565 RSC.
- S. Cicchi, G. Ghini, L. Lascialfari, A. Brandi, F. Betti, D. Berti, P. Baglioni, L. Di Bari, G. Pescitelli, M. Mannini and A. Caneschi, Soft Matter, 2010, 6, 1655–1661 RSC.
- M. Halperin-Sternfeld, M. Ghosh, R. Sevostianov, I. Grigoriants and L. Adler-Abramovich, Chem. Commun., 2017, 53, 9586–9589 RSC.
- G. Fichman, T. Guterman, L. Adler-Abramovich and E. Gazit, CrystEngComm, 2015, 17, 8105–8112 RSC.
- Z. Dzolic, K. Wolsperger and M. Zinic, New J. Chem., 2006, 30, 1411–1419 RSC.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- D. Li, Y. Shi and L. Wang, Chin. J. Chem., 2014, 32, 123–127 CrossRef CAS.
- S. Boothroyd, A. Saiani and A. F. Miller, Biopolymers, 2014, 101, 669–680 CrossRef CAS PubMed.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS PubMed.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS PubMed.
- X. Che, B. Bai, T. Zhang, C. Zhang, C. Zhang, P. Zhang, H. Wang and M. Li, New J. Chem., 2017, 41, 8614–8619 RSC.
| This journal is © The Royal Society of Chemistry 2018 |