
Fluorescent probes for the simultaneous detection of multiple analytes in biology
Jacek L.
Kolanowski†
 ab,
Fei
Liu†
c and
Elizabeth J.
New
*a
ab,
Fei
Liu†
c and
Elizabeth J.
New
*a
aSchool of Chemistry, The University of Sydney, NSW, Australia. E-mail: elizabeth.new@sydney.edu.au
bInstitute of Bio-organic Chemistry, Polish Academy of Sciences, Poznan, Poland
cState Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Institute of Microbiology, Guangdong, People's Republic of China
First published on 9th November 2017
Abstract
Many of the key questions facing cellular biology concern the location and concentration of chemical species, from signalling molecules to metabolites to exogenous toxins. Fluorescent sensors (probes) have revolutionised the understanding of biological systems through their exquisite sensitivity to specific analytes. Probe design has focussed on selective sensors for individual analytes, but many of the most pertinent biological questions are related to the interaction of more than one chemical species. While it is possible to simultaneously use multiple sensors for such applications, data interpretation will be confounded by the fact that sensors will have different uptake, localisation and metabolism profiles. An alternative solution is to instead use a single probe that responds to two analytes, termed a dual-responsive probe. Recent progress in this field has yielded exciting probes, some of which have demonstrated biological application. Here we review work that has been carried out to date, and suggest future research directions that will harness the considerable potential of dual-responsive fluorescent probes.
 Jacek L. Kolanowski | Jacek Kolanowski undertook his BSc in Biotechnology and MSc in Chemistry at AMU Poznan before completing a PhD at ENS Lyon with Professor Jens Hasserodt in 2013. He undertook his postdoctoral research in the group of Dr Elizabeth New at the University of Sydney from 2014–2017, partially funded by a postdoctoral fellowship from the French ARC Foundation for Cancer Research. In August 2017 he became a principal investigator of the Molecular Sensors and Pro-drugs group at the Institute of Bio-organic Chemistry, at the Polish Academy of Sciences (Poznan). |
 Fei Liu | Dr Fei Liu received his PhD from Dalian University of technology in 2012. He is currently an Associate Professor at Research and Development Center for Environmental Microbiology of Guangdong Institute of microbiology. In 2017 he is a visiting scholar at the University of Sydney working with Dr Elizabeth New. His research is focused on the design and synthesis of novel functional fluorescent sensors and their biological applications. |
 Elizabeth J. New | Elizabeth New undertook her BSc and MSc at the University of Sydney before completing her PhD studies in 2010 at the University of Durham (UK), working with Professor David Parker. Liz was then a Royal Commission for the Exhibition of 1851 Research Fellow at the University of California, Berkeley with Professor Chris Chang. In 2012, she returned to the University of Sydney, holding an Australian Research Council DECRA from 2012–2014, and a Westpac Research Fellowship from 2016, focussing on the development of small molecule chemical sensors for the study of oxidative stress and metal ions in biology. |
1. Introduction
Biological environments, whether body fluids such as serum and cerebrospinal fluid, intracellular organelles, or extracellular space, rely on highly controlled chemical compositions for the maintenance of health. Many physiological processes involve the coincidence of multiple chemical events in order to occur. For example, nitric oxide and hydrogen peroxide must both be present for effective pathogen killing by macrophages,1 while zinc must be colocalised with insulin in the pancreatic islet to facilitate insulin packaging and release.2 There are also numerous examples of cellular processes that require the simultaneous presence of multiple enzymes, such as topoisomerases I and IV for DNA replication,3 or prohormone convertases PC1/3 and PC2 for conversion of proinsulin to insulin.4 In order to understand cellular function, and how it is disrupted in disease, it is therefore important to be able to study multiple chemical species simultaneously.Fluorescent sensors, based on both small molecules and fluorescent proteins, have revolutionised the understanding of chemical species within biological systems, uncovering new roles for metal ions,5 anions,6 and reactive oxygen species2 amongst other chemical entities. Fluorescence microscopy offers the balance of sensitivity and resolution necessary to visualise endogenous species within their sub-cellular location.7 However, fluorescent sensor design tends to aim for selective probes for individual analytes, rather than reporting on the multiple analytes appearing concurrently. There have been far fewer probes prepared for dual sensing, which will be the focus of this review.
1.1 Criteria for simultaneous monitoring of two analytes
While it is theoretically possible to use two selective probes simultaneously to measure two analytes, even the most subtle differences in the uptake, localisation and retention of the two probes will compromise data interpretation, and it is therefore preferable to have the two sensing elements combined into a single probe. A dual-responsive probe is therefore one that has the ability to respond to two analytes (inputs), by reporting their presence in a form of one or more detectable signals (outputs); that is, it operates as a logic gate.Such a mode of response can be used for a variety of purposes in biological settings. For example, it can be useful to predict potential cellular reactions or interactions, or can be used to increase the specific of detection of cell populations – in the identification of cancerous cells, for example, it has been suggested that two or more cancer-specific parameters be used.8
Concurrent with the development of selective fluorescent sensors for biological applications has been the establishment of the field of molecular logic gates. Such systems can be described by the pattern of non-interfering inputs and outputs. A fluorescent logic gate is one for which the inputs are analytes and fluorescence changes are outputs. Molecular logic gates have been extensively reviewed elsewhere,9–11 and are commonly classed according to the dependence of the output(s) on the input signals (Fig. 1). However, not all types of gates can unambiguously confirm that two inputs (species) are both present. For example, an OR gate requires only one of the two analytes to give a positive signal, and conclusions cannot be drawn about which species is present.
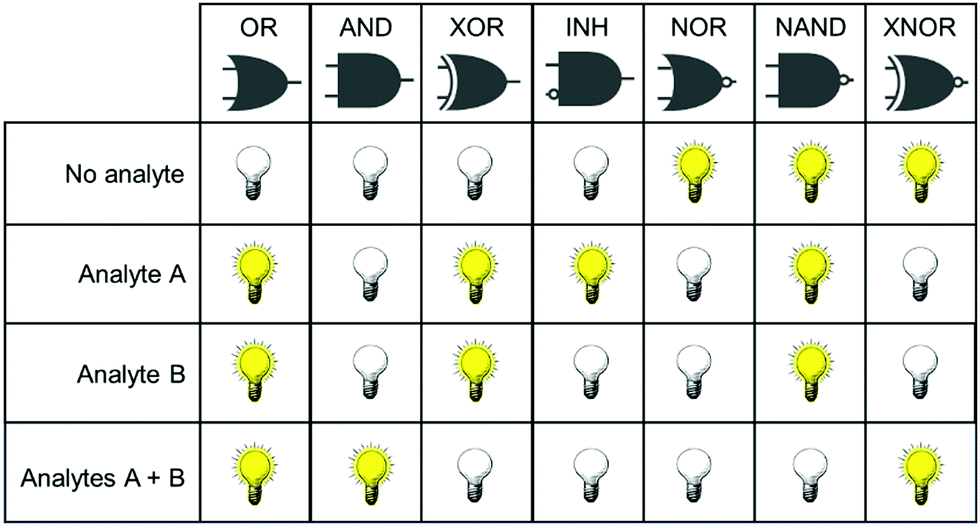 | ||
| Fig. 1 Truth tables for two input logic gates, showing the combination of inputs (analytes A and B) for which there is a measurable response in the output. XOR = exclusive OR; INH = inhibit; NAND = negative AND; XNOR = exclusive NOR. | ||
It is important for us to define clearly the sub-classes of logic gates that will enable the simultaneous study of two species. In such experiments, the co-existence of analytes A and B must be distinguishable from the absence of both analytes, or the presence of only one analyte. From Fig. 1, it is clear therefore that the only logic gates that meet this criterion are AND and NAND. This review focusses only on fluorescent sensing systems that fall into these two categories.
Fig. 1 shows only two-input one-output logic gates, for which there is a binary response, “off” or “on”. For fluorescent sensors, the output is fluorescence emission, and therefore output can vary in emission intensity, colour or lifetime. One of the challenges in biological imaging is that probe concentration can vary dramatically with incubation time and sub-cellular localisation, so emission intensity is not a sufficient parameter by which to distinguish inputs. While many reported molecular logic gates apply a threshold to distinguish between responses of different intensity,12 this cannot be reliably applied to biological imaging, as a far higher concentration of the lower-intensity response can exceed the threshold (Fig. 2a–c). In contrast, changes in emission colour can enable clear distinction of responses, as the ratio of intensities at the two emission maxima will be independent of probe concentration (Fig. 2d–f). As a result, we can envisage sets of probes for which analytes A and B independently give a different fluorescence emission from A + B (Fig. 3). The case where A, B and A + B all give different emission colours can be considered most ideal (Fig. 3(iii)), as it enables not only determination of cases where both A + B are present, but also allows identification of which of the two analytes is present alone.
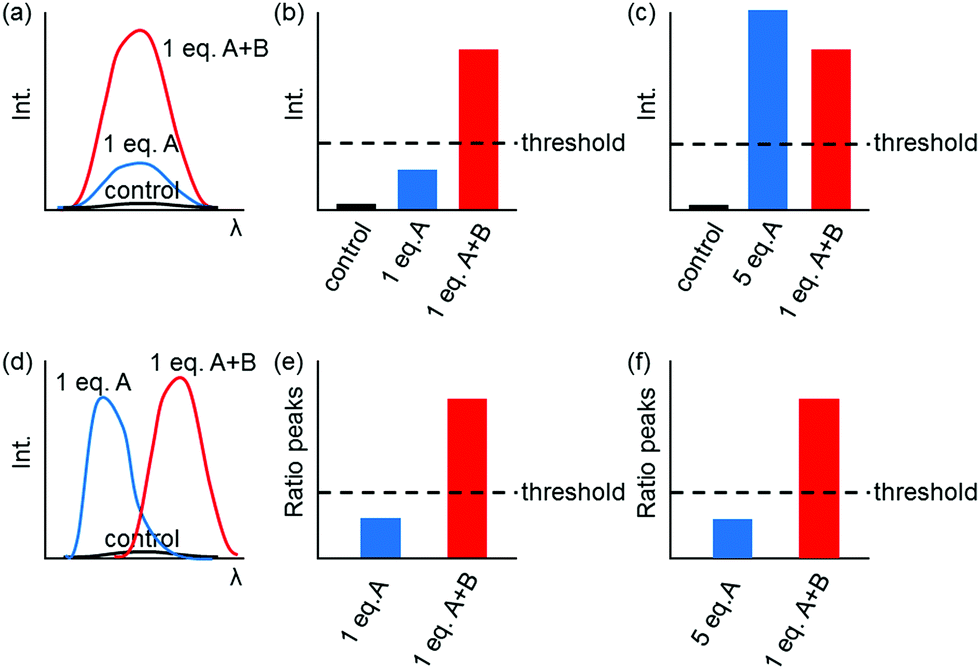 | ||
| Fig. 2 Comparison of turn-on (a–c) and ratiometric (d–f) dual-responsive fluorescent probes. Turn-on fluorescent probes undergo a change in emission intensity at a single wavelength (a), which theoretically enables determination of a threshold for distinguishing A from A + B (b). However, a higher concentration of A can exceed the threshold, compromising interpretation (c). Ratiometric probes involve changes in emission colour (d). Thresholding can be applied to the ratio of peak intensities, which is insensitive to probe concentration (e and f). | ||
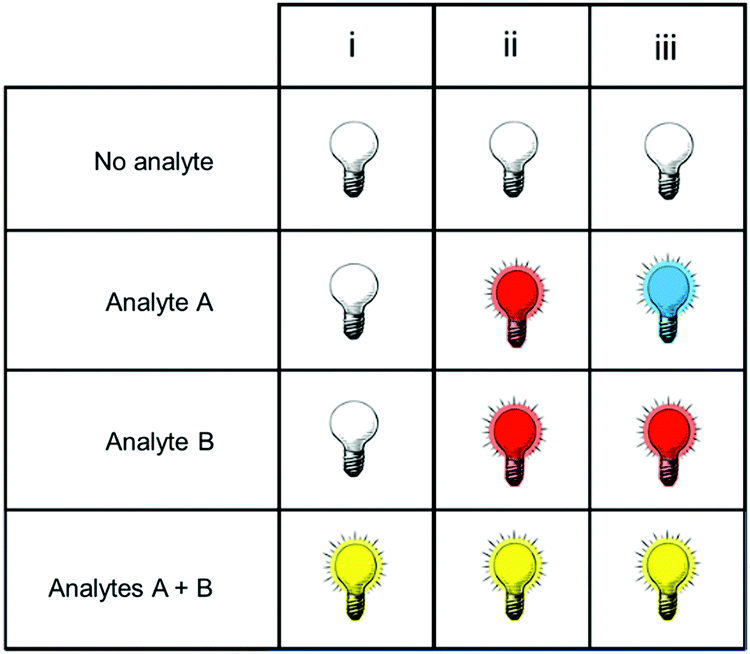 | ||
| Fig. 3 Truth tables for sensors in which the response to analytes A + B can be distinguished from the presence of no analytes, or of analyte A or B alone. | ||
1.2 Scope of this review
Since the very first reports of molecular systems that could respond to multiple analytes, there has been suggestion that they have the potential to revolutionise biological study. While significant progress has been made in genetic programing of logic gate operations,13–15 the application of small-molecule logic gates to biological study has been less fruitful. It is not uncommon in the literature to find fluorescent probes that are responsive to two or more analytes.9 However, for the overwhelming majority of these papers, either the presence of both analytes cannot be distinguished from the presence of one or both analytes alone, or the data is not available to determine response to a mixture of both analytes. As a result, such probes cannot have immediate application in biological experiments for which the primary aim is to study the simultaneous presence of two analytes. We have therefore limited our review to those with a unique response to the presence of two analytes, that is, probes that function according to the principle of the AND or NAND logic gate, in respect to the biologically-relevant inputs. These probes are summarised in Table 1.| Analytes sensed | Biological application | |
|---|---|---|
| AlP = alkaline phosphatase; PLE = porcine liver esterase; NTR = nitroreductase; β-gal = β-galactosidase; βLA = β-lactamase; β-GCD = β-glucosidase; PDE = phosphodiesterase; DTT = dithiothreitol; LAP = leucine aminopeptidase; PGA = penicillin G acylase | ||
| Reversible probes | ||
| 119 | Na+ and K+ | — |
| 220 | Ca2+ and Mg2+ | PC12 cells treated with mitochondrial uncoupler |
| 321 | Hg2+ and Ba2+ | — |
| 422 | Al3+ and Zn2+ | — |
| 523 | Fe3+ and Hg2+ | — |
| 624 | pH and cucurbit[7]uril | — |
| Reaction-based probes | ||
| 725 | H2O2 and NO | RAW264.7 macrophages stimulated with LPS |
| 826 | H2S and NO/O | L929 mouse fibroblasts treated with NaHS and DEA-NONOate separately and together |
| 927 | Hg2+ and F− | W138 lung fibroblast cells treated with Hg2+ and F− separately |
| 1028 | AlP and PLE | L929 mouse fibroblasts – endogenous enzyme activity |
| 1129 | (a) NTR and β-gal or (b) NTR and βLA | — |
| 1230 | O2˙− and polysulfides | RAW 264.7 macrophages treated with O2˙− and NaHS |
| 1331 | O2˙− and polysulfides | RAW 264.7 macrophages stimulated with PMA and LPS. Tissue sections of mice bearing murine sarcoma S180 tumours also imaged. |
| 1432 | β-GCD and PDE | Huh7 liver cell line – endogenous enzyme activity |
| Mixed probes | ||
| 1533 | Neurotransmitters and H+ | — |
| 1634 | DTT and H+ | — |
| 1737 | H+ and H2S | A549 adenocarcinoma cells treated with NaHS and PMA |
| Sequence-specific probes | ||
| 1835 | Hcy then Ga3+ | — |
| 1936 | F− then CN− | — |
| 1937 | H+ then H2S | A549 adenocarcinoma cells treated with NaHS and PMA |
| 2038 | β-Gal then LAP | C17.2 cells, C17.2 cells with leucine aminopeptidase inhibited, HeLa cells |
| 2112 | Hydrolase (PLE or PGA) then NTR | — |
| 2239 | NO then GHS/Cys | B16 cells, treated with DEA-NONOate and RAW macrophages endogenous and treated with IFN-γ/LPS/L-Arg. |
| 2340 | Biothiols then phosphate | HeLa cells treated with N-methylmaleimide or α-toxin and phosphate. |
| 2441 | Cu2+ then F− | — |
In the following section we consider the specific challenges of sensing multiple analytes in biology. We then review the small-molecule dual-responsive probes that have been reported and meet the criteria of an AND or NAND gate, before considering future directions for this field. While there are some examples of dual-analyte sensing using nanomaterials and macromolecular probes,16–18 this review will focus only on small molecule probes.
2. Classes of dual-responsive probe
The chemical structures of dual-sensing probes reported to date (Table 1) vary considerably, but they all contain key common elements: one or more fluorophore groups, and one or more sensing moieties. In general, for a probe to be dual-responsive, it should contain two distinct sensing groups, one for each analyte.We have chosen to classify probes reported to date according to the type of interaction of analyte with probe as this will not only group probes for similar target, but will also provide an indication of the types of responsive groups and strategies that can, in theory, be readily applied to preparing probes for sensing similar analytes. As with single-responsive probes,42 the sensing event for dual-responsive systems may involve the reversible binding of an analyte with the sensor, or an irreversible reaction (usually cleavage or addition). The type of interaction will determine the properties of the probe, and its most suitable application.
For dual-sensing probes, where at least two different responsive motifs are required, three classes of probes can be envisaged:
1. Reversible probes, where both analytes interact with the probe in a reversible manner;
2. Reaction-based probes, for which the two analytes react irreversibly with distinct sensing groups; and
3. Mixed reversible and reaction-based probes, which contain one reversibly-interacting group and one that respond via an irreversible change.
In addition, there is a fourth class, containing sequence-specific probes, which are only capable of sensing the second analyte following reaction with the first analyte. These probes comprise those that are triggered by a reversible interaction, reaction, or a combination. The following sections contain discussion of each class of probes.
2.1 Reversible probes
Reversible sensing is most commonly applied to the study of metal ions, but can also be used to report on anions and pH. To date, most examples of dual-responsive reversible probes seek to detect metal ions (Fig. 4). In order for a probe to give a unique response to the presence of both metal ions, it must contain two sensing groups, which are selective ligands for the two metals.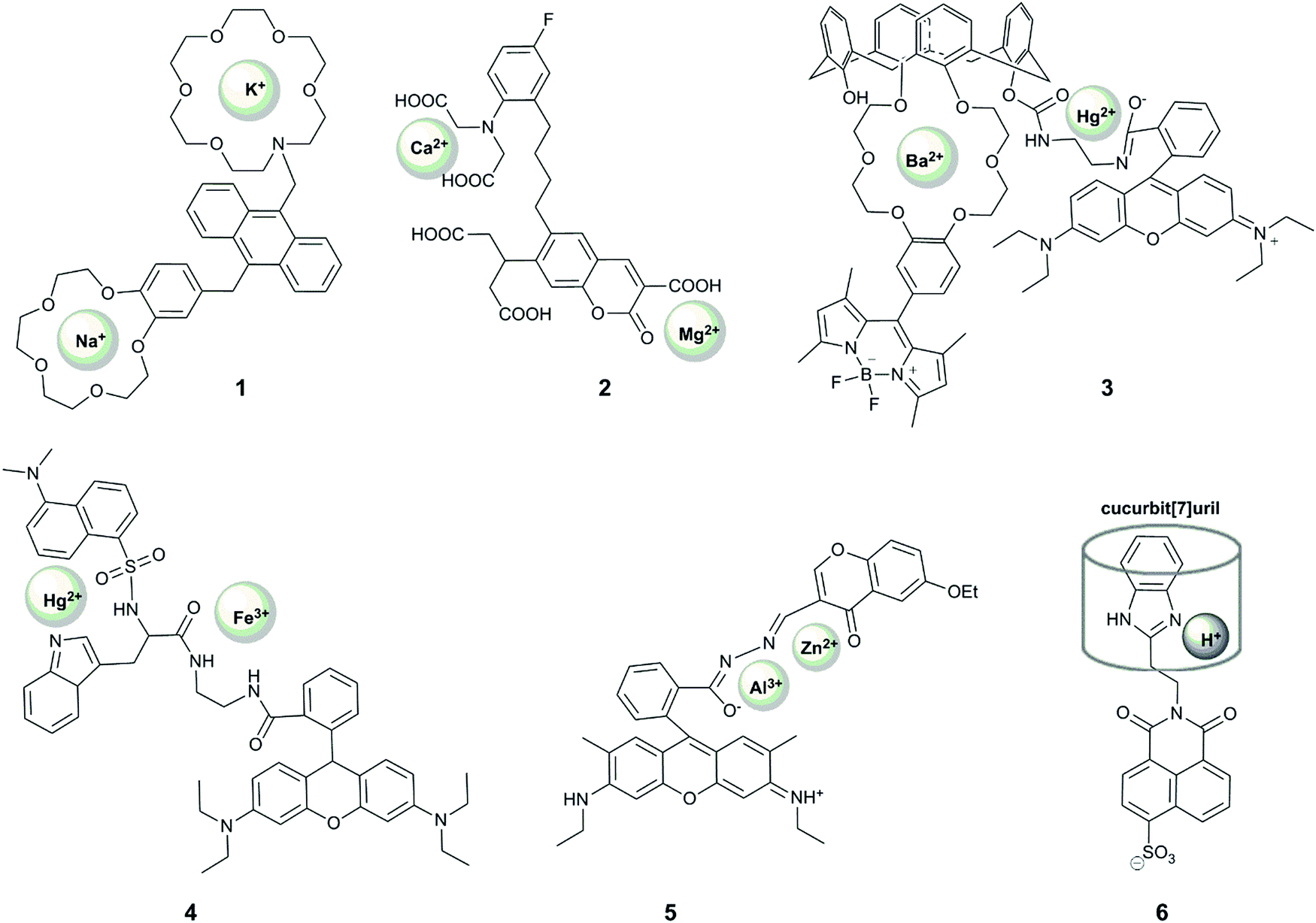 | ||
| Fig. 4 Structures of reversible dual-responsive probes showing sites of reversible interaction. | ||
Seminal work in this field by de Silva provided the first fluorescent AND gate.43 This anthracene-containing system was designed to report the presence of sodium ions and protons in organic solvent, but the presence of both analytes only gave rise to a five-fold increase in fluorescence compared to the “off” form of the probe, thus limiting its further application. Nonetheless, this work played a role in sparking further research in this field.
Xu et al. reported the first example of a probe that showed a unique response to the simultaneous presence of two metals at the same time.19 Their system (1) comprised an anthracene-based sensor with benzo-15-crown-5 and aza-18-crown-6 ethers for the binding of sodium and potassium respectively. Their motivation was that these two alkaline metal ions are crucial in physiological processes, with an increased ratio of sodium to potassium implicated as a cause and marker of cardiovascular disease.44 When the aza-crown ether remains non-protonated, potassium binds selectively to this motif, increasing the fluorescence at 436 nm due to alleviation of PET-quenching of the fluorophore by the nitrogen lone-pair. The considerable response to rubidium (up to 70% of the potassium fluorescence) is not concerning as rubidium is not biologically-abundant, but calcium can also induce up to 20% of the response observed for potassium. Interestingly, upon protonation of the aza-crown ether, the dialkoxybenzene moiety serves as a PET quencher, and the addition of sodium leads to enhancement of the fluorescence intensity at 425 nm, with some interference from potassium. Since the probe responds to each ion at different pH, the unique fluorescence response of the probe in the presence of both metal ions enables simultaneous quantification of the concentration of sodium and potassium in methanolic solution, with blue emission measured at two different pH values. The relatively wide dynamic range of the probe's response (∼0.1 mM to ∼30 mM) bodes well for its application in in vitro studies of body fluids (for example potassium levels in urine are generally above 25 mM45,46), but the interference from pH variation and a lack of documented binding in aqueous solution precludes the use of the probe in cellular studies.
Motivated by the unclear correlation between Ca2+ and Mg2+ in biology, Komatsu et al. reported KCM-1 (2), a single-molecule multianalyte sensor for calcium and magnesium, which consists of a coumarin fluorophore, the calcium binding motif BAPTA (O,O′-bis(2-aminophenyl)ethyleneglycol-N,N,N′,N′-tetraacetic acid) and a 2-carbonyl-3-carboxylate moiety as the Mg2+ binding site.20 Complexation of Ca2+ (with a binding constant of 14 μM) leads to a 45 nm blue shift in absorption and a concomittant 5 nm blue shift in the emission maximum via an ICT mechanism. On the other hand, magnesium binding (with a binding constant of 26 mM) causes a 21 nm red shift in absorption and a 5 nm red shift in emission. Simultaneous observation of the fluorescent response at three different excitation wavelengths enabled the quantification of the concentrations of both Ca2+ and Mg2+ in simulated biological conditions in vitro. The probe could also be applied to measuring Ca2+ and Mg2+ levels in the cytoplasm of PC12 cells upon treatment with the mitochondrial uncoupler FCCP, which caused a more rapid increase in the concentration of Ca2+ over Mg2+. While the selectivity of the weaker binding motif to magnesium over other metal ions should be examined, KCM-1 remains the first successful demonstration of the use of single small-molecule probe for two related analytes in a biological system, and can serve as an inspiration for the design of successful probes for other relevant pairs of analytes.
Following these examples, many other probes for the simultaneous detection of two analytes via a coordination mechanism have been reported, but only a few enabled an unambiguous identification of the simultaneous presence of two metal ions. For example, 3 is a Förster resonance energy transfer (FRET) probe containing a Hg2+-responsive rhodamine group as the FRET acceptor, which undergoes a >500 fold increase in red-emission upon Hg2+ binding, and a polyoxo-ether-calixarene-bearing BODIPY fluorophore as the FRET donor, which shows a >15-fold increase in fluorescence in the presence of 20 equivalents of Ba2+.21 This system exhibited four different fluorescence outputs depending on the combination of the targets. In particular, fluorescence intensity at 505 nm increased in the presence of Ba2+, fluorescence at 585 increased in the presence of Hg2+, with both fluorescence peaks observed when both metals were present, although the FRET effect meant that the intensity of responses was not equivalent in the presence of both metals. Despite the good selectivity of response to these analytes, with <10% interference of Cu2+ and Zn2+, the response was demonstrated only in acetonitrile, limiting its future application in biology. Nonetheless, this probe design can be potentially used very effectively in other systems.
A FRET strategy was also used for the design of probe 4 to simultaneously detect Fe3+ and Hg2+.23 In this case, a combination of two FRET events was used, between tryptophan (donor 1), dansylamide (acceptor 1, donor 2) and rhodamine B (acceptor 2). The dansylamide fluorescence at 510 nm (upon tryptophan excitation at 290 nm) is quenched upon the addition of Hg2+, while Fe3+ causes rhodamine ring-opening and therefore emission at 580 nm, which is observed upon excitation at 290, 330 or 550 nm. When both cations are bound, only emission from rhodamine is observed, and only upon excitation at 550 nm. The study of the fluorescent responses in the presence of other metal ions revealed the selectivity of the probe to Hg2+ and Fe3+, but Mn2+, Co2+/3+ and Fe2+ were notably absent from selectivity studies. In addition, while the different fluorescent response for all combinations of inputs is highly desirable, the response was only reported in ethanol, and together with the required UV excitation, suggests that it is unlikely that this probe will find application in biological studies.
A unique sequence-specific response to Zn2+ and Al3+ has been reported by Fan et al., who prepared a conjugate of rhodamine and chromone (RC), featuring a coordinatively-active linker that could accommodate both ions (5).22 Binding of Al3+ in an acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (9:1) mixture led to rhodamine ring-opening and subsequent red fluorescence. However, this response was decreased (by 30 or 50%) in the presence of Mn2+ or Cu2+, respectively. Addition of Zn2+ to a solution of the probe in ethanol:water (9:1), on the other hand, gave rise to an emission peak at 496 nm, speculated to result from the alleviation of the PET-quenching of the chromone fluorescence with a nitrogen lone pair on the linker. This response is, however, inhibited by the co-presence of Co2+. Interestingly, the addition of Al3+ to the RC–Zn complex led to the appearance of the additional rhodamine fluorescence peak, but no green chromone fluorescence could be observed when the addition of the metal ions was reversed. This unique sequence-selectivity of the RC probe opens up the way to design probes with unprecedent capacity to decipher signalling-cascades in biology (see Section 2.4), but its limited selectivity and the requirement of organic solvent may challenge future biological application of this probe.
:water (9:1) mixture led to rhodamine ring-opening and subsequent red fluorescence. However, this response was decreased (by 30 or 50%) in the presence of Mn2+ or Cu2+, respectively. Addition of Zn2+ to a solution of the probe in ethanol:water (9:1), on the other hand, gave rise to an emission peak at 496 nm, speculated to result from the alleviation of the PET-quenching of the chromone fluorescence with a nitrogen lone pair on the linker. This response is, however, inhibited by the co-presence of Co2+. Interestingly, the addition of Al3+ to the RC–Zn complex led to the appearance of the additional rhodamine fluorescence peak, but no green chromone fluorescence could be observed when the addition of the metal ions was reversed. This unique sequence-selectivity of the RC probe opens up the way to design probes with unprecedent capacity to decipher signalling-cascades in biology (see Section 2.4), but its limited selectivity and the requirement of organic solvent may challenge future biological application of this probe.
Most of the probes reported to date that use two reversible binding motifs for two different targets have been designed for the detection of metal ions. Nevertheless, the simultaneous detection of pH and a metal ion should also be quite straightforward by incorporation of a pH switch in place of one of the metal-binding groups. Similarly, the detection of anions can also be envisaged, taking inspiration from the many anion-specific probes,47 but the tight binding of anions in aqueous solutions required for applications in biology is not straightforward. Furthermore, in addition to coordination-based interactions, it is possible to envisage that other reversible interactions, including hydrogen-bond interactions and electrostatic forces, could be utilised in the design of reversible dual-responsive probes.
One notable example of a dual-responsive probe that involves reversible sensing but does not sense metal ions is 6, which senses protons and the macrocycle, cucurbit[7]uril.24 The naphthalimide-fluorophore is quenched by two mechanisms: rotation of the benzimidazole ring, which can be alleviated by binding of a macrocycle; and photoinduced electron transfer (PET) quenching by the nitrogen on the imidazole, which can be removed by protonation. Only if both modes of quenching are overcome is there a fluorescence turn-on, in the form of a 20-fold increase in emission intensity. Both events are reversible, observed by the addition of 1,5-pentanediamine (cadaverine) and base respectively. While this system is not selective for cucurbit[7]uril over other macrocycles of similar size, and only operates in a limited pH range, it represents an interesting example of the application of supramolecular principles to dual-analyte sensing.
2.2 Reaction-based probes
Irreversible reactions are commonly used in the design of single molecule probes for the detection of a variety of analytes.48 For example, H2O2 and ONOO− can rapidly react with boronate esters to induce a fluorescent response,6,42 while peptidases can be detected on the basis of the cleavage reaction that each catalyses.49 Several sensors for multiple analytes based on irreversible reactions with the probe and meeting the criterion of an AND gate have been reported. One group of such sensors contain separate reaction sites for each analyte, while another design takes advantage of a sequence of reactions, in reaction with analyte A generates an intermediate that can then react with analyte B. The latter class of probes will be considered in Section 2.4.The design of dual-responsive fluorescent probes bearing two reactive sites ensures that the order in which the probes appear does not matter. The probes that meet these criteria respond to a wide range of analytes (Fig. 5).
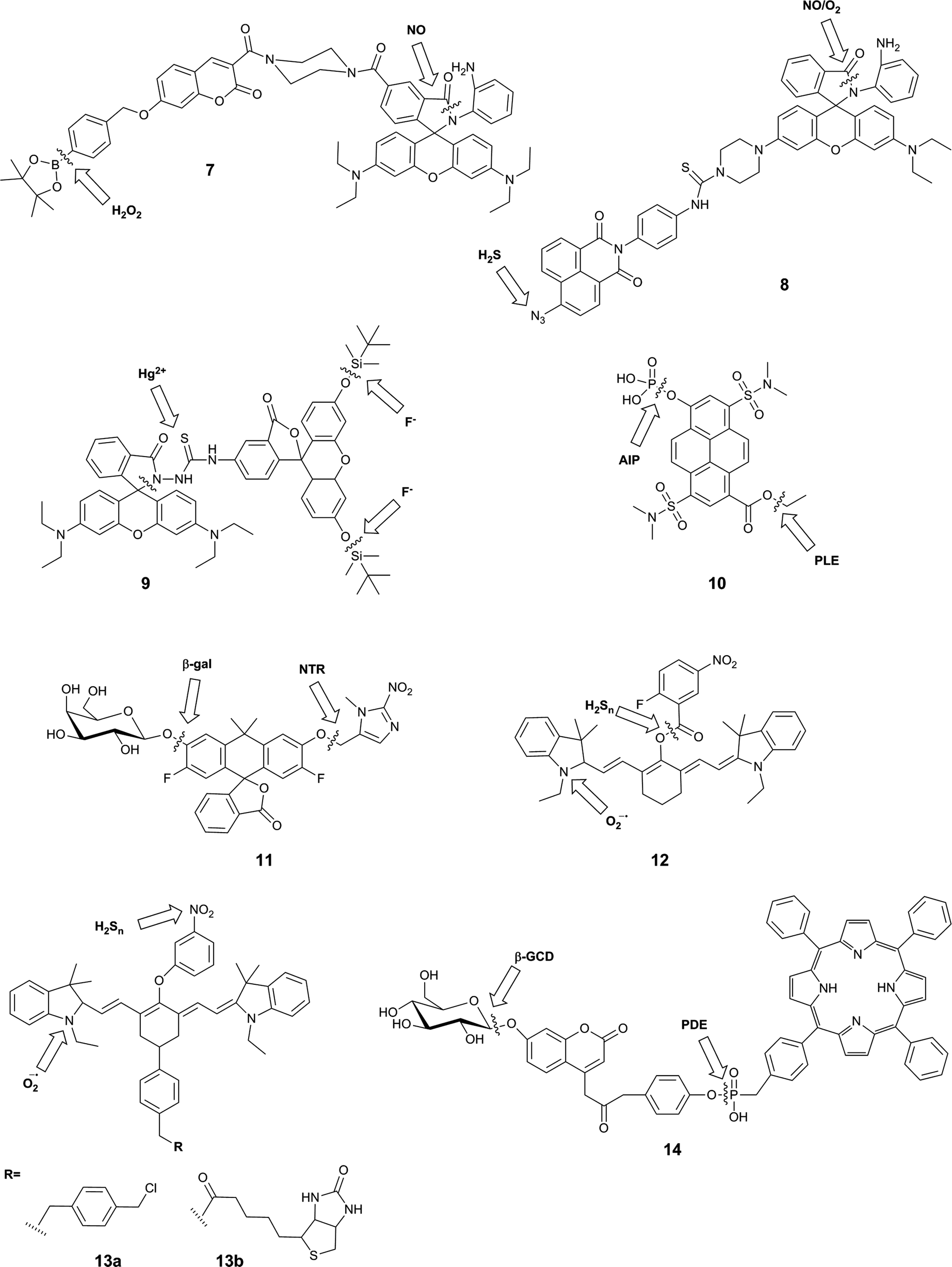 | ||
| Fig. 5 Structures of reaction-based dual-responsive probes that bear two sites of reaction, showing sites of reaction/cleavage. PLE = porcine liver esterase; AlP = alkaline phosphatase; β-gal = β-galactosidase; NTR = nitroreductase; β-GCD = β-glucosidase; PDE = phosphodiesterase. | ||
A typical example of this design is FP-H2O2-NO (7), a conjugate of a coumarin–phenylboronate, which becomes blue-fluorescent upon reaction with H2O2, and a rhodamine-(aminophenyl)-lactam, which becomes yellow-fluorescent upon NO-induced ring-opening.25 The two fluorophores are FRET partners, so both emission peaks can be observed upon excitation of the coumarin (λex = 400 nm). The good selectivity of the probe to these analytes over potential intracellular interferents and its sufficient cellular uptake enabled its use for the two-channel microscopy monitoring of H2O2 and NO in macrophages, despite the fact that all in vitro studies involved the use of 20% acetonitrile in aqueous solution. The key roles of both H2O2 and NO in redox signalling and oxidative stress50 suggests that this sensor will be particularly useful in deciphering relationships between these two species, and potentially shedding new light on their role in physiology and pathology. However, interpretation of data derived from this probe might be somewhat complicated by the fact that H2O2 and NO are known to react with each other.8,35
Zhang et al. used a similar strategy in their probe, NaphRhB (8), which was designed to sense H2S and NO. In this probe, the azide-naphthalimide was unmasked by hydrogen sulfide to become the FRET donor, while nitric oxide induced ring-opening and hence increased emission of rhodamine, the FRET acceptor. Both reactive sites showed good selectivity for their respective analyte over other reactive species. Having confirmed that the probe was not cytotoxic, the authors demonstrated that it was sensitive to exogenously-added NaHS (to generate H2S) and DEA-NONOate (an NO donor). NO and H2S are both known to play key roles as signalling molecules in the cardiovascular system,51 so it can be expected that much can be learnt using probes such as this. Again, however, H2S and NO are known to directly react,47 and therefore study of their co-existence may not be straightforward.
The fluorescein–rhodamine dyad 9 was developed for the simultaneous monitoring of Hg2+ and F−.27 Fluoride selectively promotes cleavage of the silyl ethers to unmask the FRET donor, fluorescein, while Hg2+-mediated formation of the oxadiazole leads to fluorogenic ring-opening of the FRET acceptor, rhodamine. Although all in vitro studies were performed in acetonitrile:TRIS buffer (1:1), the authors investigated the fluorescence of the probe in W138 lung fibroblast cells. Addition of F− and Hg2+ separately gave rise to green and red fluorescence respectively, but the effect of both ions simultaneously was not studied. It is not clear where in the body Hg2+ and F− might be expected to occur in measurable levels, but given the high toxicity of both of these ions, such a sensor might prove to be valuable in environmental monitoring.
In contrast to the three FRET-based probes described above, Finkler et al. described a dual-responsive probe (10) based on a single fluorophore, pyrenol.2810 was designed as a sensor of two specific enzymes, porcine liver esterase (PLE) and alkaline phosphatase (AlP). The probe bears an ester and a phosphate group as specific substrates for each enzyme respectively. Cleavage of one or both of these groups gives rise to different pyrenol structures, which exhibit different fluorescence emissions. The authors demonstrated that this probe could be used to monitor the kinetics of enzyme reactions, and also used two photon microscopy to study the activity of the two enzymes in L929 cells over time, but no inhibition or similar experiments were performed to associate the signal with enzymatic activity in the cellular environment. This probe itself is unlikely to exhibit sufficient selectivity for PLE over other esterases, or for AlP over other phosphatases, as the reactive groups used are only small: if selectivity were required, the substrate groups would need to be larger and more complex. However, this probe is a valuable first example of a dual-responsive probe for simultaneous detection of two enzymes.
The Lavis group reported a dual-responsive probe (11) based on a carbofluorescein scaffold, for the detection of two enzymatic activities with wide biological applications in complex sensing, targeting of selected cell populations and improved spatiotemporal control over fluorescent signal.29 This fluorophore requires the liberation of both phenolic groups to be fluorescent (unlike fluorescein, which becomes fluorescent upon a single unmasking event).52 By combining different protecting groups for each phenol, responsive probes of an AND type logic gate can be formed. The group succeeded in developing sensors for combinations of β-galactosidase, nitroreductase and β-lactamase, as well as a photocleavable unit. The probes were shown to have excellent sensitivity, with >100-fold increase in fluorescence, as well exhibiting stability at physiological pH, a rapid enzymatic response, and low energy excitation and emission. While the photoactivatable single-responsive system was applied in a proof-of-concept cellular experiment, neither of the dual-enzyme responsive systems was investigated in cells.
Owing to the strong emerging links between the hydrogen polysulfides and reactive oxygen species, and their roles in variety of physiological and pathological processes,30,53,54 a number of probes were developed that utilised selective reactions of the two analytes. These probes, 12,3013a and 13b,31 contain a hydrocyanine-7 conjugated to a m-nitrophenol moiety. Superoxide causes formation of an indolinium cation and therefore restoration of the push–pull mechanism critical for the far-red fluorescence of hydrocyanines. Subsequently, a further increase in fluorescence intensity can be observed upon the reduction of the nitrophenyl moiety, which causes PET quenching, to the PET-inactive aniline by hydrogen polysulfides. For 13a and b, this results in a 10 nm blue-shift in the maximum emission wavelength. While the authors did not apply their system as such, it is possible that this small spectral change could be used to positively identify the presence of both species, but it is not likely to be resolvable by current microscopy spectral imaging set-ups. As a result, it will be practically difficult to reliably distinguish the presence of higher probe/superoxide concentration from the sequential appearance of superoxide and hydrogen sulfides. Nevertheless, the authors reported the use of a mitochondrially-localisable probe 13a for investigating the links between hydrogen polysulfides and superoxide in mitochondria of RAW 264.7 and HUVECs cells and the biotinylated variant 13b was applied to study the relationship between those analytes in vivo in mouse cancerous tissues. For probe 12, nitrophenyl reduction led to a larger (170 nm) blue-shift in emission wavelength, enabling much more straight-forward distinction of the two forms of the probe. On the other hand, the presence of the unstable aryl ester linkage between the cleavable moiety and the fluorophore could leave the probe more susceptible to non-polysulfide-mediated hydrolysis. This probe was applied to image RAW 264.7 macrophages that had been treated with exogenous superoxide and NaHS.
Another attempt aiming at the development of a single probe for two enzymatic activities linked a FRET-donor, 7-β-D-glucopyranosyloxycoumarin with a FRET-acceptor meso-tetraphenylporphyrine (TPP) via a phosphodiester bond.32 The initially weak blue fluorescence of probe 14 is enhanced upon the cleavage of the sugar moiety from the phenolic oxygen of coumarin by β-D-glucosidase. The increase in the fluorescence efficiency of the coumarin was accompanied by a ∼30% decrease in the red nm fluorescence of TPP upon at 340 nm, probably due to the relative decrease in the FRET efficiency. The cleavage of the phosphodiester bond further decreased the red nm fluorescence by ∼70% as a consequence of a liberation of a free TPP, which can diffuse away from the coumarin FRET-donor, with negligible impact on the coumarin fluorescence. In the presence of both enzymes in optimal pH conditions (between the pH 7–8), the blue coumarin fluorescence increased and the red TPP fluorescence decreased, both upon 340 m excitation. A number of tested cations and anions had no effect on probe fluorescence, and the optimal pH range of 7–8 has been established to maximise the enzymatic activity and exclude fluorescent artefacts arising from the protonation equilibria. The probe was also shown to permeate cellular membranes and increased its blue and red fluorescence in Huh7 liver cells known to express both enzymes, but no additional experiments confirming the enzymatic origin of the signal were reported.
2.3 Mixed probes
Sections 2.1 and 2.2 focus on probes that respond to both analytes through reversible interaction or reaction with both analytes, respectively. It is therefore straightforward to imagine an additional set of probes for which one analyte is recognised by reversible interaction and the other by reaction. This strategy has been utilised in a phenothiazine-based probe that is designed to respond to Hg2+ by coordination and Cu+ by reaction.55 However, this probe does not fall into the scope of this review as it operates only in acetonitrile, and the presence of both Cu+ and Hg2+ cannot be spectrally identified. Since protonation is a reversible event, and we can therefore classify two probes into this grouping, which operate by a protonation/deprotonation event as well as a reaction (Fig. 6).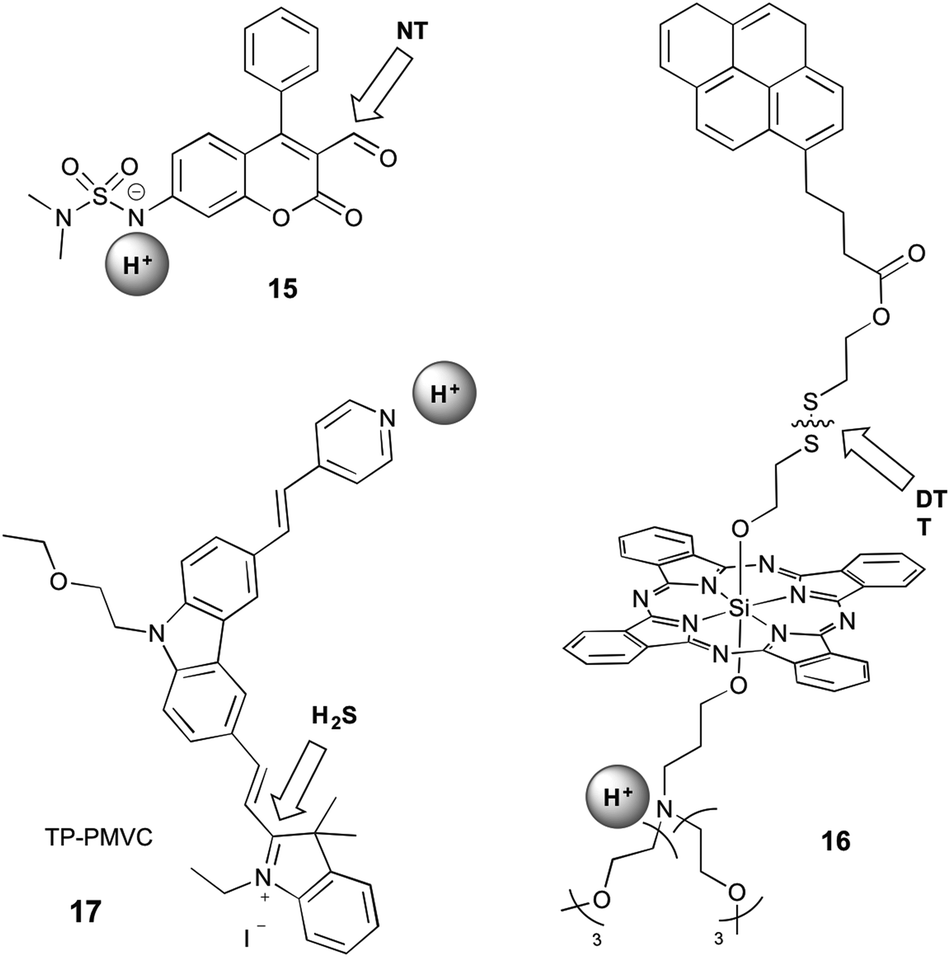 | ||
| Fig. 6 Structures of mixed dual-responsive probes, showing sites of reaction and reversible interaction. NT = amine-based neurotransmitter; DTT = dithiothreitol. | ||
Klockow et al. reported a coumarin-based sensor (15) for the study of pH and neurotransmitters, in order to visualise the exosomal transport of neurotransmitters to synapses.33 They investigated a range of sulfonamides as the pH-sensitive group, with deprotonation causing an increase in blue emission. The carbaldehyde group on the coumarin reacts with amine-based neurotransmitters (such as glutamate, norepinephrine, dopamine, and serotonin) to form an imine, leading to a shift in emission to the green. Since this event is accompanied by a shift in the absorption profile, the authors were able to selectively excite with 488 nm light to visualise only the form of the probe that was deprotonated and had reacted with the neurotransmitter. This system is not selective for a specific neurotransmitter, with the stated end-use being the study of specialised neurons known to utilise only one neurotransmitter type. The suggested value of this probe was in studying the release of neurotransmitters from acidic exosomes into the synapse, where the pH would increase, enabling a turn-on of the probe. However, this work did not extend validation studies beyond in vitro investigations.
16 contains a silicon phthalocyanine (SiPC) conjugated to a tertiary amine, which acts as a photoinduced electron transfer (PET) quencher, and a pyrene group via a disulfide bond.34 When bound, the pyrene is quenched by electronic energy transfer (EET) to the SiPC. Protonation alleviates the PET quenching, therefore restoring the red fluorescence of the SiPC, while reaction with dithiothreitol (DTT) leads to cleavage of the disulfide bond and release of the pyrene, restoring its ultraviolet emission. This compound did not have demonstrated selectivity for DTT over any other thiols. Furthermore, interpretation of data from such a probe is likely to be complicated by the fact that the two separated fluorophores might no longer be co-localised within the cell during imaging.
An excellent example of a mixed probe is 17 (TP-PMVC), which upon protonation (∼pH 4–6) at the para-pyridyl moiety exhibits a 15-fold increase in orange fluorescence.37 H2S reacts rapidly with the double bond between the indolinium and carbazole moieties, inducing a shift in fluorescence to the yellow and increase in the intensity, visible only at low pH, but the detailed responsiveness at different pH values or the importance of the sequence of the addition of the analytes were not studied. While it can be speculated, that the protonation of the probe might have some influence on the preference for the addition of H2S, the different sequence of the addition of the analytes was not further explored. The probe was selective for hydrogen sulfide over all other small molecules tested, and exhibited high photostability over an hour of irradiation. This probe can therefore be considered as a sensor for H2S concentration specifically under acidic conditions, which is highly relevant for biological study as pH is crucial in the production and metabolism of H2S.50 TP-PMVC was successfully applied to the ratiometric monitoring of changes in the levels of H2S in lysosomes of A549 cells by conventional and two-photon microscopy and in mouse liver tissue samples.
2.4 Sequence-specific reactions
Fluorescent reactive probes are based on the same strategy as pro-drugs, which are prevalent in medicinal chemistry.1 Pro-drugs are prepared by masking the active drug with a reactive group that is cleaved under specific conditions to release the active compound. In this way, the drug can be generated at the desired site of action, without causing any off-target effects. The cleavage mechanism may be enzyme-mediated56 or may involve a specific chemical reaction.57 In the translation of this concept to fluorescent sensing, a fluorescent compound is revealed upon unmasking of the pro-fluorophore. This has been applied to a number of contexts, including systems that turn on upon protein-binding,58 cobalt complexes that become fluorescent upon reduction and fluorophore release,59 and enzyme-activated systems.44The pro-fluorophore strategy lends itself well to the development of dual-responsive probes, as it is straightforward to envisage the first analyte unmasking the second analyte-recognition site, whether for subsequent reaction or reversible interaction. Such probes are particularly suited to the sensing of an analyte within a specific environment (e.g. low pH, high ROS levels), but can also be useful in reporting on the occurrence of one analyte only when it is in the presence of another analyte. It is important to note here that the sequence of events is crucial: the probe is able to report on the presence of A followed by B, but if B is presented to the probe followed by A, there will be no response. However, the simultaneous presence of A and B will also be recognised by such probes. As a result, the sequence specificity of the probe will not necessarily correlate with the biochemical reaction sequence. Probes utilising this mechanism are summarised in Fig. 7, and comprise those that operate by all three mechanisms described above: reversible, reaction and a mixture of reaction and reversible.
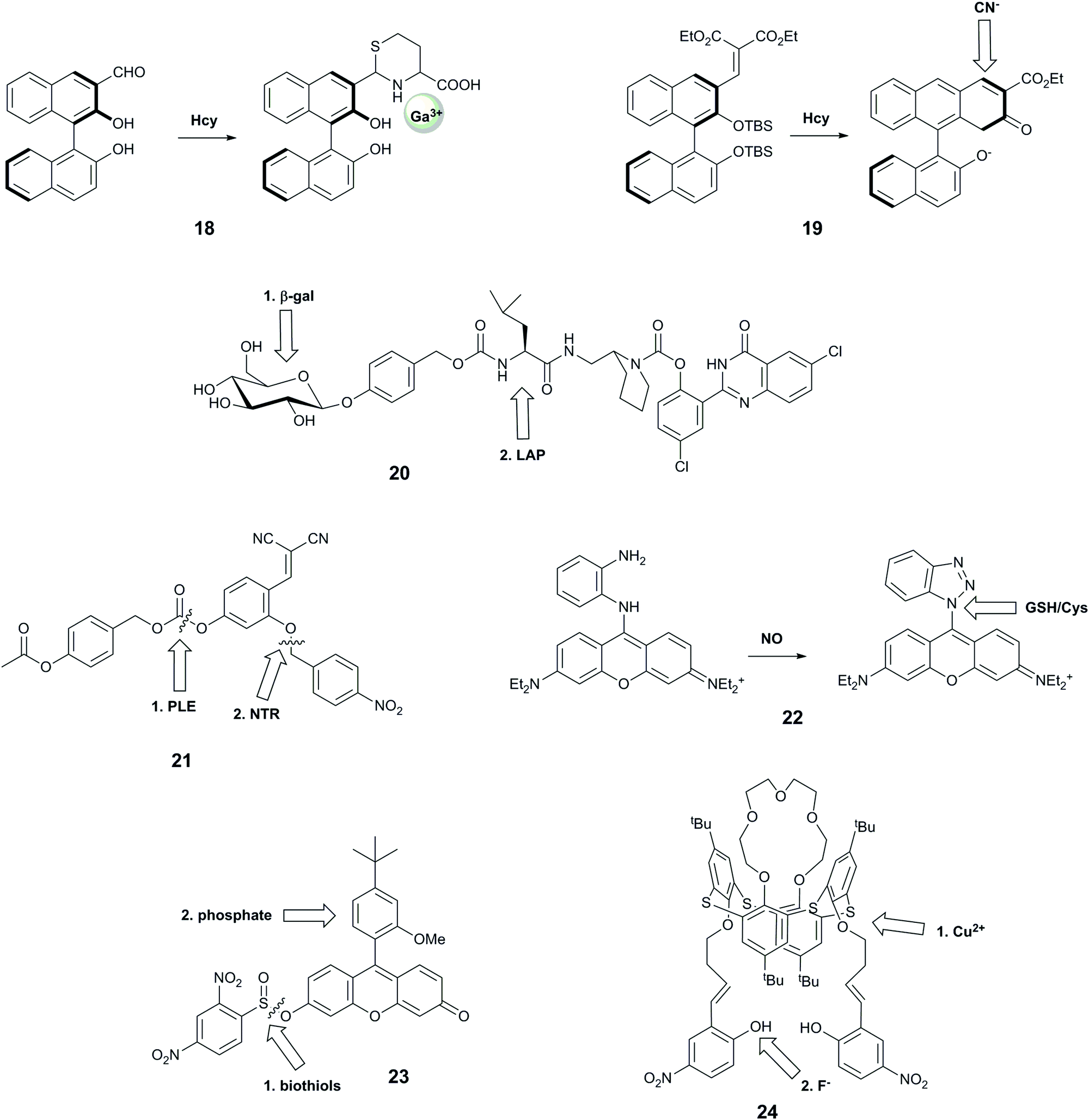 | ||
| Fig. 7 Structures of sequence-specific dual-responsive probes showing sequence of interaction. Hcy = homocysteine; β-gal = β-galactosidase; LAP = leucine aminopeptidase; PLE = pig liver esterase; NTR = nitroreductase. | ||
Wang et al. reported probe 18 based on a binaphthyl fluorophore that was capable of recognising homocysteine followed by Ga3+.35 The carbaldehyde moiety reacted selectively with homocysteine over cysteine and other amino acids, leading to a 10-fold increase in violet fluorescence. The resulting compound exhibited a new blue fluorescence peak in the presence of Ga3+ in aqueous buffer at neutral pH. This system showed selectivity for Ga3+ over other metal ions except Al3+ and In3+, but these ions could be distinguished by their ability to form an additional fluorescence peak due to excimer emission. This probe is particularly interesting due to its good selectivity for Ga3+ over Fe3+, and for homocysteine over cysteine, both of which remain challenging.48,60 Furthermore, the resulting adduct is claimed to be the first highly selective sensor for Ga3+ over Al3+, with the former having some use in nuclear medicine. While these two targets have not been linked in biology, future evaluation of this compound in biological models could potentially assist in shedding light on the impact of gallium on living systems.
Another system that comprises a binaphthyl fluorophore is 19, which uses an anion relay recognition sequence for the sequential detection of fluoride and cyanide.36 The fluorophore is decorated with tert-butyldimethylsilyl groups that are selectively cleaved by F−, promoting further ring formation, and allowing subsequent addition of a cyano group to the naphthyl core. The first transformation step leads to quenching of the emission peak at 360 nm, and an increase in fluorescence at 460 nm, while the second reaction gives rise to a further increase in 460 nm emission, with no change at 360 nm. Each form of the probe therefore has a unique spectral form, enabling ratiometric measurements. While no specific biological question for such a probe was identified, and all studies were performed in THF, this is nonetheless a useful probe design that could be harnessed for other analytes in the future.
With the aim of identifying only cells that express two enzymes, Hasserodt's group reported an elegant “pre-pro-fluorescent probe” (20).38 Their probe design incorporated a 2-(2′-hydroxyphenyl)-4(3H)-quinazolinone (HPQ) fluorophore that was masked by leucine, the substrate for leucine aminopeptidase (analyte B), which in turn was masked by a β-galactose unit, the substrate for β-galactosidase (analyte A). Only after β-galactosidase cleaved the β-galactose unit could the leucine aminopeptidase cleave the leucine group, leading to green fluorescence from precipitated HPQ. While this probe cannot report on the presence of each enzyme individually, it was applied to distinguish cell types according to their expression levels of the two enzymes.
Probe 21 was designed with a similar aim; emitting a detectable signal only when two enzymes process the probe.12 The probe is based on the covalent-assembly strategy, for which unmasking of both hydroxy groups by selective enzyme reactions unlocks the pro-sensor, leading to an intramolecular cyclisation to generate the highly fluorescent 7-hydroxy-2-iminocoumarin. Theoretically, the unmasking events can occur in either order, but the authors observed better signal when the probe was first reacted with PLE, and speculated that the substrate is too bulky in complete form to be processed by NTR. While the authors confirmed that the kinetics of the system was sufficiently rapid for biological application, they did not perform any subsequent biological experiments.
The rhodamine-o-diaminophenyl probe 22 was developed to investigate the relationship between nitric oxide, a known signalling molecule, and biologically relevant thiols.39 The well-known reaction of NO with o-dianilines leads to the formation of the benzotriazolyl–rhodamine with increased fluorescence at 616 nm (λex = 570 nm). The newly formed benzothiazole can be replaced by biologically-relevant nucleophiles in an aromatic substitution reaction, with cysteine leading to formation of a green fluorescent species and glutathione forming a product with the fluorescence colour similar to the benzothiazole–rhodamine. The selectivity towards NO over other ROS and RNS has been demonstrated, but no information on the selectivity of the benzotriazolyl–rhodamine towards other nucleophiles was reported. In addition, while the NO + Cys sequence could be distinguished by the observation of the change of colour of fluorescence from red to green, the same fluorescence wavelength of the GSH adduct and benzotriazolyl intermediate complicates the distinction process. Nevertheless, the probe has been used to monitor the exogenous and endogenous levels of NO in cells.
Motivated by reports that intracellular phosphate and biothiol levels could enable study of altered bone metabolism in disease,61 Resa et al. reported a fluorescent probe, 23, which responds to biothiols followed by phosphate.4023 bears a thiol-cleavable group that releases the fluorescein-derivative Granada green, which has previously been shown to report on changes in phosphate levels through an excited-state proton transfer (ESPT)-induced decrease in fluorescence lifetime.62 The probe does not show complete selectivity towards thiols, as the protecting group can be cleaved by other reactants including H2O2 and alanine. The origin of the reported sequence-specificity is not entirely clear, as the authors did not investigate the effect of phosphate followed by biothiol treatment, but without biothiol-induced cleavage, it is not possible to measure fluorescence lifetimes and thus observe phosphate. Fluorescence microscopy of HeLa cells showed that cells treated with N-methylmaleimide (NMM), which blocks intracellular thiols, exhibited greatly decreased fluorescence intensity. Fluorescence lifetime imaging microscopy (FLIM) studies of HeLa cells permeabilised with α-toxin to enable equilibration of ion concentration with surrounding media enabled measurement of fluorescence lifetimes for different phosphate concentrations that matched in vitro-determined values.
Probe 24 was designed as a keypad lock, responding to Cu2+ followed by F−. This system also meets our criterion of a sequence-specific AND gate for potential biological use, although there is not an immediate need to image these two analytes together in biology. The authors hypothesise that addition of F− to 24 leads to a fluorescence enhancement through deprotonation of the phenolic hydroxyl group, promoting delocalisation of the nitrophenyl π-electrons. Subsequently-added Cu2+ binds to the imino nitrogens and F− ions, causing photoinduced charge transfer and hence quenched fluorescence. Conversely, when Cu2+ is added first. F− then binds to the Cu centre, leading to the redistribution of d-energy levels, and formation of a new fluorescence peak at a longer wavelength. All studies were performed in anhydrous THF, and the probe could not be applied to biological study.
2.5 Probes that rely on competition between analytes
It is important to briefly mention here that there are a number of reported probes that rely on the competition between analytes. In such probes, each analyte has a distinctive interaction with the probe, giving a characteristic response, but the two analytes interfere with each other, whether by competing for the same reaction site or by inhibiting interaction of the other analyte with the probe. Using this strategy, probes have been reported with characteristic responses to different transition metals,63,64 ROS65 or biothiols.66–72 Theoretically, if the probe concentration, kinetics and thermodynamics of the reactions with the targets do not lead to saturation of the probe with one analyte alone, these systems will be capable of reporting on the presence of one or more analytes. However, since this cannot be reliably achieved in biological systems, we will not consider these probes further.3. Discussion
3.1 Promising strategies for probe design
One of the greatest challenges in realising the potential of dual-responsive probes for biological application is in the selection of appropriate sensing logic: only a small subset of all molecular logic gates enable the definitive identification of the co-existence of both analytes. While both AND and NAND logic gates meet this criterion, AND gates have far greater practical application: NAND gates are an example of a turn-off fluorescent probe, for which the positive, quenched, response is indistinguishable from lack of probe.While AND gates can provide information on the presence of both analytes, the ideal dual-responsive probe is one for which the presence of each analyte separately, and the co-presence of both analytes, give unique and measurable responses (Fig. 3). Such probes can provide far greater information about biological systems, including which of the two analytes appears first. As outlined in the introduction, ratiometric systems are able to provide more unambiguous information than intensity-based probes, as the latter requires a controlled probe concentration. Ratiometricity may be achieved by perturbation of FRET systems. Alternatively, alteration of probe structure in reaction-based probes can give rise to fluorophores with different structures and hence emission colours, as for probe 12. Amongst ratiometric probes, those that give a distinct colour for all four scenarios (no analyte, A alone, B alone, A + B) will provide the most readily-interpreted data. A notable example is probe 10, based on a single fluorophore alone, for which these four cases give emission at 472, 448, 558 and 536 nm respectively.28
Ratiometric response can also be achieved for a probe in which fluorescence lifetime changes with analytes, as fluorescence lifetime is independent of probe concentration. However, to date very few lifetime-based probes have been reported, even to single analytes.4,62 The only dual-responsive lifetime-based probe reported to date is 23, which exhibits a small (∼6%) change in lifetime upon the addition of phosphate, nonetheless demonstrating the value of recording lifetime changes when fluorescence emission does not change.73 This is certainly likely to be a fruitful area of probe design for the future.
This review has focussed primarily on probes that respond to two analytes, but it is possible to imagine systems that can respond to three or more analytes. To date, many of the multi-responsive systems reported are limited in applicability by the fact that two or more of the analytes interact with the same reaction site, thus requiring higher concentrations of probe than analytes.63,67,72 To overcome this limitation, it is important to have as many separate reaction or coordination sites as there are analytes to be studied, which may be most easily controlled by having a series of sequential unmasking events. Advances in spectral unmixing for microscopy set-ups74 mean that even the most ideal multi-responsive probes with a unique emission colour for each combination of analytes can be readily imaged.
It is likely that many probes that are reported as single-responsive agents do in fact have dual response, but were not tested against a sufficiently broad array of analytes to uncover such responses. There will therefore be value in investigating currently-reported single-responsive systems for their potential application in studying two analytes. In particular, many fluorescent systems reported for other analytes are pH-sensitive, so these can be used as dual probes for pH and another analyte. This may be further optimised by tuning pKa values to the biologically-relevant range pH range, and maximising the fluorescence change induced by protonation, as for 15.33
3.2 Designing dual-responsive probes for biological applications
There is no doubt that dual-responsive probes stand to contribute much to the study of biological systems. For the first time it will be possible to visualise the interplay between multiple small molecules in real time, which will enable discovery of new signalling pathways and understanding of the interaction of exogenous and endogenous species. In order to achieve this, however, it is crucial that probes are purposefully designed to answer the most pertinent biological questions, rather than just studying the two analytes that are easiest to simultaneously sense. That is, the two analytes should be known (or speculated) to be biologically-related, whether playing a role in the same disease, or having been reported to accumulate in the same sub-cellular location.The type of biological question being interrogated will determine which of the designs outlined above will be most appropriate. To observe fluxes of analytes, a reversible probe is required, and therefore the dual-reversible approach will be most useful. On the other hand, irreversible approaches enable the study of species that may be present for a time period too short or a concentration too low for rapid detection: instead, irreversible reactions enable the build-up of turned-on probe until detectible limits are reached. However, if a long incubation time is used with an irreversible probe, it is not possible to definitively conclude that the two analytes were present simultaneously: it is only certain that they both appeared at some point during the incubation. Furthermore, a reversible probe might be expected to alter the system to a lesser extent, as the target species is not consumed, but instead cycles between “off” and “on” forms.
As for all biologically-applied probes, several properties must be achieved. The excitation wavelength must be sufficiently long (>400 nm) to ensure that excitation light sources do not causes cellular damage. The probe should be non-toxic, and should not perturb cellular homeostasis. The sub-cellular localisation of the probe must be appropriate to the biological question: this may require the incorporation of small molecule or peptide-based localisation tags.
3.3 Applying dual-responsive probes to biological studies
As is the case for single-responsive probes,3 our review here has highlighted that many dual-responsive probes are reported with claims of their biological relevance, but without any supporting experiments to this end. Many of the purportedly dual-responsive probes that were examined but not included in this review reported the response of each analyte separately, but not combined: without such information, it is not possible to begin to assess the potential utility in a biological system where both analytes may be present simultaneously.In order for biological application to be feasible, in vitro tests must be carried out under biologically-relevant conditions: in aqueous solvents buffered at physiological pH, and with concentrations of analytes that might be expected within biological systems. Furthermore, selectivity studies should be performed with all interferents that are expected to also be present within the cell.
Once the in vitro behaviour is found to be compatible with cellular application, probes should then be applied to model cellular systems. Positive and negative controls will involve exogenously-added or suppressed analytes, and for dual-responsive probes it is important to test all combinations of analytes. Ideally, validation experiments should be followed by an experiment that tests the power of the probe under physiological conditions.
One of the greatest challenges in realising the potential of fluorescent probes is encouraging end users (usually biological or medical researchers) to use recently-developed probes in favour of tried-and-tested, often inferior, commercial products.3 Achieving this end will require a concerted effort by those who prepare dual-responsive probes to perform the required validation studies across a range of biological systems, and to reach out to and actively collaborate with end users. Such exertion will ensure that these powerful novel systems have great effect in a wide range of applications.
4. Conclusions
Fluorescent sensing is a topic of great current interest, with hundreds of new fluorescent probes reported each year. Dual-responsive probes comprise only a small subset of this work, but they have the potential to contribute far more powerfully to the understanding of biological systems. This review has examined a number of probes reported to date that are capable of identifying the simultaneous presence of two analytes, highlighting examples that demonstrate how powerful rigorous probe development can be. By focussing design efforts on the most pertinent sets of biological analytes, and considering the most useful combinations of fluorescent output, dual-responsive probes will achieve their potential in playing a considerable role in the understanding of health and disease.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Westpac Bicentennial Foundation for a Research Fellowship (EJN) and the Australian Research Council for funding (DP150100649).References
- A. K. Sinhababu and D. R. Thakker, Adv. Drug Delivery Rev., 1996, 19, 241–273 CrossRef CAS.
- J. L. Kolanowski, A. Kaur and E. J. New, Antioxid. Redox Signaling, 2015, 24, 713–730 CrossRef PubMed.
- E. J. New, ACS Sens., 2016, 1, 328–333 CrossRef CAS.
- J. C. Pickup, F. Khan, Z.-L. Zhi, J. Coulter and D. J. S. Birch, J. Diabetes Sci. Technol., 2013, 7, 62–71 CrossRef PubMed.
- E. J. New, Dalton Trans., 2013, 42, 3210–3219 RSC.
- T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC.
- D. J. Hare, E. J. New, M. D. de Jonge and G. McColl, Chem. Soc. Rev., 2015, 44, 5941–5958 RSC.
- S. P. Dudas, M. Chatterjee and M. A. Tainsky, Cancer Biomarkers, 2010, 6, 257–270 CrossRef PubMed.
- A. P. de Silva, Chem. – Asian J., 2011, 6, 750–766 CrossRef PubMed.
- U. Pischel, Angew. Chem., Int. Ed., 2007, 46, 4026–4040 CrossRef CAS PubMed.
- N. Renaud, M. Hliwa and C. Joachim, in Unimolecular and Supramolecular Electronics Ii: Chemistry and Physics Meet at Metal-Molecule Interfaces, ed. R. M. Metzger, Springer-Verlag Berlin, Berlin, 2012, vol. 313, pp. 217–268 Search PubMed.
- A. Romieu, Org. Biomol. Chem., 2015, 13, 1294–1306 CAS.
- T. S. Moon, C. B. Lou, A. Tamsir, B. C. Stanton and C. A. Voigt, Nature, 2012, 491, 249–253 CrossRef CAS PubMed.
- J. Bonnet, P. Yin, M. E. Ortiz, P. Subsoontorn and D. Endy, Science, 2013, 340, 599–603 CrossRef CAS PubMed.
- J. A. N. Brophy and C. A. Voigt, Nat. Methods, 2014, 11, 508–520 CrossRef CAS PubMed.
- G. Zhu, Y. Li and C.-Y. Zhang, Chem. Commun., 2014, 50, 572–574 RSC.
- C. Hao, L. Xua, C. Xing, H. Kuang, L. Wang and C. Xu, Biosens. Bioelectron., 2012, 36, 174–178 CrossRef CAS PubMed.
- X. Zhang, N. Liao, G. Chen, A. Zheng, Y. Zeng, X. Liu and J. Liu, Nanoscale, 2017, 9, 10861–10868 RSC.
- X. Xu, H. Xu and H.-F. Ji, Chem. Commun., 2001, 2092–2093 RSC.
- H. Komatsu, T. Miki, D. Citterio, T. Kubota, Y. Shindo, Y. Kitamura, K. Oka and K. Suzuki, J. Am. Chem. Soc., 2005, 127, 10798–10799 CrossRef CAS PubMed.
- M. Yuan, W. Zhou, X. Liu, M. Zhu, J. Li, X. Yin, H. Zheng, Z. Zuo, C. Ouyang, H. Liu, Y. Li and D. Zhu, J. Org. Chem., 2008, 73, 5008–5014 CrossRef CAS PubMed.
- L. Fan, J. C. Qin, T. R. Li, B. D. Wang and Z. Y. Yang, Sens. Actuators, B, 2014, 203, 550–556 CrossRef CAS.
- X. Zhou, X. Wu and J. Yoon, Chem. Commun., 2015, 51, 111–113 RSC.
- U. Pischel, V. D. Uzunova, P. Remon and W. M. Nau, Chem. Commun., 2010, 46, 2635–2637 RSC.
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed.
- P. Zhang, J. Li, B. Li, J. Xu, F. Zeng, J. Lv and S. Wu, Chem. Commun., 2015, 51, 4414–4416 RSC.
- N. R. Chereddy, P. Nagaraju, M. V. Niladri Raju, K. Saranraj, S. Thennarasu and V. J. Rao, Dyes Pigm., 2015, 112, 201–209 CrossRef CAS.
- B. Finkler, I. Riemann, M. Vester, A. Grüter, F. Stracke and G. Jung, Photochem. Photobiol. Sci., 2016, 15, 1544–1557 CAS.
- J. B. Grimm, T. D. Gruber, G. Ortiz, T. A. Brown and L. D. Lavis, Bioconjugate Chem., 2016, 27, 474–480 CrossRef CAS PubMed.
- F. Yu, M. Gao, M. Li and L. Chen, Biomaterials, 2015, 63, 93–101 CrossRef CAS PubMed.
- Y. Huang, F. Yu, J. Wang and L. Chen, Anal. Chem., 2016, 88, 4122–4129 CrossRef CAS PubMed.
- Y. Li, H. Wang, J. Li, J. Zheng, X. Xu and R. Yang, Anal. Chem., 2011, 83, 1268–1274 CrossRef CAS PubMed.
- J. L. Klockow, K. S. Hettie, K. E. Secor, D. N. Barman and T. E. Glass, Chem. – Eur. J., 2015, 21, 11446–11451 CrossRef CAS PubMed.
- E. van de Winckel, R. J. Schneider, A. de la Escosura and T. Torres, Chem. – Eur. J., 2015, 21, 18551–18556 CrossRef CAS PubMed.
- Y.-W. Wang, S.-B. Liu, W.-J. Ling and Y. Peng, Chem. Commun., 2016, 52, 827–830 RSC.
- M. Dong, Y. Peng, Y. M. Dong, N. Tang and Y. W. Wang, Org. Lett., 2012, 14, 130–133 CrossRef CAS PubMed.
- Y. Liu, F. Meng, L. He, K. Liu and W. Lin, Chem. Commun., 2016, 52, 7016–7019 RSC.
- M. Prost and J. Hasserodt, Chem. Commun., 2014, 50, 14896–14899 RSC.
- Y.-Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 10–13 Search PubMed.
- S. Resa, A. Orte, D. Miguel, J. M. Paredes, V. Puente-Muñoz, R. Salto, M. D. Giron, M. J. Ruedas-Rama, J. M. Cuerva, J. M. Alvarez-Pez and L. Crovetto, Chem. – Eur. J., 2015, 21, 14772–14779 CrossRef CAS PubMed.
- M. Kumar, A. Dhir and V. Bhalla, Org. Lett., 2009, 11, 2567–2570 CrossRef CAS PubMed.
- A. T. Aron, K. M. Ramos-Torres, J. A. Cotruvo and C. J. Chang, Acc. Chem. Res., 2015, 48, 2434–2442 CrossRef CAS PubMed.
- A. P. De Silva, H. Q. N. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42–44 CrossRef.
- A. Zadlo-Dobrowolska, M. Szczygiel, D. Koszelewski, D. Paprocki and R. Ostaszewski, Org. Biomol. Chem., 2016, 14, 9146–9150 CAS.
- B. Phakdeekitcharoen, C. Kreepala and S. Boongird, J. Med. Assoc. Thailand, 2011, 94, 1337–1345 Search PubMed.
- G. Song, R. Sun, J. Du, M. Chen and Y. Tian, Chem. Commun., 2017, 53, 5602–5605 RSC.
- M. R. Filipovic, M. Eberhardt, V. Prokopovic, A. Mijuskovic, Z. Orescanin-Dusic, P. Reeh and I. Ivanovic-Burmazovic, J. Med. Chem., 2013, 56, 1499–1508 CrossRef CAS PubMed.
- H. Peng, W. Chen, Y. Cheng, L. Hakuna, R. Strongin and B. Wang, Sensors, 2012, 12, 15907–15946 CrossRef CAS PubMed.
- O. Thorn-Seshold, M. Vargas-Sanchez, S. McKeon and J. Hasserodt, Chem. Commun., 2012, 48, 6253–6255 RSC.
- Q. Li and J. R. Lancaster, Nitric Oxide, 2013, 35, 21–34 CrossRef CAS PubMed.
- M. L. Lo Faro, B. Fox, J. L. Whatmore, P. G. Winyard and M. Whiteman, Nitric Oxide, 2014, 41, 38–47 CrossRef CAS PubMed.
- J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303–1310 CrossRef CAS PubMed.
- B. D. Paul and S. H. Snyder, Nat. Rev. Mol. Cell Biol., 2012, 13, 499–507 CrossRef CAS PubMed.
- K. Ono, T. Akaike, T. Sawa, Y. Kumagai, D. A. Wink, D. J. Tantillo, A. J. Hobbs, P. Nagy, M. Xian, J. Lin and J. M. Fukuto, Free Radical Biol. Med., 2014, 77, 82–94 CrossRef CAS PubMed.
- J. Weng, Q. Mei, B. Zhang, Y. Jiang, B. Tong, Q. Fan, Q. Ling and W. Huang, Analyst, 2013, 138, 6607 RSC.
- Y.-h. Yang, H. Aloysius, D. Inoyama, Y. Chen and L.-q. Hu, Acta Pharm. Sin. B, 2011, 1, 143–159 CrossRef CAS.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed.
- N. Yamamoto, A. K. Renfrew, B. J. Kim, N. S. Bryce and T. W. Hambley, J. Med. Chem., 2012, 55, 11013–11021 CrossRef CAS PubMed.
- V. Nikolova, S. Angelova, N. Markova and T. Dudev, J. Phys. Chem. B, 2016, 120, 2241–2248 CrossRef CAS PubMed.
- T. Yokota, S. Kinugawa, M. Yamato, K. Hirabayashi, T. Suga, S. Takada, K. Harada, N. Morita, N. Oyama-Manabe, Y. Kikuchi, K. Okita and H. Tsutsui, Diabetes Care, 2013, 36, 1341–1346 CrossRef CAS PubMed.
- J. M. Paredes, M. D. Giron, M. J. Ruedas-Rama, A. Orte, L. Crovetto, E. M. Talavera, R. Salto and J. M. Alvarez-Pez, J. Phys. Chem. B, 2013, 117, 8143–8149 CrossRef CAS PubMed.
- Y. Shiraishi, C. Ichimura, S. Sumiya and T. Hirai, Chem. – Eur. J., 2011, 17, 8324–8332 CrossRef CAS PubMed.
- V. Luxami, Renukamal, K. Paul and S. Kumar, RSC Adv., 2013, 3, 9189–9192 RSC.
- R. Zhang, J. Zhao, G. Han, Z. Liu, C. Liu, C. Zhang, B. Liu, C. Jiang, R. Liu, T. Zhao, M. Y. Han and Z. Zhang, J. Am. Chem. Soc., 2016, 138, 3769–3778 CrossRef CAS PubMed.
- X. Yang, Y. Guo and R. M. Strongin, Angew. Chem., Int. Ed., 2011, 50, 10690–10693 CrossRef CAS PubMed.
- J. Liu, Y. Q. Sun, Y. Huo, H. Zhang, L. Wang, P. Zhang, D. Song, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 574–577 CrossRef CAS PubMed.
- X.-F. Yang, Q. Huang, Y. Zhong, Z. Li, H. Li, M. Lowry, J. O. Escobedo and R. M. Strongin, Chem. Sci., 2014, 5, 2177–2183 RSC.
- X. Dai, Z. Y. Wang, Z. F. Du, J. Cui, J. Y. Miao and B. X. Zhao, Anal. Chim. Acta, 2015, 900, 103–110 CrossRef CAS PubMed.
- L.-Y. Niu, Q.-Q. Yang, H.-R. Zheng, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, RSC Adv., 2015, 5, 3959–3964 RSC.
- H. Li, W. Peng, W. Feng, Y. Wang, G. Chen, S. Wang, S. Li, K. Wang and J. Zhang, Chem. Commun., 2016, 52, 4628–4631 RSC.
- Y. Li, W. Liu, P. Zhang, H. Zhang, J. Wu, J. Ge and P. Wang, Biosens. Bioelectron., 2017, 90, 117–124 CrossRef CAS PubMed.
- S. Y. Li, L. H. Liu, H. Cheng, B. Li, W. X. Qiu and X. Z. Zhang, Chem. Commun., 2015, 51, 14520–14523 RSC.
- T. Zimmermann, J. Marrison, K. Hogg and P. O'Toole, Methods Mol. Biol., 2014, 1075, 129–148 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |