
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Swellable functional hypercrosslinked polymer networks for the uptake of chemical warfare agents†
Craig
Wilson
a,
Marcus J.
Main
b,
Nicholas J.
Cooper
b,
Michael E.
Briggs
 *a,
Andrew I.
Cooper
a and
Dave J.
Adams
*ac
*a,
Andrew I.
Cooper
a and
Dave J.
Adams
*ac
aMaterials Innovation Factory and Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
bDstl, Porton Down, Salisbury, SP4 0JQ, Wiltshire, UK
cSchool of Chemistry, College of Science and Engineering, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 1st March 2017
Abstract
The need for porous materials to function as sorbents in order to allow for bulk uptake (and potential deactivation) of chemical warfare agent (CWA) stockpiles is of significant importance in the world today. Hypercrosslinked polymers (HCPs) represent a class of such sorbents being produced using the facile and tuneable so-called “knitting” procedure. Several HCPs are reported and their properties including apparent Brunauer–Emmett–Teller surface areas (SABET) and swellability (Q) against CWA simulants are examined using two reliable swelling methods which we have developed. The HCP derived from fluorobenzene showed the greatest potential for using such materials for CWA uptake and was tested against real agents including isopropyl methylphosphonofluoridate (sarin, GB) and bis(2-chloroethyl)sulfane (sulfur mustard, HD) revealing uptakes close to 20 mL g−1.
Introduction
The Chemicals Weapons Convention, effective since 1997, has helped to reduce the proliferation of chemical warfare agents (CWAs).1 However, despite this treaty, CWAs are still being used in current conflicts such as in Syria.2 Aging stockpiles of CWAs, particularly in the United States and Russia, also present the risk of accidental release into the environment.3 The toxicity of organophosphate-based nerve agents such as isopropyl methylphosphonofluoridate (sarin, GB) and O-ethylS-[2-(diisopropylamino)ethyl] methylphosphonothioate (VX) arise from attack of these agents upon the central nervous system.4,5 Irreversible binding of the agent to the acetylcholinesterase enzyme, which is responsible for the breakdown of the neurotransmitter acetylcholine, leads to its deactivation. The resulting accumulation of acetylcholine results in the loss of respiratory muscle activity, which leads to oxygen deprivation and subsequently death by asphyxiation.6 Blister/vesicant agents including bis(2-chloroethyl)sulfane (sulfur mustard, HD) cause severe burns and blisters, respiratory tract damage, eye lesions, bone marrow depression, pneumonia, and in some cases death.7,8 The lethal toxicity of CWAs means research into decontamination methods often takes place with simulants rather than the real agents.9,10Porous materials such as activated carbons9,11,12 and metal–organic frameworks (MOFs)13–18 have been shown to be effective in the chemical decomposition of CWAs. MOFs tend to exhibit faster breakdown kinetics compared with activated carbons due to the tunability of their functionality and structure, and higher sorption capacities. However, structural instability may limit their practical application.19 The use of more robust porous networks is therefore of interest.
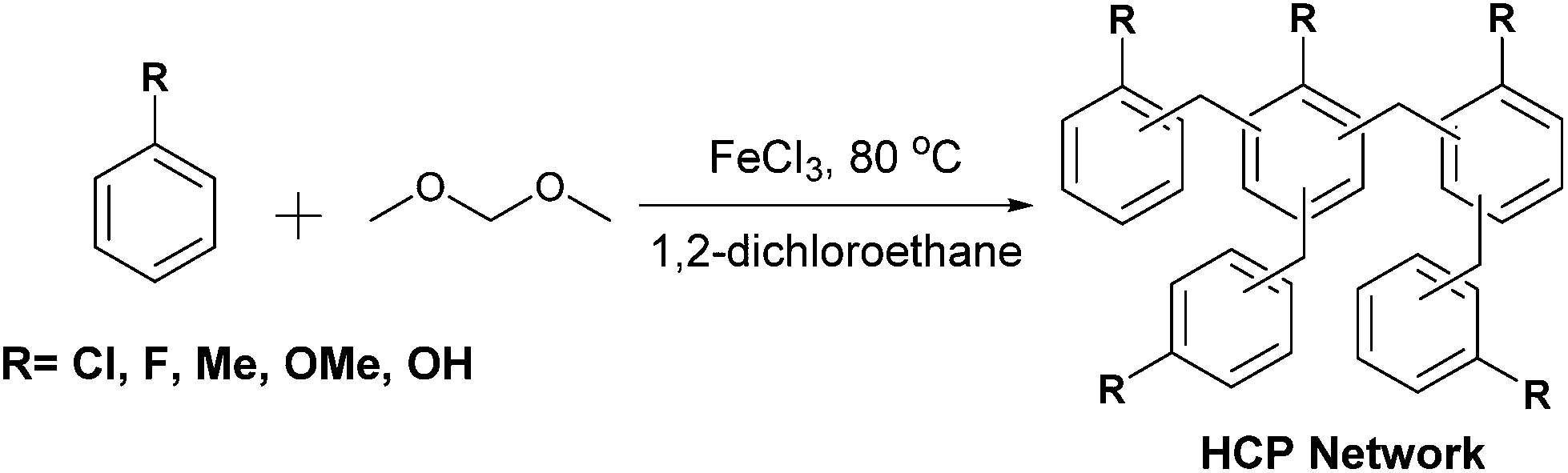
The first examples of hypercrosslinked polymers (HCPs), reported in the 1970s by Davankov and Tsyurupa,20,21 were prepared by crosslinking linear polystyrene chains using multifunctional external crosslinker molecules such as chloromethyl methyl ether. Self-condensation of small molecule monomers such as benzyl alcohol, in which the alcohol is displaced by the aromatic ring, has also resulted in the preparation of HCPs.22 Recently, a procedure known as “knitting” was introduced by Li et al.23 This involves the Friedel–Crafts alkylation of aromatic monomer units by external crosslinkers such as formaldehyde dimethyl acetal (FDA). Apparent BET surface areas (SABET) up to 1391 m2 g−1 have been reported for hypercrosslinked benzene.23 Benzene derivatives such as toluene, phenol, aniline, binaphthol, tetraphenylmethane, and also aromatic heterocycles have also been used to form HCPs using this facile and cheap approach.24–27 HCPs have been used in applications such as gas capture and storage, and heterogeneous catalysis.28 For example, we have demonstrated that a benzene derived HCP, which possesses excellent stability to strongly acidic conditions, uptakes CO2 by physical swelling of the porous network.29
The occupation of the polymeric network by relatively large organic molecules allows for the expansion of the network (i.e. swelling) thereby relieving the inner-stress which is created upon removal of the reaction solvent to form the dry polymer network.30 The swelling of HCPs is largely independent of the properties of the solvent or guest molecule. This is due to the weak inter chain interactions that exist in the polymer network as a result of the high number of bridges introduced during the crosslinking reaction.21
The swellability of HCPs can be affected by a number of parameters including the initial monomer concentration,21 the extent of monomer crosslinking and the solvating state of the growing polymer chains (nature of reaction solvent).31,32 The polarity of the polymer can also influence the swellability of the polymers. For instance, a HCP prepared via self-condensation using FeCl3 demonstrated the liquid phase adsorption of hydroxymethylfurfural (HMF).33 However, synthesis of the HCP using a Brønsted acid resulted in a material with an increased selectivity for HMF; this is thought to be due to the formation of a more hydrophobic, iron free, network.34 Similarly, modification of HCPs with imidazolium ionic liquids helped to increase polarity for enhanced adsorption of catechin from herbal plants.35
The propensity of HCPs to swell has led their use in a number of applications including the trapping of organic vapors,36 ion exchange,37,38 removal of toxic trace metals,39,40 solid phase extraction,41 and recovery of organic compounds from water.42,43 Knitting of the triptycene monomer can produce HCPs which show high adsorption for solvents including DMSO, toluene, and ethyl acetate, with a 30 times increase in the mass of the polymer observed for adsorption of chloroform.44 Functionalised β-cyclodextrins have been used to extract phenol and its derivatives from aqueous solutions.45 Gold nanoparticles have also been incorporated into hypercrosslinked β-cyclodextrin to facilitate catalytic reactions on the adsorbed materials.
An alternative polymeric material which shows good adsorption properties can be created through the polymerisation of high internal phase emulsions (PolyHIPEs).46,47 The macroporous polyHIPEs can be crosslinked to produce hierarchical porous polymer networks which have high adsorption capacities for common organic solvents,48 and can act as catalytic supports.49 In contrast to the synthesis of polyHIPEs, the “knitting” approach forms HCPs in a single step without using surfactants or inorganic silica particles, which in some cases can hinder post modification of the polyHIPEs.50
The swelling of HCPs on exposure to liquids is largely independent to the properties of the liquid, which makes them attractive candidates as universal sorbent materials for uptake of CWAs; entrapping the liquid agent reduces the risk of exposure. Maximising the swelling of HCPs allows for large volumes of agent to be uptaken with minimal amount of polymer. In this paper, we describe the preparation of a number of HCPs (Scheme 1) and measure the swelling of the polymers upon exposure to the CWA simulants triethylamine (NEt3), ethyl acetate (EtOAc), dimethylformamide (DMF), and acetyl chloride (AcCl). Two methods to determine the swelling of the HCPs on exposure to the CWA simulants are discussed, while the most promising HCPs were tested against real CWAs including GB, VX, and HD.
 | ||
| Scheme 1 Synthesis of hypercrosslinked polymer networks. | ||
Experimental
Materials and methods
All reagents were purchased from Sigma-Aldrich and used as received.Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 scans respectively.
100 scans respectively.
The remainder of the spectra were acquired at Dstl (Defence Science and Technology Laboratory). All spectra were recorded at 9.4 Tesla using a Bruker AVIII 400 MHz solid state spectrometer equipped with a 1H/X 4 mm CP/MAS probehead. All samples were loaded into zirconia MAS rotors and spun at 10 kHz at ambient temperature. 1H–13C Cross polarisation (CP) MAS spectra were recorded using a TOSS routine to eliminate spinning sidebands. Typically 1–2 K of scans were recorded and a recycle delay of 10 s used. All spectra were acquired and processed using Bruker Topspin 3.5.
Method 1. To an Eppendorf centrifuge tube (1.5 mL) a known mass of the milled polymer was added (10–30 mg). The simulant (1 mL) was added and the polymer allowed to stand in the chosen simulant for 18 hours. After this time, the height of the swollen polymer in the tube was measured. The obtained height measurements could be converted to the volume occupied by the polymer in the tube by calibrating an empty centrifuge tube (1.5 mL) with known volumes of water (Fig. S16, ESI†). The swellability (Q) of the polymer could be defined by the final volume of the swollen polymer (mL) divided by the mass of polymer used (g). In cases when sections of polymer floated to the top of the tube upon swelling (toluene derived HCP in CWA HD), the volume for the polymer at the bottom of the tube was calculated in the same manner, while the volume of the floating polymer was calculated using πr2h (r = radius of Eppendorf tube and h = height of polymer).
Method 2. The as made polymer (30 mg) was weighed into an empty vial and the simulant (5 mL) added. The polymer was allowed to stand in the simulant for 18 hours. After this time, the excess simulant was collected by filtration of the polymer/simulant suspension (using a funnel fitted with a plug of glass wool) into an empty pre-weighed vial. Measuring the amount of recovered agent allowed the amount of simulant absorbed by the polymer to be calculated. The swellability (Q) of the polymer could be defined by the amount of simulant absorbed (mL) divided by the mass of polymer used (g).
Method 3. The as made polymer (30 mg) was weighed into an empty vial and the simulant of interest (5 mL) added. The polymer was allowed to stand in the simulant for 18 hours. After this time, the vial cap was replaced with an open centred vial cap fitted with filter paper. A needle was inserted through the filter paper and the vial inverted to allow filtration of the excess simulant into an empty pre-weighed vial. Measuring the amount of recovered agent allowed the amount of simulant absorbed by the polymer to be calculated. The swellability (Q) of the polymer could be defined by the amount of simulant absorbed (mL) divided by the mass of polymer used (g).
Results and discussion
Synthesis of HCPs
The HCP polymers were prepared according to Scheme 1 and isolated as either brown or black powders, with the majority of the HCPs being obtained in yields of 90–120% (Fig. S1, ESI†). We varied both the monomer used, as well as the ratio of crosslinker to monomer, with a view to maximising the swellability of the network. Microanalysis data obtained for the HCPs showed disagreements between the theoretical and experimental values (Fig. S2, ESI†). These discrepancies became more pronounced as more of the external crosslinker, FDA was used. Deviation from the theoretical microanalysis and yields greater than 100% are a common occurrence in the synthesis of HCPs.24,25 Adsorption of atmospheric molecules, such as water, entrapped catalyst, or solvent molecules, and deviation from the idealised structure most likely due to incomplete reaction of the crosslinker, which would be more pronounced at higher loading, can help to account for these discrepancies.24,51 Thermogravimetric analysis (TGA) of the HCPs (Fig. S3, ESI†) revealed mass losses between 0.5 and 4.0 wt% for most of the polymers upon heating to 150 °C. The HCP synthesised from the more polar phenol monomer showed higher mass loss, likely due to more water adsorption as a result of hydrogen bonding between adsorbed water molecules and the hydroxyl groups in this network. XPS on the toluene HCPs prepared with 2 and 5 equivalents of FDA revealed 0 and 0.3% residual iron, respectively (Fig. S4, ESI†). An increase in the oxygen content with higher equivalents of FDA crosslinker was also observed, suggesting the presence of defects in the network caused by incomplete bridge formation when higher amounts of FDA are used.SEM micrographs obtained for the fluorobenzene, toluene, chlorobenzene, and anisole derived HCPs, formed using 2 equivalents of FDA, revealed a morphology consisting of rough, amorphous particles (Fig. S5, ESI†). The network prepared from phenol showed a fused sphere morphology, consistent with previously reported HCPs derived from phenol or binaphthol.23,25
Nitrogen adsorption–desorption isotherms for the HCPs revealed a combination of type I and type IV isotherms, with apparent SABET of between 58 and 872 m2 g−1 (Fig. S6, ESI†). Hysteresis was more significant for the most porous HCPs, which may be due to capillary condensation of nitrogen in mesopores during adsorption.52 The total pore volumes, as calculated from the nitrogen isotherms showed an increase in the total pore volume with increasing apparent SABET of the networks (Fig. S6.4, ESI†).
IR spectroscopy carried out on five of the toluene HCPs (Fig. 1) showed peaks at 1600, 1500, and 1450 cm−1 which occur due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches from the aromatic rings in the network. The peak at 3000 cm−1 can be assigned to the C–H stretching vibrations of the aromatic rings, which weakens in its intensity along with the bending vibrations (900–600 cm−1) as more FDA crosslinker is used. With increasing amounts of crosslinker, the peak occurring at ca. 1100 cm−1, usually arsing due to C–O stretches, becomes more intense. Several peaks in the 1670–1760 cm−1 region of the spectra also become more intense (Fig. 1) with increasing amounts of crosslinker, with their exact position varying from polymer to polymer (Fig. S7, ESI†).
C stretches from the aromatic rings in the network. The peak at 3000 cm−1 can be assigned to the C–H stretching vibrations of the aromatic rings, which weakens in its intensity along with the bending vibrations (900–600 cm−1) as more FDA crosslinker is used. With increasing amounts of crosslinker, the peak occurring at ca. 1100 cm−1, usually arsing due to C–O stretches, becomes more intense. Several peaks in the 1670–1760 cm−1 region of the spectra also become more intense (Fig. 1) with increasing amounts of crosslinker, with their exact position varying from polymer to polymer (Fig. S7, ESI†).
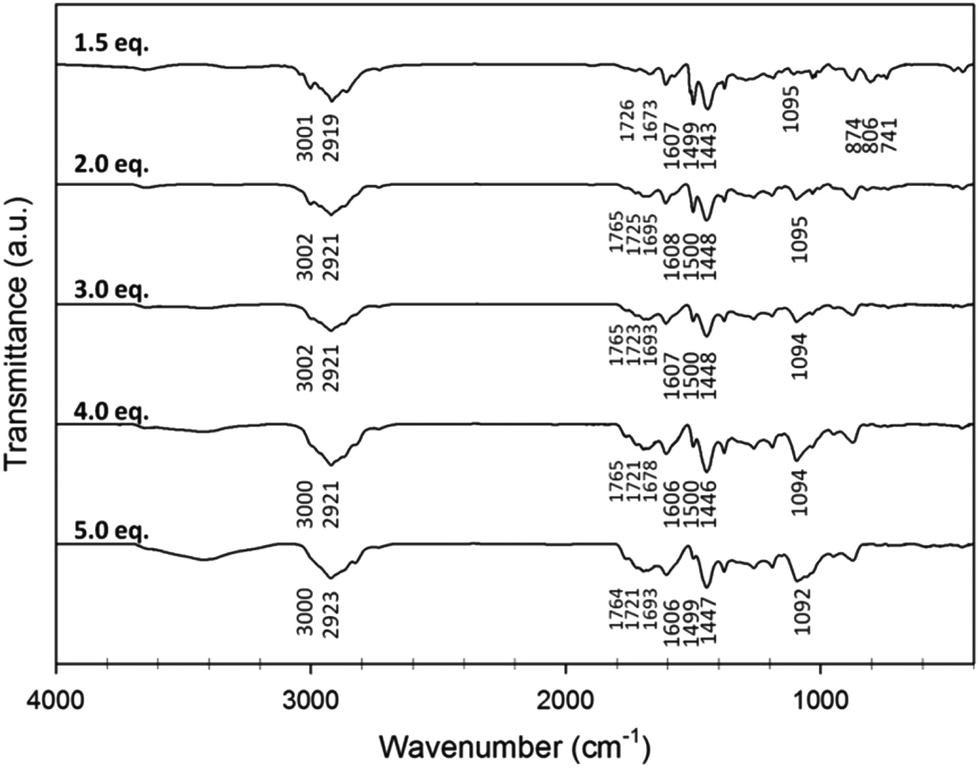 | ||
| Fig. 1 IR spectrum for toluene HCPs prepared with 1.5–5.0 equivalents of FDA. | ||
Peaks occurring in the 1670–1760 cm−1 region of the IR spectra for HCPs have previously been assigned to carbonyl groups, formed by oxidation occurring due to the mildly oxidising conditions of the reaction.53–55 Vibration overtones of substituted benzene rings can also occur as high as 1600–2000 cm−1, suggesting other assignments of these peaks are possible.56 Several test reactions were carried out to see if and how carbonyl groups could form from the monomers used in this study. Treatment of the toluene monomer or FDA crosslinker with FeCl3 did not result in carbonyl formation (Fig. S8 and S9, ESI†). The reaction of benzyl methyl ether with FeCl3 was also carried out to establish if oxidation of any unreacted methoxy groups during HCP formation could give rise to aldehyde or carboxylic acid groups. The expected porous solid produced via self-condensation of the monomer in the absence of crosslinker had an apparent SABET of 529 m2 g−1.57 The FTIR spectrum for the solid produced did show peaks at 1657 and 1709 cm−1 (Fig. S10, ESI†), however, further evidence to support these peaks occurring due to the presence of carbonyl groups proved difficult due to the insoluble nature of the porous solid.
13C cross-polarization magic-angle spinning NMR was carried out on the toluene HCP networks prepared with varying equivalents of FDA (Fig. S11a, ESI†). The spectra showed peaks at 135 and 129 ppm for the HCP prepared with 1.5 equivalents of FDA, which can be assigned to substituted and non-substituted aromatic carbons respectively. The peak assigned to the substituted carbon predominates for the HCPs prepared with greater than 2 equivalents of FDA. The peak at 36 ppm corresponding to the carbon of the methylene linker was present for all HCPs, along with the peak at 20 ppm, which corresponds to the methyl carbon of the toluene monomer. The presence of this peak confirms the methyl group survives the crosslinking process and is unlikely to undergo oxidation. This is in agreement with toluene HCPs prepared previously.23 Very low intensity peaks at 192 and 168 ppm also occurred in the spectrum for the HCP prepared with 5 equivalents of FDA. This indicates that very few carbonyl groups may exist in the polymer network. It is also worth noting the existence of peaks at 59 and 69 ppm in the spectrum of the HCP prepared with 5 equivalents of FDA, which is absent from the spectra of the HCPs prepared with fewer equivalents. These peaks are consistent with the aliphatic carbons present in benzyl methyl ether (Fig. S12, ESI†), which supports the theory that incomplete crosslinking is more likely with higher equivalents of FDA due to incomplete bridge formation between aromatic monomer units; this observation has previously been reported for carbazole based HCPs.58
The CP MAS NMR for the HCPs prepared with monomers other than toluene showed the same characteristic peaks (Fig. S11b, ESI†). The spectra obtained for HCPs derived from fluorobenzene, anisole, and phenol have a peak at 160 ppm (150 ppm for the phenol derived HCP) as a result of the aromatic carbon directly attached to the fluorine, methoxy, or hydroxyl group, respectively. In addition, the anisole derived HCP also exhibited a peak at 60 ppm from the methyl carbon attached directly to the oxygen. Lower intensity peaks are present in the 180–200 ppm region for the spectra of the HCPs derived from phenol and anisole, similar to the toluene spectra with 5 equivalents of FDA. A peak at 15 ppm also features in the phenol and anisole based spectra, which may be due to methylation of the aromatic rings by the acetal crosslinker.29
In order for the HCPs to be used for CWA decontamination, the polymers must possess good stability towards basic and acidic conditions for a prolonged period of time. This was demonstrated by subjecting the HCP derived from fluorobenzene to strongly acidic (1 M aqueous sulfuric acid) or basic (1 M aqueous sodium hydroxide) solutions. Exposure to the acid or base for 24 hours had no noticeable effect on the porosity of the network (as determined from the apparent SABET before and after treatment), while no chemical change was observed by IR spectroscopy and elemental analysis (Fig. S18, ESI†).
Swelling methods
In contrast to polymers such as styrene-divinylbenzene, prepared via free radical copolymerisation,21 the powdery nature of HCPs can make it difficult to obtain a quantitative measure for their swelling in liquids. In order to allow for testing of the HCPs with CWA simulants and real agents, reliable, reproducible and scalable methods needed to be developed. In order to achieve this we developed and evaluated three methods. Eppendorf tubes were used in Method 1 to determine the increase in polymer volume on swelling. Methods 2 and 3 both involved recovery of the excess agent by filtration after mixing with the polymer, which allowed the amount of simulant absorbed by the polymer to be calculated.Methods 1–3 were tested using the toluene and fluorobenzene derived HCPs with the CWA simulants NEt3, EtOAc, and DMF with differences between the methods assessed (Fig. S13, ESI†). Method 3 resulted in the largest error between measurements due to loss of simulant during the filtration stage, while the powdery nature of the HCPs can lead to clogging of the filter paper, preventing all of the excess simulant from being recovered. In addition, Method 3 has an increased exposure risk due to the time taken to perform the measurement. Methods 1 and 2 give a better match; however, notably higher Q values were obtained using Method 2. Filtration of a series of polymer free simulants with a range of volumes using Method 2 revealed on average 0.5 mL of simulant is lost during the filtration stage in Method 2 (Fig. S14, ESI†). Retention of the simulant by the glass wool plug and adhesion of simulant to the walls of the sample vial is likely to give rise to this loss. Taking the loss into account results in an approximate halving of the initially determined Q values, which affords a closer agreement between Methods 1 and 2 (Fig. 2).
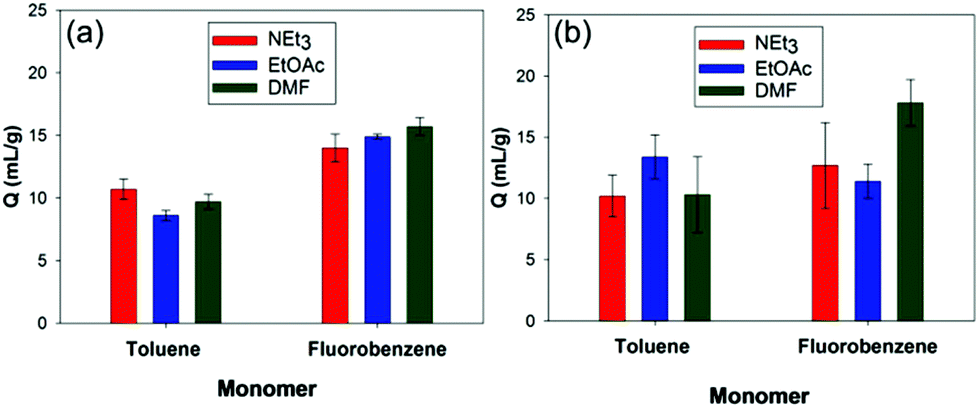 | ||
| Fig. 2 Comparison of the swellability (Q) for toluene and fluorobenzene derived HCPs (prepared using 2 equivalents of FDA) against the three simulants triethylamine (NEt3), ethyl acetate (EtOAc), and dimethylformamide (DMF) using (a) Method 1 and (b) Method 2. | ||
Method 1 does not result in any transfer of the polymer/simulant mixture and therefore no simulant is lost during the measurement. As a consequence this method limits potential exposure to the simulant or CWA. However, the scale-up of this method for increased quantities of polymer may be more difficult due to the size of the Eppendorf tubes. As a result, we chose to test the scalability of Method 2 using the toluene and fluorobenzene derived HCPs against ethyl acetate as simulant (Fig. S15, ESI†).
Addition of both polymers to a fixed volume of ethyl acetate (5 mL) required 0.5–0.6 g of polymer for complete uptake of ethyl acetate, with the toluene derived HCP requiring a larger mass for complete adsorption (Fig. S15, ESI†). This supports the results from the small scale experiments (Fig. 2) in which the fluorobenzene derived HCP has a higher uptake. No release of the sequestered liquid was observed following complete uptake of the ethyl acetate. Due to good correlation between Methods 1 and 2, all further swelling was calculated using Method 1.
Varying synthesis conditions upon porosity and swellability
The effect of varying the amount of FDA crosslinker on the apparent SABET was investigated for toluene derived HCPs (Fig. 3). Increasing of the amount of FDA resulted in a gradual increase in the apparent SABET of the polymers as more crosslinking between monomer units occurred. The surface area peaked for the HCP prepared using 2 equivalents of FDA, and is consistent with previously reported literature values.23 Further increases in the amount of crosslinker used in the Friedel–Crafts reaction resulted in little change to the apparent SABET of the networks. An increase in the steric hindrance around the central aromatic ring with increasing crosslinking likely prevents formation of the theoretical structure, in which an increasing number of aromatic positions are crosslinked. Consequently, the resulting networks exhibit a similar structure that is obtained when 2 equivalents of the crosslinker are used. This is likely to explain why excessive equivalents of FDA are often not used for the synthesis of HCPs via this method.23,24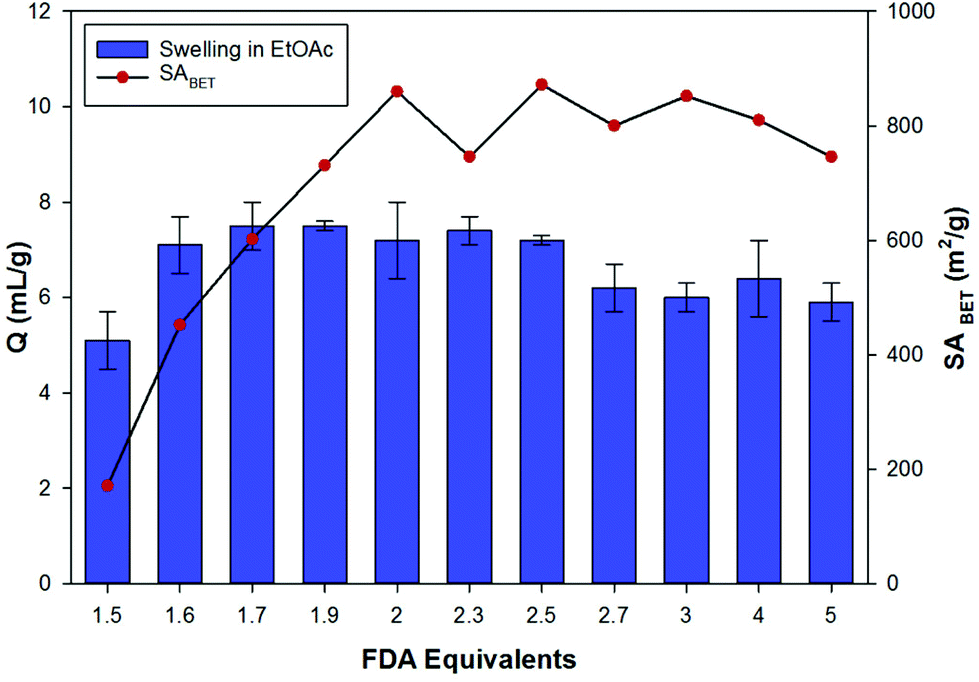 | ||
| Fig. 3 Variation of the apparent BET surface area (SABET) and swellability (Q) in ethyl acetate, for the toluene derived HCPs. | ||
Evaluation of the swellability and hence uptake of the toluene derived networks was carried out using ethyl acetate as a CWA simulant (Fig. 3). Almost all of the toluene derived HCPs have uptakes of 6–8 mL g−1 upon exposure to ethyl acetate. The HCPs prepared with less than 2 equivalents of FDA still showed a volume increase in line with the more highly porous HCPs. While the more porous networks (greater than or equal to 2 equivalents of FDA) are likely to have greater inner-stress in their dry state, the increased flexibility of the networks produced when using lower amounts of crosslinker (1.5 equivalents) is likely to result in a constant trend in the swelling behaviour across the FDA series.
Changing the nature of the monomer allowed for a series of HCPs to be produced (Table 1). The monomers vary in their reactivity towards the Friedel–Crafts alkylation reaction, with all the HCPs prepared using fixed amounts of FDA (2 equivalents).
| Monomer | SABET (m2g−1) | Swellability Q (mL g−1) | |||
|---|---|---|---|---|---|
| NEt3 | EtOAc | DMF | AcCl | ||
| Q values calculated as an average of three measurements, with error (shown in brackets) calculated as the standard deviation. | |||||
| Chlorobenzene | 88 | 7.3 (±0.5) | 6.8 (±0.1) | 7.1 (±0.3) | 6.9 (±0.5) |
| Fluorobenzene | 483 | 14.0 (±1.1) | 14.9 (±0.2) | 15.7 (±0.7) | 16.1 (±0.8) |
| Toluene | 860 | 10.7 (±0.8) | 8.6 (±0.4) | 9.7 (±0.6) | 11.5 (±0.6) |
| Anisole | 627 | 7.6 (±1.6) | 7.5 (±1.7) | 8.5 (±1.6) | 8.5 (±1.3) |
| Phenol | 58 | 4.0 (±0.2) | 4.4 (±0.3) | 6.4 (±1.1) | 4.3 (±0.1) |
The HCP derived from the chlorobenzene monomer exhibited a much lower apparent SABET compared with the fluorobenzene derived HCP. The lower reactivity of the chlorobenzene monomer towards the Friedel–Crafts reaction likely results in less crosslinking and therefore a lower apparent SABET when prepared using 2 equivalents of FDA. In addition, there are a number of factors which could affect the apparent SABET including the molecular weight and relative size of the substituent attached to the aromatic ring; both of which would result in less nitrogen being adsorbed for the chlorobenzene derived HCP. Interestingly, when 3 equivalents of FDA were used to crosslink chlorobenzene, a HCP network with a 319 m2 g−1 apparent SABET was produced (Fig. 4a), indicating incomplete crosslinking occurred with 2 equivalents of FDA. HCPs formed using chlorobenzene (3 equivalents) and phenol (2 equivalents) have previously been reported with apparent SABET of 438 m2 g−1 and 400 m2 g−1, respectively.23 Increasing the amount of FDA used to 3 equivalents for the other monomers showed the change in surface area is more pronounced for the two least reactive monomers, chlorobenzene and fluorobenzene (Fig. 4a). There is however very little change in the surface area for the other HCPs with no distinct increase in the surface area for the phenol derived HCP.
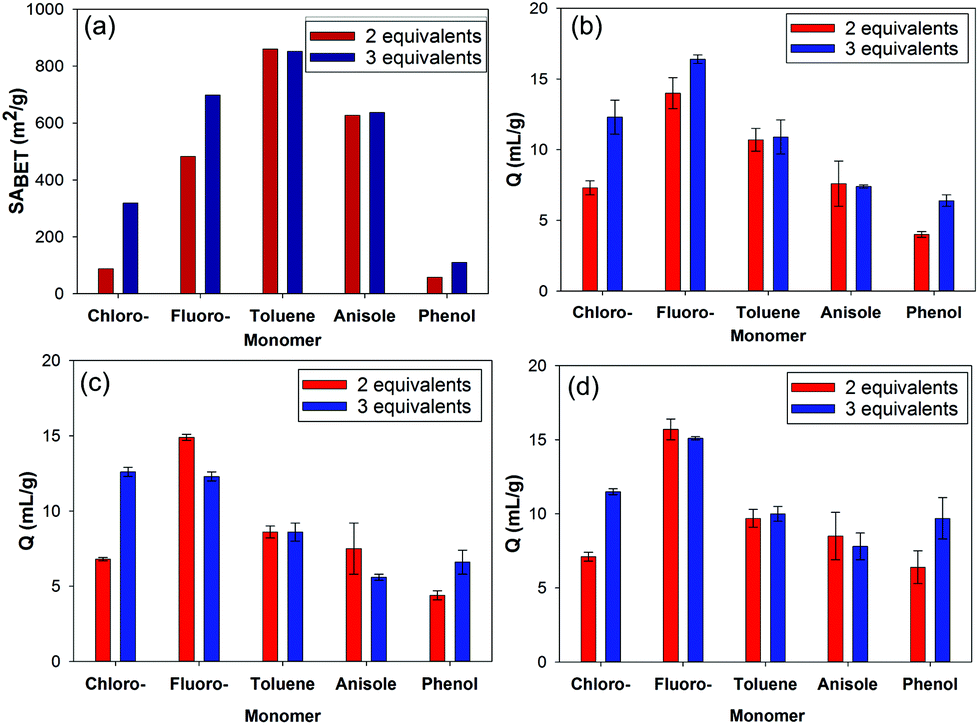 | ||
| Fig. 4 (a) Apparent SABET and swellability (Q) in; (b) triethylamine, (c) ethyl acetate, and (d) DMF for the HCPs prepared using 2 equivalents and 3 equivalents of FDA as crosslinker. | ||
Our group has consistently found a low porosity polymer network is produced when using phenol to form the HCPs.59 A similar case is also observed when using the monomer aniline, which has enhanced reactivity towards the Friedel–Crafts reaction compared to phenol.24 The low porosity observed when using monomers such as phenol and aniline may arise due to the interaction of the hydroxyl or amine units of the monomers with the iron(III) catalyst resulting in partial deactivation of the monomers. As shown here, using anisole as the monomer still gives rise to a porous HCP network but with lower apparent SABET compared with the toluene derived network, despite the anisole monomer being more reactive.
The swellability of the HCPs in a range of simulants confirmed that the swelling of the polymers is largely solvent independent, as expected for hypercrosslinked polymers.21 The greatest degree of swelling was obtained for the fluorobenzene derived HCP, despite it not having the largest apparent SABET. HCPs which have the largest apparent SABET are likely to exhibit a higher degree of crosslinking which results in less pore collapse in the network on drying.60 While increasing the degree of crosslinking can enable an increase in the swelling of the network, as demonstrated by the increased swelling of the fluorobenzene derived HCP compared with the chlorobenzene derived HCP (Table 1), high levels of crosslinking may hinder expansion of the rigid polymer network. This may account for the fluorobenzene derived HCP having a larger degree of swelling compared to the HCP with the largest apparent SABET, derived from the more reactive toluene monomer. Despite the lower apparent SABET obtained for the chlorobenzene and phenol based networks, they still show a reasonable swelling of between 4 and 7 mL g−1.
While both the chlorobenzene and fluorobenzene derived HCPs both show an increase in their apparent SABET on increasing to 3 equivalents of FDA, only the chlorobenzene HCP shows a large increase in its swelling with the simulants (Fig. 4b–d). The swelling of this network increased to roughly that observed for the fluorobenzene derived HCP.
While increasing crosslinking, when using 3 equivalents of FDA can allow for more inner-stress of the HCP network in the dry state, it is likely that this may also lead to a reduction in the mobility of the network, preventing further expansion (swelling) for the fluorobenzene derived HCP, prepared using 3 equivalents of FDA. The swelling of the phenol derived HCP can also benefit from increasing the amount of crosslinker to 3 equivalents, despite the apparent SABET not increasing to a great extent.
Other porous materials including activated carbon, y-zeolite, molecular sieves and CMP-1 were tested against the simulants used in this study (Fig. S17, ESI†). These materials gave much lower swelling values (Q) compared with the majority of the HCPs prepared here, especially the higher swelling HCPs derived from fluorobenzene and toluene, highlighting the advantage HCPs possess for CWA uptake.
Real agent testing
The two most promising HCPs derived from fluorobenzene and toluene, both synthesised with 2 equivalents of FDA crosslinker, were tested against the CWAs GB, VX, and HD (Fig. 5). Both HCPs were found to be effective sorbents for all three agents, despite differences in the chemical structure and physical properties of the agents.61 The fluorobenzene derived HCP shows an enhanced performance against HD and GB, with values close to 20 mL g−1. This is in agreement with the small scale experiments carried out with the CWA simulants, which showed a higher swellability for the fluorobenzene derived HCP. This validates the work with the simulants, and also shows the potential of the HCPs as sorbents for CWAs.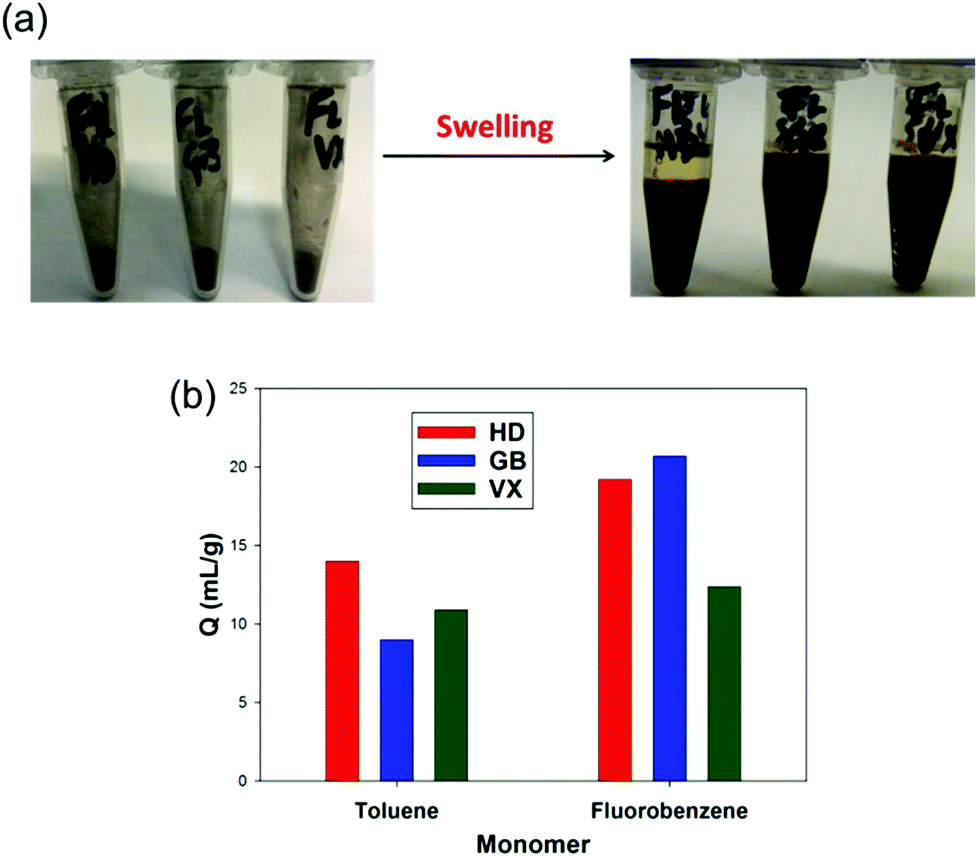 | ||
| Fig. 5 (a) Before and after swelling of the fluorobenzene derived HCP against CWAs; HD, GB, and VX. (b) Swellability (Q) for the toluene and fluorobenzene derived HCPs against the three CWAs. | ||
Conclusions
We have developed two methods for the determination of the swellability of HCP type polymers. While the first method is clearly the most suitable method for the smaller scale testing of the polymers against CWAs and their simulants, the good comparison between Method 1 and 2 enables larger scale testing to be carried out using Method 2.While varying the synthesis conditions by increasing the amount of the external crosslinker, FDA, can result in a clear change in the apparent SABET of the networks, the swellability of the HCPs in ethyl acetate remains independent of the porosity of the network. Increasing the amount of crosslinker to greater than 2 equivalents serves no real benefit with regards to increasing the surface area or the swellability of the networks in most cases. This works also suggests the existence of peaks that occur in the carbonyl region of the IR spectra are not due to the presence of carbonyl groups, but are likely the CC stretching from substituted aromatic rings, which grow in intensity as a higher degree of substituted aromatic rings are formed with increasing amounts of FDA. These findings support recent work upon the assignment of these peaks in HCP networks,62,63 and explains why the position of these peaks are dependent upon the monomer used to form the HCP.
The small amount of mass lost from the polymers and the minimal levels of iron residues in the polymers as measured by TGA and XPS, respectively, indicate the discrepancies in the microanalysis data and greater than 100% yields are more a consequence of non-ideal network formation rather than the presence of contaminants in the polymer. The solid state NMR data suggests the formation of benzyl methyl ether carbons when the greatest amounts of crosslinker were used. This indicates incomplete crosslinking has taken place with larger amounts of FDA, which is not accounted for when calculating the theoretical mass and CHN data.
Several HCPs were produced using the knitting approach with a fixed amount of external crosslinker (2 equivalents). The swelling of the HCPs in a range of CWA simulants is largely independent of the nature of the simulant and appears only to be restricted by the porosity of the networks when HCPs are formed with low apparent SABET. Increasing the amount of external crosslinker to 3 equivalents shows only an increase in the apparent SABET for the two least reactive monomers (chlorobenzene and fluorobenzene). By contrast, chlorobenzene is the only HCP which shows an increase in swelling upon increasing to 3 equivalents. The HCP derived from fluorobenzene is also seen to have the larger swellability despite not having the highest apparent SABET. This work suggests the swellability of the HCPs is not directly proportional to the apparent SABET of the polymers. As a result, maximising the surface area to increase the swelling of the HCPs is likely to be effective only in cases were low porosity HCPs are initially produced. HCPs derived from toluene and fluorobenzene are also effective for the uptake of real agents including HD, GB, and VX.
The ability of HCPs to uptake large volumes of liquid by swelling of the porous networks, as demonstrated here, is a property which could be utilised in many other applications, for instance in medical applications in the uptake of wound exudate. Maximising the swellability of HCPs, alongside the incorporation of catalytic groups into the networks to allow for chemical decontamination of agents will be the next point of focus, in order to work towards the development of a universal polymeric material which is able to uptake and destroy stockpiles of CWAs.
Acknowledgements
The authors would like to thank Defence Science and Technology Laboratory (Dstl) for funding, as well as testing of the polymers against the real agents and solid-state NMR services provided by Dr James T. A. Jones. DJA thanks the EPSRC for a Fellowship (EP/L021978/1). We thank the UK National Solid-state NMR Service at Durham for solid-state NMR services. SEM imaging was obtained by Rob Clowes and Dr Tom Mitra. XPS measurements were obtained by Dr David Hesp and Dr Vin Dhanak at the Stephenson Institute for Renewable Energy at the University of Liverpool. CMP-1 was provided by Ammar Alahmed.Notes and references
- P. Convention on the Prohibition of the Development, Stockpiling and Use of Chemical Weapons and on Their Destruction, Available at: https://www.opcw.org, Accessed 17th October 2016.
- M. Enserink and J. Kaiser, Science, 2013, 341, 1050–1051 CrossRef CAS PubMed.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- F. R. Sidell and J. Borak, Ann. Emerg. Med., 1992, 21, 865–871 CrossRef CAS PubMed.
- A. Watson, D. Opresko, R. Young, V. Hauschild, J. King and K. Bakshi, Organophosphate Nerve Agents, Elsevier Academic Press Inc, San Diego, 2009 Search PubMed.
- A. N. Bigley and F. M. Raushel, Biochim. Biophys. Acta, 2013, 1834, 443–453 CrossRef CAS PubMed.
- K. G. Davis and G. Aspera, Ann. Emerg. Med., 2001, 37, 653–656 CrossRef CAS PubMed.
- K. Kehe and L. Szinicz, Toxicology, 2005, 214, 198–209 CrossRef CAS PubMed.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed.
- Y. Liu, A. J. Howarth, N. A. Vermeulen, S.-Y. Moon, J. T. Hupp and O. K. Farha, Coord. Chem. Rev., 2016 DOI:10.1016/j.ccr.2016.11.008.
- J. V. Romero, J. W. H. Smith, B. M. Sullivan, M. G. Mallay, L. M. Croll, J. A. Reynolds, C. Andress, M. Simon and J. R. Dahn, ACS Comb. Sci., 2011, 13, 639–645 CrossRef CAS PubMed.
- J. W. H. Smith, J. V. Romero, T. R. Dahn, K. Dunphy, L. M. Croll and J. R. Dahn, J. Hazard. Mater., 2012, 235, 279–285 CrossRef PubMed.
- Y. Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS PubMed.
- E. Lopez-Maya, C. Montoro, L. M. Rodriguez-Albelo, S. D. A. Cervantes, A. A. Lozano-Perez, J. L. Cenis, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2015, 54, 6790–6794 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley, P. Ghosh, P. L. Liao, W. Bury, G. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- S. Y. Moon, Y. Y. Liu, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 6795–6799 CrossRef CAS PubMed.
- G. W. Peterson, S. Y. Moon, G. W. Wagner, M. G. Hall, J. B. DeCoste, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 9684–9686 CrossRef CAS PubMed.
- S. S. Mondal and H. J. Holdt, Angew. Chem., Int. Ed., 2016, 55, 42–44 CrossRef CAS PubMed.
- G. W. Peterson, G. W. Wagner, A. Balboa, J. Mahle, T. Sewell and C. J. Karwacki, J. Phys. Chem. C, 2009, 113, 13906–13917 CAS.
- V. A. Davankov, S. V. Rogozhin and M. P. Tsyurupa, USSR Pat299165, 1969 Search PubMed.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- Y. L. Luo, S. C. Zhang, Y. X. Ma, W. Wang and B. Tan, Polym. Chem., 2013, 4, 1126–1131 RSC.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- R. Dawson, T. Ratvijitvech, M. Corker, A. Laybourn, Y. Z. Khimyak, A. I. Cooper and D. J. Adams, Polym. Chem., 2012, 3, 2034–2038 RSC.
- R. Dawson, L. A. Stevens, T. C. Drage, C. E. Snape, M. W. Smith, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 10741–10744 CrossRef CAS PubMed.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- Y. L. Luo, B. Y. Li, W. Wang, K. B. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Snape, T. C. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028–9035 CrossRef CAS PubMed.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193–203 CrossRef CAS.
- X. S. Tang, W. Jia and J. Yan, J. Appl. Polym. Sci., 2004, 94, 2041–2049 CrossRef CAS.
- X. Zhao, X. H. Wang and J. Yan, J. Appl. Polym. Sci., 2004, 92, 997–1004 CrossRef CAS.
- C. Detoni, C. H. Gierlich, M. Rose and R. Palkovits, ACS Sustainable Chem. Eng., 2014, 2, 2407–2415 CrossRef CAS.
- K. Schute and M. Rose, ChemSusChem, 2015, 8, 3419–3423 CrossRef CAS PubMed.
- X. Y. Wu, Y. Liu, Y. F. Liu, D. L. Di, M. Guo and L. Zhao, RSC Adv., 2015, 5, 72601–72609 RSC.
- V. V. Podlesnyuk, J. Hradil and E. Kralova, React. Funct. Polym., 1999, 42, 181–191 CrossRef CAS.
- M. G. Kiseleva, L. V. Radchenko and P. N. Nesterenko, J. Chromatogr. A, 2001, 920, 79–85 CrossRef CAS PubMed.
- P. N. Nesterenko, P. A. Kebets and Y. V. Volgin, J. Anal. Chem., 2001, 56, 715–720 CrossRef CAS.
- N. A. Penner and P. N. Nesterenko, J. Chromatogr. A, 2000, 884, 41–51 CrossRef CAS PubMed.
- M. H. Tai, B. Saha and M. Streat, React. Funct. Polym., 1999, 41, 149–161 CrossRef CAS.
- N. Fontanals, M. Galia, P. A. G. Cormack, R. M. Marce, D. C. Sherrington and F. Borrull, J. Chromatogr. A, 2005, 1075, 51–56 CrossRef CAS PubMed.
- B. C. Pan, Y. Xiong, Q. Su, A. M. Li, J. L. Chen and Q. X. Zhang, Chemosphere, 2003, 51, 953–962 CrossRef CAS PubMed.
- A. M. Li, H. S. Wu, Q. X. Zhang, G. C. Zhang, C. Long, Z. H. Fei, F. Q. Liu and J. L. Chen, Chin. J. Polym. Sci., 2004, 22, 259–267 CAS.
- C. Zhang, P. C. Zhu, L. Tan, J. M. Liu, B. Tan, X. L. Yang and H. B. Xu, Macromolecules, 2015, 48, 8509–8514 CrossRef CAS.
- H. Y. Li, B. Meng, S. H. Chai, H. L. Liu and S. Dai, Chem. Sci., 2016, 7, 905–909 RSC.
- S. D. Kimmins and N. R. Cameron, Adv. Funct. Mater., 2011, 21, 211–225 CrossRef CAS.
- N. R. Cameron, Polymer, 2005, 46, 1439–1449 CrossRef CAS.
- X. J. Yang, L. X. Tan, L. L. Xia, C. D. Wood and B. Tan, Macromol. Rapid Commun., 2015, 36, 1553–1558 CrossRef CAS PubMed.
- I. Pulko, J. Wall, P. Krajnc and N. R. Cameron, Chem. – Eur. J., 2010, 16, 2350–2354 CrossRef CAS PubMed.
- R. T. Woodward, D. W. H. Fam, D. B. Anthony, J. D. Hong, T. O. McDonald, C. Petit, M. S. P. Shaffer and A. Bismarck, Carbon, 2016, 101, 253–260 CrossRef CAS.
- V. A. Davankov and M. P. Tsyurupa, in Hypercrosslinked Polymeric Networks and Adsorbing Materials: Synthesis, Properties, Structure, and Applications, Elsevier Science Bv, Amsterdam, 2011, vol. 56 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- N. Grassie, C. Schoff and J. G. Cunningham, Eur. Polym. J., 1976, 12, 647–650 CrossRef CAS.
- R. V. Law, D. C. Sherrington, C. E. Snape, I. Ando and H. Kurosu, Macromolecules, 1996, 29, 6284–6293 CrossRef CAS.
- M. Streat and L. A. Sweetland, React. Funct. Polym., 1997, 35, 99–109 CrossRef CAS.
- K. Nakanishi, Infrared Absorption Spectroscopy, Practical, Nankodo Company Ltd, Tokyo, 1962 Search PubMed.
- C. D. Wood, B. Tan, A. Trewin, H. J. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- J. H. Zhu, Q. Chen, Z. Y. Sui, L. Pan, J. G. Yu and B. H. Han, J. Mater. Chem. A, 2014, 2, 16181–16189 CAS.
- R. Dawson, T. Ratvijitvech and M. Corker, Unpublished work.
- L. J. Abbott and C. M. Colina, Macromolecules, 2014, 47, 5409–5415 CrossRef CAS.
- B. Singh, G. K. Prasad, K. S. Pandey, R. K. Danikhel and R. Vijayaraghavan, Def. Sci. J., 2009, 60, 428–441 CrossRef.
- M. P. Tsyurupa, Z. K. Blinnikova, Y. A. Davidovich, S. E. Lyubimov, A. V. Naumkin and V. A. Davankov, React. Funct. Polym., 2012, 72, 973–982 CrossRef CAS.
- M. P. Tsyurupa, Y. A. Borisov, Z. K. Blinnikova, N. P. Platonova, A. V. Ul'yanov, A. K. Buryak and V. A. Davankov, Prot. Met. Phys. Chem. Surf., 2014, 50, 59–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py00040e |
| This journal is © The Royal Society of Chemistry 2017 |