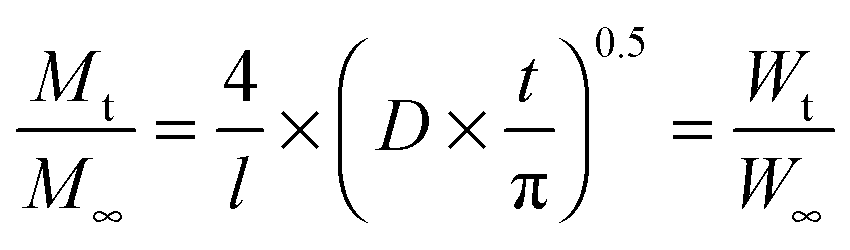DOI:
10.1039/C6QM00243A
(Research Article)
Mater. Chem. Front., 2017,
1, 767-779
Dual-responsive hybrid thermoplastic shape memory polyurethane†
Received
29th September 2016
, Accepted 14th October 2016
First published on 3rd November 2016
Abstract
A urethane-based dual-responsive shape memory polymer (SMP) comprising poly(ethylene glycol) (PEG), poly(ε-caprolactone) (PCL) and poly(dimethylsiloxane) (PDMS) was prepared. Water-responsive SME was achieved through the solvation and recrystallization of PEG switching segments. Thermo-responsive SME of the copolymer at physiological temperature was also demonstrated. The dual-responsive SMP shows promise for biomedical applications in which the physiological environment ensures constant exposure to water for water-triggered actuation with a suitable response time, while the thermally-triggered SME of the copolymer may be utilised to further augment shape recovery.
1. Introduction
Shape-memory materials are a class of intelligent materials1,2 – materials that respond to external stimuli3,4 – that can be deformed and fixed into a temporary shape, and subsequently recovered to their original “memorized” permanent shape when subjected to certain external stimuli. The shape memory phenomena in polymers arise from the co-existence of two distinct structural phases; a soft segment that is highly elastic, and a switching segment that experiences a reduction in stiffness upon exposure to stimuli. In the original macroscopic shape, the polymer chains in the SMP arrange in conformations associated with maximum entropy.
According to Boltzmann's equation,
| | S = kB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnW lnW | (1) |
where
S is the entropy,
kB is the Boltzmann's constant, and
W represents the probability associated with a certain macrostate, the coiled conformation is the state of maximum entropy. Exposure to stimuli then activates the micro-Brownian motion of the switching segment, increasing the mobility of the polymer chains, and allowing the material to be deformed with a corresponding loss in entropy.
5 The SMP can then be fixed into a new temporary shape
via network chain immobilization,
6 by reversing or removing the external stimuli. This programming process stores the mechanical energy used for deformation.
5,7 To recover their original permanent shape, these materials have to be subjected to external stimuli, for example, by immersion in solvent,
8,9 exposure to light,
10,11 electricity,
12–14 magnetic fields,
15,16 pH,
17,18 or most commonly, by heating.
19–21 This re-mobilizes the kinetically ‘frozen’ segments, allowing the polymer chains to return to their most probable configuration, causing a macroscopic shape change back to its original “permanent” shape – dictated by chemical or physical crosslinks, or even entanglements.
22
In recent years, research into SMPs has gained much traction, hugely due to the vast applications of the said technology. Some examples include implants for minimally invasive surgery,23,24 smart breathable fabrics,25 intelligent medical devices with drug delivery functions,26 and self-deployable sun sails in a spacecraft.27
Currently, thermo-responsive SMPs are the most common and most studied class of SMPs.28,29 Associated with the switching segment is the switching transition temperature (Ttrans), above which shape recovery is triggered. By increasing the temperature beyond Ttrans, the switching segment melts or becomes rubbery, and allows the deformation of the SMP. Ttrans can either be associated with the glass transition or melting process, although a melting process is favoured as it translates into a better defined shape recovery temperature.1,30
In the current literature, a large majority of reported SMP systems involve thermal actuation via a local rise in temperature. While shape recovery in thermally actuated systems can be easily triggered, it could cause thermal tissue damage to various extents.31 Water-triggered SMPs thus present an interesting alternative to circumvent such problems, and make attractive propositions as devices for in vivo deployment where shape recovery can be triggered by the presence of water in physiological environments – such as sutures, stents and scaffolds with an adjustable pore size.32,33 Water-responsive SMPs have been of interest, after reports of the effect of water molecules on the lowering of Tg were published.34 When a water-sensitive SMP is immersed, water molecules act as plasticizers by penetrating the polymer matrix. This improves the mobility of the switching segments and causes the observed lowering of Tg as reported.35 Shape recovery can then occur if the surrounding temperatures are higher than the new Tg. However, the current literature reporting the shape memory response that occurs via this mechanism is rather slow, with a recovery time of at least 140 minutes.36
Alternatively, water-responsive SMPs can also be achieved by the incorporation of hydrophilic or water-swellable components in a system containing hydrophobic components. When exposed to water, hydrophilic components in the system will swell and the crystalline structure is disrupted. The SMP in the swollen state becomes compliant, and significant deformation can be achieved for shape-programming. The temporary shape can be fixed by recovering the crystalline structure through a drying process.37 Through this mechanism, the shape recovery times can be reduced significantly. Utilizing a “wet-fixing” shape memory cycle and reducing the diffusion distance of water also serve to speed up the actuation.36 Yang et al. reported water-driven SMP systems with recovery times of between 75 to 140 minutes from a folded shape,38 while Yong et al. reported a water-aided SMP with recovery ratios of 70% and a recovery time of 5 minutes in water at 35 °C.39
In our previous work, we demonstrated the shape memory capabilities of a thermoplastic polyurethane which exhibits fast-response shape recovery and a Ttrans in the body temperature range, and its potential for biomedical applications such as bone tissue engineering.19,20,24 While chemically crosslinked thermoset SMPs generally display good shape recovery and shape fixity,40,41 the use of covalent crosslinkers in thermoset SMPs render such SMPs unsuitable for many post-synthesis processing techniques.42 We have demonstrated the key advantages of physically crosslinked SMPs in prior work, which include fast-response shape recovery20 as well as the ease and ability to process physically crosslinked SMPs using various methods such as heat processing (e.g. melt processing, extrusion, thermoforming, injection moulding etc.) and solvent processing.1 Herein, we extend the study by incorporating poly(ethylene glycol) (PEG) as a hydrophilic component and report a polyurethane-based dual-responsive shape memory copolymer that responds to both water and heat. We hypothesise that the incorporation of hydrophilic PEG in a system that comprises hydrophobic components such as polycaprolactone (PCL) and polydimethylsiloxane (PDMS) could yield water-sensitive SMPs with a range of acceptable actuation times that range between several minutes to a few hours for biomedical applications. At the same time, the thermo-responsiveness of the copolymer would be retained to afford dual-responsive SMPs that actuate in the presence of water and temperature close to body temperature. The thermo-responsiveness of the SMP could help to augment the recovery of the programmed shape. Alternatively, the SMP could be programmed into two temporary shapes using both wet-fixing and temperature cycles.
PEG is widely used in biomedical applications because it does not harm active proteins and is nontoxic, non-immunogenic and nonantigenic.43 PCL has a very low degradation rate in vivo – taking more than 2 years for complete degradation,44 has high drug permeability, and good processability.45 PDMS is inert and stable to oxygen plasma treatment and sterilization (steam autoclave and ethanol) – common procedures in biomedical applications.46 PDMS has a very low Tg of −125 °C, and thus exists in the rubbery state in the operating range of temperatures. In the proposed SMP, PDMS serves as the soft segment, and its presence in segmented copolymers generally augments their overall flexibility (elongation at failure), elasticity and stretchability.47,48 Moreover, the addition of semi-inorganic siloxane may provide benefits like improved biocompatibility, gas permeability, anti-adhesion properties, anti-thrombogenicity, and oxidative, thermal, and chemical stability.49,50 In this system, PEG would serve as the hydrophilic switching segment, while PCL and PDMS serve as the soft segments, and the urethane hard segments memorize the original shape. The choice of using urethane linkages also enables phase separation, which is essential for shape memory. The water-triggered shape actuation would ideally function by the formation and dissolution of the crystalline PEG phase from dehydration and immersion in water at room temperature, while the heat-triggered shape actuation results from the melting and formation of PCL crystallites.
2. Experimental section
Materials
Poly(ethylene glycol) (PEG) (99%) with an Mn of ca. 2000, poly(ε-caprolactone)-diol (PCL-diol) with an Mn of ca. 2000, dibutyltin dilaurate (DBT) (95%) and 1,6-hexamethylene diisocyanate (HDI) (98%) were purchased from Sigma-Aldrich. Dihydroxyl-terminated polydimethylsiloxane (PDMS-diol) with an of Mn of 2000 was purchased from Scientific Polymer Products Inc. The solvents used, including chloroform, toluene, and n-hexane, were of ACS grade. All the materials were used as received unless otherwise specified.
Synthesis of poly(PEG/PCL/PDMS urethane)s
Random multi-block poly(PEG/PCL/PDMS urethane) copolymers were produced via step-growth polymerisation with various weight ratios of PEG/PCL/PDMS using HDI as a coupling reagent. The number average molecular weight (Mn) and the weight average molecular weight (Mw) of the polymers were investigated using gel permeation chromatography (GPC). The Mn and Mw of PEG were found to be 1750 and 2740 g mol−1, respectively. The Mn and Mw of PCL-diol were found to be 2470 and 4720 g mol−1, respectively. The Mn and Mw of PDMS-diol were found to be 4050 and 6860 g mol−1, respectively.
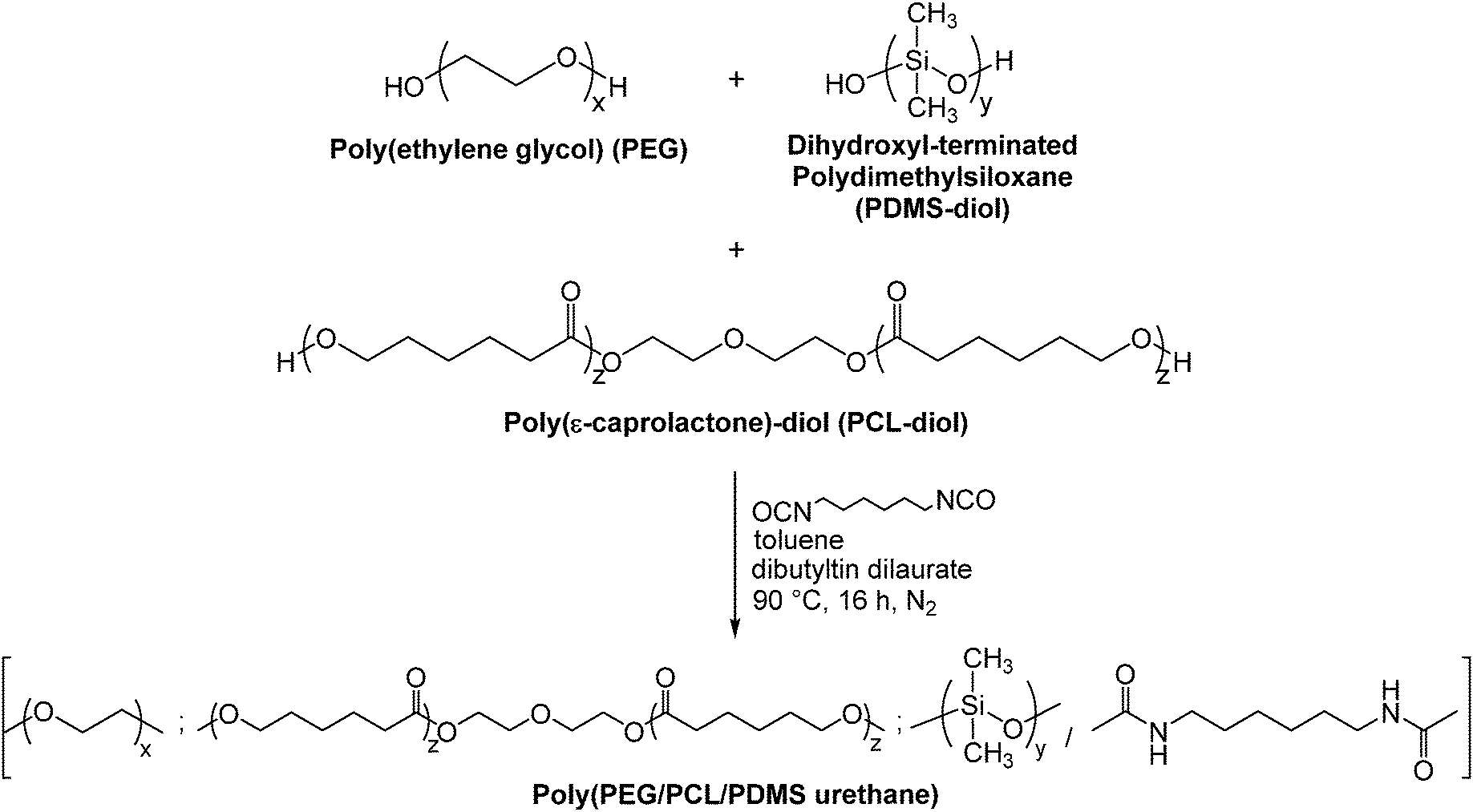
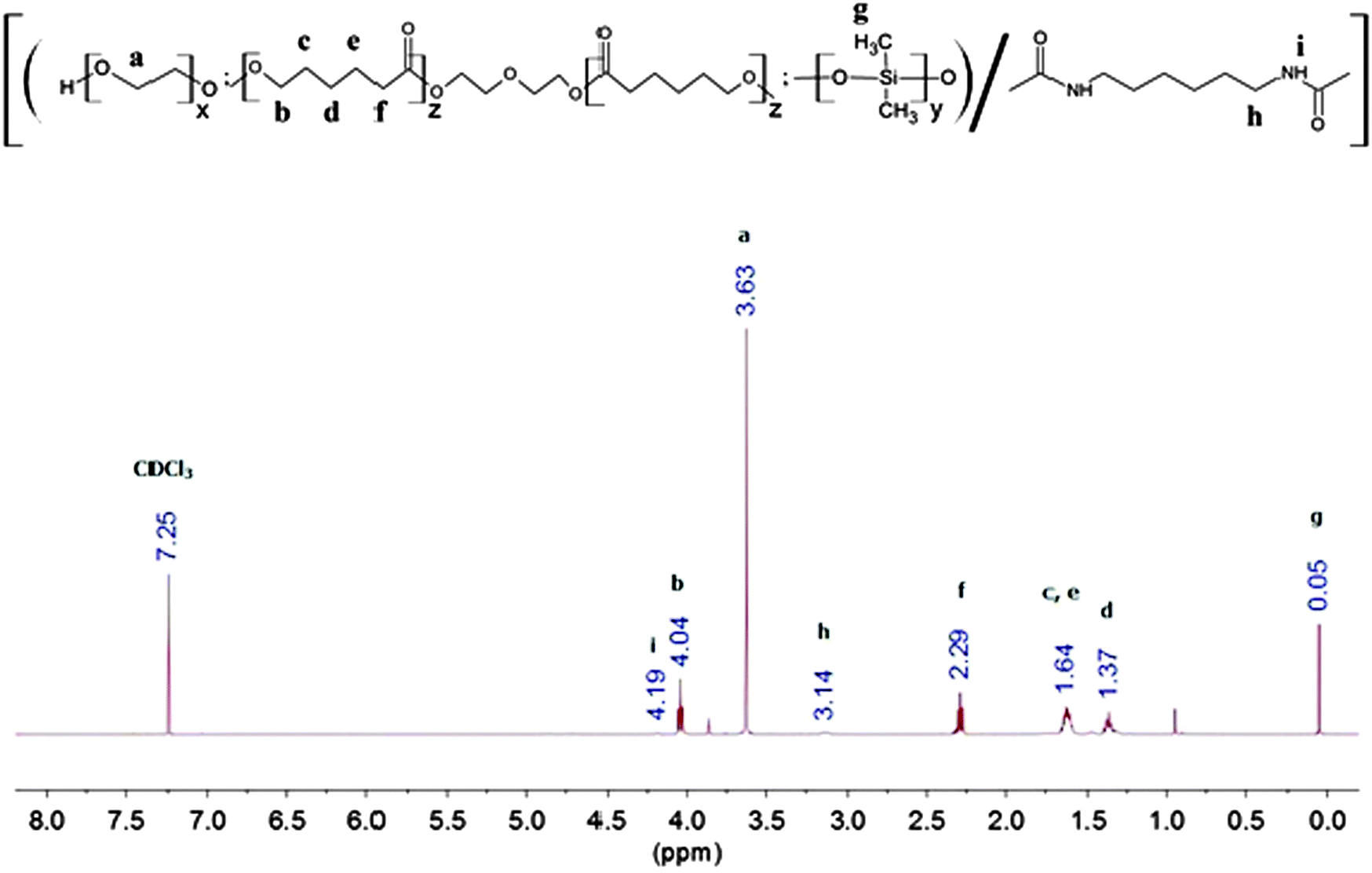
The synthesis method is generally similar to that of previous work done by our group.20,51–53 In a typical set-up, 4 g of PEG (Mn = 1750, 2.3 × 10−3 mol), 14 g of PCL-diol (Mn = 2470, 5.7 × 10−3 mol), and 2 g of PDMS-diol (Mn = 4050, 4.9 × 10−4 mol) were dissolved in a round-bottom flask in 100 mL of anhydrous toluene at 90 °C. Water was removed from the system via azeotropic distillation using a Buchi Rotavapor R-210 rotary evaporator until only about 1 mL of toluene was left in the flask. This process was repeated twice. Following which, 1.35 mL (8.4 × 10−3 mol) of HDI and 2 drops of dibutyltin dilaurate (DBT) (∼8 × 10−3 g) were added sequentially. The reaction mixture was then stirred at 90 °C under a nitrogen atmosphere for ca. 16 h. The resulting copolymer was then precipitated from a six-fold excess volume of n-hexane, and dried in a vacuum oven for 24 h. The yield was 70% and above after isolation and purification. This process is illustrated in Fig. 1. 1H NMR (CDCl3) of poly(PEG/PCL/PDMS urethane)s: δ (ppm) 0.05 (OSi(C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3)2), 1.37 (OCH2CH2C2CH2CH2CO), 1.64 (OCH2C2CH2C2CH2CO), 2.29 (OC2CH2CH2CH2CH2CO), 3.14 (OOCNHC2CH2CH2CH2CH2C2NHCOO), 3.63 (OC2C2O), 4.04 (OC2CH2CH2CH2CH2CO), 4.19 (OOCNCH2CH2CH2CH2CH2CH2NCOO).
3)2), 1.37 (OCH2CH2C2CH2CH2CO), 1.64 (OCH2C2CH2C2CH2CO), 2.29 (OC2CH2CH2CH2CH2CO), 3.14 (OOCNHC2CH2CH2CH2CH2C2NHCOO), 3.63 (OC2C2O), 4.04 (OC2CH2CH2CH2CH2CO), 4.19 (OOCNCH2CH2CH2CH2CH2CH2NCOO).
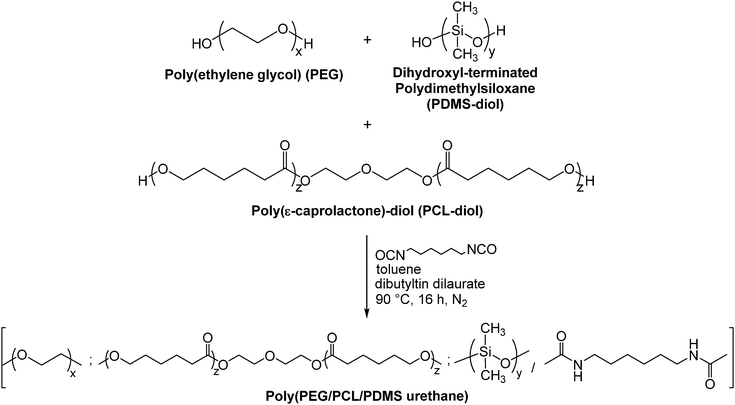 |
| | Fig. 1 Schematic diagram depicting the synthesis of poly(PEG/PCL/PDMS urethane)s. | |
Molecular characterisation
Gel permeation chromatography (GPC) analysis was performed using a Viscotek GPCmax module equipped with two Phenogel 103 Å and 105 Å columns (size: 300 × 7.80 mm) in series and a Viscotek TDA 305 triple detector. HPLC grade tetrahydrofuran (THF) was used as an eluent at a flow rate of 1.0 mL min−1 at a column temperature of 40 °C. Monodispersed polystyrene standards were used to obtain a calibration curve.
1H nuclear magnetic resonance (NMR) spectra were recorded on a JEOL 500 MHz NMR spectrometer at room temperature. Measurements were done with 16 scans, and chemical shifts were referenced to the solvent peak (δ = 7.26 ppm for CDCl3).
Fourier transform infrared (FTIR) spectra were recorded on an Agilent Technologies Cary 660 FT-IR Spectrometer. Polymers were dissolved in THF, then coated and dried on potassium bromide pellets. 16 scans were signal-averaged with a resolution of 4 cm−1 at room temperature.
Thermal analysis
Differential scanning calorimetry (DSC) measurements were performed using a TA Instruments Q100 differential scanning calorimeter equipped with an autocool accessory and calibrated using indium. Measurements were performed under continuous nitrogen purge (flow rate = 50 mL min−1). Samples were heated from room temperature to 90 °C at 20 °C min−1 (first heating run) before they were cooled to −30 °C at 10 °C min−1 (first cooling run). The samples were then re-heated to 170 °C at 10 °C min−1 (second heating run). Values for glass transition temperatures (Tg), melting temperatures (Tm), and enthalpies of melting (ΔHm) were obtained from the second heating run, with the melting temperatures taken as the peak maxima. In order to study the mechanism behind the water-actuated SME and for the deconvolution of peaks to study the degree of crystallinity of PEG and PCL separately, DSC measurements were also performed for samples that were immersed in water to solvate the hydrophilic PEG segments.
Thermogravimetric analyses (TGA) were performed on a TA Instruments TGA Q500. Samples were heated from room temperature to 700 °C at a rate of 20 °C min−1 in a dynamic nitrogen atmosphere (flow rate = 40 mL min−1).
Water uptake
The water swelling behaviour of the copolymers was studied gravimetrically. To determine equilibrium swelling, copolymer films (dimensions = 10 mm × 10 mm) were immersed in deionised water and kept at room temperature. Each sample was retrieved at regular time intervals, and excess surface moisture was removed, and weighed. The average of three measured values was taken for each sample. The weight-swelling ratio (W) was calculated using wet weight (mwet) and dry weight (mdry) of the copolymer films using the following equation:36,51| | 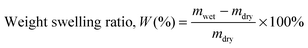 | (2) |
Static uniaxial tensile test
The tensile properties of wet and dry copolymer films were studied using an Instron 5569 uniaxial tensile testing system equipped with a video extensometer. Tensile tests were performed using a 100 N load cell at room temperature. Samples were prepared into ASTM D638 Type IV tensile bars in accordance with ASTM D638 standards. Samples in the wet state were immersed into water for ca. 2 h prior to testing. All samples were deformed extensionally with a crosshead speed of 10 mm min−1 until a strain (ε) of 700% was reached. Young's modulus was calculated from the initial slope (0 < ε < 5%) of the stress–strain curves by utilising linear regression. Ultimate tensile stress (UTS) values were reported as being greater than the maximum stress attained if the sample did not fracture. Elongation-at-break (εb) values were reported as the strain at which the applied force dropped to zero. An average of the three measured values was taken for each sample.
Water-triggered shape memory characterisation
For water-triggered SMPs, the swelling of the SMPs is often significant and makes recovery difficult to quantify using cyclic thermomechanical tests due to the competing effects of swelling and strain recovery, as well as the need for immersion in water and drying. As such, the water-triggered SME was characterised using a fold-deploy test.54 Here, angular variations of shape recovery and fixity ratios that measure the extent of shape recovery (esr) and the extent of shape fixity (esf) were adopted to characterise the SME, using the following equations:| | 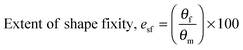 | (3) |
| | 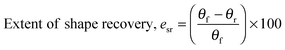 | (4) |
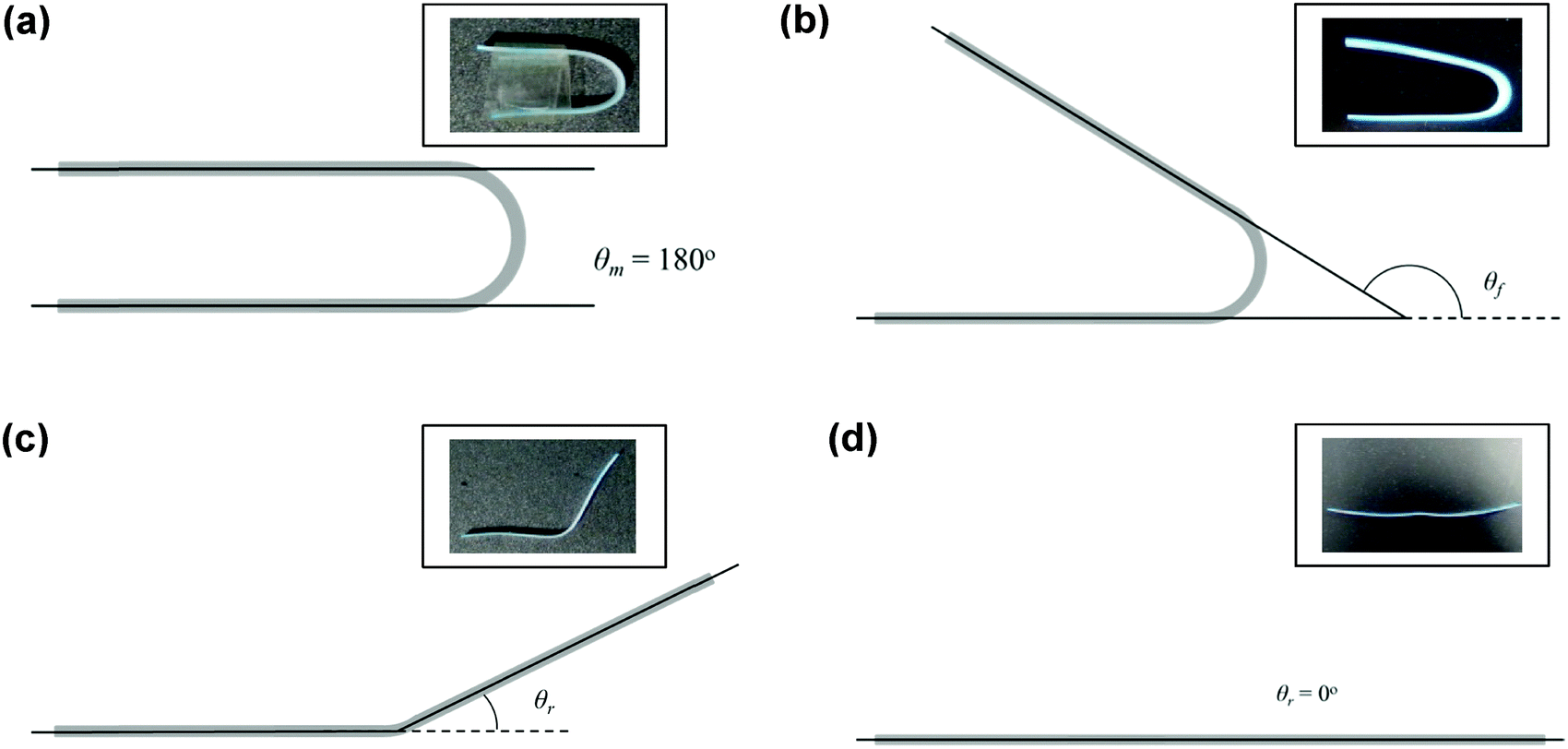
Copolymer films were prepared by casting a ca. 7 × 10−2 g mL−1 chloroform solution onto a poly(tetrafluoroethylene) (PTFE) dish. After slow evaporation of the chloroform, the casted copolymer films were dried under a high vacuum at 40 °C for a day to remove any remaining solvent. Rectangular strips of dimensions ca. 50 mm (length) × 5.0 mm (width) × 0.7 mm (thickness) were cut from the cast films. Samples were immersed in deionised water for 2 h before they were taken out and deformed into a ‘U’-shape. The maximum angle of deformation, θm, was measured by extrapolating the straight ends of the strips until they intersected (unless they were parallel, in which case θm = 180°) (Fig. 2). The strips were held in this deformed state for 1 h and air-dried under ambient conditions. Following which, the applied stress was released, and the angle of shape fixity (θf) was measured. The deformed samples were immersed in deionised water for 2 h for shape recovery and the shape recovery angle (θr) was measured. This process is illustrated in Fig. 2.
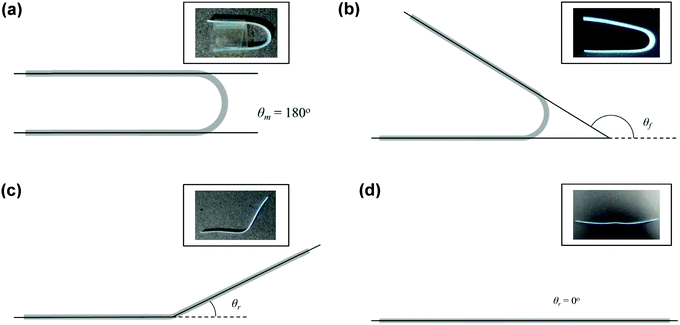 |
| | Fig. 2 Schematic diagram depicting the water-triggered SME characterisation. (a) Measurement of the maximum angle of deformation, (b) measurement of the fixity angle, (c) and (d) measurement of the recovery angle. | |
Heat-triggered shape memory characterisation
Heat-triggered shape memory properties of the materials were determined via dynamic mechanical analysis (DMA) using a TA Instruments DMA Q800 in a controlled force mode with a preload force of 0.001 N. Copolymer films were prepared by casting a ca. 7 × 10−2 g mL−1 chloroform solution onto a poly(tetrafluoroethylene) (PTFE) dish. After slow evaporation of the chloroform, the casted copolymer films were dried under a high vacuum at 40 °C for a day to remove any remaining solvent. Rectangular strips of dimensions ca. 50 mm (length) × 5.0 mm (width) × 0.7 mm (thickness) were cut from the cast films. In a typical cycle, samples elongated to a force of 8.0 N at a rate of 1.00 N min−1 at room temperature (this strain was recorded as εm), cooled to 0 °C at a rate of 3.0 °C min−1, with an isothermal time of 15.0 min, unloaded to 0.001 N at a rate of 1.0 N min−1 (this strain was recorded as εu), then heated to 35 °C at a rate of 3.0 °C min−1, with an isothermal duration of 20.0 min to observe the recovery (this strain was recorded as εr). This process was repeated seven more times on the same sample in order to obtain multiple cycles. The shape recovery ratio (Rr) and the shape fixity ratio (Rf) were calculated using the following equations:5,20,55| | 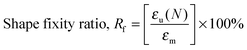 | (5) |
| | 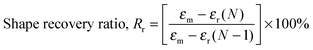 | (6) |
3. Results and discussion
Synthesis and characterisation of poly(PEG/PCL/PDMS urethane)s
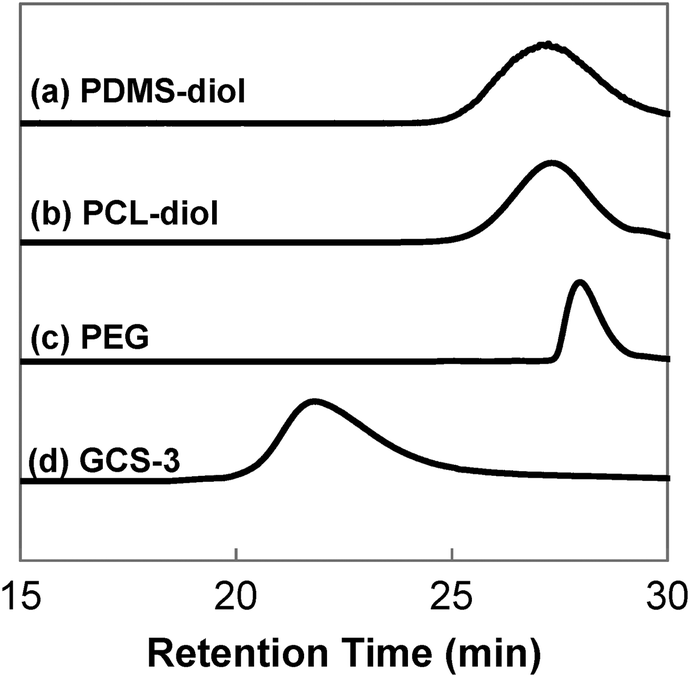
GPC was performed to obtain the molecular weights and molecular weight distributions of the samples, and the results are tabulated in Table 1. The GPC chromatographs of all the samples showed unimodal peaks, which had no overlaps with the chromatographs of their constituents, PEG, PCL-diol and PDMS-diol. This absence of unreacted precursors is indicative of complete polymerisation. The typical GPC profile of GCS-3, alongside those of its precursors is shown in Fig. 3.
Table 1 Molecular characterisation of the poly(PEG/PCL/PDMS urethane) samples
| Sample |
Actual compositiona (wt%) |
Molecular weight characteristicsb |
| PEG |
PCL |
PDMS |
M
n (×103) |
M
w (×104) |
Đ
M
|
|
Actual composition was determined from the relative peak intensities in the 1H NMR spectra.
As determined by GPC.
Dispersity, ĐM = Mw/Mn.
|
| GCS-1 |
7.0 |
87.7 |
5.3 |
49.1 |
11.1 |
2.25 |
| GCS-2 |
18.3 |
73.1 |
8.6 |
36.2 |
7.72 |
2.13 |
| GCS-3 |
29.8 |
63.6 |
6.6 |
30.4 |
6.06 |
1.99 |
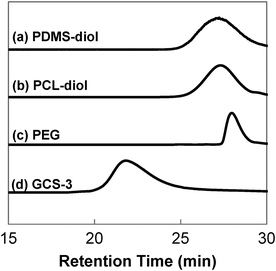 |
| | Fig. 3 GPC chromatographs of poly(PEG/PCL/PDMS urethane), GCS-3, and its precursors. | |
FTIR and 1H NMR spectroscopy was used to study the chemical structure of the poly(PEG/PCL/PDMS urethane)s. The 1H NMR spectra of GCS-3 in CDCl3 is shown in Fig. 4, with all proton signals of the components identified and attributed according to several references.20,56–59 The relative intensities of the NMR peaks were used to calculate the weight ratios of PEG, PCL and PDMS in the samples, and were in good agreement with the feed ratios of the precursors.
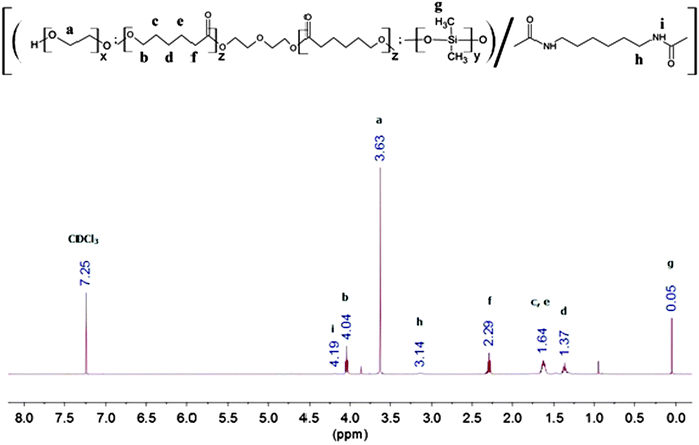 |
| | Fig. 4 500 Hz 1H NMR spectrum of GCS-3 in CDCl3. | |
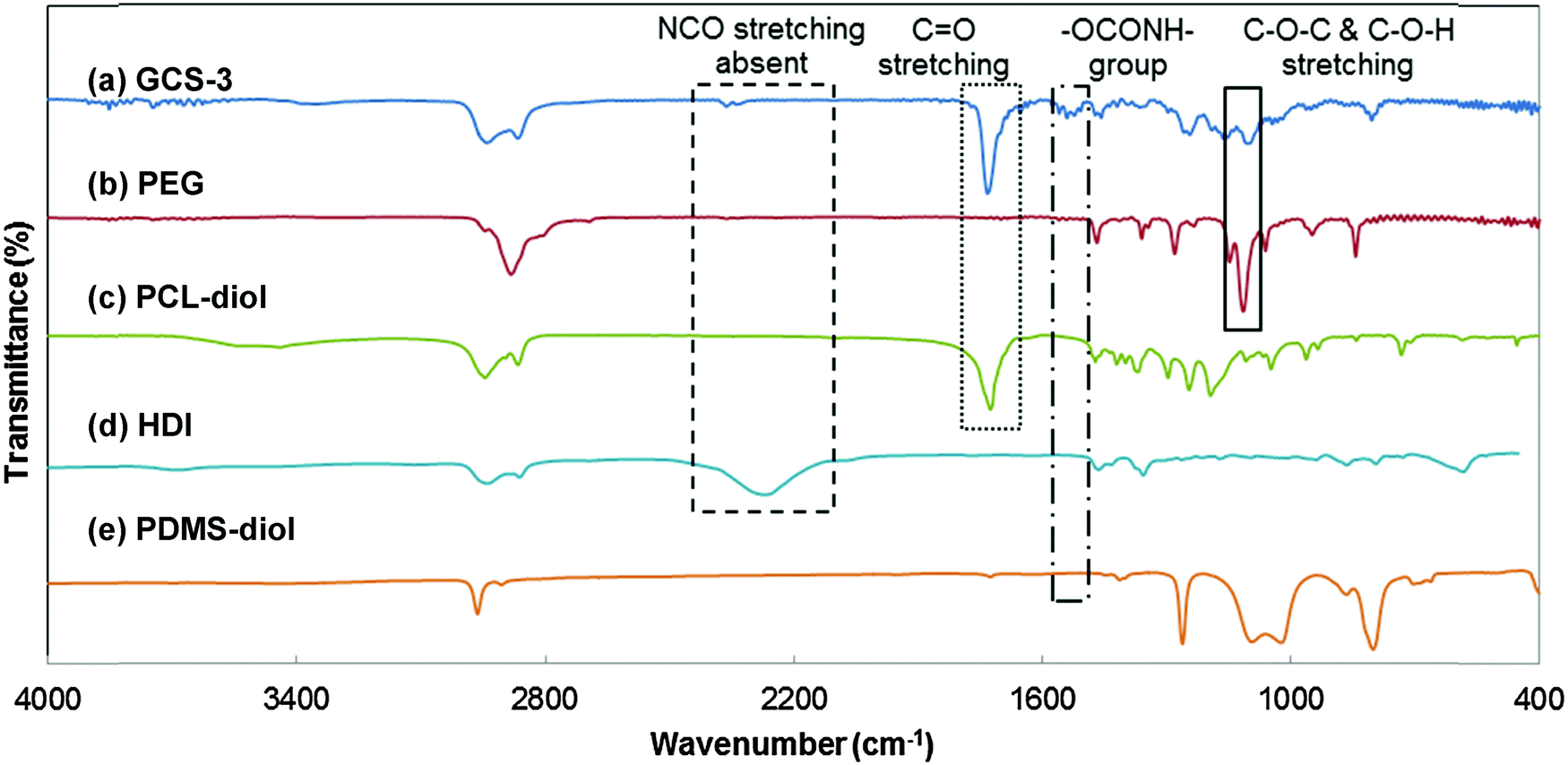
The chemical structures of the copolymers were further validated by the identification of functional groups using FTIR spectroscopy (Fig. 5). The characteristic C–O–C stretching vibration attributed to the OCH2CH2 units of PEG is observed at 1108 cm−1, and the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band due to the carbonyl group in PCL is observed at 1731 cm−1; both were observed in the spectrum of GCS-3.20,59 The broad stretching peak at around 2200 cm−1 was attributed to –NCO– stretching from HDI and was observed to be absent in the copolymer. Instead, the spectrum of GCS-3 exhibited a peak attributed to –NH bending (in –OCONH–) at 1530 cm−1,20,56 which indicates that the HDI had completely reacted to form the urethane bonds present in the copolymer.
O stretching band due to the carbonyl group in PCL is observed at 1731 cm−1; both were observed in the spectrum of GCS-3.20,59 The broad stretching peak at around 2200 cm−1 was attributed to –NCO– stretching from HDI and was observed to be absent in the copolymer. Instead, the spectrum of GCS-3 exhibited a peak attributed to –NH bending (in –OCONH–) at 1530 cm−1,20,56 which indicates that the HDI had completely reacted to form the urethane bonds present in the copolymer.
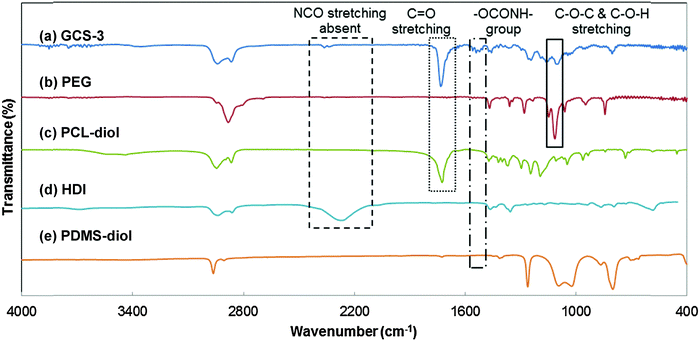 |
| | Fig. 5 FTIR spectra of poly(PEG/PCL/PDMS urethane), GCS-3, and its precursors. | |
Water swelling behaviour
Water uptake plays a core role in the water-actuated shape memory effect. Therefore, there is a need for the investigation of the swelling behaviour of immersed samples. Pure PEG is hydrophilic and interacts favourably with water, dissolving spontaneously. On the other hand, hydrophobic PCL and PDMS are insoluble and immiscible with water, respectively. For the synthesised copolymers, water was expected to permeate through the hydrophilic domains in the copolymer selectively, resulting in an increase in the total weight of the sample. The hydrophobic domains and chain entanglements in the copolymer act as physical crosslinks or net points to maintain the structural integrity of the copolymer, preventing dissolution and limiting the extent of swelling.
The Peppas and Korsmeyer equation (eqn (7)) was used to model the water-swelling behaviour of the synthesised SMPs:
| | 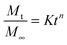 | (7) |
where
Mt is the total mass of water taken up in time
t,
M∞ is the total mass of water taken up after an infinite duration,
K is a geometric constant of the polymer, and
n is a constant related to the mode of transport of the penetrant.
n = 0.5 indicates Fickian kinetics, where diffusion is limited by the rate of diffusion of the solvent; otherwise, non-Fickian processes might be present. In general, the diffusion of the solvent (deionised water) into the polymer matrix can be described in two main processes, namely, polymer relaxation which shifts the swollen–unswollen boundary and the in-diffusion of the solvent into the already swollen matrix. In this procedure, it is assumed that no leaching or dissolution of the samples occurred, and that the polymers swelled to their fullest extent after 7 days. Both the time the samples took to take up 60% of the final amount of water,
tf, and the time taken for the swelling process to stabilise and enter into the solvent diffusion-limited state,
ti, are recorded in
Table 2.
Table 2 Water-swelling behaviour and diffusion parameters of the samples
| Sample |
W
(%) |
t
i
(h) |
t
f
(h) |
N
|
D
(×107) (cm2 s−1) |
|
Water swelling ratio calculated using eqn (2).
Time taken for the swelling process to stabilize and enter into the solvent diffusion-limited state.
Time taken for samples to take up 60% of the final amount of water.
Collective diffusion coefficient.
|
| GCS-1 |
10.7 ± 0.42 |
2.00 |
24.00 |
0.490 |
0.25 |
| GCS-2 |
21.7 ± 0.40 |
0.17 |
2.00 |
0.424 |
3.03 |
| GCS-3 |
43.0 ± 0.42 |
0.42 |
1.00 |
0.427 |
3.76 |
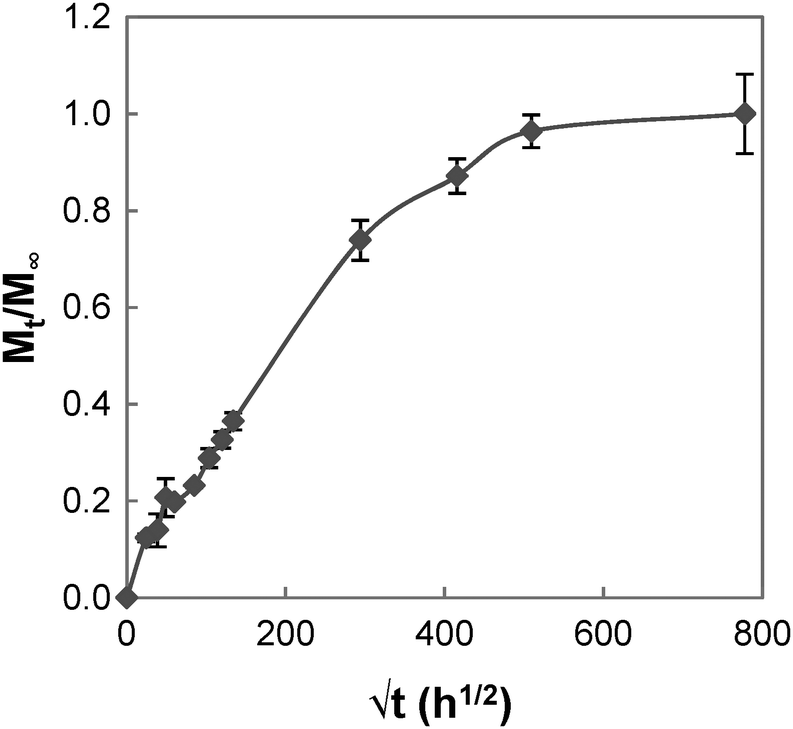
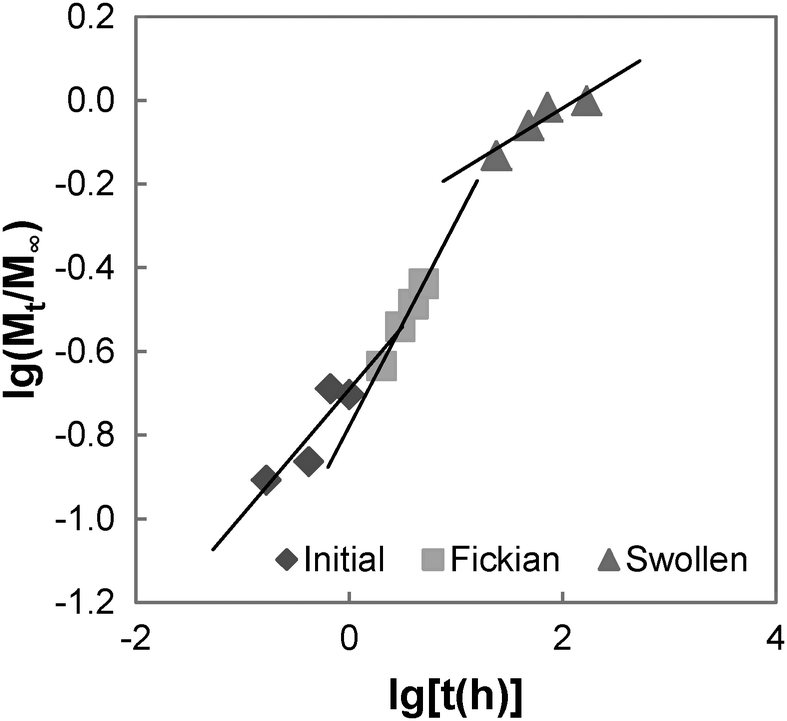
The swelling process of GCS-1 is illustrated in Fig. 6 and 7. The shape of the plot of Mt/M∞ against 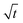 in Fig. 7 appears to comprise a two-step sorption process60 comprising an initial Fickian stage, followed by a structural relaxation stage.
in Fig. 7 appears to comprise a two-step sorption process60 comprising an initial Fickian stage, followed by a structural relaxation stage.
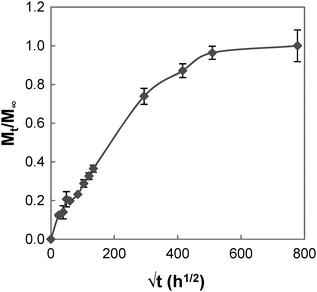 |
| | Fig. 6 Graph of Mt/M∞vs. √t for GCS-1. | |
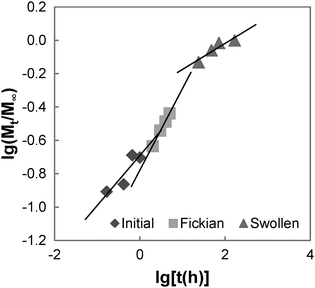 |
| | Fig. 7 Graph of lg(Mt/M∞) vs. lg(t) for GCS-1. | |
In the initial sorption regime in Fig. 7, n is 0.3 (<0.5), and this is indicative of the presence of other processes causing a deviation from Fickian diffusion. These deviations may be time dependent, as in the case of a relaxing medium, or structure dependent, as in the case of an inhomogeneous medium.61 From the DSC data, we note that the poly(PEG/PCL/PDMS urethane) copolymers synthesised are semi-crystalline. The glassy domains of PEG and PCL hinder the swelling process, thereby decreasing the sorption rate; unlike the rubbery regions, they are not able to readjust as easily for the entry of water molecules. On the other hand, non-uniformity in the form of porosity or inhomogeneous density may be present in the sample due to the use of a slow solvent casting step for sample preparation. Once the sample had swelled sufficiently, diffusion became characterised by typical Fickian kinetics, where n is 0.5. For GCS-1 in the Fickian regime, n is 0.49 (≈0.5). Here, the inward diffusion of water is facilitated by the shifting of the mobile chain segments between inter-chain bonds, which occurs more rapidly than Fickian diffusion and the rate-determining step is the Fickian diffusion of water.
By the structural relaxation stage, the initial Fickian diffusion is terminated, and a second rate-determining step is observed. This is likely attributed to the larger-scale rearrangement of the polymer matrix that can only occur following the breakdown of the inter-chain interactions to allow water to diffuse inward, as opposed to the more energetically accessible micro rearrangements in the previous stage.62 The diffusion thus hinges on the slower process – the rate at which structural relaxation occurs, which is why Fickian diffusion was not observed in the final stages of the swelling process. For the case of GCS-1, this is indicated by n decreasing to 0.16 in the structural relaxation stage.
To quantify the diffusion rates, the data were also fitted with the equation:63
| | 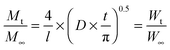 | (8) |
where
l is the initial thickness of the swollen polymer film, and
D is the collective diffusion coefficient of the polymer. The values for
D are also tabulated in
Table 2. The relatively high values of the diffusion coefficients were generally found to have a positive correlation with the amount of PEG present in the synthesised copolymers. The favourable interactions between the water molecules and the hydrophilic PEG drew water in at a faster rate and allowed for greater degrees of swelling of the polymer.
Thermal properties
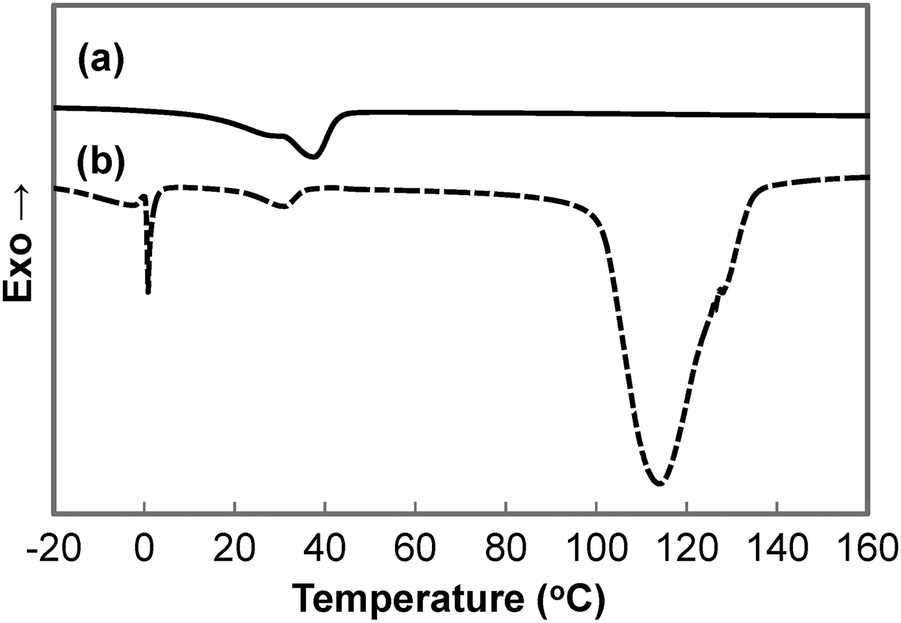
A summary of the thermal properties of the copolymers is summarised in Table 3. DSC thermograms of the second heating run for dry and wet GCS-3 samples are depicted in Fig. 8. The temperature of 90 °C was chosen as the limit for the first heating run to preserve the water content and to match the highest temperature obtained during synthesis to erase the thermal history. This allows for the investigation of the intrinsic thermal properties of the copolymers. The endothermic peak at 0 °C in the DSC thermogram of the wet sample is attributed to the melting of the ice crystals formed by the free water that was trapped in the polymer matrix, forming water that re-solvates the polymer matrix. The broader endothermic peak that occurs at around 100 to 140 °C is associated with the evaporation and freeing of the bound water that is tightly attached to the hydrophilic segments of the polymer matrix. This is indicative of the presence of bound water in the immersed samples due to the overall hygroscopic nature of the synthesised copolymers.
Table 3 Thermal properties of the poly(PEG/PCL/PDMS urethane) samples
| Sample |
T
m,PEG
(°C) |
T
m,PCL
(°C) |
ΔHm,totalb (J g−1) |
ΔHm,PEGb,c (J g−1) |
ΔHm,PCLb,c (J g−1) |
X
c,PEG
(%) |
X
c,PCL
(%) |
T
d
(°C) |
|
Melting point determined in the DSC second heating run for the dry samples.
Enthalpy change during melting obtained by integrating the endothermic peak at Tm during the second heating run.
ΔHm,PCL was obtained by dissolving the crystalline PEG domains by immersing the samples in water for 30 days prior to executing DSC. Based on a comparison with samples without PEG, it was noted that water played a role in influencing the crystallinity of PCL, and led to a reduction in ΔHm,PCL by 6.0 J g−1. ΔHm,PEG is the minimum enthalpy change attributed to the melting of crystalline PEG segments, which was estimated by subtracting ΔHm,PCL and the maximum contribution of water from ΔHm,total.
Crystallinity calculated based on melting enthalpies. Xm = ΔHm/ΔH0m, where ΔHm was determined from the integration of the endothermic peaks from the DSC results. ΔH0m was taken to be 139.5 J g−1 for completely crystallised PCL,64 and 196.8 J g−1 for completely crystallised PEG.65
Temperature at which a 10% mass loss occurred from the TGA curves. Measured from the dry samples.
|
| GCS-1 |
— |
36.3 |
36.0 |
1.5 |
28.5 |
0.8 |
20.4 |
344.5 |
| GCS-2 |
— |
37.1 |
32.9 |
2.7 |
24.2 |
1.4 |
17.3 |
334.0 |
| GCS-3 |
20.8 |
38.3 |
39.0 |
13.0 |
20.0 |
6.6 |
14.3 |
328.5 |
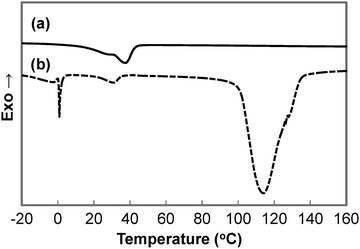 |
| | Fig. 8 DSC thermograms of (a) dry and (b) wet GCS-3. | |
Two endothermic peaks could be identified in most thermograms of the dry samples. However, the endothermic peaks occurred in close proximity to each other, resulting in peak overlap. On the contrary, the wet samples only showed one distinct endothermic peak between 20 and 50 °C, which is attributed to PCL. This confirms that the crystalline hydrophilic PEG domains turned into amorphous compliant domains as they were solvated in water. Partial water-triggered shape recovery could then occur by virtue of the net points established by the urethane linkages, entanglements and the PCL segments. The water-triggered shape recovery was only partial as some PCL segments were also deformed in the shape fixing process. Further shape recovery is possible through the melting of PCL crystallites, thus accounting for the thermo-responsiveness of the copolymer. The water-responsive temporary shape of the SMP can be programmed by recrystallizing the hydrophilic PEG switching segments.
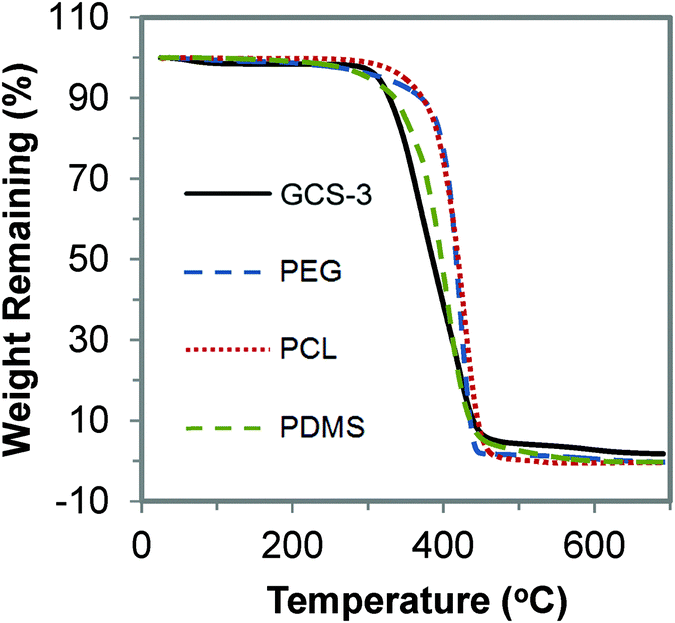
The thermal stability of the copolymers was studied using TGA. The TGA curves of GCS-3 and its precursors are shown in Fig. 9. It was observed that the copolymers undergo a single-step thermal degradation and are thermally stable up to 320 °C, indicating their suitability for sterilization at high temperatures during the processing stage and for usage at body temperature.
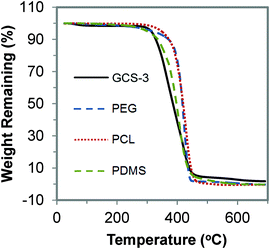 |
| | Fig. 9 TGA curve of poly(PEG/PCL/PDMS urethane), GCS-3, and its precursors. | |
Degradation studies
From the DSC results, an interesting observation is the shift in the endothermic PCL melting peak to lower temperatures and a slight reduction in ΔHm,PCL for the immersed samples. GPC was thus performed on the samples after immersion in water for 30 days to investigate their degradation behaviour. The results, listed in Table 4, reveal that all the samples underwent slight hydrolytic degradation, resulting in a decrease in their molecular weights and an increase in the dispersity of the copolymers. This reduction in molecular weight led to the observed decrease in Tm due to the hydrolytic cleavage of the ester bonds in PCL producing more chain ends, introducing crystalline defects in the PCL domains and causing the reduction of ΔHm,PCL as well, according to Flory's analysis.66
Table 4 Comparison of molecular weights for the dry and wet samples
| Samples |
Dry Mn (×103) |
Dry Mw (×104) |
Dry ĐMa |
WetbMn (×103) |
WetbMw (×104) |
WetbĐMa |
|
Dispersity, ĐM = Mw/Mn.
GPC for the wet samples were done after 30 days of immersion in water.
|
| GCS-1 |
49.1 |
11.1 |
2.25 |
44.2 |
10.6 |
2.41 |
| GCS-2 |
36.2 |
7.72 |
2.13 |
33.5 |
7.99 |
2.39 |
| GCS-3 |
30.4 |
6.06 |
1.99 |
26.2 |
5.50 |
2.10 |
Mechanical properties
Macroscopic mechanical properties of the copolymers were studied using the Instron uniaxial tensile test. Certain samples were elastic, capable of being deformed by up to 700%, indicating the possibility of recovering large deformations for applications in minimally invasive surgeries. It was observed that the mechanical properties of the copolymers (Table 5) decreased with increasing PEG content. This is likely attributed to the disruption of the crystalline PCL domains by the PEG chains, as the semi-crystalline PCL is the phase that imparts mechanical strength to the copolymers. The reduced crystallinity of PCL was also supported by the DSC results. The moduli of the synthesised copolymers also match well with those of human blood vessels (Table 5),67 which make the copolymers potential materials for use as biomedical stents in terms of mechanical properties matching.
Table 5 Mechanical properties of the synthesised copolymers and natural blood vessels
| Samples |
Young's modulus (MPa) |
UTS (MPa) |
ε
b (%) |
|
Results from a single sample after repeated errors on the Instron failed to capture the full range of data.
|
| Dry GCS-1 |
58.8 ± 23.4 |
≥10.3 |
≥700 |
| Dry GCS-2 |
37.0 ± 18.3 |
≥6.30 |
≥700a |
| Wet GCS-2 |
26.2 ± 1.3 |
≥7.78 |
≥700 |
| Dry GCS-3 |
18.9 ± 3.1 |
3.79 ± 0.02 |
598 ± 65 |
| Wet GCS-3 |
16.5 ± 2.3 |
2.74 ± 0.04 |
107 ± 23 |
| Natural tissue |
Young's modulus, longitudinal (MPa) |
Young's modulus, transverse (MPa) |
| Aorta |
5.22 |
3.59 |
| Carotid |
1.47 |
1.61 |
| Vena cava |
4.57 |
13.9 |
Water-triggered shape memory behaviour
Water-triggered SME of the copolymers was investigated by utilizing the fold-deploy test to quantify esf and esr. These were repeated for 10 cycles and the behaviours of the samples were studied in order to determine the repeatability of the shape recovery. esf and esr values of the copolymers for each cycle are tabulated in Table 6.
Table 6 Extents of shape recovery and shape fixity of the poly(PEG/PCL/PDMS urethane)s
| Cycles |
GCS-1 |
GCS-2 |
GCS-3 |
|
e
sf (%) |
e
sr (%) |
e
sf (%) |
e
sr (%) |
e
sf (%) |
e
sr (%) |
| 1 |
65.0 |
47.0 |
72.8 |
59.9 |
87.2 |
70.7 |
| 2 |
62.2 |
30.8 |
71.7 |
34.9 |
75.0 |
62.2 |
| 3 |
75.0 |
38.5 |
74.4 |
31.3 |
76.7 |
59.4 |
| 5 |
77.8 |
29.3 |
86.1 |
41.9 |
93.9 |
59.8 |
| 10 |
83.3 |
21.3 |
88.9 |
35.6 |
100.0 |
50.0 |
Good extents of shape recovery were observed, with esf values generally observed to be 65% and above. A positive correlation between PEG composition and esf was present, confirming the role of PEG crystallites in the water-triggered shape memory behaviour of the copolymers. We also note the trend of decreasing extent of shape recovery over the number of shape recovery cycles. This was attributed to the reduction of chain relaxation capability over repeated tests,54 wherein some destructive processes of the polymer chain inevitably occur in each cycle that led to the observed hysteresis. The average time taken for shape recovery to complete in the first cycle, tsr, was more than 2 hours for GCS-1, while it was 64 minutes and 17 minutes for GCS-2 and GCS-3, respectively.
Fig. 10 depicts the recovery of GCS-3 at 2 minute intervals. The experimental results obtained provide reason to believe that the shape recovery hinges upon the dissolution and crystallisation of the PEG phase. Firstly, from DSC data and the results of the fold-deploy test, improved extents of both shape recovery and fixity were observed with higher degrees of crystallinity of PEG. With increased PEG segments available to form crystallised domains to fix the macroscopic shape during programming, higher extents of shape fixity and shape recovery can be achieved. Additionally, tsr was observed to decrease with larger diffusion coefficients obtained from the mass swelling test, showing that the inward diffusion of water was required for the solvation of the crystalline PEG domains for shape memory actuation to take place. The faster these domains were solvated, the faster the shape recovery could occur. From Table 6, we also note that none of the samples achieved a high extent of shape recovery. This partial recovery could be attributed to the deformation of the unsolvated PCL domains which were observed in the DSC runs, which could be recovered by exposure to elevated temperatures as demonstrated in the following section.
 |
| | Fig. 10 Frame-by-frame analysis of the water-triggered SME of the poly(PEG/PCL/PDMS urethane), GCS-3. | |
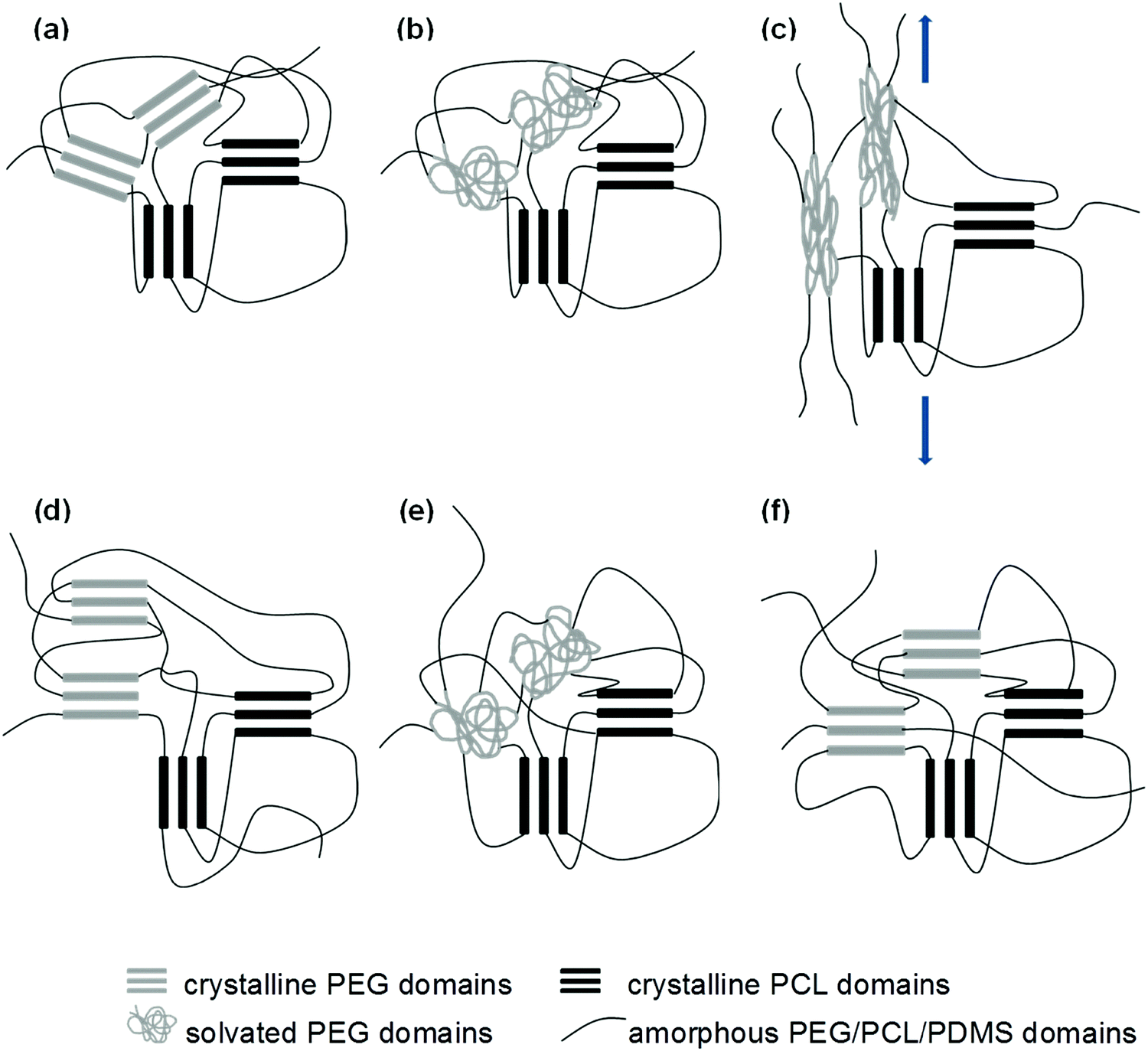
From the uniaxial tensile tests, it was observed that the wet samples have lower elastic moduli than their dry counterparts. This supports our proposed hypothesis that the inward diffusion of water transforms the crystalline PEG domains into amorphous and compliant regions, allowing for easier deformation of the SMP before the temporary shape is fixed. The microstructural changes that take place through the entire deformation, fixation, and recovery process are illustrated in Fig. 11. Future improvements to the system should therefore take this concept into consideration, and it should serve as the starting point for the fine tuning of the water-triggered SME.
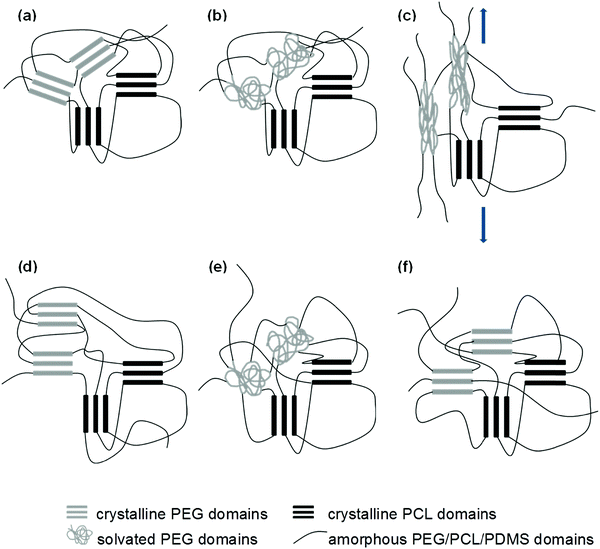 |
| | Fig. 11 Schematic diagram to illustrate microstructural changes during the shape memory process, (a) → (b): immersion in water, (b) → (c): deformation, (c) → (d): fixing, (d) → (e): re-immersion in water (water-triggered SME), (e) → (f): drying. | |
Heat-triggered shape memory behaviour
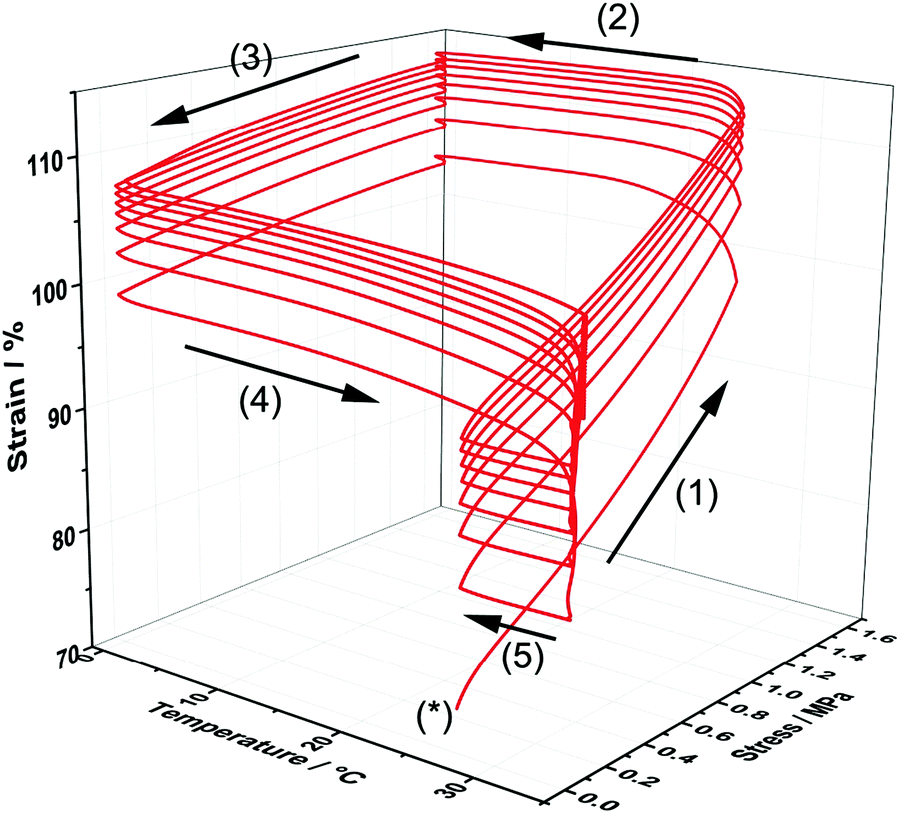
In order to demonstrate the thermo-responsive shape memory behaviour of the poly(PEG/PCL/PDMS urethane) samples, heat-triggered SME of the copolymer GCS-3 was studied by means of thermocyclic shape memory experiments using dynamic mechanical analysis (DMA). Fig. 12 depicts an eight-cycle thermocyclic experiment for the copolymer GCS-3 in which the arrows denote the stages of the cycles: (*) marks the start of a thermocycle; (1) loading and elongation at 25 °C; (2) shape fixation and cooling to 0 °C; (3) unloading and evaluation of Rf; (4) heating to 35 °C to induce shape recovery and evaluation of Rr; and (5) cooling to 25 °C for the beginning of the next cycle.
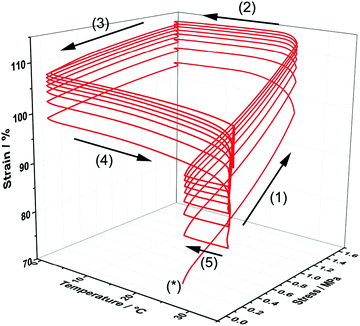 |
| | Fig. 12 Stress–strain–temperature curve for poly(PEG/PCL/PDMS urethane), GCS-3, in an eight-cycle thermocyclic test after initial pre-conditioning. | |
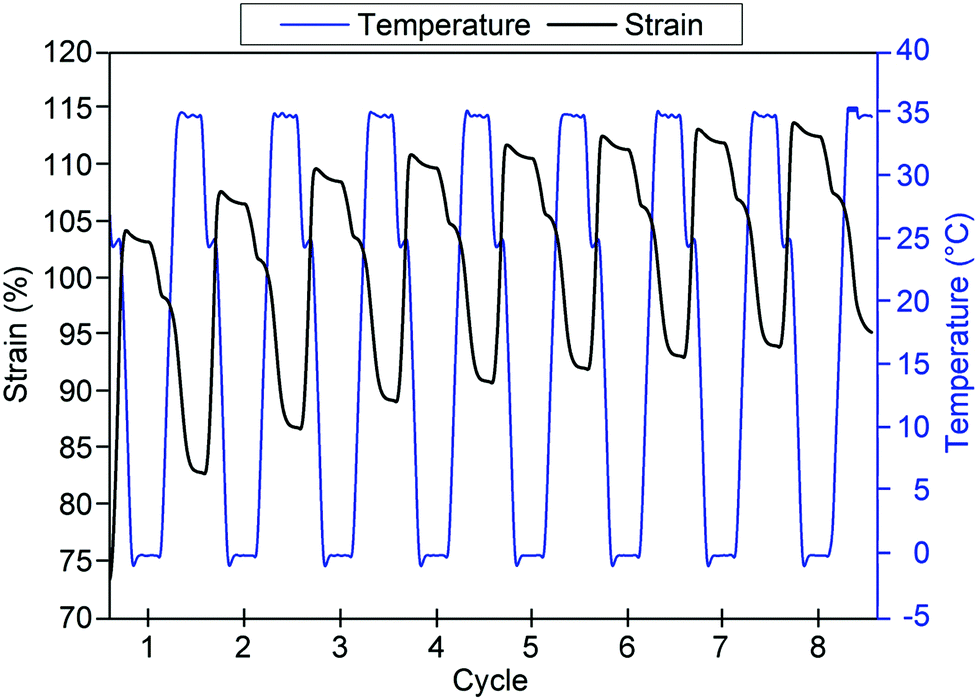
Shape fixity and shape recovery ratios across multiple thermocycles are tabulated in Table 7. Shape fixity was observed to be low in the first cycle but increased in subsequent cycles to 92% and above for GCS-3. Low Rf in the first cycle could be attributed to the relatively low degree of PCL crystallinity (∼14.3%) due to the interference caused by the crystallisation of neighbouring PEG segments68 as PCL forms the thermo-responsive switching segments responsible for the heat-triggered SME. As shape recovery was performed at 35 °C, which is below the Tm,PCL of GCS-3, partial melting of the PCL crystallites occurs. Non-melted crystallites serve as seeds that facilitate recrystallisation during subsequent cooling cycles.69 With subsequent cycles, improved shape fixity could arise from increased PCL crystallinity caused by controlled cooling in each cycle as well as self-nucleation-assisted recrystallisation due to partial melting of PCL crystallites.70,71 The copolymer also exhibited good shape recovery, with Rr generally observed to be above 80% after initial pre-conditioning. Furthermore, the shape recovery ratio of the copolymer was observed to have an increasing trend (Fig. 13), converging at ∼95% after multiple cycles. The improvement of Rf and Rr with pre-conditioning is in good agreement with prior studies,40,41,72 demonstrating the need for pre-deployment conditioning prior to use in order to fully utilize the shape memory potential of this material.
Table 7 Shape fixity and shape recovery ratios of the poly(PEG/PCL/PDMS urethane), GCS-3, over eight thermocycles
| |
Cycle |
| 1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
R
f (%) |
95.3 |
95.6 |
95.5 |
95.6 |
96.4 |
95.5 |
95.5 |
95.6 |
|
R
r (%) |
68.4 |
83.2 |
89.0 |
92.0 |
93.8 |
94.5 |
95.3 |
94.1 |
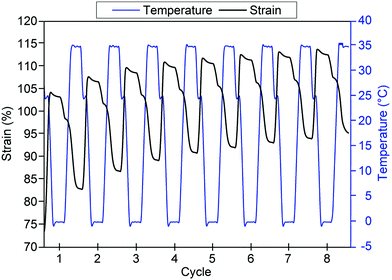 |
| | Fig. 13 Time-dependent temperature and strain profiles for poly(PEG/PCL/PDMS urethane), GCS-3, after initial pre-conditioning. | |
4. Conclusion
Water-triggered shape memory has been demonstrated in PEG–PCL–PDMS thermoplastic polyurethanes (TPUs), achieving good extents of shape recovery and shape fixity of up to 71% and 100%, respectively, with recovery occurring within a span of 20 minutes for GCS-3. The copolymers were also shown to possess heat-triggered shape memory capabilities, with excellent shape fixity (<96%) and shape recovery (<95%) at physiological temperature. Such dual-responsive SMPs show much promise as potential smart materials for biomedical applications for deployment in physiological environments where shape recovery can be triggered by the presence of water and further augmented by a brief heat stimulus to prevent tissue damage from prolonged exposure to elevated temperatures. Alternatives to PEG with greater mechanical strength could be explored to serve as the hydrophilic switching segment to improve the overall mechanical properties of the system. Water-responsive shape memory performance of the copolymers could be improved by processing the copolymers into porous structures to provide a greater surface area and porosity for the faster inward diffusion of water. Future work includes in vitro and in vivo studies to evaluate and optimise the biocompatibility of the material and the evolution of its mechanical properties over extended periods of time.
Acknowledgements
The authors acknowledge the financial support from the Institute of Materials Research and Engineering, A*STAR, Singapore (IMRE/13-2P0806). The authors thank S. Y. Chan for proofreading the manuscript. B. Q. Y. Chan would like to acknowledge the A*STAR Graduate Scholarship.
References
- B. Q. Y. Chan, Z. W. K. Low, S. J. W. Heng, S. Y. Chan, C. Owh and X. J. Loh, ACS Appl. Mater. Interfaces, 2016, 8, 10070–10087 CAS.
-
C. A. Rogers, in Intelligent Structures, ed. K. P. Chong, S. C. Liu and J. C. Li, Elsevier, London, 1990, pp. 3–41 Search PubMed.
- P. T. Mather, X. Luo and I. A. Rousseau, Annu. Rev. Mater. Res., 2009, 39, 445–471 CrossRef CAS.
- H. Meng, H. Mohamadian, M. Stubblefield, D. Jerro, S. Ibekwe, S.-S. Pang and G. Li, Smart Mater. Struct., 2013, 22, 093001 CrossRef.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- J. Leng, X. Lan, Y. Liu and S. Du, Prog. Mater. Sci., 2011, 56, 1077–1135 CrossRef CAS.
- C. Liu, S. B. Chun, P. T. Mather, L. Zheng, E. H. Haley and E. B. Coughlin, Macromolecules, 2002, 35, 9868–9874 CrossRef CAS.
- H. Lu, Y. Liu, J. Leng and S. Du, Eur. Polym. J., 2010, 46, 1908–1914 CrossRef CAS.
- H. Lu, J. Appl. Polym. Sci., 2012, 123, 1137–1146 CrossRef CAS.
- Q. Shou, K. Uto, M. Iwanaga, M. Ebara and T. Aoyagi, Polym. J., 2014, 46, 492–498 CrossRef CAS.
- H. Lu, Y. Yao, W. M. Huang, J. Leng and D. Hui, Composites, Part B, 2014, 62, 256–261 CrossRef CAS.
- X. Luo and P. T. Mather, Soft Matter, 2010, 6, 2146–2149 RSC.
- J. T. Kim, H. J. Jeong, H. C. Park, H. M. Jeong, S. Y. Bae and B. K. Kim, React. Funct. Polym., 2015, 88, 1–7 CrossRef CAS.
- S. S. Mahapatra, S. K. Yadav, H. J. Yoo, M. S. Ramasamy and J. W. Cho, Sens. Actuators, B, 2014, 193, 384–390 CrossRef CAS.
- F. H. Zhang, Z. C. Zhang, C. J. Luo, I. T. Lin, Y. Liu, J. Leng and S. K. Smoukov, J. Mater. Chem. C, 2015, 3, 11290–11293 RSC.
- M. Y. Razzaq, M. Behl, U. Nöchel and A. Lendlein, Polymer, 2014, 55, 5953–5960 CrossRef CAS.
- H. Meng, J. Zheng, X. Wen, Z. Cai, J. Zhang and T. Chen, Macromol. Rapid Commun., 2015, 36, 533–537 CrossRef CAS PubMed.
- Y. Hu, C.-H. Lu, W. Guo, M. A. Aleman-Garcia, J. Ren and I. Willner, Adv. Funct. Mater., 2015, 25, 6867–6874 CrossRef CAS.
- D. Kai, M. P. Prabhakaran, B. Q. Y. Chan, S. S. Liow, S. Ramakrishna, F. Xu and X. J. Loh, Biomed. Mater., 2016, 11, 015007 CrossRef PubMed.
- B. Q. Y. Chan, S. S. Liow and X. J. Loh, RSC Adv., 2016, 6, 34946–34954 RSC.
- D. A. Collins, C. M. Yakacki, D. Lightbody, R. R. Patel and C. P. Frick, J. Appl. Polym. Sci., 2016, 133, 42903 CrossRef.
- X. Gu and P. T. Mather, Polymer, 2012, 53, 5924–5934 CrossRef CAS.
- A. Metcalfe, A.-C. Desfaits, I. Salazkin, L. H. Yahia, W. M. Sokolowski and J. Raymond, Biomaterials, 2003, 24, 491–497 CrossRef CAS PubMed.
- D. Kai, M. J. Tan, M. P. Prabhakaran, B. Q. Y. Chan, S. S. Liow, S. Ramakrishna and X. J. Loh, Colloids Surf., B, 2016, 148, 557–565 CrossRef CAS PubMed.
- S. Mondal and J. L. Hu, Indian J. Fibre Text. Res., 2006, 31, 66–71 CAS.
- H. M. Wache, D. J. Tartakowska, A. Hentrich and M. H. Wagner, J. Mater. Sci.: Mater. Med., 2003, 14, 109–112 CrossRef CAS PubMed.
-
C. Douglas, L. Mark, S. Mark, N. Emmett and S. Randal, 46th AIAA/ASME/ASCE/AHS/ASC Structures, Structural Dynamics and Materials Conference, American Institute of Aeronautics and Astronautics, 2005.
-
J. Xu and J. Song, Orthopedics and Physical Rehabilitation Publications and Presentations, 2011 Search PubMed.
- S. Mondal, Mini-Rev. Org. Chem., 2009, 6, 114–119 CrossRef CAS.
-
J. Hu, Shape Memory Polymers and Textiles, Woodhead Publishing Limited, Cambridge CB21 6AH, England, 2007 Search PubMed.
-
P. Dubruel and S. Van Vlierberghe, Biomaterials for Bone Regeneration: Novel Techniques and Applications, Elsevier Science, 2014 Search PubMed.
- W. M. Huang, B. Yang, Y. Zhao and Z. Ding, J. Mater. Chem., 2010, 20, 3367–3381 RSC.
- J. Mendez, P. K. Annamalai, S. J. Eichhorn, R. Rusli, S. J. Rowan, E. J. Foster and C. Weder, Macromolecules, 2011, 44, 6827–6835 CrossRef CAS.
- B. Yang, W. M. Huang, C. Li, C. M. Lee and L. Li, Smart Mater. Struct., 2004, 13, 191 CrossRef CAS.
-
T. Hatakeyama and F. X. Quinn, Thermal Analysis: Fundamentals and Applications to Polymer Science, Wiley, 1999 Search PubMed.
- X. Gu and P. T. Mather, RSC Adv., 2013, 3, 15783–15791 RSC.
- G. Niu and D. Cohn, Science, 2013, 3, 49–50 Search PubMed.
- W. M. Huang, B. Yang, L. An, C. Li and Y. S. Chan, Appl. Phys. Lett., 2005, 86, 114105 CrossRef.
- C. J. Yong, H. S. Hyang and W. C. Jae, J. Macromol. Sci., Part B: Phys., 2006, 45, 453–461 CrossRef.
- C. A. Schoener, C. B. Weyand, R. Murthy and M. A. Grunlan, J. Mater. Chem., 2010, 20, 1787–1793 RSC.
- D. Zhang, K. M. Petersen and M. A. Grunlan, ACS Appl. Mater. Interfaces, 2013, 5, 6 Search PubMed.
-
A. Cervenka, in Mechanics of Composite Materials and Structures, ed. C. A. M. Soares, C. M. M. Soares and M. J. M. Freitas, Springer, Netherlands, 1999, vol. 361, pp. 289–298 Search PubMed.
-
J. M. Harris, Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications, Springer, US, 2013 Search PubMed.
- H. Sun, L. Mei, C. Song, X. Cui and P. Wang, Biomaterials, 2006, 27, 1735–1740 CrossRef CAS PubMed.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed.
- A. Mata, A. Fleischman and S. Roy, Biomed. Microdevices, 2005, 7, 281–293 CrossRef CAS PubMed.
- C.-C. M. Ma, J.-T. Gu, W.-C. Fang, J.-C. Yang and L.-D. Tsai, J. Appl. Polym. Sci., 1997, 66, 67–75 CrossRef CAS.
- D. J. Buckwalter, D. L. Inglefield, J. S. Enokida, A. G. Hudson, R. B. Moore and T. E. Long, Macromol. Chem. Phys., 2013, 214, 2073–2082 CrossRef CAS.
-
R. G. Jones, W. Ando and J. Chojnowski, Silicon-containing polymers: the science and technology of their synthesis and applications, Springer Science & Business Media, 2013 Search PubMed.
-
S. J. Clarson and J. A. Semlyen, Siloxane Polymers (Ellis Horwood Series in Polymer Science and Technology), Prentice Hall, 1993 Search PubMed.
- X. J. Loh, K. B. C. Sng and J. Li, Biomaterials, 2008, 28, 10 Search PubMed.
- X. Li, X. J. Loh, K. Wang, C. B. He and J. Li, Biomacromolecules, 2005, 6, 8 Search PubMed.
- X. J. Loh, S. H. Goh and J. Li, Biomacromolecules, 2007, 8, 9 CrossRef PubMed.
- M. R. Ramdas, K. S. Kumar and C. R. Nair, J. Mater. Chem. A, 2015, 3, 11596–11606 Search PubMed.
-
X. J. Loh and J. Li, US Pat., US 2013/0018111A1, 2013 Search PubMed.
- X. J. Loh, K. B. Colin Sng and J. Li, Biomaterials, 2008, 29, 3185–3194 CrossRef CAS PubMed.
- X. J. Loh, Y.-L. Wu, W. T. Joseph Seow, M. N. Irzuan Norimzan, Z.-X. Zhang, F.-J. Xu, E.-T. Kang, K.-G. Neoh and J. Li, Polymer, 2008, 49, 5084–5094 CrossRef CAS.
- X. J. Loh, B. J. H. Yee and F. S. Chia, J. Biomed. Mater. Res., Part A, 2012, 100A, 2686–2694 CrossRef CAS PubMed.
- X. J. Loh, S. H. Goh and J. Li, Biomacromolecules, 2007, 8, 585–593 CrossRef CAS PubMed.
- D. De Kee, Q. Liu and J. Hinestroza, Can. J. Chem. Eng., 2005, 83, 913–929 CrossRef CAS.
- J. H. Petropoulos, M. Sanopoulou and K. G. Papadokostaki, diffusion-fundamentals.org, 2009, 11, 5 Search PubMed.
- E. Bagley and F. Long, J. Am. Chem. Soc., 1955, 77, 2172–2178 CrossRef CAS.
- X. J. Loh, G. Roshan Deen, Y. Y. Gan and L. H. Gan, J. Appl. Polym. Sci., 2001, 80, 268–273 CrossRef CAS.
- V. Crescenzi, G. Manzini, G. Calzolari and C. Borri, Eur. Polym. J., 1972, 8, 449–463 CrossRef CAS.
- K. Pielichowski and K. Flejtuch, Polym. Adv. Technol., 2002, 13, 690–696 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1949, 17, 223–240 CrossRef CAS.
- F. H. Silver, P. B. Snowhill and D. J. Foran, Ann. Biomed. Eng., 2003, 31, 793–803 CrossRef PubMed.
- L. Peponi, I. Navarro-Baena, J. E. Báez, J. M. Kenny and A. Marcos-Fernández, Polymer, 2012, 53, 4561–4568 CrossRef CAS.
- J. Zhou, S. A. Turner, S. M. Brosnan, Q. Li, J.-M. Y. Carrillo, D. Nykypanchuk, O. Gang, V. S. Ashby, A. V. Dobrynin and S. S. Sheiko, Macromolecules, 2014, 47, 1768–1776 CrossRef CAS.
- B. Fillon, J. C. Wittmann, B. Lotz and A. Thierry, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1383–1393 CrossRef CAS.
- J. Xu, Y. Ma, W. Hu, M. Rehahn and G. Reiter, Nat. Mater., 2009, 8, 348–353 CrossRef CAS PubMed.
- D. Zhang, M. L. Giese, S. L. Prukop and M. A. Grunlan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00243a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Sylvester Jun Wen
Heng‡
ab,
Sing Shy
Liow
a,
Kangyi
Zhang
a and
Xian Jun
Loh
*abc
ab,
Sylvester Jun Wen
Heng‡
ab,
Sing Shy
Liow
a,
Kangyi
Zhang
a and
Xian Jun
Loh
*abc
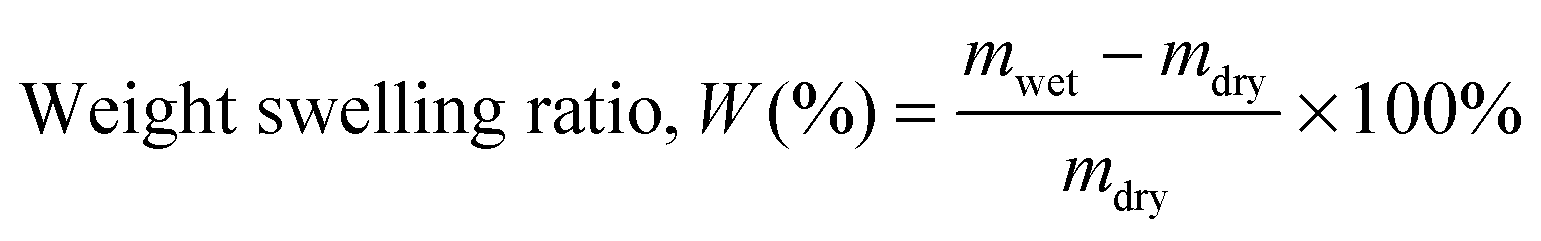
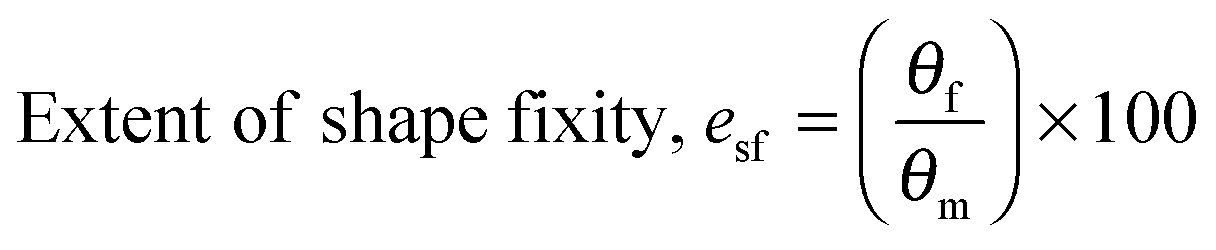
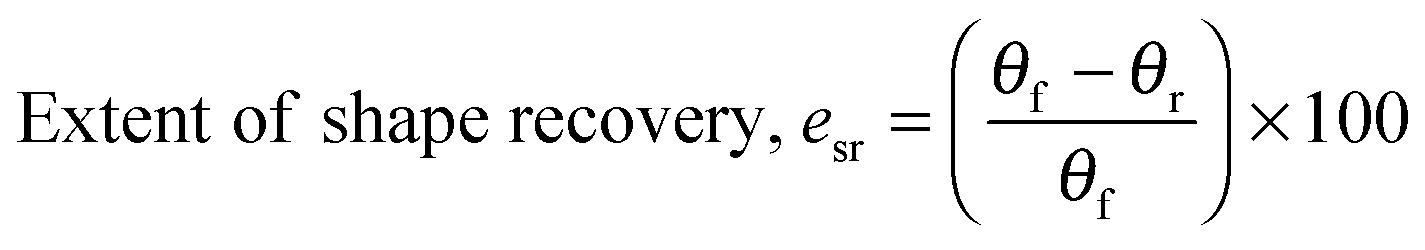
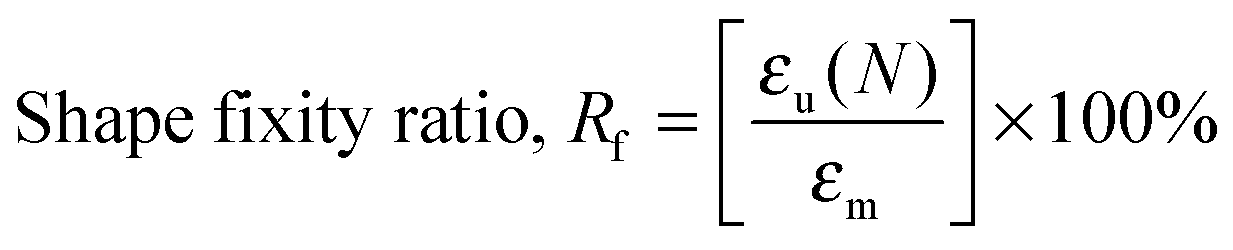
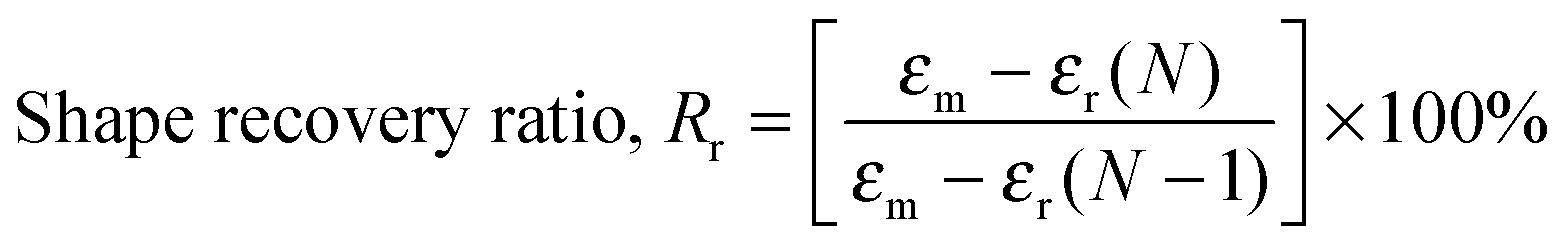
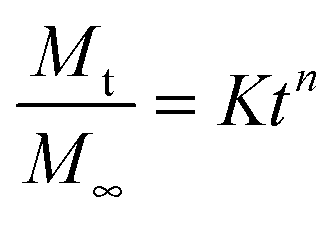
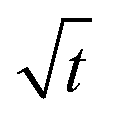 in Fig. 7 appears to comprise a two-step sorption process60 comprising an initial Fickian stage, followed by a structural relaxation stage.
in Fig. 7 appears to comprise a two-step sorption process60 comprising an initial Fickian stage, followed by a structural relaxation stage.