DOI:
10.1039/C6QM00375C
(Research Article)
Mater. Chem. Front., 2017,
1, 1514-1519
Two-dimensional metal–organic frameworks for selective separation of CO2/CH4 and CO2/N2†
Received
21st December 2016
, Accepted 12th January 2017
First published on 13th January 2017
Abstract
Two two-dimensional (2D) microporous metal–organic frameworks, [Zn2(TMTA)(DMF)2]·NO3·2H2O·3DMF (1, DMF = N,N-dimethylformamide) and [Zn3(TETA)2(DMF)4]·3DMF (2), were synthesized via a solvothermal reaction, where H3TMTA and H3TETA are 4,4′,4′′-(2,4,6-trimethylbenzene-1,3,5-triyl)tribenzoic acid and 4,4′,4′′-(2,4,6-triethylbenzene-1,3,5-triyl)tribenzoic acid, respectively. Crystallographic studies, including powder and single crystal X-ray diffraction (XRD) analyses, reveal that compound 1 features a 2D microporous framework composed of paddle-wheel Zn2(RCOO)3 clusters and TMTA3− ligands, while the 2D double-layered structure 2 is assembled by discrete trinuclear Zn3(RCOO)6 clusters and TETA3− ligands. Both activated frameworks of 1 and 2 show permanent porosities with Brunauer–Emmett–Teller (BET) surface areas of 788 and 421 m2 g−1, respectively. Desolvated 1 exhibits a considerable carbon dioxide adsorption of 94.2 cm3 g−1 at 1 bar and 273 K, which is higher than that of 42.0 cm3 g−1 for 2. Furthermore, both materials show selective carbon dioxide adsorption over methane and nitrogen at ambient temperature.
Introduction
Carbon dioxide capture and separation have gained considerable attention due to environmental, energy and health concerns.1,2 Among different technologies, physical sorption using adsorbents such as metal–organic frameworks (MOFs, also known as porous coordination polymers) is more energy-efficient.3,4 Considering different CO2 capture applications such as biogas upgrading, flue gas treatment, trace CO2 removal in confined spaces, and air capture,5 the ideal sorbents are those that can be easily modified to show tunable sorption performance.
As a new generation of porous materials, MOFs are unparalleled for their uniform pore structure, high porosity, designable structures and tunable pore size.6–11 Over the past two decades, MOFs have attracted much attention owing to their potential in gas storage,12–14 separation,15–18 sensing,19–26 catalysis,27–30 and biomedicine.31,32 By virtue of rational selection of metal ions and design of organic ligands, the pore volume and pore size can be optimized for different host–guest interactions, resulting in customizable performance.33–35 In fact, MOFs have been demonstrated as good candidates for CO2 capture.3,36,37 MOFs with open metal sites (OMSs),38–40 organic amines,41–44 specific functional groups (–NH2, –OH, etc.)45,46 and narrow pore size47,48 are effective for CO2 sorption over a wide concentration range. Benefitting from these designable and modifiable MOFs, significant progress has been made in the realm of selective CO2 capture.43,48–50 However, there is still a big gap between these discoveries and making MOFs practical for industrial CO2 capture. Also, some key aspects should be taken into account during the design of MOFs for CO2 capture, including adsorption capacity and kinetics, selectivity, regeneration conditions, material stability, impurity tolerance, and economic cost.5
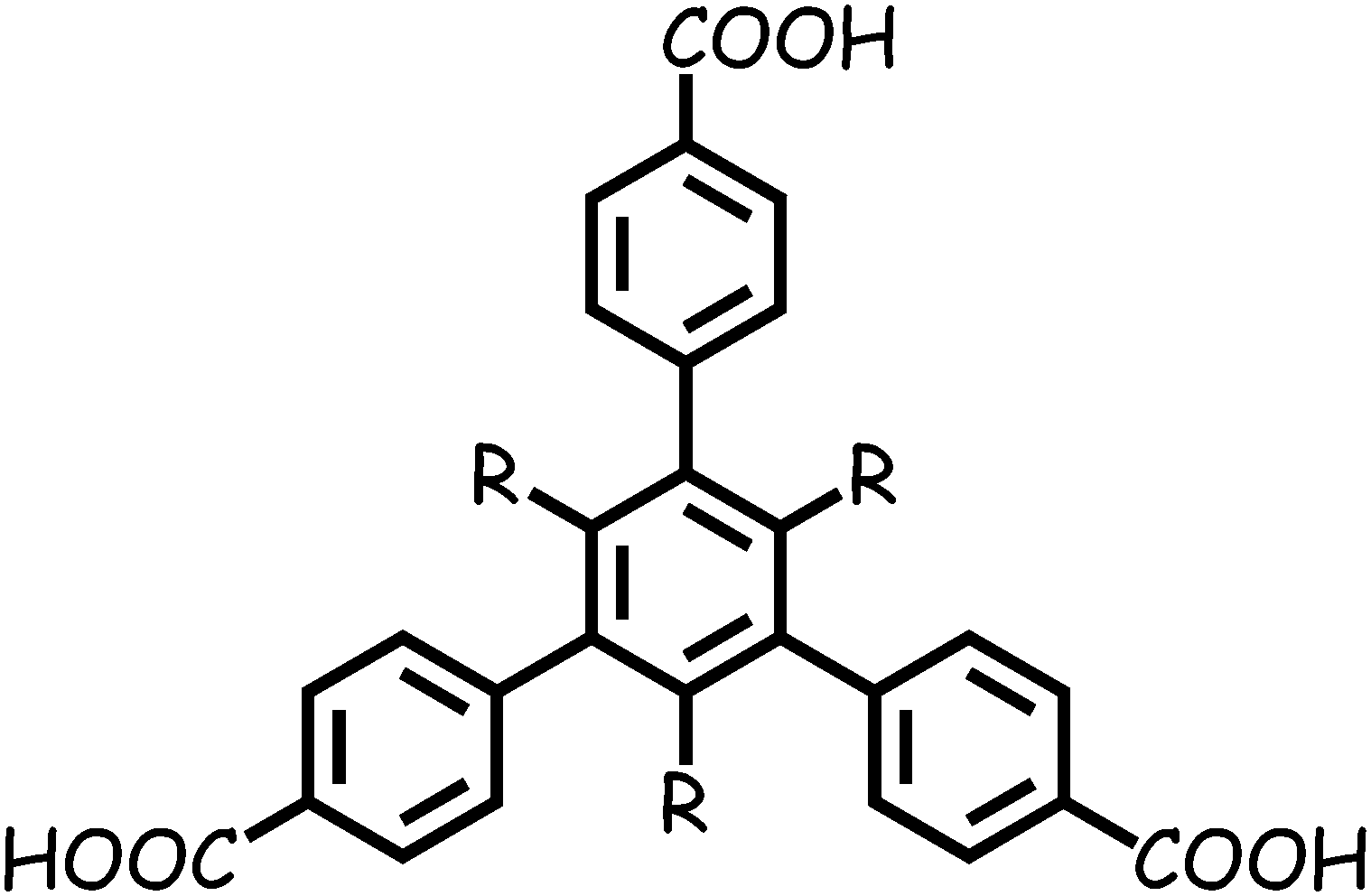
Herein, based on two substituted 1,3,5-tris(4-carboxyphenyl)-benzene ligands TMTA3− and TETA3− (Scheme 1), we synthesized two microporous MOFs, [Zn2(TMTA)(DMF)2]·NO3·2H2O·3DMF (1) and [Zn3(TETA)2(DMF)4]·3DMF (2), both of which are 2D layered structures with permanent porosity, showing porosity-dependent carbon dioxide uptake and selective carbon dioxide adsorption over methane and nitrogen.
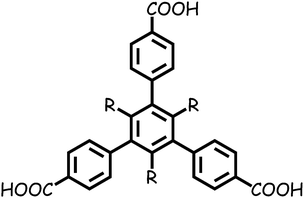 |
| | Scheme 1 Schematic molecular structure of H3TMTA (R = Me) and H3TETA (R = Et). | |
Experimental section
Materials and methods
All reagents and solvents were used directly as received from the chemical suppliers. The organic linker of H3TMTA was prepared according to the literature procedure.51 Also, H3TETA was prepared using the same method as H3TMTA (Scheme 2). Thermogravimetric analysis (TGA) was carried out using a Shimadzu TGA-50 analyzer under a nitrogen atmosphere at a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) patterns were collected using a Rigaku Ultima IV diffractometer operated at 40 kV and 44 mA with a scan rate of 1.0 deg min−1. 1H NMR spectra were measured using a Varian INOVA 500 MHz spectrometer in CDCl3 and DMSO-d6 at room temperature. The spectra were referenced internally using the residual solvent resonance (CDCl3, δ = 7.26 and DMSO-d6, δ = 2.50) relative to SiMe4. An internal standard of δ = 39.5 ppm was used for the 13C NMR spectrum, recorded in DMSO-d6 solution.
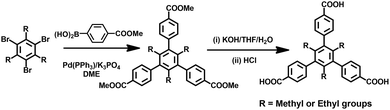 |
| | Scheme 2 Schematic synthesis of the organic linkers H3TMTA and H3TETA. | |
Synthesis of trimethyl-4,4′,4′′-(2,4,6-triethylbenzene-1,3,5-triyl)tribenzoate (Me3TETA)
A mixture of (3-(methoxy-carbonyl)phenyl)boronic acid (5.0 g, 26.6 mmol), 1,3,5-tribromo-2,4,6-triethylbenzene (3.0 g, 7.5 mmol), potassium carbonate (6.9 g, 50.0 mmol), and palladium tetrakis(triphenylphosphine) (0.25 g, 0.225 mmol) was added to a 250 mL three-necked flask and stirred under nitrogen for 30 min at room temperature, and then dry 1,2-dimethoxyethane (80 mL) was added. The reaction mixture was stirred at 100 °C for 3 days. After cooling to room temperature, the organic solvent was removed under vacuum, and 60 mL water was added. The solution with gray suspension was extracted with chloroform (100 mL × 3). The combined organic phase was evaporated under vacuum, and the residue was repeatedly purified by chromatography on silica using hexane/CH2Cl2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent, giving Me3TETA 2.2 g. 1H NMR (CDCl3): δ 8.10 (d, J = 5 Hz, 6H), 7.40 (d, J = 5 Hz, 6H), 3.94 (s, 9H), 2.04 (d, J = 5 Hz, 6H), 3.93 (t, J = 5 Hz, 9H). 13C NMR (DMSO-d6): 167.21, 146.05, 139.57, 138.52, 130.40, 129.38, 128.85, 52.27, 24.73, 15.25.
:1) as the eluent, giving Me3TETA 2.2 g. 1H NMR (CDCl3): δ 8.10 (d, J = 5 Hz, 6H), 7.40 (d, J = 5 Hz, 6H), 3.94 (s, 9H), 2.04 (d, J = 5 Hz, 6H), 3.93 (t, J = 5 Hz, 9H). 13C NMR (DMSO-d6): 167.21, 146.05, 139.57, 138.52, 130.40, 129.38, 128.85, 52.27, 24.73, 15.25.
Synthesis of the H3TETA ligand
A mixture of Me3TETA 2.2 g, KOH 4.0 g, and 150 mL of tetrahydrofuran/H2O (1:1) was stirred and refluxed for 48 hours. The organic solvent was removed under vacuum, and the mixture was filtered. The pH value of the filtrate was adjusted to ∼3–4 with concentrated HCl. The resulting white solid was collected by filtration, washed with water, and then dried under vacuum. Yield: 1.9 g, 92%. 1H NMR (DMSO-d6): δ 12.95 (b, 3H), 8.00 (d, J = 5 Hz, 6H), 7.43 (d, J = 5 Hz, 6H), 1.95 (dd, J = 5 Hz, 6H), 0.57 (t, J = 5 Hz, 9H). 13C NMR (DMSO-d6): 167.16, 144.78, 138.73, 138.23, 130.15, 129.46, 129.05, 30.42, 14.86.
Synthesis of 1 and 2
A mixture of Zn(NO3)2·6H2O (0.040 g, 0.3mmol) and H3L (L3− = TMTA3− or TETA3−, 0.01 mmol) was dissolved in DMF (1.5 mL) in a 20 mL vial, and tetrafluoroboric acid (0.05 mL) was added. The resultant mixture was sealed and placed in an oven at 90 °C for 48 hours. After cooling to room temperature, the colorless block-shaped crystals were collected, washed with DMF, and dried under vacuum, with yields of 80 and 84% for 1 and 2, respectively.
Single crystal X-ray crystallography measurement and refinement
Single crystals were selected and mounted in Paratone oil onto a glass fiber and were frozen under a cold nitrogen stream for X-ray diffraction. The data were collected at 98(2) K using a Rigaku AFC12/Saturn 724 CCD fitted with Mo Kα radiation (λ = 0.71073 Å). Data collection and unit cell refinement were conducted using the Crystal Clear software.52 Data processing and absorption correction were performed using Crystal Clear and ABSCOR,53 respectively, to generate minimum and maximum transmission factors. The structure of 2 was solved using direct methods and refined on F2 by full-matrix, least-squares techniques using SHELXL-97.54 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atom positions were added to their mother carbon atoms by geometry and refined by a riding model. Due to the serious disorder of DMF guests, the ‘SQUEEZE’ command was used for 2. Crystal data and details of data collection and refinements are summarized in Table S1, ESI.† CCDC 1522317 for 2 contains supplementary crystallographic data.
Gas sorption measurements
Prior to the gas sorption measurements, the as-synthesized crystals of 1 and 2 were exchanged with dry acetone six times and then evacuated at room temperature for 24 hours and then at 100 °C for another 24 hours, giving two desolvated MOFs (1a and 2a). A Micromeritics ASAP 2020 surface area analyzer was employed to determine gas adsorption isotherms. The temperature was kept at 77 K using a liquid nitrogen bath, 273 K using an ice-water bath, and 296 K using a water bath in an air-conditioned 23 °C laboratory.
IAST calculation
In order to calculate the selective sorption performance of 1a and 2a toward the separation of binary mixed gases, the fitting of the single-component CO2, CH4, and N2 adsorption isotherms was carried out based on the dual-site Langmuir–Freundlich (DSLF) model.55 The fitting parameters of the DSLF equation are displayed in Table S3, ESI.† Adsorption isotherms and gas selectivities of mixed CO2/CH4 (50%:50%) and CO2/N2 (15%:85%) at 273 and 296 K for 1a and 2a were predicted using the ideal adsorbed solution theory (IAST).56 The results are shown in Fig. 4, Fig. S8, S9 and Table S2, ESI.†
Results and discussion
Crystal structures of 1 and 2
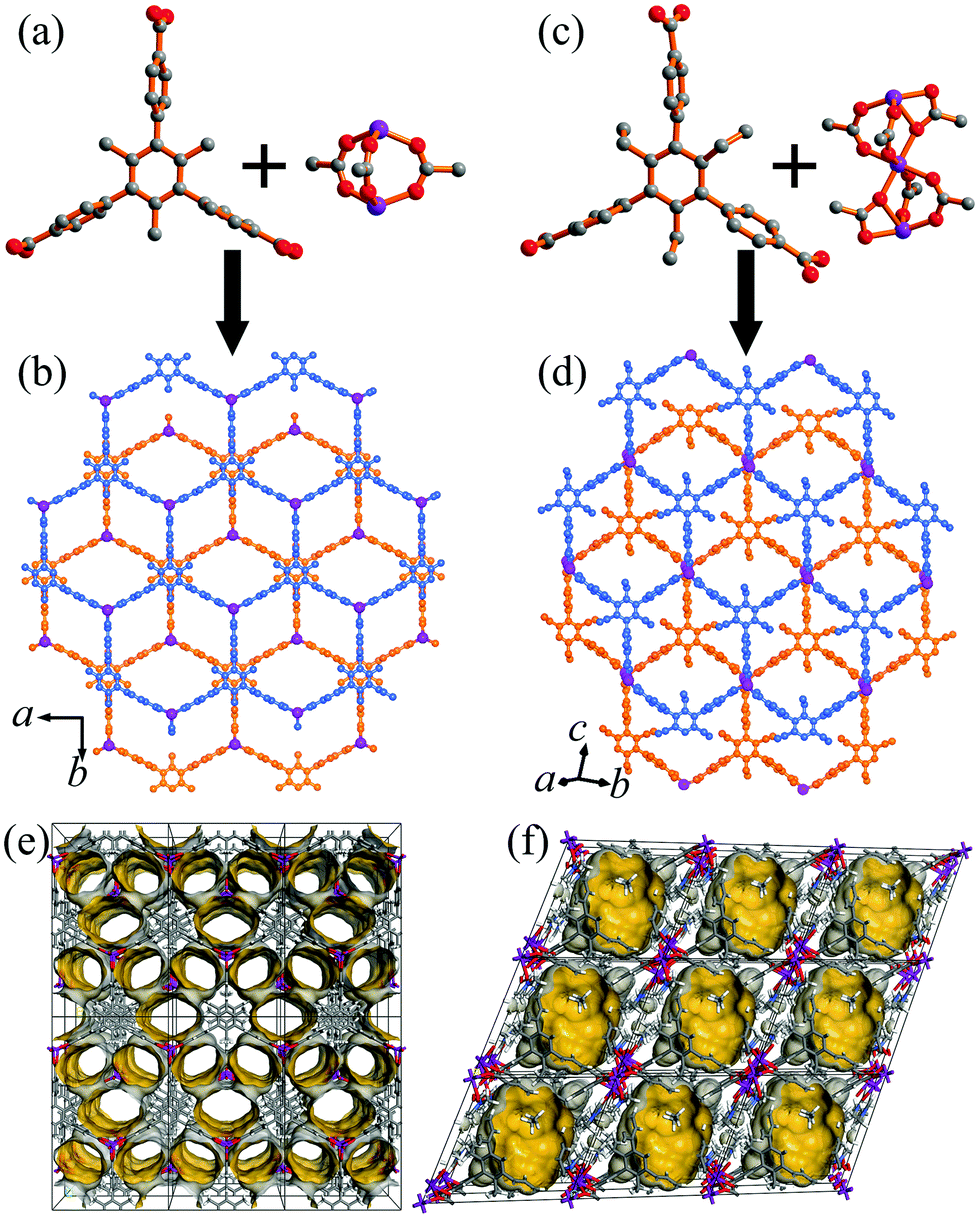
The colorless block-shaped crystals of 1 and 2 were synthesized by the solvothermal reaction of zinc nitrate hexahydrate, H3TMTA/H3TETA, and tetrafluoroboric acid in DMF solution at 90 °C for 48 hours. The crystals of 1 were not suitable for determining the structure due to the quick loss of solvent after removal from the mother liquid. On the basis of the powder X-ray diffraction (XRD) analysis of 1 (Fig. S1, ESI†), it is isostructual with the reported compound [Zn2(TMTA)(H2O)2]·NO3·6H2O·DEF (α-1).51 The structure is formed from TMTA3− linkers and rare paddle-wheel Zn2(RCOO)3 clusters, exhibiting a 2D single-layer honeycomb structure (Fig. 1a and b). The adjacent layers are further stacked into a 3D supramolecular architecture with a large pore cavity size (12.8 × 12.8 × 6.2 Å3) but a small aperture (4.5 × 7.6 Å2) (Fig. 1e). The single crystal XRD revealed that 2 crystallizes in the triclinic system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S1, ESI†). There are one and half Zn2+ ions, one TETA3− ligand, and one DMF molecule in the asymmetric unit of 2. One Zn2+ ion locates on the inverse center of 2, and trinuclear Zn3(RCOO)6 clusters are formed to coordinate with six carboxylic groups (Fig. 1c). The central Zn2+ ion is coordinated by six oxygen atoms from different carboxylic groups, and the outer zinc ions are coordinated by three oxygen atoms originating from three TETA3− ligands and two oxygen atoms of DMF molecules. The Zn3 clusters are connected by six carboxylic groups of TETA3− ligands to form a 2D double-layered framework (Fig. 1d). Comparing the two crystal structures reveals that the neighbouring layers of 1 and 2 stack in a similar yet not identical way (Fig. 1b and d). The single layers of 1 stack on top of each other with the help of a π–π interaction, while the bilayer of 2 is organized primarily by coordination interactions. The pore surface in 2 is totally different from that in 1 (Fig. 1f), while its pore cavity size (12.8 × 12.8 × 6.9 Å3) is similar to that of 1. As shown in Fig. S3 and S4 (ESI†), thermogravimetric analysis (TGA) curves display approximately 29.3 and 39.7% weight loss in the temperature range from 23 to 300 °C for 1 and 2, respectively, which are assigned to the release of two coordinated DMF molecules, two guest water molecules and three guest DMF molecules for 1 per [Zn2(TMTA)]NO3 unit. There are four coordinated DMF molecules and three guest DMF molecules for 2 in each [Zn3(TETA)2] unit. Above 300 °C, the coordination layers started to decompose. As a consequence of the above provided data, the chemical formulae for 1 and 2 were calculated to be [Zn2(TMTA)(DMF)2]·NO3·2H2O·3DMF and [Zn3(TETA)2(DMF)4]·3DMF, on the basis of the crystallographic X-ray diffraction studies and TGA. The potential void calculated using the PLATON software occupies 52.3% and 45.6% of the total volume of 1 and 2, respectively. The phase purities for the bulk materials of 1 and 2 have been confirmed by the PXRD analysis (Fig. S1 and S2, ESI†).
space group (Table S1, ESI†). There are one and half Zn2+ ions, one TETA3− ligand, and one DMF molecule in the asymmetric unit of 2. One Zn2+ ion locates on the inverse center of 2, and trinuclear Zn3(RCOO)6 clusters are formed to coordinate with six carboxylic groups (Fig. 1c). The central Zn2+ ion is coordinated by six oxygen atoms from different carboxylic groups, and the outer zinc ions are coordinated by three oxygen atoms originating from three TETA3− ligands and two oxygen atoms of DMF molecules. The Zn3 clusters are connected by six carboxylic groups of TETA3− ligands to form a 2D double-layered framework (Fig. 1d). Comparing the two crystal structures reveals that the neighbouring layers of 1 and 2 stack in a similar yet not identical way (Fig. 1b and d). The single layers of 1 stack on top of each other with the help of a π–π interaction, while the bilayer of 2 is organized primarily by coordination interactions. The pore surface in 2 is totally different from that in 1 (Fig. 1f), while its pore cavity size (12.8 × 12.8 × 6.9 Å3) is similar to that of 1. As shown in Fig. S3 and S4 (ESI†), thermogravimetric analysis (TGA) curves display approximately 29.3 and 39.7% weight loss in the temperature range from 23 to 300 °C for 1 and 2, respectively, which are assigned to the release of two coordinated DMF molecules, two guest water molecules and three guest DMF molecules for 1 per [Zn2(TMTA)]NO3 unit. There are four coordinated DMF molecules and three guest DMF molecules for 2 in each [Zn3(TETA)2] unit. Above 300 °C, the coordination layers started to decompose. As a consequence of the above provided data, the chemical formulae for 1 and 2 were calculated to be [Zn2(TMTA)(DMF)2]·NO3·2H2O·3DMF and [Zn3(TETA)2(DMF)4]·3DMF, on the basis of the crystallographic X-ray diffraction studies and TGA. The potential void calculated using the PLATON software occupies 52.3% and 45.6% of the total volume of 1 and 2, respectively. The phase purities for the bulk materials of 1 and 2 have been confirmed by the PXRD analysis (Fig. S1 and S2, ESI†).
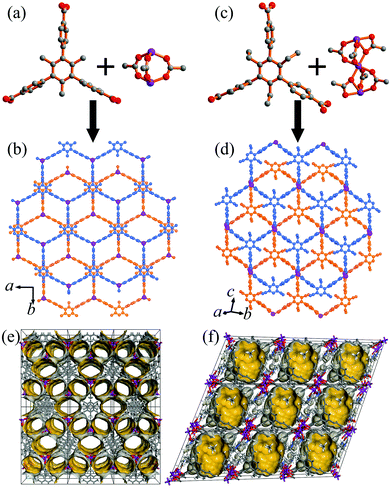 |
| | Fig. 1 Structure of 1 and 2 indicating (a and c) the organic ligands and Zn clusters, (b and d) comparison of 2D structures of 1 (b) and 2 (d), and the pore surface structures of 1 (e, along the c axis) and 2 (f, along the a axis) (carbon: gray, oxygen: red, zinc: pink, hydrogen atoms and guest molecules are omitted for clarity). | |
Establishment of permanent porosity
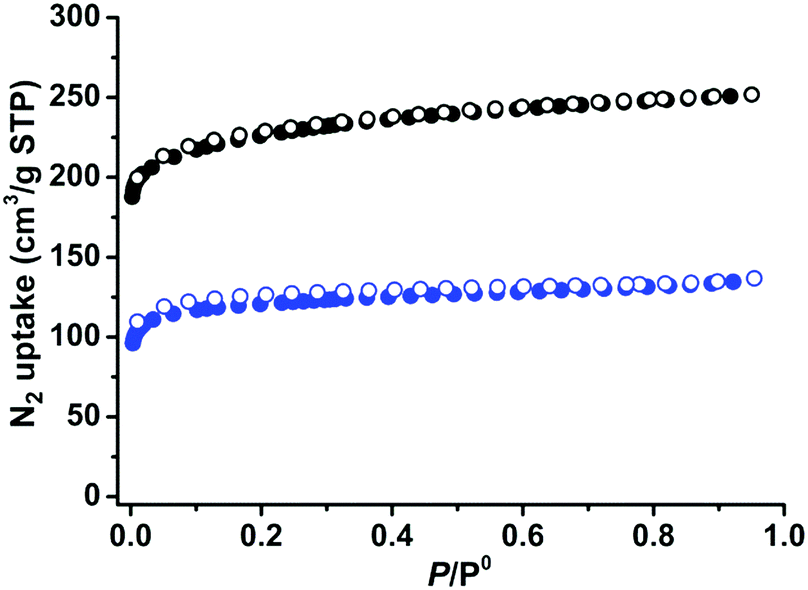
The establishment of permanent porosities is one of the important goals in MOF research. The as-synthesized crystals of 1 and 2 were solvent exchanged five times with dry acetone. The acetone-exchanged samples were degassed at 296 K for 24 hours and then at 373 K for another 24 hours, until the outgas rate was 6 μm Hg min−1, to produce activated phases 1a and 2a for the gas sorption measurements. As can be seen in Fig. S1 and S2 (ESI†), the active phases are highly crystalline. During desolvation, the structural transformations of 1 and 2 are different as indicated by their activated PXRD with different shifts. Pawley refinement of the PXRD pattern for compound 1a showed that its unit cell underwent a significant change (Fig. S5, ESI†), while 2a remained almost the same as its as-synthesized phase. This phenomenon might be attributed to different steric effects of different functional groups, or the diverse stacking modes of the two-dimensional coordination compounds.57,58 The N2 gas sorption curves of 1a and 2a at 77 K were recorded to check their porosities, Fig. 2. Both 1a and 2a show type-I N2 adsorption isotherms at 77 K, suggesting permanent microporosity. 1a and 2a show N2 gas uptake of 251 and 137 cm3 g−1, respectively, at 77 K and 1 bar. The pore volumes were calculated as 0.39 and 0.21 cm3 g−1 for 1a and 2a, respectively, smaller than the theoretical pore volumes, 0.60 cm3 g−1 for 1 and 0.50 cm3 g−1 for 2, which are typical of the flexible MOFs because of the structural contractions during activation. The Brunauer–Emmett–Teller (BET) surface areas were calculated as 788 and 421 m2 g−1, respectively.
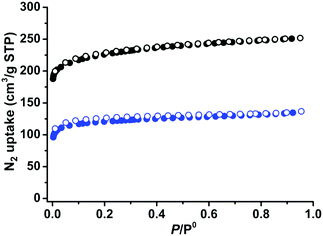 |
| | Fig. 2 N2 sorption isotherms of 1 (black) and 2 (blue) at 77 K (solid symbols: adsorption, open symbols: desorption). | |
Gas sorption and separation
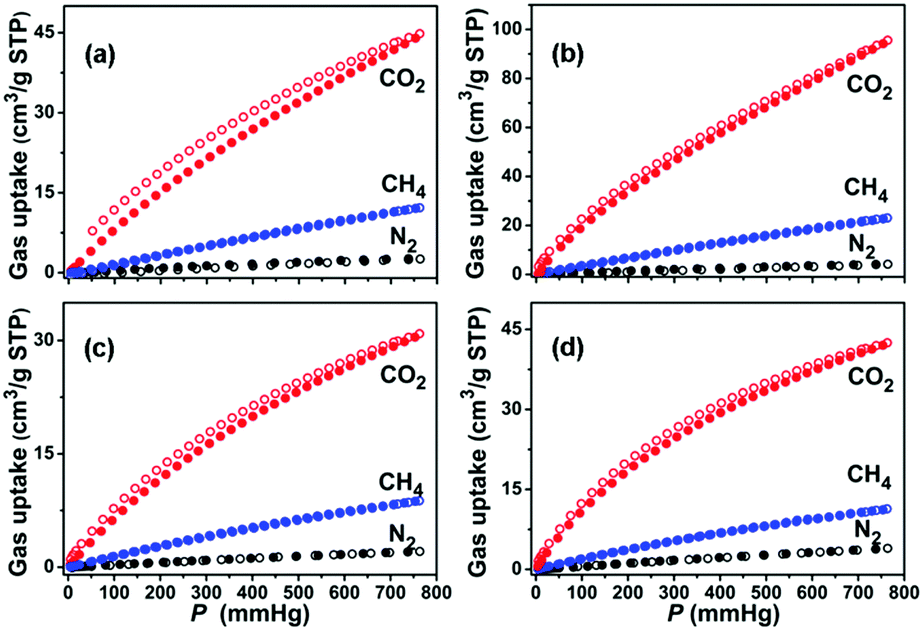
Gas storage and separation are inherent advantages of MOFs due to their unique and well-defined pore sizes and channel environments. In particular, the gas separation functionality of MOFs shows much promise for the large-scale production of ultra-pure gases as industrial materials and fuels. Microporous MOFs have been intensively studied for their capabilities in gas separation, suggesting that pore diameters of about 5.0 Å are desirable for exploiting the size effect.59 Particularly, those MOFs with large cavity size but small apertures make it possible to have both large sorption capacity and good separation performance. Given the facts that compounds 1a and 2a are highly porous, and their pore sizes are greatly affected by the functionality, we used single component CO2, CH4 and N2 gases to predict the performance of 1a and 2a in selective gas separation at different temperatures. As shown in Fig. 3, 1a shows different capacities for CO2 (44.0 cm3 g−1), CH4 (12.1 cm3 g−1), and N2 (2.7 cm3 g−1) at 1 bar and 296 K. Following a temperature decrease to 273 K, the uptake of CO2, CH4, and N2 increases to 94.2, 22.7, and 4.1 cm3 g−1, respectively. Similar to 1a, 2a also shows a temperature-dependent gas sorption behavior for these gases (at 1 bar and 273 K, CO2: 42.0 cm3 g−1, CH4: 11.2 cm3 g−1, N2: 3.8 cm3 g−1; and at 1 bar and 296 K, CO2: 30.4 cm3 g−1, CH4: 8.7 cm3 g−1, N2: 2.0 cm3 g−1). Most noticeably, the adsorption amounts for CO2 at 1 bar are several times higher than that of CH4, and 10–15 times higher than that of N2 for these two desolvated MOFs at the same temperatures, which indicates an opportunity to explore their capabilities in CO2/CH4 and CO2/N2 separation. Also, the coverage-dependent adsorption enthalpies of 1a and 2a towards CO2, CH4, and N2 were calculated based on the virial method to correlate the gas sorptions and pore structures of these two MOFs (Fig. S6 and S7, ESI†). The enthalpies of 1a towards CO2, CH4, and N2 were 28.8, 15.7, and 13.8 kJ mol−1, respectively, at zero coverage. The zero coverage enthalpies of 2a over CO2, CH4, and N2 were 20.0, 14.7, and 12.1 kJ mol−1, respectively. The enthalpies of CO2 for 1a and 2a are comparable with other microporous MOFs that have open metal sites, such as HKUST-1 (26 kJ mol−1).60 To ensure the repeatability of gas adsorption, multi-cycle sorption measurements were carried out, which showed that there was no noticeable loss in adsorption capacity after several cycles (Fig. S10, ESI†).
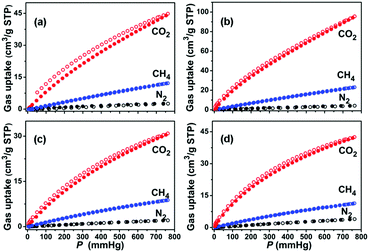 |
| | Fig. 3 Gas sorption isotherms of 1a (a and b) and 2a (c and d) at 296 and 273 K, respectively. (CO2: red, CH4: blue, N2: black, solid symbols: adsorption, open symbols: desorption). | |
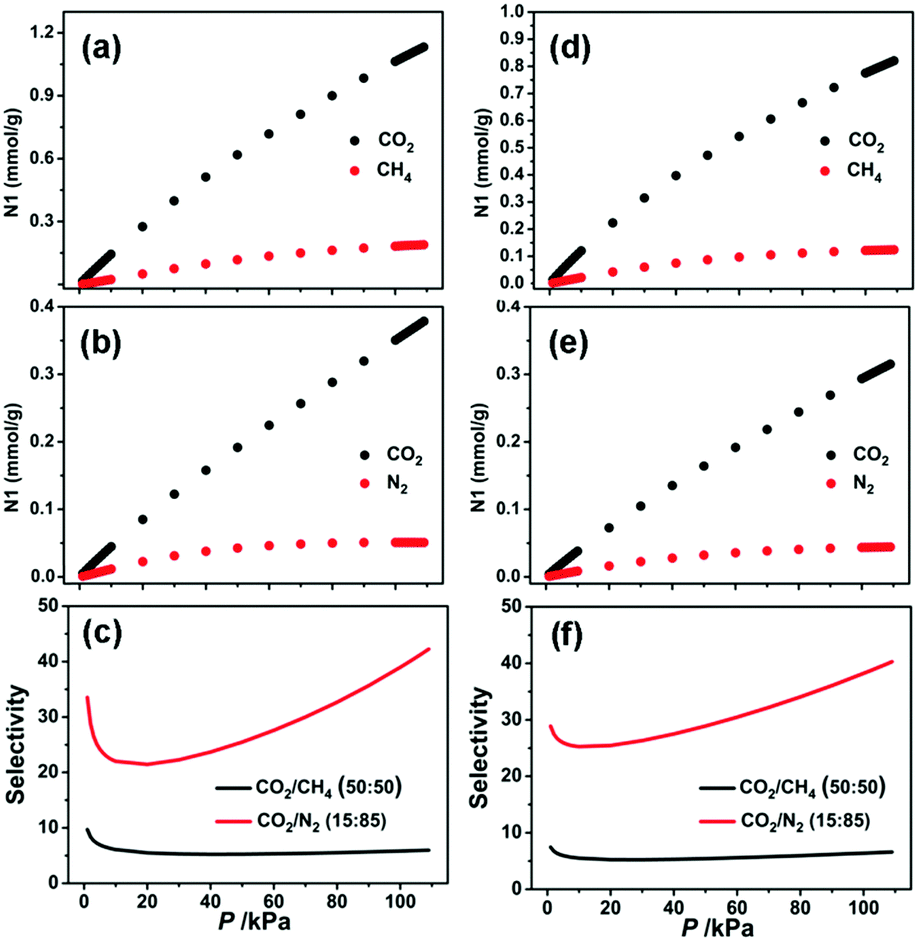
For the purpose of predicting the CO2/CH4 and CO2/N2 selectivities at different pressures and temperatures, the single component adsorption isotherms of CO2, CH4, and N2 were fitted on the basis of the dual-site Langmuir–Freundlich (DSLF) model. The obtained adsorption parameters were further used to predict the selectivity of the CO2/CH4 (50%:50%) and CO2/N2 (15%:85%) mixtures by using the popular ideal adsorbed solution theory (IAST) method.56 As shown in Fig. 4, the pressure-dependent isotherm and selectivity profiles show the CO2/CH4 and CO2/N2 separation behaviors at 296 K. The predicted selectivities of CO2/N2 and CO2/CH4 at 296 K under 1 bar have been calculated to be 39.3 and 5.8 for 1a and 38.4 and 6.4 for 2a, respectively. It should be noted that these values are lower than those for HKUST-1 (CO2/N2: 101),61 Mg-MOF-74 (CO2/N2: 182, CO2/CH4: 105),49 UTSA-16 (CO2/N2: 315, CO2/CH4: 30),49 and SIFSIX-3-Zn (CO2/N2: 1818, CO2/CH4: 231),48 but still higher than those for Ni-MOF-74 (CO2/N2: 30),61 ZIF-79 (CO2/N2: 23.2, CO2/CH4: 5.4),62 SIFSIX-2-Cu (CO2/N2: 13.7, CO2/CH4: 5.3),48 and PCN-88 (CO2/N2: 18.1, CO2/CH4: 5.3),63 making these MOFs qualified candidates for CO2 capture and separation.
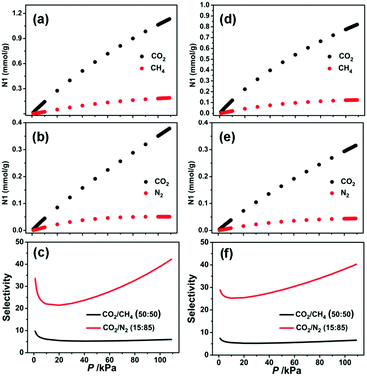 |
| | Fig. 4 Mixture adsorption isotherms and selectivities predicted by IAST of 1a (a–c) and 2a (d–f) for CO2/CH4 (50%:50%) and CO2/N2 (15%:85%) at 296 K. | |
Conclusions
In summary, by virtue of substituted 1,3,5-tris(4-carboxyphenyl)-benzene ligands, we successfully obtained two 2D microporous MOFs. Their microporosities enable these MOFs to selectively adsorb CO2 over CH4 and N2, as shown by single component gas sorption and selectivity calculations. The high selectivity of CO2 over CH4 and N2 indicates the potential application of these materials for the industrial separation of CO2/CH4 and CO2/N2. Benefitting from the straightforward design of MOFs with modifiable performance, we are now developing customizable adsorbents for specific gas separations.
Acknowledgements
This work was supported by the Welch Foundation (grant AX-1730 to B. C.).
Notes and references
-
WMO, Greenhouse Gas Bulletin, 2016, No. 12 Search PubMed.
- K. S. Lackner, S. Brennan, J. M. Matter, A. H. A. Park, A. Wright and B. van der Zwaan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13156–13162 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- H. Wang, B. Li, H. Wu, T.-L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chemistry, 2016, 1, 557–580 CrossRef CAS.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Science, 2016, 353, 141 CrossRef CAS PubMed.
- H. Zhang, P. Lin, E. Chen, Y. Tan, T. Wen, A. Aldalbahi, S. M. Alshehri, Y. Yamauchi, S. Du and J. Zhang, Chem. – Eur. J., 2015, 21, 4931–4934 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- R.-B. Lin, S.-Y. Liu, J.-W. Ye, X.-Y. Li and J.-P. Zhang, Adv. Sci., 2016, 3, 1500434 CrossRef PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed.
- W.-G. Lu, L. Jiang, X.-L. Feng and T.-B. Lu, Inorg. Chem., 2009, 48, 6997–6999 CrossRef CAS PubMed.
- A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420–1425 RSC.
- J.-H. Wang, M. Li and D. Li, Chem. Sci., 2013, 4, 1793–1801 RSC.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- L. Cao, Z. Lin, F. Peng, W. Wang, R. Huang, C. Wang, J. Yan, J. Liang, Z. Zhang, T. Zhang, L. Long, J. Sun and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 4962–4966 CrossRef CAS PubMed.
- Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS PubMed.
- M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed.
- R.-B. Lin, T.-Y. Li, H.-L. Zhou, C.-T. He, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2015, 6, 2516–2521 RSC.
- Q.-G. Zhai, X. Bu, C. Mao, X. Zhao, L. Daemen, Y. Cheng, A. J. Ramirez-Cuesta and P. Feng, Nat. Commun., 2016, 7, 13645 CrossRef CAS PubMed.
- T.-L. Hu, H. Wang, B. Li, R. Krishna, H. Wu, W. Zhou, Y. Zhao, Y. Han, X. Wang, W. Zhu, Z. Yao, S. Xiang and B. Chen, Nat. Commun., 2015, 6, 7328 CrossRef CAS PubMed.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 CAS.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC.
- F. Luo, C. Yan, L. Dang, R. Krishna, W. Zhou, H. Wu, X. Dong, Y. Han, T.-L. Hu, M. O'Keeffe, L. Wang, M. Luo, R.-B. Lin and B. Chen, J. Am. Chem. Soc., 2016, 138, 5678–5684 CrossRef CAS PubMed.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650 CrossRef CAS PubMed.
- R.-B. Lin, D. Chen, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Inorg. Chem., 2012, 51, 9950–9955 CrossRef CAS PubMed.
- M. Wriedt, J. P. Sculley, A. A. Yakovenko, Y. Ma, G. J. Halder, P. B. Balbuena and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 9804–9808 CrossRef CAS PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- H. He, J. A. Perman, G. Zhu and S. Ma, Small, 2016, 12, 6309–6324 CrossRef CAS PubMed.
- X. Zhao, J. Dou, D. Sun, P. Cui, D. Sun and Q. Wu, Dalton Trans., 2012, 41, 1928–1930 RSC.
-
Crystal Clear, Rigaku/MSC, Inc., Rigaku Corporation, The Woodlands, TX, 2005 Search PubMed.
-
T. Higashi, ABSCOR, Rigaku Corporation, Tokyo, Japan, 1995 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- S. J. Caldwell, B. Al-Duri, N. Sun, C.-g. Sun, C. E. Snape, K. Li and J. Wood, Energy Fuels, 2015, 29, 3796–3807 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- Y. Hijikata, S. Horike, M. Sugimoto, M. Inukai, T. Fukushima and S. Kitagawa, Inorg. Chem., 2013, 52, 3634–3642 CrossRef CAS PubMed.
- D. Chen, Y.-J. Liu, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, CrystEngComm, 2011, 13, 3827–3831 RSC.
- C. Zhang, R. P. Lively, K. Zhang, J. R. Johnson, O. Karvan and W. J. Koros, J. Phys. Chem. Lett., 2012, 3, 2130–2134 CrossRef CAS PubMed.
- P. Aprea, D. Caputo, N. Gargiulo, F. Iucolano and F. Pepe, J. Chem. Eng. Data, 2010, 55, 3655–3661 CrossRef CAS.
- B. Li, H. Wang and B. Chen, Chem. – Asian J., 2014, 9, 1474–1498 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, crystallographic tables and selectivity calculation results. CCDC 1522317. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qm00375c |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.