
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective cross-dehydrogenative C–O coupling of N-hydroxy compounds with pyrazolones. Introduction of the diacetyliminoxyl radical into the practice of organic synthesis†
Igor B.
Krylov
 a,
Stanislav A.
Paveliev
a,
Boris N.
Shelimov
a,
Boris V.
Lokshin
b,
Irina A.
Garbuzova
b,
Viktor A.
Tafeenko
c,
Vladimir V.
Chernyshev
cd,
Alexander S.
Budnikov
ae,
Gennady I.
Nikishin
a and
Alexander O.
Terent'ev
*a
a,
Stanislav A.
Paveliev
a,
Boris N.
Shelimov
a,
Boris V.
Lokshin
b,
Irina A.
Garbuzova
b,
Viktor A.
Tafeenko
c,
Vladimir V.
Chernyshev
cd,
Alexander S.
Budnikov
ae,
Gennady I.
Nikishin
a and
Alexander O.
Terent'ev
*a
aN. D. Zelinsky Institute of Organic Chemistry of the Russian Academy of Sciences, 47 Leninsky prosp., Moscow 119991, Russian Federation. E-mail: alterex@yandex.ru
bA. N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, 28 Vavilova St., Moscow 119991, Russian Federation
cDepartment of Chemistry, M.V. Lomonosov Moscow State University, 1-3 Leninskie Gory, Moscow 119991, Russian Federation
dA. N. Frumkin Institute of Physical Chemistry and Electrochemistry RAS, 31 Leninsky prospect, Moscow 119071, Russian Federation
eHigher Chemical College of the Russian Academy of Sciences, Mendeleev University of Chemical Technology of Russia, 9 Miusskaya sq., Moscow 125047, Russian Federation
First published on 18th July 2017
Abstract
Oxidative C–O coupling of pyrazolones with N-hydroxy compounds of different classes (N-hydroxyphthalimide, N-hydroxybenzotriazole, oximes) was achieved; both one-electron oxidants (Fe(ClO4)3, (NH4)2Ce(NO3)6) and two-electron oxidants (PhI(OAc)2, Pb(OAc)4) are applicable, and the yields reach 91%. Apparently, the coupling proceeds via the formation of N-oxyl radicals from N-hydroxy compounds. One of the N-oxyl intermediates, the diacetyliminoxyl radical, was found to be exclusively stable in solution in spite of being sterically unhindered; it was isolated from an oxidant and used as a new reagent for the synthesis and mechanism study. The products of C–O coupling of pyrazolones with N-hydroxyphthalimide can be easily transformed into aminooxy compounds, valuable substances for combinatorial chemistry.
Introduction
The development of C–C and C–heteroatom cross-dehydrogenative coupling (CDC) methods is one of the major trends in modern organic synthesis and green chemistry. Such methods avoid prefunctionalization of coupling partners (with -Hal, -OTf, -SnBu3, -B(OH)2, and other groups) and thus afford high atom and step economy (Scheme 1).1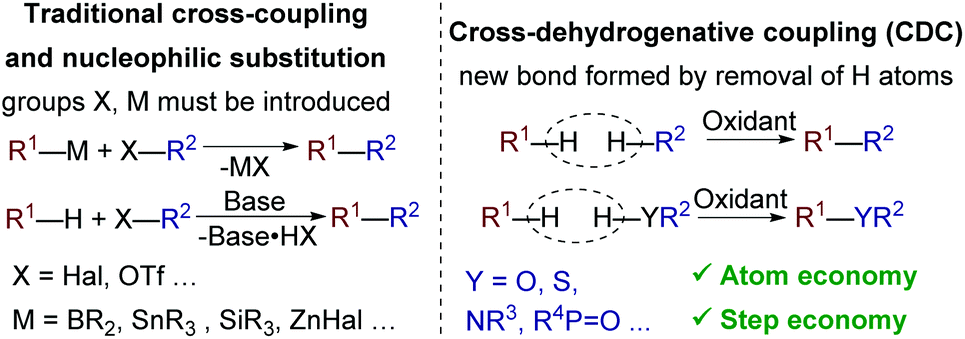 | ||
| Scheme 1 Traditional and cross-dehydrogenative coupling. | ||
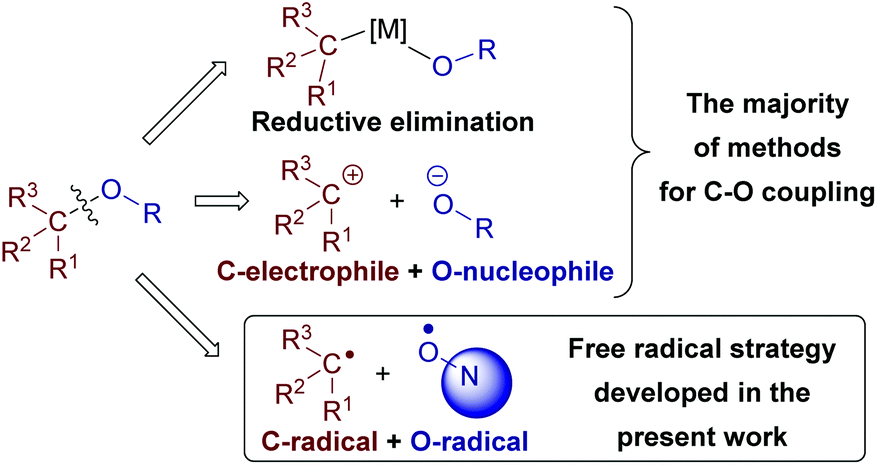
C–O bonds are abundant in natural and synthetic organic compounds, which makes the development of C–O cross-dehydrogenative coupling (C–O CDC) desirable. Nevertheless, C–O CDC remains one of the most challenging types of oxidative couplings1c–f due to the ease of side oxidation processes. Usually a new C–O bond between two molecules is formed via reductive elimination in a metal catalyzed process or as a result of the reaction of an O-nucleophile with a C-electrophile (Scheme 2).1c O-Reagents are frequently used in excess amounts to maintain the selectivity, which limits the scope of O-reagents to simple molecules. To overcome the mentioned limitations and open new coupling possibilities, we focused our attention on O-radicals as intermediates for C–O bond formation. N-Oxyl radicals derived from the N-hydroxy compounds proved to be useful for intramolecular cyclizations,2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond functionalization,3 oxidation,4 C–O CDC with alkylarenes,5a,b β-dicarbonyl compounds,5c,d and aldehydes.5e,f
C bond functionalization,3 oxidation,4 C–O CDC with alkylarenes,5a,b β-dicarbonyl compounds,5c,d and aldehydes.5e,f
 | ||
| Scheme 2 Strategies for C–O coupling. | ||
Nevertheless, structural diversity of C-reagents for the coupling and N-oxyl intermediates remains limited. In the present study we demonstrated the applicability of N-oxyl radicals for oxidative coupling with heterocyclic compounds. Pyrazolones were chosen as heterocycle representatives because they are both challenging substrates for radical coupling due to the easiness of their oxidation and oxidative dimerization6 and important compounds for medicinal chemistry.
Pyrazolin-5-ones and pyrazolidine-3,5-diones are known as anti-inflammatory drugs (Chart 1), neuroprotectants and antioxidants (Edaravone), antiviral,7 antitumor,8 fungicidal and bactericidal9 compounds, HNO donors,10 agonists of farnesoid X receptor,11a AT1 angiotensin II receptor antagonists,11b and Dyrk1A11c and UDP-N-acetylenolpyruvyl glucosamine reductase11d inhibitors.
 | ||
| Chart 1 Examples of bioactive compounds and drugs with pyrazolone moiety. | ||
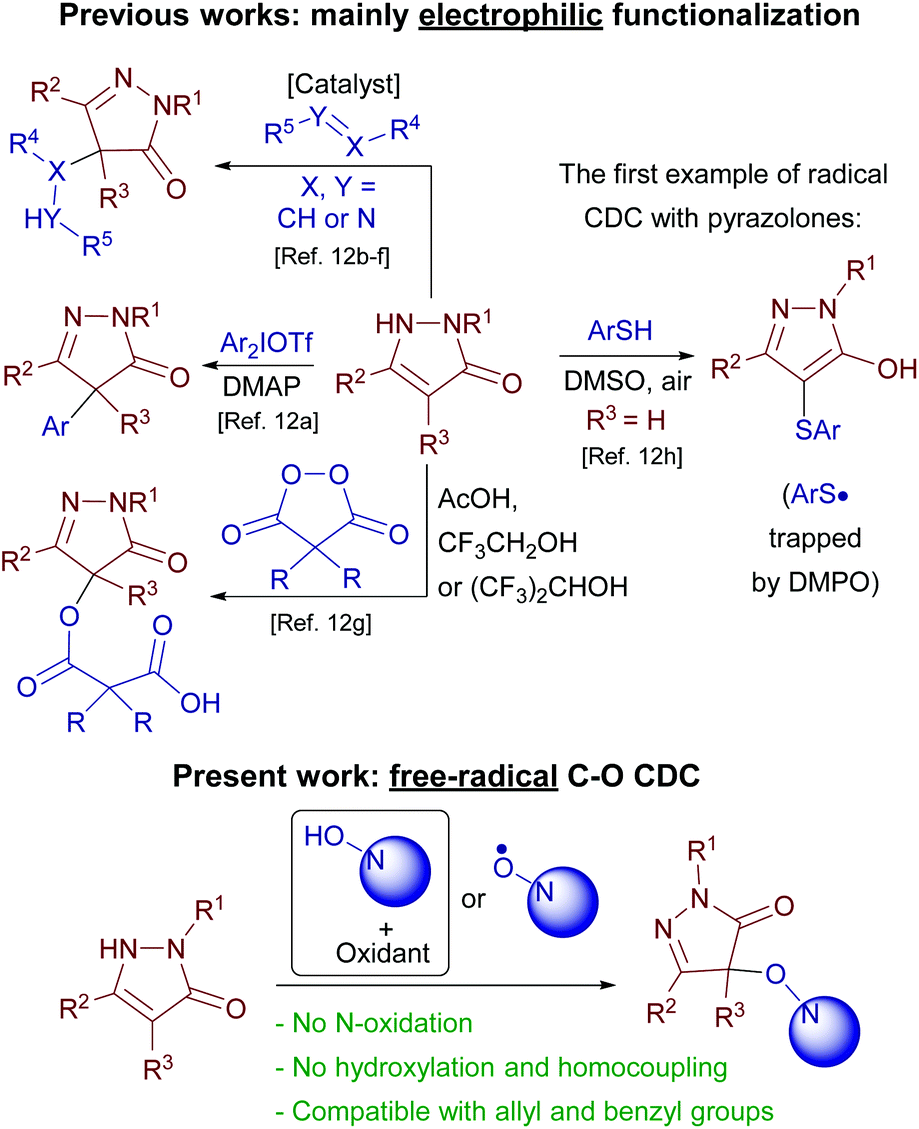
Methods for pyrazolone functionalization have been intensively developed in the last few years, but almost all of them are conceptually based on the same principle, namely, electrophilic attack on position 4 of the heterocycle (Scheme 3).
 | ||
| Scheme 3 Methods for oxidative functionalization of pyrazolones. | ||
Diaryliodonium salts,12a nitroalkenes,12b 4-oxo-4-arylbutenoates,12c alkynones,12d azodicarboxylates,12e isatin-derived N-Boc ketimines12f and diacyl peroxides12g were used as electrophiles. A rare example of free-radical oxidative C–S coupling of pyrazolones with thiophenols was reported recently (Scheme 3).12h In the present study free-radical oxidative C–O coupling of pyrazolones with N-hydroxy compounds is reported (Scheme 3). Typical problems for O-centered radicals, harsh generation conditions and low selectivity, were successfully circumvented. A substantial insight into the nature of a free-radical coupling mechanism was achieved by the discovery of a new free-radical reagent, the diacetyliminoxyl radical, which previously was known as the only plausible intermediate.5d
Results and discussion
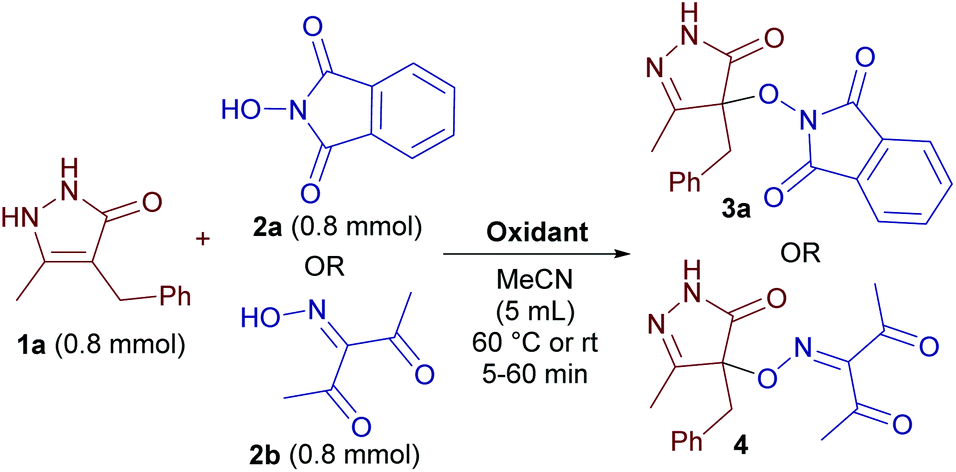
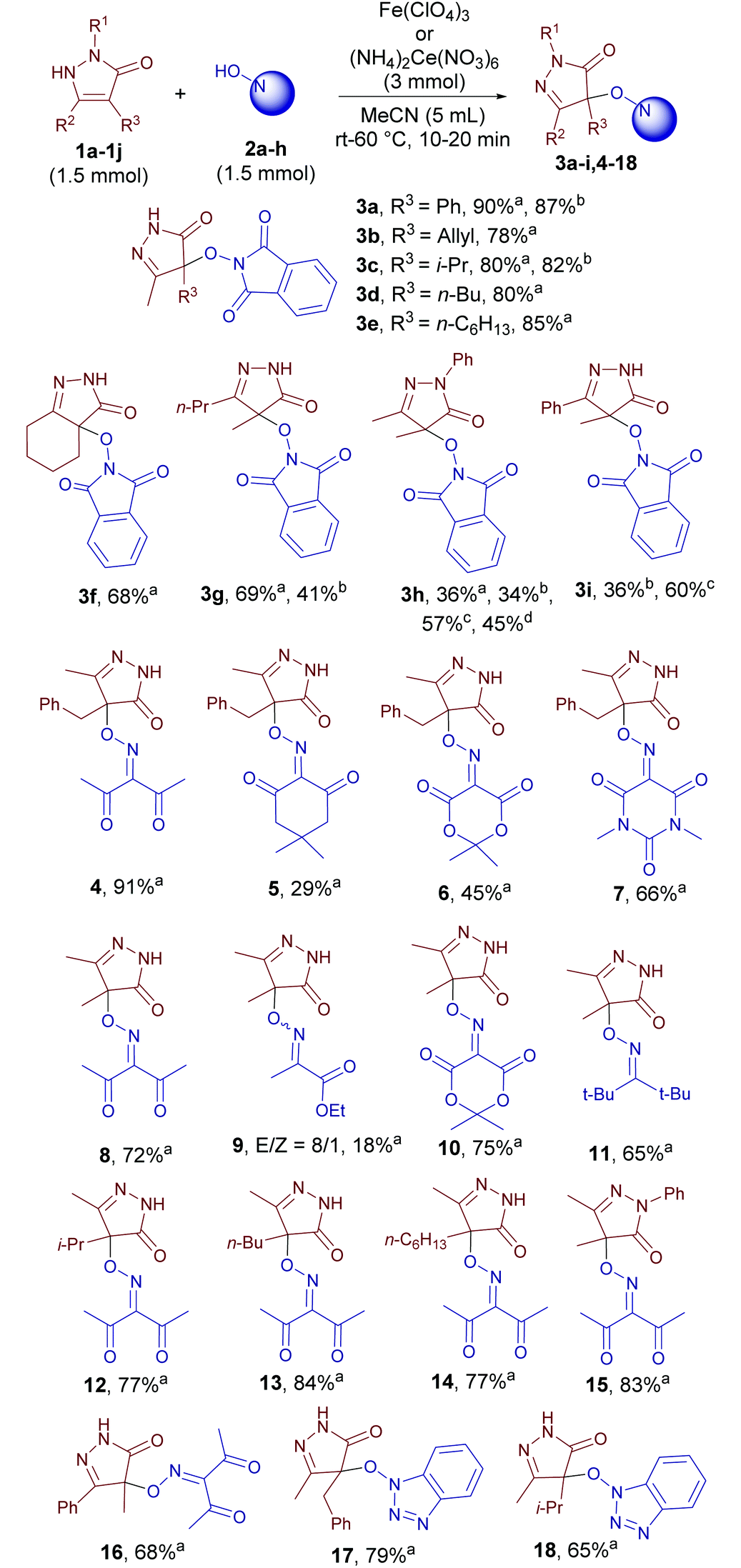
With 4-benzyl-3-methylpyrazolin-5-one 1a, N-hydroxyphthalimide (NHPI) 2a and 3-(hydroxyimino)-2,4-pentanedione 2b as the model substrates, the influence of reaction parameters on the yield of C–O coupling products 3a and 4 was studied (Table 1).|
|
||||
|---|---|---|---|---|
| Run | Oxidant (mol/mol of 1a) | Time (min) | T (°C) | Yield (%) |
| a AcOH was used as the solvent. | ||||
| Oxidative C–O coupling of 1a with NHPI 2a | ||||
| 1 | Fe(ClO4)3·nH2O (2) | 10 | 60 | 90 |
| 2 | Fe(ClO4)3·nH2O (2) | 20 | rt | 72 |
| 3 | Fe(NO3)3·9H2O (2) | 20 | 60 | <5 |
| 4 | FeCl3 (2) | 20 | 60 | 15 |
| 5 | (NH4)2Ce(NO3)6 (2) | 20 | rt | 87 |
| 6 | Pb(OAc)4 (1) | 20 | 60 | 84 |
| 7 | PhI(OAc)2 (1) | 20 | 60 | 69 |
| 8 | Cu(ClO4)2·6H2O (2) | 20 | 60 | 24 |
| 9 | Mn(OAc)3·2H2O (2)a | 20 | 60 | 9 |
| 10 | KMnO4 (0.4)a | 20 | 60 | 35 |
| Oxidative C–O coupling of 1a with oxime 2b | ||||
| 11 | Fe(ClO4)3·nH2O (2) | 10 | 60 | 91 |
| 12 | Fe(ClO4)3·nH2O (2) | 20 | rt | 90 |
| 13 | Fe(ClO4)3·nH2O (2) | 5 | rt | 69 |
| 14 | KMnO4 (0.4)a | 10 | 60 | 85 |
| 15 | Mn(OAc)3·2H2O (2)a | 10 | 60 | 52 |
| 16 | Mn(OAc)3·2H2O (2) | 60 | 60 | 20 |
| 17 | Cu(ClO4)2·6H2O (2) | 10 | 60 | 36 |
| 18 | (NH4)2Ce(NO3)6 (2) | 10 | 60 | 24 |
| 19 | Pb(OAc)4 (1) | 10 | 60 | 43 |
| 20 | PhI(OAc)2 (1) | 10 | 60 | 30 |
In contrast to the previously reported coupling of NHPI with β-dicarbonyl compounds,5c the reaction with pyrazolones proceeds under the action of either single-electron oxidants (Fe(ClO4)3, (NH4)2Ce(NO3)6, runs 1, 2, and 5) or two-electron oxidants (Pb(OAc)4, PhI(OAc)2, runs 6 and 7). The highest yield was obtained with Fe(ClO4)3 (run 1, 90%), whereas iron(III) chloride and nitrate were inefficient (entries 3 and 4). Low yields were observed with Cu(ClO4)2 and manganese based oxidants (runs 8–10).
When oxime 2b was used instead of NHPI 2a, a different order of oxidant efficacy was observed (runs 11–20), Fe(ClO4)3 being still the best. In the case of (NH4)2Ce(NO3)6, the low yield of 4 can be attributed to the instability of iminoxyl radicals derived from oxime 2b in the presence of (NH4)2Ce(NO3)6.5d
With the optimized conditions in hand we tested the scope of the discovered coupling (Table 2). Under universal reaction conditions (Fe(ClO4)3 as the oxidant, 60 °C, 10 min) pyrazolin-5-ones 1 reacted smoothly with N-hydroxy compounds of different classes: NHPI (products 3a–3i), oximes (products 4–16), and N-hydroxybenzotriazole (products 17–18). Lower yields were obtained in the reaction of NHPI with pyrazolones containing a phenyl substituent (products 3h and 3i). We proposed that these pyrazolones are oxidized faster than NHPI with the formation of side products. Indeed, when the reagent addition order was changed and NHPI was mixed with an oxidant to generate N-oxyl radicals before the addition of pyrazolones, the yields of products 3h and 3i substantially increased (Table 2, yields with notes c and d).
| a Fe(ClO4)3 was added to a stirred mixture of pyrazolone and N-hydroxy compound at 60 °C, reaction time was 10 min b (NH4)2Ce(NO3)6 was added to a stirred mixture of pyrazolone and NHPI at room temperature, reaction time was 20 min c Mixing order changed: pyrazolone 1h–i was added portion wise to the stirred mixture of NHPI and (NH4)2Ce(NO3)6 in MeCN at room temperature. d Mixing order changed: pyrazolone 1h was added portion wise to the stirred mixture of NHPI and Fe(ClO4)3 in MeCN at 60 °C. |
|---|
|
In the row of N-hydroxy compounds the yield depends on the stability of the corresponding N-oxyl radicals. The lowest yield was obtained with the oxime of ethyl pyruvate (18%, product 9).
Pyrazolidine-3,5-dione 19, known as the anti-inflammatory drug phenylbutazone, reacts with NOH-compounds 2 analogously to pyrazolin-5-ones 1 (Scheme 4).
 | ||
| Scheme 4 The oxidative C–O coupling of pyrazolidine-3,5-dione 19 with N-hydroxy compounds 2a, b, and h. | ||
A plausible mechanism of the oxidative coupling of pyrazolones with N-hydroxy compounds is depicted in Scheme 5. N-Oxyl radicals are generated from N-hydroxy compounds under the action of an oxidant. Then two sequences are possible: the attack of an N-oxyl radical on pyrazolone (A) followed by oxidation or oxidation of pyrazolone (B) followed by the addition of the radical.
 | ||
| Scheme 5 Possible pathways of the oxidative C–O coupling of pyrazolones with N-hydroxy compounds. | ||
The formation of N-oxyl radicals from NHPI under the action of used oxidants was confirmed by EPR spectroscopy (Scheme 6 and ESI†). The formation of iminoxyl radicals from oxime 2b under analogous conditions was reported earlier.5d
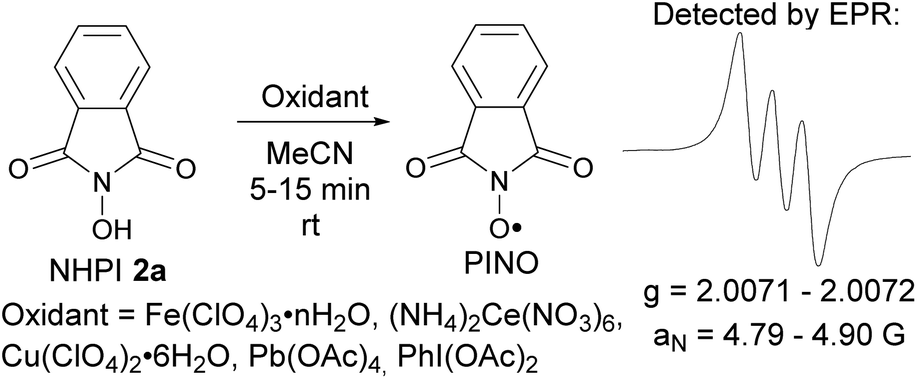 | ||
| Scheme 6 Generation of phthalimide-N-oxyl radicals (PINO) from NHPI. | ||
Diacetyliminoxyl free radical
The detection of a free radical under the reaction conditions does not prove its participation in the process and does not reveal its exact role. It is desirable to directly observe the “individual” reactivity of radicals in the absence of other reagents, such as oxidants used for their generation, which is usually impossible due to the high reactivity of free radicals, including sterically unhindered N-oxyl radicals with acceptor groups. To solve this problem, a method for the synthesis of diacetyliminoxyl radical 21,13 a plausible intermediate, was developed (Scheme 7).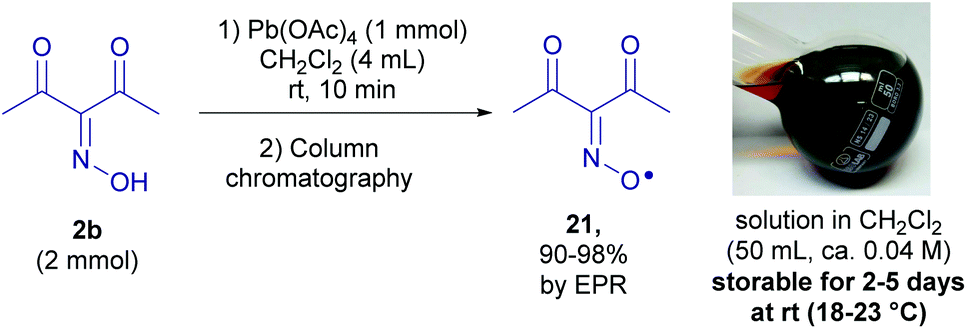 | ||
| Scheme 7 Synthesis of diacetyliminoxyl radical 21. | ||
Oxidation of 2b with Pb(OAc)4 gave rise to oxime radical 21 with almost quantitative yield based on EPR (see the ESI†). Radical 21 turned out to be surprisingly stable despite being sterically unhindered; it tolerated column chromatography on silica gel and the resulting dark red solution of 21 in CH2Cl2 (ca. 0.04 M) was stored at room temperature for 2–5 days without a significant decomposition detectable by EPR or FTIR spectroscopy. As far as we know it is record stability for the unhindered oxime radical that was not reported previously.13
Oxime radical 21 reacted with pyrazolones 1a, c, i, and h giving C–O coupling products 4, 12, 15, and 16, respectively, and oxime 2b (Scheme 8). Apparently, one equivalent of 21 formed the product and another one played the role of the oxidant. The yields are close to that obtained with in situ generation of iminoxyl radicals using Fe(ClO4)3 (see Table 2). These results are convincing evidence in favor of the mechanism depicted in Scheme 5.
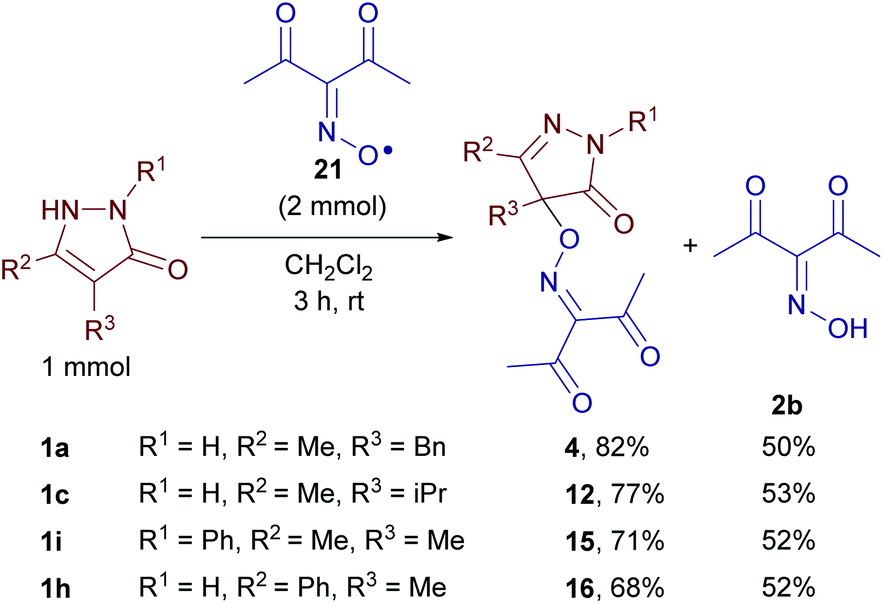 | ||
| Scheme 8 The reaction of diacetyliminoxyl radical 21 with pyrazolin-5-ones. | ||
It should be noted that the structure of the synthesized radical 21 has little in common with known stable N-oxyl radicals (Chart 2). The majority of the stable N-oxyl radicals are amine-N-oxyl radicals. Only some representatives of this extensive type of radicals are depicted. This class includes both cyclic structures (TEMPO,14 AZADO,15 ABNO,16 IAPNO,17 nitronyl nitroxides18) and acyclic structures (Fremy's salt,19 bis(trifluoromethyl)nitroxide,20 TIPNO and others21). These N-oxyl radicals found wide use in various fields22a,b including oxidation processes,15b,16a,17,22a–d “living” radical polymerization,21b,c,22a spin-labeling22e and synthesis of magnetic materials.18b,c,22f Stable oxime radicals (imine-N-oxyl type) are very rare and highly hindered, examples are di-tert-butyliminoxyl radical and di(1-adamantyl)iminoxyl radical (Chart 2).23 An important feature of radical 21 is its synthetic accessibility: the parent oxime can be prepared in one simple step from acetylacetone, NaNO2 and H2SO4.24
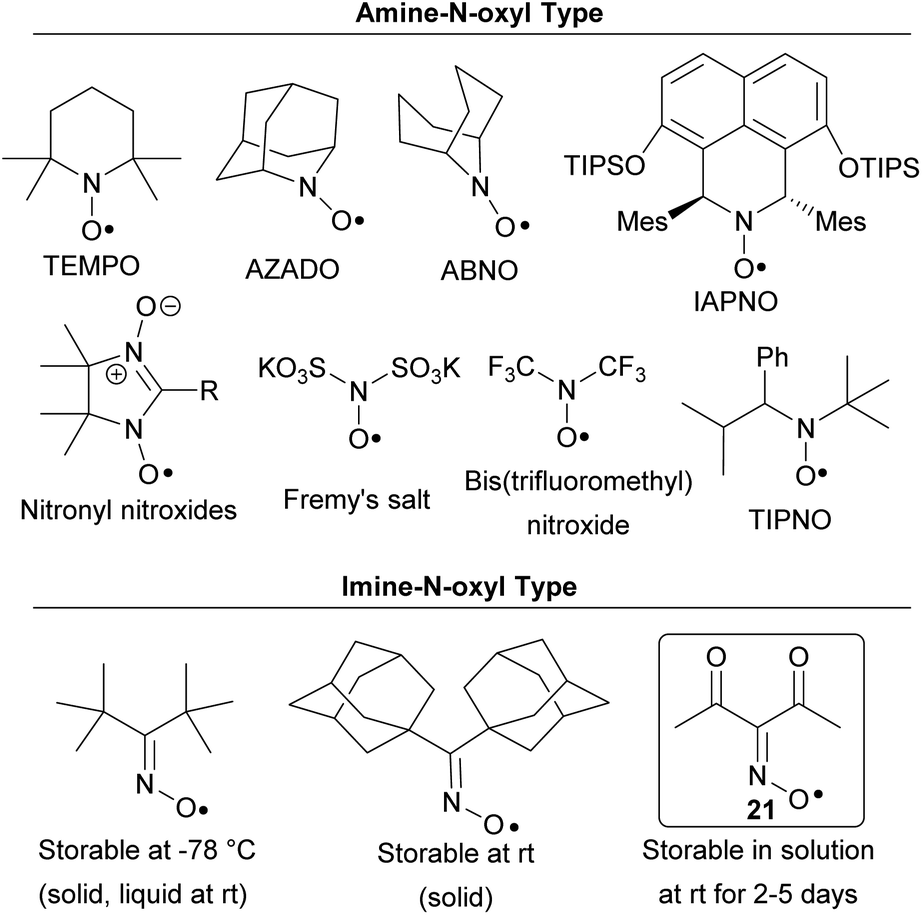 | ||
| Chart 2 Examples of known stable N-oxyl radicals and synthesized radical 21. | ||
Synthetic application of the coupling products
Finally, the synthetic utility of some of the synthesized products was tested (Scheme 9). Novel O-substituted hydroxylamines 24a, c, d, and f were synthesized from products 3a, c, d, and f without the need for chromatographic purification. In the case of the product 3a one-pot deprotection/condensation sequence was demonstrated to obtain oxime ether 25.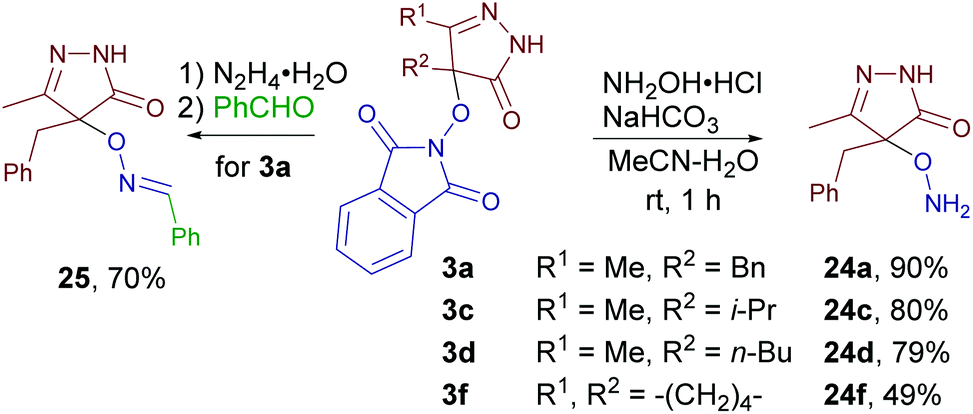 | ||
| Scheme 9 The synthetic utility of the synthesized oxidative C–O coupling products. | ||
Conclusions
In conclusion, a new type of oxidative C–O coupling was realized, the method was applied to a wide range of N-hydroxy compounds and pyrazolones. N-Oxyl radicals are identified as key intermediates that selectively add to position 4 of the pyrazolone ring. The first method for the synthesis of the diacetyliminoxyl radical in solution was proposed. This radical can be used as an easily available reagent and a model radical for mechanistic studies.Experimental
Iron(III) perchlorate hydrate reagent grade (Alfa Aesar, anhydrous basis purity ca. 65%), Fe(NO3)3·9H2O 99+%, FeCl3 98% anhydrous, (NH4)2Ce(NO3)6 99%, Pb(OAc)4 95%, PhI(OAc)2 98%, Cu(ClO4)2·6H2O 98%, Mn(OAc)3·2H2O 95%, KMnO4 99%, N-hydroxyphthalimide 98%, N-hydroxybenzotriazole hydrate 98% (11–26% H2O), 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) 98%, benzaldehyde 98+%, N2H4·H2O (64% hydrazine), 4-butyl-1,2-diphenyl-3,5-pyrazolidinedione (phenylbutazone) 99+%, NH2OH·HCl 99%, and NaHCO3 99% were used as is from commercial sources. CH2Cl2 was distilled prior to use. MeCN and EtOAc were distilled over P2O5. Glacial acetic acid was used as is from commercial sources. Preparation of the starting pyrazolones and oximes is described in the ESI.†General reaction conditions for oxidative C–O coupling of 1a with NHPI 2a (Table 1)
To a mixture of 4-benzyl-3-methylpyrazolin-5-one 1a (150 mg, 0.797 mmol), N-hydroxyphthalimide 2a (130 mg, 0.797 mmol) and solvent (5 mL) stirred at given temperature, an oxidant (50.4–874 mg, 0.4–2 mol/mol of 1a) was added for 5–20 seconds; stirring was continued at the same temperature for 20 minThe reaction mixture was cooled to room temperature, diluted with CH2Cl2 (10 mL) and water (20 mL) and shaken. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL), and all organic extracts were combined. In the case of an intensive color of extract indicative of the presence of metal complexes, it was additionally washed with an aqueous solution of Na2S2O4 (200 mg in 20 mL of water). Organic extract was washed with water (2 × 20 mL), dried over Na2SO4, and rotary evaporated under water-jet vacuum. C–O coupling product 3a was isolated by column chromatography on silica gel using the EtOAc/CH2Cl2 eluent; the volume part of EtOAc was gradually increased from 0 to 20%.
General reaction conditions for oxidative C–O coupling of 1a with oxime 2b (Table 1)
To a mixture of 4-benzyl-3-methylpyrazolin-5-one 1a (150 mg, 0.797 mmol), 3-(hydroxyimino)-2,4-pentanedione 2b (103 mg, 0.797 mmol) and solvent (5 mL) stirred at 60 °C, an oxidant (50.4–874 mg, 0.4–2 mol/mol 1a) was added for 5–20 seconds; stirring was continued at 60 °C for 10 min. The coupling product 4 was isolated as described above for 3a.General reaction conditions for Table 2 and Scheme 4
General procedure a (all experiments in Scheme 4 and experiments in Table 2 with note a): to a mixture of pyrazolone (1.5 mmol), N-hydroxy compound (1.5 mmol) and MeCN (5 mL) stirred at 60 °C, Fe(ClO4)4·nH2O (3 mmol) was added; stirring was continued for 10 min at 60 °C.General procedure b (experiments in Table 2 with note b): to a mixture of pyrazolone (1.5 mmol), N-hydroxy compound (1.5 mmol) and MeCN (5 mL) stirred at room temperature, (NH4)2Ce(NO3)6 (3 mmol) was added; stirring was continued for 20 min at room temperature.
General procedure c (experiments in Table 2 with note c): to a mixture of N-hydroxyphthalimide (1.5 mmol) and MeCN (5 mL) stirred at room temperature, (NH4)2Ce(NO3)6 (3 mmol) was added for 5–10 seconds, stirring was continued for 4 min, and then pyrazolone (1.5 mmol) was added portion wise for 7–10 min; after the complete addition of pyrazolone, stirring was continued for 5 min at room temperature.
General procedure d (experiments in Table 2 with note d): to a mixture of N-hydroxyphthalimide (1.5 mmol) and MeCN (5 mL) stirred at 60 °C, Fe(ClO4)4·nH2O (3 mmol) was added for 5–10 seconds, and then pyrazolone (1.5 mmol) was added portion wise for 1 min; stirring was continued for 5 min at 60 °C.
The products 3a–i, 4–18, 20a, b, and h were isolated as described for 3a in experiment in Table 1.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3–CN–O), 1.89 (s, 3H, 3–CN–NH), 1.40 (s, 3H, 3–C–O–N), 1.21 (t, J = 7.1 Hz, 3H, 3–CH2); minor Z isomer δ: 11.02 (bs, 1H, NH), 4.37–4.24 (m, 2H, OCH2), 1.95 (s, 3H, 3–CN–O), 1.88 (s, 3H, 3–CN–NH), 1.27 (t, J = 7.1 Hz, 3H, 3–CH2), 1.26 (s, 3H, 3–C–O–N). 13C NMR (75.47 MHz, DMSO-d6): major E isomer δ: 174.6 (HN–O), 162.4 (O–O), 159.2 (N–NH), 152.0 (N–O), 82.6 (–O–N), 61.5 (OH2), 17.4, 13.9, 12.5, 11.6 (CH3); minor Z isomer δ: 17.1, 16.2, 12.4. IR (KBr) ν(cm−1): 3222, 1717, 1432, 1374, 1329, 1308, 1204, 1178, 1151, 1124, 1006, 932, 863, 754, 673, 570. Elemental analysis calcd (%) for C10H15N3O4: C, 49.79; H, 6.27; N, 17.42. Found: C, 49.71; H, 6.25; N, 17.40.
3–CN–O), 1.89 (s, 3H, 3–CN–NH), 1.40 (s, 3H, 3–C–O–N), 1.21 (t, J = 7.1 Hz, 3H, 3–CH2); minor Z isomer δ: 11.02 (bs, 1H, NH), 4.37–4.24 (m, 2H, OCH2), 1.95 (s, 3H, 3–CN–O), 1.88 (s, 3H, 3–CN–NH), 1.27 (t, J = 7.1 Hz, 3H, 3–CH2), 1.26 (s, 3H, 3–C–O–N). 13C NMR (75.47 MHz, DMSO-d6): major E isomer δ: 174.6 (HN–O), 162.4 (O–O), 159.2 (N–NH), 152.0 (N–O), 82.6 (–O–N), 61.5 (OH2), 17.4, 13.9, 12.5, 11.6 (CH3); minor Z isomer δ: 17.1, 16.2, 12.4. IR (KBr) ν(cm−1): 3222, 1717, 1432, 1374, 1329, 1308, 1204, 1178, 1151, 1124, 1006, 932, 863, 754, 673, 570. Elemental analysis calcd (%) for C10H15N3O4: C, 49.79; H, 6.27; N, 17.42. Found: C, 49.71; H, 6.25; N, 17.40.
Generation of phthalimide-N-oxyl radical from N-hydroxyphthalimide (experimental details for Scheme 6)
An oxidant (quantities are given below) was added to a 0.002 M solution of N-hydroxyphthalimide in MeCN (20 mL) at room temperature (18–23 °C), and the mixture was shaken until the complete dissolution of the oxidant; the EPR spectrum of the solution was registered 5–15 min after mixing. Following oxidants were used: (NH4)2Ce(NO3)6 (21.9 mg, 0.04 mmol), Fe(ClO4)3·nH2O (ca. 35% H2O, 21.8 mg, 0.04 mmol), Cu(ClO4)2·6H2O (14.8 mg, 0.04 mmol), Pb(OAc)4 (8.9 mg, 0.02 mmol), PhI(OAc)2 (6.4 mg, 0.02 mmol). The triplet EPR spectrum characteristic of the phthalimide-N-oxyl radical was observed in all cases (see the ESI† for details).Generation and characterization of diacetyliminoxyl radical 21 (experimental details for Scheme 7)
All experiments with diacetyliminoxyl radical 21 were conducted at room temperature (18–23 °C).Diacetyl oxime 2b (258 mg, 2 mmol) was dissolved in CH2Cl2 (4 mL) at 18–23 °C, and then Pb(OAc)4 (467 mg, 1 mmol) was added with vigorous stirring. The mixture immediately turned dark red, stirring was continued for 10 min, and then the mixture was transferred to the chromatographic column, prepared by suspending the silica gel (12 g) in excess of CH2Cl2. CH2Cl2 was used as an eluent, and the fraction corresponding to the dark-red spot was collected, so that the volume of the fraction was 50 mL. The obtained solution of diacetyliminoxyl radical 21 in CH2Cl2 (50 mL, C ≈ 0.04 mmol mL−1 according to quantitative EPR measurement, see the ESI†) was used for experiments described below. The stability and purity of 21 in solution was confirmed by EPR, FT-IR spectroscopy and ICP-MS (to confirm separation from the lead compounds); for spectral data and discussion, see the ESI.†
Reactions of diacetyliminoxyl radical 21 with pyrazolin-5-ones 1a, 1c, 1h, and 1i (experimental details for Scheme 8)
To a stirred solution of diacetyliminoxyl radical 21 in CH2Cl2 (50 mL, ca. 0.04 mol L−1, ≈2 mmol, prepared as described above), pyrazolin-5-one (1 mmol; 1a: 188.2 mg; 1c: 140.2 mg; 1h: 188.2 mg; 1i: 174.2 mg) was added at room temperature (18–23 °C). Stirring was continued for 3 h, and gradual dissolution of pyrazolin-5-one and the decrease in the intensity of the red color of the solution were observed. The mixture was rotary evaporated under water-jet vacuum, an aliquot (20 mg) of the residue was analyzed by 1H and 13C NMR, and the rest was transferred to a silica gel chromatographic column and eluted with EtOAc/CH2Cl2 (EtOAc content was increased gradually from 0 to 30 vol%) to isolate the reaction products. In the case of pyrazolin-5-one 1h, an additional experiment was performed with a reaction time of 24 h (instead of 3 h), and the same product yields were observed.The 1H and 13C NMR spectra of the reaction mixtures of diacetyliminoxyl radical 21 with pyrazolones 1a, c, h, and i are given in the ESI.† Signals were assigned to the coupling products (4, 12, 15 and 16) and oxime 2b by comparing the spectra of reaction mixtures with the spectra of individual compounds. No significant impurity signals were observed.
Experimental details for Scheme 9
N bond was performed with the aid of 2D NMR experiments, HMBC and NOESY (see the ESI†). Mp = 120–121 °C. 1H NMR (300.13 MHz, CDCl3) δ: 10.88 (bs, 1H, NH), 8.46 (s, 1H, HCN), 7.58–7.50 (m, 2H, ArH), 7.47–7.37 (m, 3H, ArH), 7.33–7.17 (m, 5H, ArH), 3.21 (d, J = 12.9 Hz, 1H, CH2), 3.09 (d, J = 12.9 Hz, 1H, CH2), 1.97 (s, 3H, CH3). 13C NMR (75.47 MHz, CDCl3) δ: 174.2 (CONH), 158.1 (CN–N), 151.8 (CN–O), 132.3, 130.9, 130.7, 129.9, 128.9, 128.2, 127.2 (Ph), 86.0 (C–O–N), 37.2 (CH2), 13.7 (CH3). IR (KBr) ν(cm−1): 3417, 3221, 3065, 1732, 1702, 1455, 1377, 1161, 1076, 1016, 921, 758, 749, 697, 628, 570, 519, 510. Elemental analysis calcd (%) for C18H17N3O2: C, 70.34; H, 5.58; N, 13.67. Found: C, 70.31; H, 5.62; N, 13.59.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Grant 15-29-05828). Authors would like to thank Dr Roman Novikov for 2D NMR experiments and the ZIOC-Interlab-Analytik Jena International Analytical Center for ICP-MS analysis by Analytik Jena PlasmaQuant MS Elite.Notes and references
- (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) I. B. Krylov, V. A. Vil’ and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92 CrossRef PubMed; (d) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (e) R. Samant, K. Match and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769 CrossRef; (f) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed.
- (a) B. C. Giglio and E. J. Alexanian, Org. Lett., 2014, 16, 4304 CrossRef CAS PubMed; (b) R. K. Quinn, V. A. Schmidt and E. J. Alexanian, Chem. Sci., 2013, 4, 4030 RSC; (c) F. Chen, X.-L. Yang, Z.-W. Wu and B. Han, J. Org. Chem., 2016, 81, 3042 CrossRef CAS PubMed; (d) Y.-Y. Liu, X.-H. Yang, J. Yang, R.-J. Song and J.-H. Li, Chem. Commun., 2014, 50, 6906 RSC; (e) X.-X. Peng, Y.-J. Deng, X.-L. Yang, L. Zhang, W. Yu and B. Han, Org. Lett., 2014, 16, 4650 CrossRef CAS PubMed; (f) B. Han, X.-L. Yang, R. Fang, W. Yu, C. Wang, X.-Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8816 CrossRef CAS PubMed; (g) X. Zhu, Y.-F. Wang, W. Ren, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214 CrossRef CAS PubMed; (h) B. C. Lemercier and J. G. Pierce, Org. Lett., 2015, 17, 4542 CrossRef CAS PubMed.
- (a) X.-F. Xia, S.-L. Zhu, Z. Gu, H. Wang, W. Li, X. Liu and Y.-M. Liang, J. Org. Chem., 2015, 80, 5572 CrossRef CAS PubMed; (b) R. Bag, D. Sar and T. Punniyamurthy, Org. Biomol. Chem., 2016, 14, 3246 RSC; (c) X.-F. Xia, Z. Gu, W. Liu, H. Wang, Y. Xia, H. Gao, X. Liu and Y.-M. Liang, J. Org. Chem., 2015, 80, 290 CrossRef CAS PubMed; (d) J. Zhang and Y. Tang, Adv. Synth. Catal., 2016, 358, 752 CrossRef CAS; (e) X.-F. Xia, S.-L. Zhu, Y.-N. Niu, D. Zhang, X. Liu and H. Wang, Tetrahedron, 2016, 72, 3068 CrossRef CAS; (f) B. C. Giglio, V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 13320 CrossRef CAS PubMed; (g) R. Bag, D. Sar and T. Punniyamurthy, Org. Lett., 2015, 17, 2010 CrossRef CAS PubMed.
- (a) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800 CrossRef CAS PubMed; (b) L. Melone and C. Punta, Beilstein J. Org. Chem., 2013, 9, 1296 CrossRef CAS PubMed; (c) Y. Amaoka, S. Kamijo, T. Hoshikawa and M. Inoue, J. Org. Chem., 2012, 77, 9959 CrossRef CAS PubMed; (d) R. Lin, F. Chen and N. Jiao, Org. Lett., 2012, 14, 4158 CrossRef CAS PubMed.
- (a) J. M. Lee, E. J. Park, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2008, 130, 7824 CrossRef CAS PubMed; (b) A. O. Terent'ev, I. B. Krylov, M. Y. Sharipov, Z. M. Kazanskaya and G. I. Nikishin, Tetrahedron, 2012, 68, 10263 CrossRef; (c) A. O. Terent'ev, I. B. Krylov, V. P. Timofeev, Z. A. Starikova, V. M. Merkulova, A. I. Ilovaisky and G. I. Nikishin, Adv. Synth. Catal., 2013, 355, 2375 CrossRef; (d) I. B. Krylov, A. O. Terent'ev, V. P. Timofeev, B. N. Shelimov, R. A. Novikov, V. M. Merkulova and G. I. Nikishin, Adv. Synth. Catal., 2014, 356, 2266 CrossRef CAS; (e) M. Dinda, C. Bose, T. Ghosh and S. Maity, RSC Adv., 2015, 5, 44928 RSC; (f) H. Yao, Y. Tang and K. Yamamoto, Tetrahedron Lett., 2012, 53, 5094 CrossRef CAS.
- (a) X. Sheng, J. Zhang, H. Yang and G. Jiang, Org. Lett., 2017, 19, 2618 CrossRef CAS PubMed; (b) S. Veibel, Acta Chem. Scand., 1972, 26, 3685 CrossRef CAS.
- (a) V. Hadi, Y.-H. Koh, T. W. Sanchez, D. Barrios, N. Neamati and K. W. Jung, Bioorg. Med. Chem. Lett., 2010, 20, 6854 CrossRef CAS PubMed; (b) R. Ramajayam, K.-P. Tan, H.-G. Liu and P.-H. Liang, Bioorg. Med. Chem. Lett., 2010, 7849 CrossRef CAS PubMed; (c) R. Srinivasan, B. Narayana, B. K. Sarojini, V. Bhanuprakash, C. G. D. Raj and P. S. Nayak, Lett. Drug Des. Discovery, 2016, 13, 149 CrossRef CAS.
- (a) X.-Y. Zhang, Y.-F. Gu, T. Chen, D.-X. Yang, X.-X. Wang, B.-L. Jiang, K.-P. Shao, W. Zhao, C. Wang, J.-W. Wang, Q.-R. Zhang and H.-M. Liu, MedChemComm, 2015, 6, 1781 RSC; (b) S. Wu, Y. Li, G. Xu, S. Chen, Y. Zhang, N. Liu, G. Dong, C. Miao, H. Su, W. Zhang and C. Sheng, Eur. J. Med. Chem., 2016, 115, 141–147 CrossRef CAS PubMed.
- A. Dandia and A. K. Jain, J. Heterocycl. Chem., 2013, 50, 104 CrossRef CAS.
- (a) S. Nourian, R. P. Lesko, D. A. Guthrie and J. P. Toscano, Tetrahedron, 2016, 72, 6037 CrossRef CAS; (b) S. Nourian, Z. A. Zilber and J. P. Toscano, J. Org. Chem., 2016, 81, 9138 CrossRef CAS PubMed; (c) D. A. Guthrie, N. Y. Kim, M. A. Siegler, C. D. Moore and J. P. Toscano, J. Am. Chem. Soc., 2012, 134, 1962 CrossRef CAS PubMed.
- (a) G. Deng, W. Li, J. Shen, H. Jiang, K. Chen and H. Liu, Bioorg. Med. Chem. Lett., 2008, 18, 5497 CrossRef CAS PubMed; (b) B. Le Bourdonnec, E. Meulon, S. Yous, J.-F. Goossens, R. Houssin and J.-P. Hénichart, J. Med. Chem., 2000, 43, 2685 CrossRef CAS PubMed; (c) K. A. Koo, N. D. Kim, Y. S. Chon, M.-S. Jung, B.-J. Lee, J. H. Kim and W.-J. Song, Bioorg. Med. Chem. Lett., 2009, 19, 2324 CrossRef CAS PubMed; (d) M. Gilbert, A. Failli, J. Shumsky, Y. Yang, A. Severin, G. Singh, W. Hu, D. Keeney, P. J. Petersen and A. H. Katz, J. Med. Chem., 2006, 49, 6027 CrossRef PubMed.
- (a) S. Mao, X. Geng, Y. Yang, X. Qian, S. Wu, J. Han and L. Wang, RSC Adv., 2015, 5, 36390 RSC; (b) Y.-H. Liao, W.-B. Chen, Z.-J. Wu, X.-L. Du, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Adv. Synth. Catal., 2010, 352, 827 CrossRef CAS; (c) Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928 CrossRef CAS PubMed; (d) Z. Wang, Z. Chen, S. Bai, W. Li, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2012, 51, 2776 CrossRef CAS PubMed; (e) Z. Yang, Z. Wang, S. Bai, X. Liu, L. Lin and X. Feng, Org. Lett., 2011, 13, 596 CrossRef CAS PubMed; (f) X. Bao, B. Wang, L. Cui, G. Zhu, Y. He, J. Qu and Y. Song, Org. Lett., 2015, 17, 5168 CrossRef CAS PubMed; (g) A. O. Terent'ev, V. A. Vil’, E. S. Gorlov, O. N. Rusina, A. A. Korlyukov, G. I. Nikishin and W. Adam, ChemistrySelect, 2017, 2, 3334 CrossRef; (h) P. Sun, D. Yang, W. Wei, L. Jiang, Y. Wang, T. Dai and H. Wang, Org. Chem. Front., 2017, 4, 1367 RSC.
- (a) C. Lagercrantz and K. Torssell, Ark. Kemi, 1967, 29, 203 Search PubMed; (b) S. Hoffman, A. Jezierski and B. Jezowska-Trzebiatowska, Bull. Pol. Acad. Sci., Chem., 1986, 34, 251 CAS.
- O. L. Lebelev and S. N. Kazarnovskii, Zh. Obshch. Khim., 1960, 30, 1631 Search PubMed.
- (a) R.-M. Dupeyre and A. Rassat, Tetrahedron Lett., 1975, 16, 1839 CrossRef; (b) M. Shibuya, M. Tomizawa, I. Suzuki and Y. Iwabuchi, J. Am. Chem. Soc., 2006, 128, 8412 CrossRef CAS PubMed.
- (a) M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612 CrossRef CAS; (b) R.-M. Dupeyre and A. Rassat, J. Am. Chem. Soc., 1966, 88, 3180 CrossRef CAS; (c) M. Shibuya, M. Tomizawa, Y. Sasano and Y. Iwabuchi, J. Org. Chem., 2009, 74, 4619 CrossRef CAS PubMed.
- S. Bar, J. N. Kumar, M. Amar, H. Toledo, R. J. Batrice and A. M. Szpilman, ChemCatChem, 2015, 7, 1129 CrossRef CAS.
- (a) J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 1078 CrossRef CAS; (b) C. Hirel, K. E. Vostrikova, J. Pecaut, V. I. Ovcharenko and P. Rey, Chem. – Eur. J., 2001, 7, 2007 CrossRef CAS PubMed; (c) E. V. Tretyakov and V. I. Ovcharenko, Russ. Chem. Rev., 2009, 78, 971 CrossRef CAS.
- H. Zimmer, D. C. Lankin and S. W. Horgan, Chem. Rev., 1971, 71, 229 CrossRef CAS.
- W. D. Blackley and R. R. Reinhard, J. Am. Chem. Soc., 1965, 87, 802 CrossRef CAS.
- (a) V. A. Reznikov, I. A. Gulorov, Yu. V. Gatilov, T. V. Rybalova and L. B. Volodarsky, Russ. Chem. Bull., 1996, 45, 384 CrossRef; (b) D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904 CrossRef CAS; (c) S. Grimaldi, J.-P. Finet, F. Le Moigne, A. Zeghdaoui, P. Tordo, D. Benoit, M. Fontanille and Y. Gnanou, Macromolecules, 2000, 33, 1141 CrossRef CAS.
- (a) E. G. Bagryanskaya and S. R. A. Marque, Chem. Rev., 2014, 114, 5011 CrossRef CAS PubMed; (b) I. B. Krylov, M. O. Kompanets, K. V. Novikova, I. O. Opeida, O. V. Kushch, B. N. Shelimov, G. I. Nikishin, D. O. Levitsky and A. O. Terent'ev, J. Phys. Chem. A, 2016, 120, 68 CrossRef CAS PubMed; (c) J. M. Bobbitt, C. BrüCkner and N. Merbouh, in Organic Reactions, ed. S. E. Denmark, et al., John Wiley and Sons, 2010, ch. 2, vol. 74, DOI:10.1002/0471264180.or074.02; (d) R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245 CrossRef CAS; (e) E. S. Babaylova, A. A. Malygin, A. A. Lomzov, D. V. Pyshnyi, M. Yulikov, G. Jeschke, O. A. Krumkacheva, M. V. Fedin, G. G. Karpova and E. G. Bagryanskaya, Nucleic Acids Res., 2016, 44, 7935 CrossRef CAS PubMed; (f) M. V. Fedin, S. L. Veber, E. G. Bagryanskaya and V. I. Ovcharenko, Coord. Chem. Rev., 2015, 289–290, 341 CrossRef CAS.
- (a) J. L. Brokenshire, G. D. Mendenhall and K. U. Ingold, J. Am. Chem. Soc., 1971, 93, 5278 CrossRef CAS; (b) K. U. Ingold, in Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, John Wiley & Sons, Ltd, Chichester, UK, 2010, ch. 6, pp. 231–244. DOI:10.1002/9780470666975.ch6.
- P. A. Nikitina, L. G. Kuz'mina, V. P. Perevalov and I. I. Tkach, Tetrahedron, 2013, 69, 3249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR, ESR, FTIR spectral data (PDF), compound 3h (CIF). CCDC 1411623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00447h |
| This journal is © the Partner Organisations 2017 |