
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The workings of ferritin: a crossroad of opinions
Wilfred R.
Hagen
 *a,
Peter-Leon
Hagedoorn
a and
Kourosh
Honarmand Ebrahimi
*b
*a,
Peter-Leon
Hagedoorn
a and
Kourosh
Honarmand Ebrahimi
*b
aDelft University of Technology, Department of Biotechnology, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: w.r.hagen@tudelft.nl
bUniversity of Oxford, Department of Chemistry, Inorganic Chemistry Laboratory, South Parks Road, Oxford OX1 3QR, UK. E-mail: kourosh.honarmandebrahimi@chem.ox.ac.uk
First published on 31st May 2017
Abstract
Biochemistry of the essential element iron is complicated by radical chemistry associated with Fe(II) ions and by the extremely low solubility of the Fe(III) ion in near-neutral water. To mitigate these problems cells from all domains of life synthesize the protein ferritin to take up and oxidize Fe(II) and to form a soluble storage of Fe(III) from which iron can be made available for physiology. A long history of studies on ferritin has not yet resulted in a generally accepted mechanism of action of this enzyme. In fact strong disagreement exists between extant ideas on several key steps in the workings of ferritin. The scope of this review is to explain the experimental background of these controversies and to indicate directions towards their possible resolution.
Introduction
Transition-metal ions are an essential ingredient of terrestrial life, but the intrinsic reactivity of the aqua ions, either as Lewis-acid catalyst or as redox catalyst, is also a matter of challenge for a cell working towards physiological metal homeostasis.1 Iron is a rather extreme case in point with the ever present danger of radical chemistry such as the single-electron reduction of molecular oxygen to superoxide anion by the ferrous aqua ion, with the concomitant and equally disturbing threat of trading-in iron availability, and getting structurally compromising rust in return, due to the extremely low solubility of the ferric aqua ion around neutral pH. The cell's preparation for this walk on the wild site is ferritin: a protein complex for the reversible storage of iron ions that acts by taking up Fe(II), then catalytically, in a controlled manner, oxidizing the Fe(II), using dioxygen or peroxide, to Fe(III), and ultimately internally storing, under the label ‘core’, as a mineral with approximate ferrihydrite composition. The structural framework for this functioning, as schematically outlined in Fig. 1A, is a single (or a few different types of) small (circa 20 kDa) helical protein monomer(s) that apparently spontaneously assemble(s) into regular 24-mers in the form of a hollow nano-football with inner and outer diameter of circa 8 and 12 nm, respectively (Fig. 1B). While mitochondrial, microbial and plant ferritins are homomultimers, vertebrate cytosolic ferritins can consist of two, or three different subunits. Their slight difference in mass is used to name them as H (higher), L (lower), and M (medium) mass subunit, with the L-subunit always lacking the catalytic ferroxidase activity. For example, human ferritin is made up of H- and L-subunits with mass 21 and 20 kDa, respectively, with the percentage L-subunit being organ dependent. The L-subunit is thought to contribute to overall stability and to function in nucleation of the core, but its full physiological relevance remains to be established (cf.ref. 3). The physiological advantage of having an M-subunit present in the HML-ferritin of, e.g. frog species, is also unknown. The oxidative catalysis takes place in an active site, that goes under the name of ‘ferroxidase center’,4 in the interior of all, or some, of the 24 subunits. Structural genes for ferritin appear to occur ubiquitously in the three domains of life.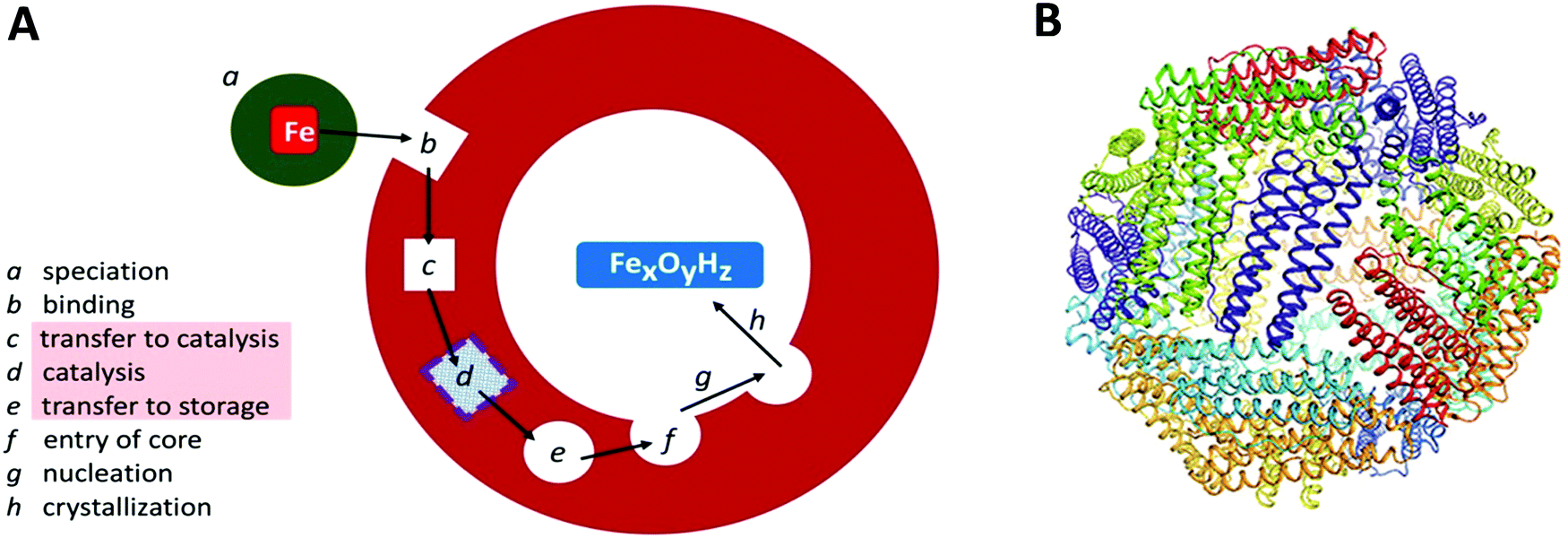 | ||
| Fig. 1 (A) Schematic outline of phases involved in the uptake, oxidation, and storage of Fe(II) by ferritin; (B) the conserved nanocage structure of 24-meric ferritin (taken from ref. 2). | ||
Ferritins exhibit unusual resistance versus denaturation especially by elevated temperature. For example, a ferritin from the hyperthermophilic archaeon Pyrococcus furiosus is apparently structurally and functionally unaffected by boiling for 24 hours.5 Ferritins of eukaryotic origin are somewhat less stable, but typically can be purified using a heat step at 80 °C, and they probably survive a short period in boiling water. This property may well have been instrumental in affording a very long-term biochemical research tradition. The first report on ferritin purification to perhaps near homogeneity dates back to an 1894 paper by Schmiedeberg reporting on ‘Ferrialbuminsäure’ or ‘Ferratin’ with 6% iron per dry mass obtained from pig liver by a procedure including heating ‘a few minutes’ in boiling water.6 Ferratin was re-named ferritin by Laufberger who crystallized the protein from horse spleen in 1937.7 Since then a slowly picking up pace but ever increasing and expanding research effort has been under way, and one would expect in all reasonableness to have available by now a comprehensive mechanistic picture of how ferritin does what it does. Nothing could be further from the truth: to date we find ourselves in the middle of a heated global debate not only on many partial aspects of ferritin structure-function relations, but even on the ontological question whether there is such a thing as ‘the’ ferritin. The goal of the present review is to delineate the hotspots of the battlefield (c–e in Fig. 1) in terms of conflicting arguments on proposed elements of the working mechanism, and to try to indicate possible ways out to their resolution.
The fundamental problem of variability
Structural genes of proteins are the toy of evolution. Even on a relatively short geological timescale evolution has, for example, produced a wide variety of hemoglobin sequences. Yet common opinion has it that there is such a thing as ‘the’ mechanism of hemoglobin: we fully expect that a newly discovered vertebrate species will carry a hemoglobin functionally characterized by reversible, non-redox binding of O2 subject to inter-subunit positive cooperativity. This principle of functional oneness appears to have been originally formulated by Kluyver in 19268 in the lofty phrase: “From the elephant to butyric acid bacterium – it is all the same”. Of course, this unity in biochemistry does not mean that Escherichia coli drinks with a trunk nor that the elephant moves with a flagellum; what it does imply is that we do not necessarily have to scrutinize all hemoglobin-carrying species on the globe to make biochemically meaningful statements about ‘the’ hemoglobin. To get the ‘hemoglobin’ idea it suffices to study only a handful of species to double-check proposed models and to establish extent of variability. The principle of unity also implies that it is perfectly all right to choose a species of study on practical grounds such as availability of biomass, e.g., slaughterhouse material, favorable doubling time of microbial growth, or heterologous expressibility of structural genes. Remarkably, this established concept of unity has recently become a hotly debated controversy for ferritin, or rather: ferritins, where an observed, or perceived, significant variability between proteins that all go under the name of ‘ferritin’ has led to the repeated proposal that structural and functional diversity is an inherent property of the ferritin superfamily of proteins.9–12Before we look at some of the details of ferritin biochemistry it is of importance to clearly delineate the boundaries of this unity-versus-diversity discourse. To simplify mechanistic analyses, and also to avoid complications with heterologous expression of heteromultimers, the majority of recent functional studies has been concerned with recombinant homomultimers. In other words, proposals on ferritin diversity do not usually refer to subunit heterogeneity, and the latter theme is therefore excluded from the present review. Furthermore, to keep the discussion as focussed as possible, also (largely) excluded from this review are heme-containing bacterioferritins (cf.ref. 13), 12-subunit mini-ferritins or Dps (DNA-binding proteins from starved cells) proteins (cf.ref. 14) and the iron-induced assembling archaeoferritin.15 In summary, the reviewers do not strive for comprehensivity; rather we focus on a limited number of key controversies that feature prominently in the acute debate on the working mechanism of ferritin, i.e. the 24-meric non-heme protein that is believed to be the major iron-storage protein in all domains of life.
Our deliberate choice for a limited enzymological vantage point is not to be taken to imply unawareness of broader relevance of ferritin as for example frequently reviewed in the columns of this journal in terms of iron homeostasis16–18 and of human health and pathology.19–23 However, the central thread of our argumentation below is that the basic biochemistry of ferritin is presently in a state of considerable uncertainty; therefore, its resolution should precede consideration of mechanistic implications in wider frameworks.
Key controversy-1: the concept of a transferable dinuclear iron cluster
In the reaction of molecular oxygen with Fe(II)-loaded ferroxidase centers a blue intermediate is observable on a sub-second time scale.24,25 The intermediate is generally assigned to a diferric peroxo species, but the exact oxygen binding mode is presently still not clear. In 2000 a seminal publication on this peroxodiferric intermediate a multidisciplinary research collaboration of several groups reported an EXAFS spectroscopy study whose main result was the identification of an unusually short Fe–Fe distance of 2.53 Å compared to the 3–4 Å typically found for peroxodiferric complexes. This plain fact was then audaciously interpreted to suggest that in ferritin the diiron complex can act as a moving substrate in sharp contrast to the function of stable cofactor in other O2-activating enzymes.26Fig. 2 is a pictorial representation of this idea reproduced here from a later paper from a different group.27 The model implies that an Fe–O(H)–Fe unit can spontaneously transfer out of the ferroxidase center without disintegration into monomeric units, to the interior, where it becomes part of the core mineral. The proposal is called here audacious because it would require unprecedented coordination chemistry to stepwise transfer a dinuclear iron–oxo cluster while maintaining coordinative saturation. The required chemistry was not addressed in the paper of the original proposal, nor has it been in any detail ever since.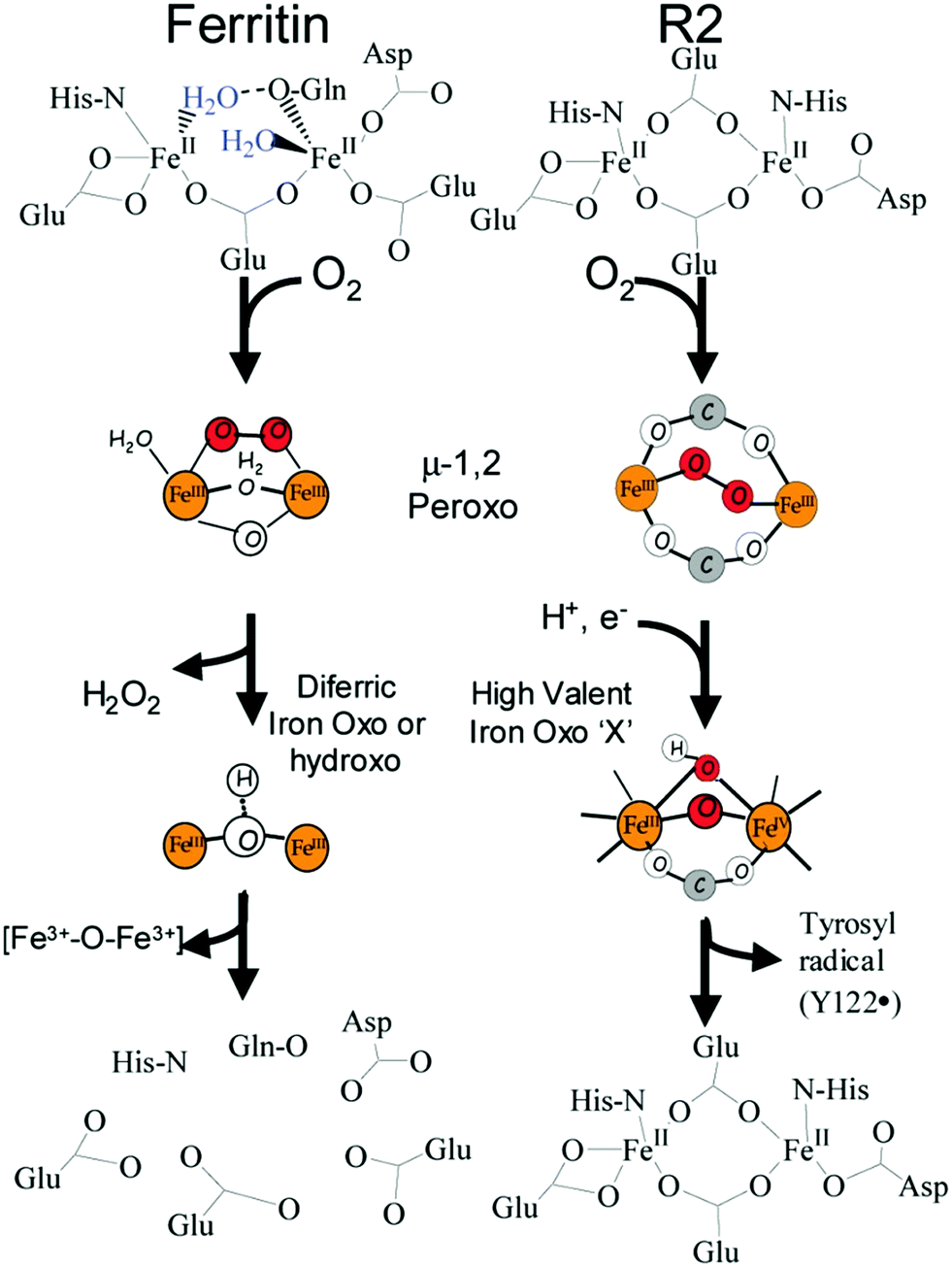 | ||
| Fig. 2 Comparison of reaction pathways for a ferritin and a ribonucleotide reductase. Figure reprinted with permission from: J. K. Schwartz, X. S. Liu, T. Tosha, E. C. Theil and E. I. Solomon, J. Am. Chem. Soc. 2008, 130, 9441–9450. Copyright 2008 American Chemical Society. | ||
The proposal of the transferable Fe–O(H)–Fe unit was put in doubt with the observation that the loaded ferroxidase center in P. furiosus ferritin could be reversibly converted from Fe(III)–Fe(III) to Fe(III)–Fe(II) to Fe(II)–Fe(II) in slow, equilibrium redox titrations by observation of the mixed-valence Fe(III)–Fe(II) dimer with EPR spectroscopy, suggesting the possibility that also here the iron dimer would actually be a stable prosthetic group.28–30 This alternative proposal was later relaxed to the concept of metastability in which the iron in the ferroxidase is stable until new individual (i.e.: not pairs of) Fe(II) ions arrive from the environment to replace individual Fe(III).31,32 The latter model, which was proposed based on parallel experiments on P. furiosus archaeal ferritin and human recombinant H-chain ferritin to check for inter-domain unity, is obviously incompatible with the concept of transferable Fe dimers.
Against this proposal are the conclusions drawn from a series of combined crystallographic, NMR-spectroscopic, and magnetic-susceptibility studies on recombinant M-chain ferritin from bullfrog, R. catesbeiana (the wild type is an HML-ferritin with ferroxidase centers in both the H and M subunits)33,34 as reaffirmed in several recent reviews.11,12,35–37 The working hypothesis for these studies was to assume that increased loading of ferritin under aerobic conditions would first produce a fully Fe(III)-dimer loaded ferroxidase center in each subunit and subsequently a set of well-defined intermediates when the dinuclear Fe–O(H)–Fe cluster as a unit would move on its way to the core from the ferroxidase site to a set of sequential binding sites thus producing locally a paramagnetic transmitter that should be monitorable as paramagnetic shifts in the NMR of specific amino-acid residues at increasing distance from the ferroxidase center. Indeed, paramagnetic broadening was observed in 13C–13C NOESY spectra particularly from side-chain carbons of isoleucine residues. When apo-ferritin was given 2 Fe(II) per subunit to process under aerobic conditions broadening occurred at Ile-144, a residue at some distance from the ferroxidase center with a side chain ‘protruding towards the interior’.33 Another residue, Ala-26, is also affected; Ala-26 is also near the ferroxidase center and also has a side chain ‘pointing towards the interior of the bundle’. Contrarily, Ile-141, which is ‘adjacent’ to the ferroxidase center and with a side chain ‘directed towards the exterior of the bundle’, was unaffected. The obvious conclusion is that following its formation the Fe(III) pair as a whole has spontaneously left the ferroxidase center to bind at a side near Ile-144. The situational geometry is outlined in the upper panel of Fig. 3.
 | ||
| Fig. 3 Situational geometry of the ferroxidase center in R. catesbeiana M-chain ferritin based on a crystal structure of protein loaded with Mg (upper panel38) or with Fe (lower panel34). | ||
The catch-22 is in the crystallographic data that form the basis of the interpretation of the NMR data, namely a structure of M-ferritin with 2 Mg(II) in the ferroxidase center published in 1999 (1MFR.pdb)38 and re-determined on a different crystal type at higher resolution shortly after the completion of the NMR study, affording a structure that “is essentially identical to that observed previously’ (3KA3.pdb).39 Later, subsequent to the NMR analysis, iron-preloaded M-ferritin with 2 Fe(III) per subunit was eventually successfully crystallized. The structure (3RBC.pdf ref. 26) turned out to be different from the Mg-protein in several, essential aspects, some of which are schematically illustrated in the lower panel of Fig. 3. The Fe(III)-loaded ferroxidase center is now found in between Ile-144 and Ala-26 and both residues have their side chain pointing towards the active center. Ile-141 is now a bit further away from the center. This rearrangement in the iron-loaded protein has the important implication of reversing one of the main conclusions of the NMR study: the Fe(III) does not spontaneously leave the ferroxidase center. And since the broadening at Ile-144 and Ala-26 does not disappear during subsequent aerobic incubations with Fe(II) it is clear that the center is always paramagnetic (i.e. filled) during iron uptake. So how does the Fe(III) leave the ferroxidase center? The revised-structure paper ends with a single suggestive sentence: “New incoming iron(II) may push the di-iron(III) moiety towards the cavity”.34 No experimental data are provided to support this suggestion in any way. And the di-iron(III) is still considered to move as a ‘moiety’ (i.e. as an Fe(III)–O(H)–Fe(III) dimer).
In subsequent years internalization of these observations has led to a discourse of remarkable nebulosity on the part of the original authors of the NMR study and the structure-revision paper. In two reviews Theil and collaborators choose to completely ignore the problems of ferroxidase-center stability and of what structure actually leaves the ferroxidase center.11,35 In a review on the paramagnetic NMR study of ferritin the suggestion of “the release of products as di-iron clusters under the effect of new incoming iron(II) ions” is repeated without giving any proof for this ‘effect’. And the products are still, without comment, presented as ‘di-iron clusters’.36 In a subsequent review on paramagnetic protein NMR in general Turano et al. cite the ferritin NMR study as having shown that: “we could establish that the diferric products remains stably bound at the catalytic center after the oxidation reaction has occurred”,37 which is a statement that is obviously in contrast to what was concluded in the original paper.33
As an aside, we briefly turn our attention to the heme-containing bacterioferritin. While Carrondo proposed the ferroxidase center in this system from Desulfovibrio desulfuricans to act as an oxidation and transfer site,40 Le Brun and collaborators,41,42 and more recently Solomon et al.43 maintain that the bacterioferritin from E. coli holds a stable Fe-dimeric prosthetic group. We note that none of these papers describe direct evidence for iron non-exchangeability in the ferroxidase center of bacterioferritin such as the iron isotope exchange studies performed with ferritin.31 Later structural studies on bacterioferritin from Pseudomonas aeruginosa44 and from Rhodobacter sphaeroides45 are again interpreted in terms of the ferroxidase center acting as a substrate site. In summary, as of today the matter remains unsettled.
So where are we now? More than a decade after the original observation of Tatur et al. on a stable, redox-titratable cluster in the ferroxidase center of P. furiosus ferredoxin28 it appears that most workers in the field have finally abandoned the idea of an Fe(III) cluster that magically leaves the center without any clear thermodynamic incentive to do so. However, as evidenced either by explicit statement or by implicitly ignoring other possibilities, several workers apparently still consider the leaving group to be an Fe(III)–O(H)–Fe(III) dimer, be it with the modifier that the act of leaving may be ‘under the effect of new incoming iron(II) ions’. Both concepts, i.e. the transferable dinuclear iron cluster and the push by Fe(II) ions are open for experimentation and have indeed been explored as will be discussed below. But first let us turn our attention briefly to another highly controversial matter: is the ferroxidase center a coordinative structure for two irons or for more than two irons?
Key controversy-2: the concept of a third iron-binding site, the C-site, associated with the ferroxidase center
The first high-resolution structure of an iron-loaded ferritin was obtained with the Escherichia coli protein after aerobic soaking in ferrous sulfate for several hours.46,47 Two irons were found approximately where they were expected on the basis of previously determined structures of human H-ferritin with cadmium48 and R. catbesiana M-ferritin with magnesium38 in two sites labelled A and B, together forming the ferroxidase center. A surprise was the detection of a third iron in a site labeled C close to the ferroxidase center with a glutamate bridging between FeC3+ and FeB3+.46,47 A possible physiological function of site C was extensively discussed (Fe(II) entry to, or Fe(III) exit from the ferroxidase center), but at that point in time there did not appear to be a C site either in human or in frog ferritin and the authors’ puzzlement was expressed in the complaint that “Evolutionary pressure for the loss of site C in eukaryotic ferritins is not obvious”.47 In subsequent years a considerable body of gained crystallographic knowledge indicated the occurrence of C-sites in many other ferritins, e.g., the archaeal ferritin from P. furiosus (Fig. 4A).49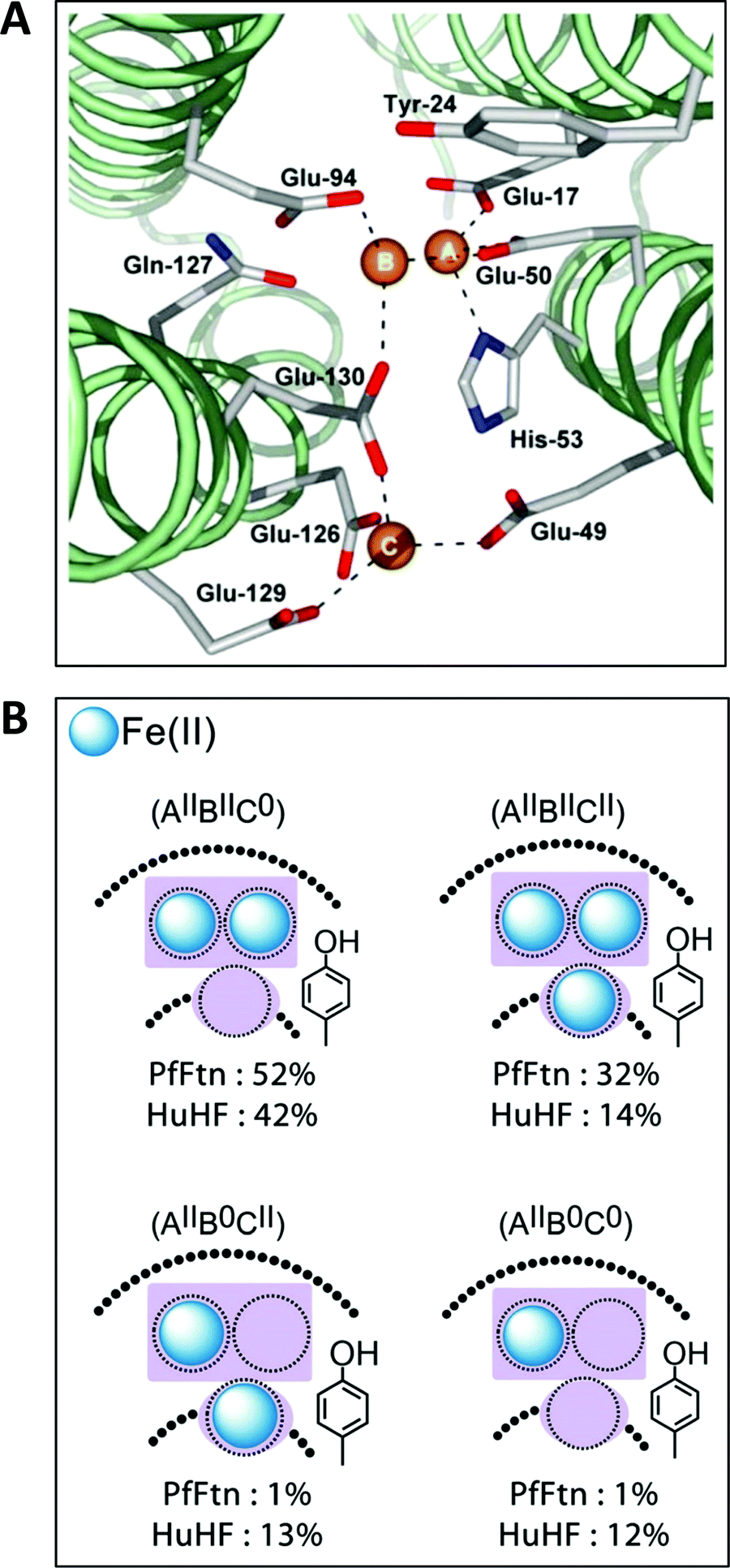 | ||
| Fig. 4 (A) The ferroxidase center (A and B) in P. furiosus ferritin with a third iron-binding site (the C site) nearby (taken from ref. 41). (B) Percentage occurrence distribution of species with different iron loading of the ferroxidase center and the C site in P. furiosus ferritin (PfFtn) and human H-chain ferritin (HuHF) following anaerobic incubation with Fe(II) as determined with Mössbauer spectroscopy (figure taken from ref. 2). | ||
These data have been extensively reviewed by us recently50 and need not be repeated here. However, discussion of the matter in the ensuing secondary literature again proves to be quite ambiguous. Theil et al. decided to not comment on the subject at all.11,35,51 LeBrun et al. rather chose to report on different C-sites in an encyclopaedic manner except on one occasion when they conclude that “Fe3+ ions at the C site and the ferroxidase center are transferred to the internal cavity”, without providing any reference to experiments that could support this mechanistic view.52
Since recently the C site has become prominent in the ongoing discussion on the variability of ferritins,9,12 it is of importance to clearly delineate the present state of knowledge. Here, we face a problem of definition: what exactly is a C site? In the original E. coli structure the C site harbours an Fe(III) ion that is carboxylate-bridged to FeB, and this pattern has been corroborated in several other microbial ferritins. However, the concept of unity, although it excludes the possibility that elephants move by means of a flagella, does not set any boundary conditions on the length of an elephant's trunk. Likewise a C site in ferritin is not necessarily definable by its distance from the ferroxidase center, nor by the details of its coordination chemistry. Rather, its nature should be pinned down in its physiological functioning. This now happens to be very much an open question. One reason for this is the experimental problem of determining and interpreting the occupancy of the site. In P. furiosus ferritin the binding affinities for Fe(II) decrease in the order: A-site, B-site, C-site as determined by isothermal titration calorimetry under anaerobic conditions.31 In human H-chain ferritin the same method also identifies three binding sites, however, the affinities of the A- and the B-site are an order of magnitude weaker, making that of the B-site essentially equal to that of the C-site,31 which may or may not be related to the lack of a bridging carboxylate between FeB and FeC. The observed variation in affinities of the Fe(II) binding sites in ferritins (in solution; not in crystals) goes some way towards explaining the considerable variation in occupancy of divalent metal binding sites within ferritins observed in their crystallography,50 although one should perhaps be careful about physiological relevance of binding metal ions to crystallized apo-protein and of binding non-iron ions to iron proteins. Site-directed-mutagenesis studies are incomplete: mutation of three out of the four Glu C-site ligands in E. coli ferritin have only a minor effect on overall iron oxidation rate;53 mutation of the fourth Glu C-site ligand in P. furiosus ferritin drastically reduces this rate.31
Theoretically, the presence of a C-site is the source of an extra reducing equivalent, and the idea has been put forth that by combination with a cation-radical-forming, highly-conserved tyrosine nearby one would have a convenient machine to reduce molecular oxygen in a single, four-electron step without formation of reactive oxygen species.2 Experimental exploration of this straightforward concept turns out to be an involved matter of complex spectral deconvolution due to the general sub-stoichiometric occupancy in the first few turnovers of the iron binding sites involved as illustrated in Fig. 4B.
New impetus to the discussion on the existence and nature of the C site has recently come forth from time-lapse crystallographic studies of ferritin incubated with iron ions, and this is our next topic.
Key controversy-3: a ‘bucket brigade’ of Fe(II) binding sites from environment to the ferroxidase center
Wouldn’t it be great if we could ‘film’ an enzyme-catalyzed reaction in detail by determination of the electronic-molecular structures of all the relevant intermediates with atomic resolution? It so happens that beggars can’t be choosers, so let us go for what is probably the next best thing presently available: a slowed-down series of snapshots, namely, the X-ray structure determination of crystals of apo-ferritin incubated for a finite set of times with the substrate Fe(II) and semi-instantaneously cryo-frozen in a nitrogen-gas flow. From the onset let us be on guard for the risks that we run: crystals are not proteins in solution, crystallized proteins may be restricted in their conformational adjustability, and reaction rates of crystallized enzymes are slowed down. Mangani and collaborators have recently published crystallographic studies on R. catesbeiana M-chain ferritin10,54 and on human H-chain ferritin,55 employing the slowed-down snapshot approach to test the hypothesis that Fe(II) ions from the environment end up in the ferroxidase center via an intramolecular conduit consisting of more or less well-defined transient iron binding sites, also known as a ‘bucket brigade’.11 There are several major problems associated with these studies, and since these largely remained unnoticed in a very recent review,12 here, we go through some length to spell them out explicitly.In this series of studies the authors choose for a highly unusual approach: iron loading is performed aerobically by allowing ‘free diffusion’ of iron from solid ammonium iron(II) sulfate, since loading with pre-dissolved Fe(II) does not work. No control experiments are reported on the operational concentration and the redox fate of iron ‘freely diffusing’ from Mohr's salt crystals in the presence of oxygen on a time scale of 1–60 minutes. The final pH in the protein crystallization procedure is circa 8, and apparently the incubation of ferritin crystals with iron crystals is also done at this pH (supplementary material to ref. 10) although the buffer is not specified, nor is the temperature of the experiment. In conventional iron-oxidation activity measurements of dissolved ferritin the pH, the buffer, the temperature, and the iron-ion concentration are relevant parameters because the biological activity must compete with the inorganic oxidation whose rate increases exponentially with increasing pH. If the latter would predominate (which is not particularly unlikely at pH 8), then the crystallographic snapshots may actually be showing, or partially showing, how Fe(III), rather than Fe(II) ions move through ferritin. This problem is particularly acute in the study of R. catesbeiana ferritin, since it was crystallized in the presence of circa 1 M Mg(II) as precipitant.10 How much of this ends up in the final incubation medium with Fe(II) is not clear (in particular since this medium is not specified by the authors), but the reported apo-structure (4LQH.pdb) of the ferritin contains 9 Mg(II) ions in a single subunit. Stopped-flow control experiments with dissolved protein show an overall reduction of Fe(II) oxidation rate by a factor of 450 when in the presence of 0.1 M Mg(II), although for unexplained reasons these experiments were done at pH 7.10
Crystals of apo-ferritin were incubated with Mohr's salt crystals as described above, and the reactions were quenched at six different time points from initiation: 1, 2, 5, 15, 30, 60 minutes.10 Crystallographic results afford a remarkably inconsistent picture: in five out of the six crystals iron is only found in the A and B site of the ferroxidase center, always in substoichiometric occupancy. These sites are labelled Fe1 and Fe2. The only exception to this bucket-brigade variant of Dirac's delta function is the t = 15 minutes crystal structure in which an Fe3 is detected (0.4 occupancy) very close to Fe2 (0.7 occupancy) but with a poorly defined coordination: since it appears to dangle on a single bond to the Nδ1 of His54 the authors conclude that “Fe2 and Fe3 cannot be simultaneously bound to RcMf”, and one cannot help but wonder whether Fe2 and Fe3 are not one and the same beast. Another iron, indexed as Fe4, is found this time with a bona fide coordination of two bidentate carboxylates, Glu57 and Asp140, plus a water molecule. These referees would call that a C site. The pdb file for the unique t = 15 minutes configuration (4LYU.pdb) actually features not four but five Fe spheres at/near the ferroxidase center with occupancies 0.7, 0.7, 0.4, 0.5, 0.5; remarkably, the fifth iron is not alluded to in the text of the paper nor in the supplementary material.10
Obviously more meaningful results could be expected when the substrate O2 would be left out of the reaction equation by running the Fe diffusion under anaerobic conditions. Unfortunately, for unknown reasons also the strictly anaerobic incubation with pre-dissolved Fe(II) does not afford iron-loaded R. catesbeiana M-chain ferritin crystals, and one has to turn once more to the Mohr's salt dissolution method.10 However, with this approach the crystals are found to crack a few minutes after adding the Fe(II) salt grain. The authors believe that this “emphasizes the osmotic pressure that builds up from Fe2+ diffusing into the crystal in the absence of enzymatic turnover”. These referees find the novel notion of a protein operating as a semi-permeable membrane difficult to grasp, and they propose the alternative explanation of a significant conformational change (or sequence of changes) upon Fe(II) binding at/near the ferroxidase center. Some indication for the latter may be found in the crystal structure obtained after 3 minutes anaerobic incubation, just before cracking: although the authors note “the presence of four iron ions in the oxidoreductase site that are located, within experimental error, at the same positions as observed in the aerobic crystals after 15 min of treatment with Mohr's salt”, the detailed coordination exhibits several differences, e.g., Asp140 is now bridging between Fe4 and Fe2, Fe4 gets a new monodentate ligand from Glu136, a water is now bridging between Fe4 and Fe2, Fe4 gets a new water ligand, Fe3 gets a new water ligand, Glu57 is now bridging between Fe4 and Fe3, Glu103 (in one of its conformations) is now bridging between Fe3 and Fe2, and His54 may (in a second conformation) now perhaps also bind to Fe2. The poorly defined separation between Fe2 and Fe3 in the 15 min aerobic crystal has its counterpart in the 3 minutes anaerobic crystal as “a continuous stretch of anomalous density elongating from Fe3 and reaching the Fe2 maximum”, and, combined with their substoichiometric occupancy of 0.5 Fe2 and 0.4 Fe3, their separate indexing may again be put into question: alternative indexing of Fe2 + Fe3 as a flexible B site would make Fe4 (occupancy 0.25) a C site.
The method of letting iron ions diffuse, under aerobic conditions, from Mohr's salt crystals to a ferritin crystal has also been applied to human H-chain ferritin.55 Four structures have been determined, namely, after 1, 5, 15, 30 minutes of incubation. There is no mention of anaerobic incubations, although one experiment is described in which an anaerobically grown crystal of apo-ferritin is aerobically incubated with iron ions for 8 minutes. No structure is reported. The rationale given for this latter experiment is that it would provide “a check for the oxidation state of iron in the aerobically grown crystals”, which is beyond these reviewers’ comprehension unless there would be a hitherto unnoticed interaction of molecular oxygen with the apo-protein. Again, the ferritin is crystallized in the presence of Mg(II), and there are five magnesium ions in the pdb file for a single subunit. After 15 minutes two of these were found to be replaced by iron ions. All in all the caveat pronounced for the R. catesbeiana ferritin also applies here, and the necessity for future gathering of information on the redox state of iron is equally pressing. Compared to the R. catesbeiana ferritin ‘film’ of iron topology in time this is one of increased, be it not full, consistency: all four structures show four protein-bound irons. Two are in the A and B site of the ferroxidase center; a third one close by is initially a bit blurred but after 5 minutes it becomes well defined with a 6 O coordination from Glu61, Gln58, and four water molecules. Note that this is not too far off the previously predicted C site for human ferritin.50 At 5 Å from this site a fourth iron is found, and the two are bridged by Glu61 (see Fig. 5).
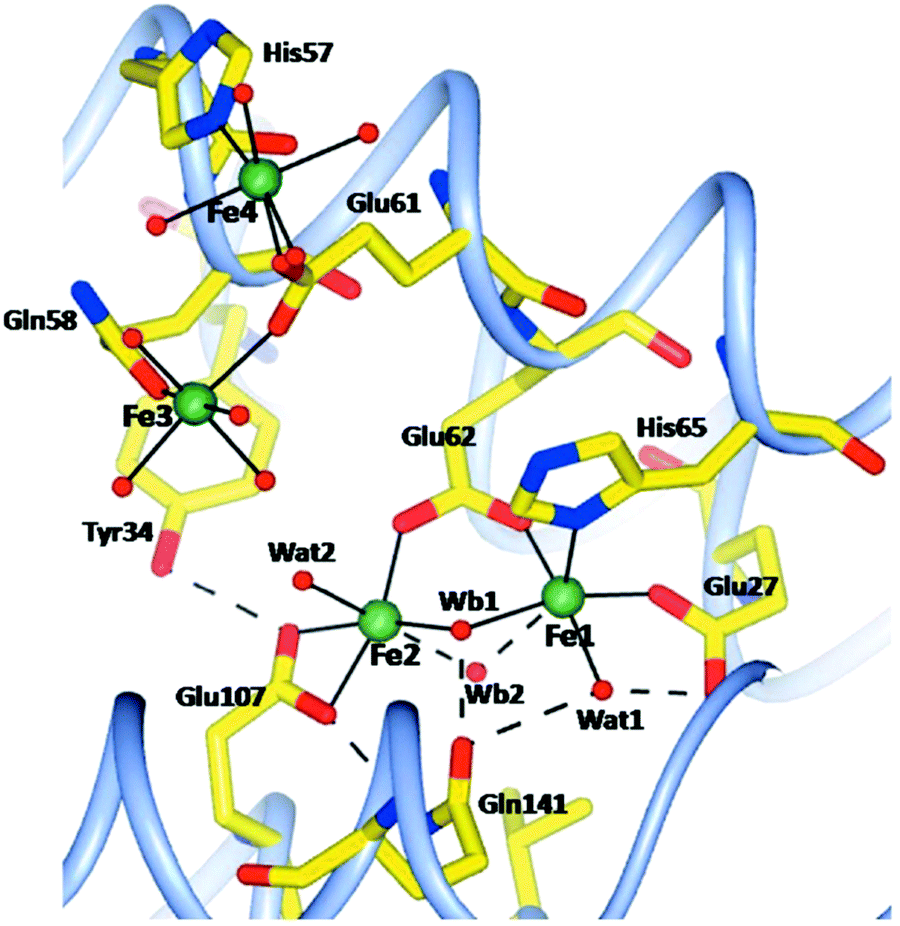 | ||
| Fig. 5 Schematic view of the ferroxidase center and nearby iron binding sites in human H-chain ferritin after 5 minutes aerobic incubation of a crystal of apo-ferritin with solid Mohr's salt. Dotted lines indicate hydrogen bonds. Figure reprinted from C. Pozzi, F. Di Pisa, C. Bernacchioni, S. Ciambellotti, P. Turano and S. Mangani, Acta Crys., 2015, D71, 1909–1920. | ||
Occupancies of these ABCD sites are (0.7; 0.2; 0.3; 0.3) after 1 minute, and (1.0; 0.5; 0.5; 0.5) after 5 minutes. In the 15 and 30 minute structures these occupancies do not change any more, which leads the authors to conclude that “after about 5 min a dynamic equilibrium has been reached in the protein crystals”. The conclusion is not altogether convincing, since ‘dynamic equilibrium’ in (bio)chemistry is typically reserved for a reversible reaction. The paper ends with a summarizing statement that “the postulated third iron-binding site, which is a common characteristic of all ferritins, does not exist as a specific, well defined metal-coordination site, but is rather a region extending up to about 10 Å from the Fe1 and Fe2 sites where heterogeneous iron-anchoring points in ferritins of different origin are present”. This appears to be no less than a paradigmatic change from the opinion that “there is no site C in vertebrate ferritins” (as very recently concluded in a review12 on basis of the work just discussed). These referees interpret as follows: at this time the distribution in space and time of iron incoming in vertebrate ferritins, or for that matter in ferritins in general, is not well defined, but the statement, above, of an extended region of binding next to the ferroxidase center can serve as a practical null hypothesis in future studies with, hopefully, rigorous control of iron oxidation state.
Is the distance between FeA and FeB in the ferroxidase center not an indication for the oxidation state of the iron? Strictly speaking this is not the case where the range of values in model compounds overlap for Fe(III)–Fe(III), Fe(III)–Fe(II), and Fe(II)–Fe(II),56 however, a trend of decreasing distance should be observable with increasing oxidation state for a single compound. The average distance for the R. catesbeiana time-series crystals is 3.64 Å. In a previous crystallographic study on oxidized R. catesbeiana protein an average distance of 3.07 Å was found (range over subunits: 2.89–3.22).34 In that same study a crystal incubated aerobically for 1 minute with Fe(II) solution (at that time the impossible was apparently still possible) afforded an average distance of 3.96 (range over subunits: 3.05–4.76). The crystal was lighter in color than the fully oxidized one and, therefore, presumably contained a mixture of Fe(II) and Fe(III). The authors suggest that “oxidation requires changes in the coordination environment of iron(II) at site 2 and parallel reduction in the metal–metal distance”. True as this may be, it does raise the question whether rearrangements coupled to oxidation are not hindered or blocked in the crystalline state. Furthermore, one should not forget that all the reported crystallographic data, cited above, result from averaging each subunit over the whole crystal10,34,55 with the additional complication of partial occupation of metal sites. For completion we note that the average distance in the time series of human ferritin was 3.49 Å,55 and that in a recent report on E. coli ferritin (see below) a distance was reported of circa 3.35 Å for a preparation that should have its iron predominantly fully oxidized.49 The bottom line is that we do not know whether the average Fe–Fe distance under all conditions might be a reliable indication of oxidation state. Spectroscopy, in particular EPR and/or Mössbauer, would be a possible means to obtain less equivocal information on the redox state of the iron. In a very recent paper describing iron loading of human ferritin crystals, Pozzi et al. apply optical spectroscopy on crystals of L-chain versus H-chain crystals, but no quantitative analysis of redox state is attempted.58 Interestingly, this paper also shows that aerobic solutions of Fe(II) at pH 7 immediately produce Fe(III) ions at a quasi-first-order rate, which implies that the crystals never see pure Fe(II).
Key controversy-4: a bucket-pair brigade of Fe(III) dimer binding sites from the ferroxidase center to the core
This concept is really a ‘child’ of the concept, treated above, of the transferable dinuclear iron cluster (key controversy-1), and falsification of the latter obviously will also put the former to rest. However, the idea that Fe(III) hops, in the form of an indestructible Fe(III)–O(H)–Fe(III) unit, from specific binding site to specific binding site until it is accommodated in the core, has been prominently present in the literature for nearly two decades, and recent reviews on ferritin do not explicitly send it to the realms of fantasy. A few pertinent remarks are, therefore, in order.We have done a series of experiments to test the hypothesis of Fe(III) leaving the ferroxidase center as a dimer.31 In Fig. 6, we have re-drawn the outcome of some of these in schematic form. Experiment A uses iron enriched in isotope 57Fe (red color) whose nuclear spin can be detected with Mössbauer spectroscopy versus iron with a natural isotopic abundance, which (with only 2% in 57Fe) remains undetected. The starting point is a sample of P. furiosus ferritin prepared with two antiferromagnetically coupled ferric ions (solid spheres) in the ferroxidase center (as borne out by the lack of any EPR signal). To this we add two ferrous ions (solid squares) under aerobic conditions. After sufficient reaction time the iron-transport protein transferrin is added to remove two Fe(III) specifically from the ferroxidase center (vertical arrow down). After separation of the ferritin from the transferrin the 57Fe remaining anywhere in the ferritin is quantitatively determined from its Mössbauer spectrum. The experiment is done two ways: initial loading with 57Fe and subsequent loading with NATFe and the other way around. Independent of the question whether the Fe(III) leaves the ferroxidase spontaneously or is pushed out by the incoming Fe(II), the bucket-pair brigade model predicts 2.0 57Fe to remain in the ferritin in experiment A1 and 0.0 57Fe to remain in experiment 2. Instead, we find the indicated broken numbers, and since no mononuclear Fe(III) is observed in the EPR, the conclusion is drawn that Fe(II) displaces Fe(III) in the ferroxidase center one-by-one. One could object that the presence of a C site (or even a range of C sites) would complicate the picture, but the lack of a mononuclear iron EPR signal after full oxidation rebuts this objection.
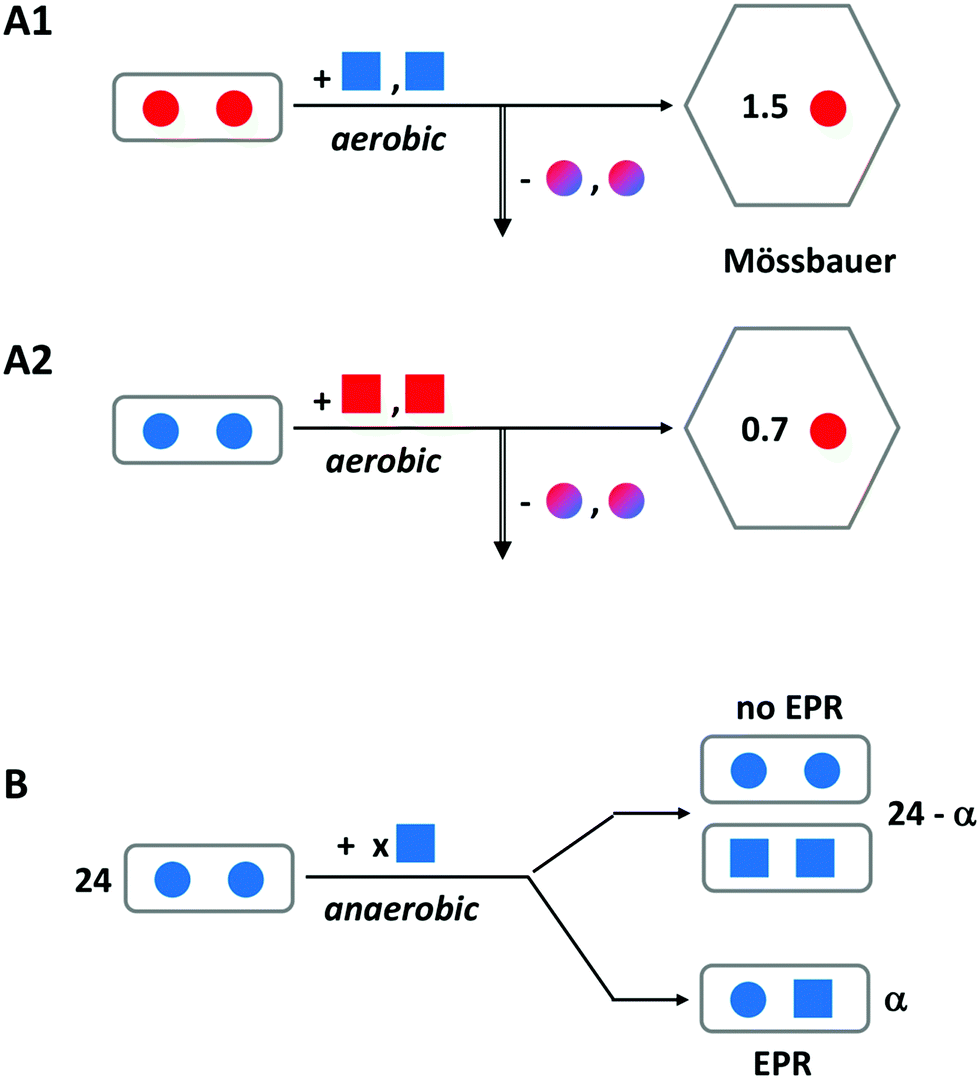 | ||
| Fig. 6 Schematic outline of two experiments to disprove the hypothesis of an Fe(III)-pair leaving the ferroxidase center of ferritin. A solid circle represents Fe(III) and a solid square is Fe(II). Red encodes 57Fe and blue is natural-abundance Fe; mixed colors means a mixture of 57Fe and NATFe. Experiment A is for P. furiosus ferritin preloaded with two Fe(III) per ferroxidase center and subsequently aerobically loaded with two Fe(II) per center. The vertical arrow indicates removal of iron from the center by transferrin. Remaining iron is Mössbauer detected. Experiment B uses anaerobic loading and detection of mixed valence pair formation with EPR. See text for further explanation. Adapted from ref. 31. | ||
In experiment B a different approach is taken, although the starting point is again ferritin, in which all ferroxidase centers are initially loaded with two antiferromagnetically coupled, EPR silent ferric ions. This is done with P. furiosus ferritin versus human H-chain ferritin in comparison. We now titrate in ferrous ions under anaerobic conditions and we monitor the formation of mixed-valence pairs Fe(III)–O(H)–Fe(II) by means of EPR spectroscopy. The bucket-pair brigade model would predict no formation of mixed-valence pairs at all. Instead, we found a maximum of circa 10 mixed-valence pairs per ferritin 24-mer when circa 37 Fe(II) were added, and thus we conclude that Fe(III) does not move in pairs but rather is displaced one-by-one with Fe(II).
Our conclusion is at variance with the interpretation of magnetic-susceptibility measurements carried out in parallel with the 13C NMR study cited above33 involving aerobic addition of 2n (n = 1–10) Fe(II) to R. catesbeiana M-chain ferritin (Fig. 3D in ref. 33). The conclusion drawn from this experiment, in combination with the NMR study, is that a dimer of Fe(III) is formed in the ferroxidase center which then spontaneously moves to the next bucket-pair brigade binding site; addition of another two Fe(II) leads to formation of a four-iron cluster down the bucket line; addition of another 4 Fe(II) leads to an eight-iron cluster one more position down the bucket line. The idea of ‘increasingly large clusters’ is repeated in a later review.37 These reviewers are troubled by many misgivings. Clusters of higher nuclearity, such as the M-cluster and the P-cluster in nitrogenase, have a rich redox chemistry and typically exhibit characteristic EPR signals.59,60 Those signals have never been reported for any ferritin. The argument of the authors that the decreasing effective magnetic moment per iron is in support of larger cluster formation does not convince because this would also hold for iron in a ferrihydrite core. In this respect note that the iron incubation “followed the procedure described for the 13C–13C NOESY”; the latter procedure involves incubation at 25 °C for two days. The employed Evans method of proton shifts of inert reference molecules induced by paramagnetic molecules in the same solution has been tried on metalloproteins for some time in the seventies but it was superseded by the SQUID (superconducting quantum interference device) which is not only characterized by increased sensitivity but also by the possibility to measure susceptibility over a very wide temperature range (typically down to some 1.5 K) contrast to the limitation to room temperature of the NMR-shift method. Temperature-dependent measurements are mandatory in particular for biological samples where, e.g., small amounts of contaminating high-spin iron can swampingly contribute to the overall susceptibility. In any case, presenting susceptibility data of metalloproteins without a check by, e.g., EPR spectroscopy of the structures involved is uncommon, and we consider the proposal of four- and eight-iron clusters an overinterpretation.
Key controversy-5: the nature of the ferroxidase center reaction intermediate(s)
Finally, we return full circle to the 2000 paper initially promoting the transferable dinuclear iron cluster.26 The leading theme of the paper was the question: what (if any) is the difference between ferritin and other iron oxo dimer containing enzymes such as methane monoxygenase. The answer of the authors then was: ferritin has a peroxo-intermediate with unusually short Fe–Fe distance, which confers decreased stability, and that explained why this dimer leaves the ferroxidase center as a whole spontaneously.The structure of the intermediate was proposed to be μ-1,2-peroxo on the basis of Mössbauer61 and resonance Raman62 spectroscopy in comparison to data from methane monooxygenase and ribonucleotide reductase. However, recently a range of other possible structures has been considered for methane monooxygenase (e.g., ref. 63), and also in our own recent Mössbauer studies on P. furiosus and human H-chain ferritins a spectroscopic comparison to other enzymes and models led us to propose the alternative μ-η1:η2 binding mode.2
Once more, it would be great if one could make a structural snapshot of this unusual intermediate, and that is precisely what has been reported recently for a kinetically impaired mutant (S20A) of E. coli ferritin by Kim and collaborators.57 These authors claim to have identified the structure of the ferroxidase center in three forms: the regular one with O(H) bridging, one with molecular oxygen bridging, and one with peroxo bridging. In a very recent review the work has been cited without critical comment.12 We tend to remain reluctant in the face of this exciting news because all three structures were determined from one and the same crystal. In narrative terms this would imply that the ferritin crystal can tell molecular oxygen to go to a particular 1/3 of all subunits in all molecules of the crystal to bind and remain indifferent, to go to another particular 1/3 to react to a peroxo form, and to go to the last particular 1/3 to fully react to the (hydr)oxo end product. It should be noted that the paper is not particularly clear about how the crystal was prepared. From the experimental procedures we read that one starts by preparing apoferritin and that this is “termed low-iron bound state”. This is then incubated with an unspecified amount of Fe(II) at pH 7.0, presumably under aerobic conditions (not specified) to afford something that is called “the low-iron state protein”, which now “has approximately three irons per protein”. This preparation is then crystallized for a period of one week to two months by hanging drop vapor diffusion, and the resulting crystal exhibits the ferroxidase center in the three states (two of which would be transients). While awaiting follow-up studies the present reviewers have their hats within reach to be eaten. The paper also reports on parallel studies on Helicobacter pylori ferritin, but the pdb files have not been released to date.
We have recently discussed possibilities for the oxygen binding mode, including non-bridging ones, in the blue peroxo intermediate in comparison with dioxygen activating enzymes and synthetic model compounds in particular based on Mössbauer and resonance Raman spectroscopic data,50 and we will not be repetitive except for concluding that the exact molecular nature of this intermediate in ferritin is still to be unambiguously determined.
Of course, the peroxo structure is not the only intermediate in the ferroxidase reaction. A conserved tyrosine close by has been repeatedly implicated in catalysis (e.g., ref. 24 and 64), and a role as one-electron donor to molecular oxygen, in combination with three electrons from the ferroxidase center and from the third Fe(III) C site has been proposed, with a tyrosyl cationic radical formed as a second intermediate directly after the peroxo one.65 The enzymological question as to how many identifiable intermediates stage a reaction mechanism may be an opera aperta by definition. Rather than claiming completeness of the present inventory, we re-address the question of any difference between ferritin and the oxygen-activation iron–oxo enzymes from a more general perspective. Since we do no longer believe that a dinuclear cluster spontaneously leaves the ferroxidase center, and since we sincerely doubt that, when the Fe(III) is pushed out, this would happen in the form of an Fe–O(H)–Fe dimer, can we say something about other mechanistic properties of iron–oxo enzymes that have not yet been identified in ferritin? High-valent iron chemistry, i.e. transient formation of a ferryl intermediate, is a general property of iron–oxo enzymes. The possibility of a (di)ferryl intermediate of ferritin has, in fact, been proposed in the past,61 but it has never been tested experimentally. We offer this as a putative outlook to approach the question of how ferritins works from a fresh perspective.
Concluding remarks
Ferritin research goes back a long way. In the last decades it has developed in a very active field of research, whose primary reports have been sided by a constant and sizable stream of reviews. The spirit of these review has generally been one of: we may not be there yet, but we are making steady progress. The present review takes a different approach in putting forth the notion that the field is in turmoil with opposing views clashing on several key controversies regarding details of the machinery of ferritin catalysis such as: how does Fe(II) reach the catalytic center, what is the catalytic center, what are the intermediates of the oxidation reaction, what is the Fe(III) product and how is it transferred to the core area. And how general or particular are these mechanistic attributes in ferritins of different origin and in comparison to other enzymes with similar active centers. The extent to which we will be able to find resolution to these controversies will determine whether the field will stall or jump ahead.Acknowledgements
KHE acknowledges financial support from the European Molecular Biology Organization via a long-term fellowship ALTF 157-2015.References
- J. J. R. Fraústo da Silva and R. J. P. Williams, The biological chemistry of the elements, Clarendon Press, Oxford, 1991 Search PubMed.
- K. Honarmand Ebrahimi, E. Bill, P.-L. Hagedoorn and W. R. Hagen, Mol. BioSyst., 2016, 12, 3576–3588 RSC.
- D. Finazzi and P. Arosio, Arch. Toxicol., 2014, 88, 1787–1802 CrossRef CAS PubMed.
- S. Levi, A. Luzzago, G. Cesareni, A. Cozzi, F. Franceschinelli, A. Albertini and P. Arosio, J. Biol. Chem., 1988, 263, 18086–18092 CAS.
- J. Tatur, P.-L. Hagedoorn and W. R. Hagen, Extremophiles, 2006, 10, 139–148 CrossRef CAS PubMed.
- O. Schmiedeberg, Arch. Exp. Pathol. Pharmakol., 1894, 33, 101–116 Search PubMed.
- V. Laufberger, Bull. Soc. Chim. Biol., 1937, 19, 1575–1582 CAS.
- H. C. Friedmann, Perspect. Biol. Med., 2004, 47, 47–66 CrossRef PubMed.
- J. M. Bradley, G. R. Moore and N. E. Le Brun, J. Biol. Inorg. Chem., 2014, 19, 775–785 CrossRef CAS PubMed.
- C. Pozzi, F. Di Pisa, D. Lalli, C. Rosa, E. Theil, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 941–953 CAS.
- E. C. Theil, T. Tosha and R. K. Behera, Acc. Chem. Res., 2016, 49, 784–791 CrossRef CAS PubMed.
- J. M. Bradley, G. R. Moore and N. E. Le Brun, Curr. Opin. Chem. Biol., 2017, 37, 122–128 CrossRef CAS PubMed.
- J. M. Bradley, N. E. Le Brun and G. R. Moore, J. Biol. Inorg. Chem., 2016, 21, 13–28 CrossRef CAS PubMed.
- K. Zeth, Biochem. J., 2012, 445, 297–311 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P.-L. Hagedoorn, L. van der Weel, P. D. E. M. Verhaert and W. R. Hagen, J. Biol. Inorg. Chem., 2012, 17, 975–985 CrossRef PubMed.
- P. Cornelis, Q. Wei, S. C. Andrews and T. Vinckx, Metallomics, 2011, 3, 540–549 RSC.
- L. C. Kühn, Metallomics, 2015, 7, 232–243 RSC.
- E. C. Theil, Metallomics, 2015, 7, 15–24 RSC.
- O. Han, Metallomics, 2011, 3, 103–109 RSC.
- G. Sebastiani and K. Pantopoulos, Metallomics, 2011, 3, 971–986 RSC.
- E. Gammella, P. Buratti, G. Cairo and S. Recalcati, Metallomics, 2014, 6, 1336–1345 RSC.
- D. B. Kell and E. Pretorius, Metallomics, 2014, 6, 748–773 RSC.
- L. Ma, A. Terwilliger and A. W. Maresso, Metallomics, 2015, 7, 1541–1554 RSC.
- G. S. Waldo, J. Ling, J. Sanders-Loehr and E. C. Theil, Science, 1993, 259, 796–798 CAS.
- A. Treffry, Z. Zhao, M. A. Quail, J. R. Guest and P. M. Harrison, Biochemistry, 1995, 34, 15204–15213 CrossRef CAS PubMed.
- J. Hwang, C. Krebs, B. H. Huynh, D. E. Edmonson, E. C. Theil and J. E. Penner-Hahn, Science, 2000, 287, 122–125 CrossRef CAS PubMed.
- J. K. Schwartz, X. S. Liu, T. Tosha, E. C. Theil and E. I. Solomon, J. Am. Chem. Soc., 2008, 130, 9441–9450 CrossRef CAS PubMed.
- J. Tatur and W. R. Hagen, FEBS Lett., 2005, 579, 4729–4732 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P.-L. Hagedoorn, J. A. Jongejan and W. R. Hagen, J. Biol. Inorg. Chem., 2009, 14, 1265–1274 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, J. Biol. Inorg. Chem., 2010, 15, 1243–1253 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, E. Bill, P.-L. Hagedoorn and W. R. Hagen, Nat. Chem. Biol., 2012, 8, 941–948 CrossRef CAS PubMed.
- K. Honarmand Ebbrahimi, P.-L. Hagedoorn and W. R. Hagen, FEBS Lett., 2013, 587, 220–225 CrossRef PubMed.
- P. Turano, D. Lalli, I. C. Felli, E. C. Theil and I. Bertini, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 545–550 CrossRef CAS PubMed.
- I. Bertini, D. Lalli, S. Mangani, C. Pozzi, C. Rosa, E. C. Theil and P. Turano, J. Am. Chem. Soc., 2012, 134, 6169–6176 CrossRef CAS PubMed.
- E. C. Theil, R. K. Behera and T. Tosha, Coord. Chem. Rev., 2013, 257, 579–586 CrossRef CAS PubMed.
- D. Lalli and P. Turano, Acc. Chem. Res., 2013, 46, 2676–2685 CrossRef CAS PubMed.
- M. Piccioli and P. Turano, Coord. Chem. Rev., 2015, 284, 313–328 CrossRef CAS.
- Y. Ha, D. Shi, G. W. Small, E. C. Theil and N. M. Allewell, J. Biol. Inorg. Chem., 1999, 4, 243–256 CrossRef CAS PubMed.
- T. Tosha, H.-L. Ng, O. Bhattasali, T. Alber and E. C. Theil, J. Am. Chem. Soc., 2010, 132, 14562–14569 CrossRef CAS PubMed.
- M. A. Carrondo, EMBO J., 2003, 22, 1959–1968 CrossRef CAS PubMed.
- S. Baaghil, A. Lewin, G. R. Moore and N. E. Le Brun, Biochemistry, 2003, 42, 14047–14056 CrossRef CAS PubMed.
- A. Crow, T. L. Lawson, A. Lewin, G. R. Moore and N. E. Le Brun, J. Am. Chem. Soc., 2009, 131, 6808–6813 CrossRef CAS PubMed.
- Y. Kwak, J. K. Schwartz, V. W. Huang, E. Boice, D. M. Kurtz Jr. and E. I. Solomon, Biochemistry, 2015, 54, 7010–7018 CrossRef CAS PubMed.
- S. K. Weeratunga, S. Lovell., H. Yao, K. P. Battaile, C. J. Fisher, C. E. Gee and M. Rivera, Biochemistry, 2010, 49, 1160–1175 CrossRef CAS PubMed.
- K. H. Nam, Y. Xu, S. Piao, A. Priyadarshi, E. H. Lee, H.-Y. Kim, Y. H. Jeon, N.-C. Ha and K. Y. Hwang, Biochem. Biophys. Res. Commun., 2010, 391, 990–994 CrossRef CAS PubMed.
- P. D. Hempstead, A. J. Hudson, P. J. Artymiuk, S. C. Andrews, M. J. Banfield, J. R. Guest and P. M. Harrison, FEBS Lett., 1994, 350, 258–262 CrossRef CAS PubMed.
- T. J. Stillman, P. D. Hempstead, P. J. Artymiuk, S. C. Andrews, A. J. Hudson, A. Treffy, J. R. Guest and P. M. Harrison, J. Mol. Biol., 2001, 307, 587–603 CrossRef CAS PubMed.
- D. M. Lawson, P. J. Artymiuk, S. J. Yewdall, J. M. A. Smith, J. C. Livingstone, A. Treffy, A. Luzzago, S. Levi, P. Arosio, G. Cesareni, C. D. Thomas, W. V. Shaw and P. M. Harrison, Nature, 1991, 349, 541–544 CrossRef CAS PubMed.
- J. Tatur, W. R. Hagen and P. M. Matias, J. Biol. Inorg. Chem., 2007, 12, 615–630 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, Chem. Rev., 2015, 115, 295–326 CrossRef CAS PubMed.
- X. Liu and E. C. Theil, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- N. E. Le Brun, A. Crow, M. E. P. Murphy, A. G. Mauk and G. R. Moore, Biochim. Biophys. Acta, 2010, 1800, 732–744 CrossRef CAS PubMed.
- A. Treffry, Z. Zhao, M. A. Quail, J. R. Guest and P. M. Harrison, FEBS Lett., 1998, 432, 213–218 CrossRef CAS PubMed.
- C. Bernacchiono, C. Pozzi, F. Di Pisa, S. Mangani and P. Turano, Chem. – Eur. J., 2016, 22, 1–8 CrossRef.
- C. Pozzi, F. Di Pisa, C. Bernacchioni, S. Ciambellotti, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1909–1920 CAS.
- E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS PubMed.
- S. Kim, J.-H. Lee, J. H. Seok, Y.-H. Park, S. W. Jung, A. E. Cho, C. Lee, M. S. Chung and K. H. Kim, J. Mol. Biol., 2016, 428, 5007–5018 CrossRef CAS PubMed.
- C. Pozzi, S. Ciambellotti, C. Bernacchioni, F. Di Pisa, S. Mangani and P. Turano, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2580–2585 CrossRef CAS PubMed.
- W. R. Hagen, H. Wassink, R. R. Eady, B. E. Smith and H. Haaker, Eur. J. Biochem., 1987, 169, 457–465 CrossRef CAS PubMed.
- A. J. Pierik, H. Wassink, H. Haaker and W. R. Hagen, Eur. J. Biochem., 1993, 212, 51–61 CrossRef CAS PubMed.
- A. S. Pereira, W. Small, C. Krebs, P. Tavares, D. E. Edmondson, E. C. Theil and B. H. Huynh, Biochemistry, 1998, 37, 9871–9876 CrossRef CAS PubMed.
- P. Moenne-Loccoz, C. Krebs, K. Herlihy, D. E. Edmondson, E. C. Theil, B. H. Huynh and T. M. Loehr, Biochemistry, 1999, 38, 5290–5295 CrossRef CAS PubMed.
- M. H. Sazinsky and S. J. Lippard, Met. Ions Life Sci., 2015, 15, 205–256 Search PubMed.
- S. Sun and D. Chasteen, Biochemistry, 1994, 33, 15095–15102 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, ChemBioChem, 2013, 14, 1123–1133 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |