
Integration of fluorescence imaging with proteomics enables visualization and identification of metallo-proteomes in living cells†
Yau-Tsz
Lai‡
a,
Ya
Yang‡
a,
Ligang
Hu
a,
Tianfan
Cheng
a,
Yuen-Yan
Chang
a,
Mohamad
Koohi-Moghadam
a,
Yuchuan
Wang
b,
Jiang
Xia
c,
Junwen
Wang
de,
Hongyan
Li
a and
Hongzhe
Sun
 *a
*a
aDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China. E-mail: hsun@hku.hk; Fax: +852 28571586
bMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou, P. R. China
cDepartment of Chemistry, The Chinese University of Hong Kong, Hong Kong, P. R. China
dCenter for Individualized Medicine & Department of Health Sciences Research, Mayo Clinic, Scottsdale, AZ 85259 USA
eDepartment of Biomedical Informatics, Arizona State University, Scottsdale, AZ 85259 USA
First published on 25th October 2016
Abstract
Metalloproteins account for nearly one-third of proteins in proteomes. To date, the identification of metalloproteins relies mainly on protein purification and the subsequent characterization of bound metals, which often leads to losses of metal ions bound weakly and transiently. Herein, we developed a strategy to visualize and subsequently identify endogenous metalloproteins and metal-binding proteins in living cells via integration of fluorescence imaging with proteomics. We synthesized a “metal-tunable” fluorescent probe (denoted as Mn+-TRACER) that rapidly enters cells to target proteins with 4–40 fold fluorescence enhancements. By using Ni2+-TRACER as an example, we demonstrate the feasibility of tracking Ni2+-binding proteins in vitro, while cellular small molecules exhibit negligible interference on the labeling. We identified 44 Ni2+-binding proteins from microbes using Helicobacter pylori as a showcase. We further applied Cu2+-TRACER to mammalian cells and found 54 Cu2+-binding proteins. The strategy we report here provides a great opportunity to track various endogenous metallo-proteomes and to mine potential targets of metallodrugs.
Significance to metallomicsIdentification of metals in proteins relies exclusively on protein purification and characterization in vitro thus often leads to the loss of weakly- or transiently-bound metals. We herein present a strategy to visualize and subsequently identify endogenous metal-binding proteins in situ in living cells through integrating fluorescence imaging with proteomics. We developed a novel sensor which rapidly enters living cells to target proteins with significant fluorescence enhancement, while the sensor could anchor on targeted proteins to promote subsequent protein identification. This strategy provides huge potentials to track metal-binding proteins at a proteome-wide scale and to mine important targets of metallodrugs. |
Introduction
Metals exist in diverse coordination geometries and multiple oxidation states, which allows them to promote complex biochemical events and participate in specialized and unique functions in proteins.1,2 Indeed, many diseases (e.g. neurodegenerative diseases) are known to be closely associated with dysregulation of metal ions.3–5 Understanding of metal homeostasis under physiological conditions thus has significant impacts on human health. Accumulated data demonstrate that around 30% of proteins encoded by human genomes are metalloproteins and/or metal-binding proteins, with the intrinsic metal ions playing critical roles in protein functions.6,7 Identification of a metal ion in a protein can significantly facilitate the functional assignment of the protein and helps to find its appropriate position in the context of known cellular pathways. Metal ions are also frequently incorporated into pharmaceuticals for therapeutic and diagnostic purposes.1,8–11 Proteins and enzymes have been known to either serve as direct targets of metallodrugs or play key roles in their pharmacology.12Currently there are numerous methods developed for identification of metals in proteins including fluorescence spectroscopy, inductively coupled plasma with either atomic emission spectroscopy (ICP-AES) or mass spectrometry (ICP-MS), or synchrotron-based techniques such as (extended) X-ray absorption spectroscopy and X-ray fluorescence spectroscopy.13,14 A new approach through the combination of high-throughput tandem MS/MS-based proteomics with ICP-MS analyses revealed that microbial metallo-proteomes are around twice as extensive as previously recognized.15 However, all of these methods rely exclusively on the purification of proteins and subsequent metal identification, which often leads to losses of weakly or transiently bound metal ions.
Alternatively, metalloproteins are identified by in vitro investigations of specific metal ions that bind to overexpressed proteins, thus false information may be received since a protein might bind a series of metal ions in vitro owing to the properties of the metal ions. However, selection of a metal ion by a protein in cells not only depends on specific features of the protein, but also depends on metal ion availability and cellular location.6 Metal selectivity of metalloproteins might be distinct inside living cells from those in in vitro experiments. For example, human copper, zinc superoxide dismutase 1 (hSOD1) selects Zn2+ only but not Cu2+ in its native binding site in cells; different from in vitro experiments in which hSOD1 exhibits no discrimination between Zn2+ and Cu2+ in this site, and binds two equivalents of Zn2+ per subunit at near physiological conditions.16,17 Therefore, it is highly desirable to develop appropriate approaches to study metal–protein interactions in situ, in particular when such interactions are transient (e.g. metallochaperones) or/and weak. Moreover, unveiling of potential protein targets for metallodrugs in cellular systems would improve our understanding of their mechanism of action,18 in turn facilitating the design of more effective metallodrugs.11 Unfortunately, to date, there appear to be no approaches enabling visualization and identification of metallo-proteomes inside live cells. Although in-cell NMR has been recently utilized to investigate metal–protein interactions under physiologically relevant conditions,17,19,20 this method is limited to certain proteins that give rise to good NMR signals and is not suitable for general application.
Here, we develop a “metal-tunable” fluorescent reporter (Mn+-TRACER), which rapidly enters cells (ca. 20 min) and binds to intracellular proteins with a significant enhancement in fluorescence (4–40 fold). The targeted proteins (i.e. proteins of interest, POI) can be further covalently anchored via photo-activation of the arylazide within the reporter, facilitating subsequent protein separation and identification through proteomics analysis. The strategy was first validated by tracking of a Ni2+-binding protein HpHspA overexpressed in Escherichia coli and then applied to Helicobacter pylori cells to unveil the Ni2+-proteome. Subsequently, using Cu2+-TRACER as a showcase, we demonstrate its feasibility to visualize and identify the Cu2+-proteome in mammalian cells (HeLa). The methodology can be readily extended to visualize different metallo-proteomes in different types of cells and mine potential targets of metals or metallodrugs at a proteome-wide scale.
Results and discussion
Design, synthesis and characterization of a fluorescent reporter Mn+-TRACER
We sought to develop a new strategy to identify intracellular metalloproteins or metal-binding proteins inside live cells. We have previously synthesized a fluorescent ligand NTA–AC by conjugating a coumarin fluorophore with nitrilotriacetate (NTA) and arylazide, which can cross cell membranes rapidly to image His6-tagged proteins in living cells when the ligand is coordinated to nickel(II).21 By taking advantage of arylazide within the probe, which can form a covalent linkage between the probe and targeted proteins through photo-activation, we further extended the strategy to visualize endogenous metal-associated proteins in living cells and subsequently to identify these proteins using high-throughput proteomics techniques (Scheme 1). Given the versatility of NTA in binding to metal ions, it is feasible to develop a “metal-tunable” fluorescent probe by chelating NTA–AC with different metal ions to target specific endogenous metalloproteins. We thus rename NTA–AC as TRACER (Tunable Reagent of Arylazide-Conjugated fluorescER). The probe, denoted as Mn+-TRACER, where Mn+ stands for metal ions with a strong chelation to the NTA moiety of the agent (e.g. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K > 10), can be classified as a metal-tunable fluorescent reporter and allows endogenous metallo-proteomes to be tracked.
K > 10), can be classified as a metal-tunable fluorescent reporter and allows endogenous metallo-proteomes to be tracked.
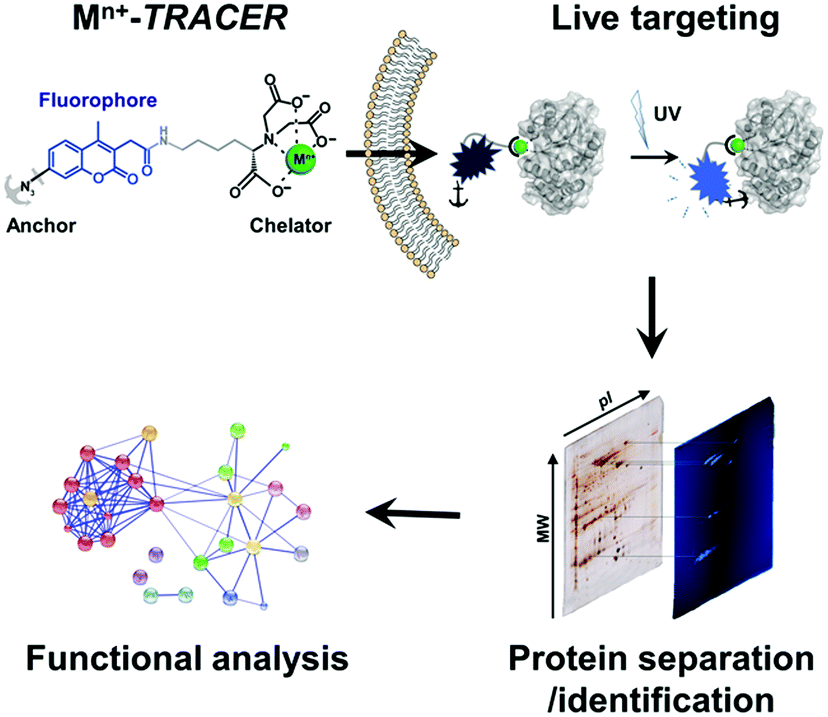 | ||
| Scheme 1 Schematic representation of the integrative strategy used to image and identify metallo-proteomes in live cells. | ||
TRACER itself exhibits a maximum absorption at 342 nm (ε = 1.11 × 104 M−1 cm−1) and emits at 448 nm (Φ = 0.056).21 Its fluorescence was quenched by 60–99% upon coordination to paramagnetic ions such as Cu2+, Ni2+ and Co2+, whereas it was almost unchanged upon binding to the diamagnetic ion Zn2+. Using Cu2+ as a proof of concept, we further investigated the binding affinity and stoichiometry of Mn+ with the TRACER. By monitoring the relative fluorescence intensity changes with Cu2+ concentration, the dissociation constant (Kd) of Cu2+-TRACER was determined to be 30 ± 15 nM via fitting the titration data to the Ryan–Weber equation22 (Fig. 1A), while the dissociation kinetics (koff) was determined to be 0.018 ± 0.002 s−1 through monitoring the displacement of Cu2+ from the NTA-moiety of Cu2+-TRACER upon the addition of NTA (Fig. S1, ESI†). Such strong binding ensures that the metal ions will not readily dissociate from the TRACER and is essential for tracking intracellular metal-binding proteins. Indeed, we have previously observed the Ni2+-TRACER–protein ternary complex using MALDI-TOF mass spectrometry,21 providing direct evidence that the metal ions will not be readily removed upon binding of proteins. A Job's plot constructed by monitoring the fluorescence changes in TRACER and Cu2+ gave rise to a maximum fluorescence change at a molar ratio of [Cu2+] to [TRACER] of 0.5, indicative of the formation of a 1:1 complex, i.e. Cu2+-TRACER (Fig. 1B). The metal-tunable fluorescent reporter Mn+-TRACER was thus generated by incubation of equimolar amounts of metal ions with TRACER in buffered aqueous solution (pH 7.2). Subsequent ESI-MS data further corroborated the formation of 1:1 complexes between different metal ions, i.e. Ni2+, Cu2+, Co2+, Zn2+ and Ga3+, and TRACER (Fig. S2, ESI†).
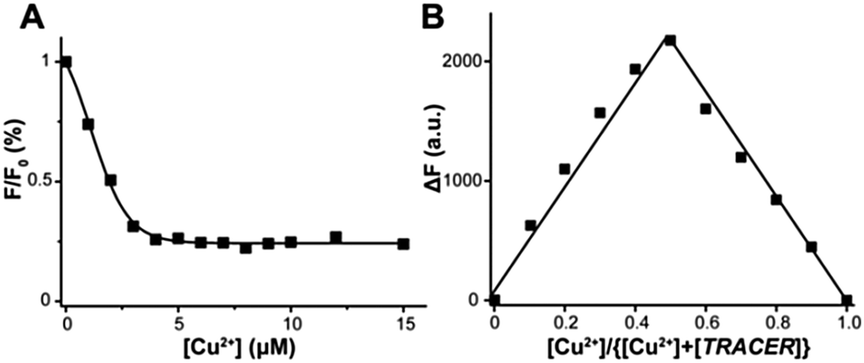 | ||
| Fig. 1 Characterization of binding affinity and stoichiometry of Mn+-TRACER using Cu2+-TRACER as an example. (A) Dependence of relative fluorescence intensities of TRACER (5 μM) on Cu2+ concentration in 20 mM Tris–HCl buffer (pH 7.2). The data are nonlinearly fitted using the Ryan–Weber equation, giving rise to a dissociation constant (Kd) of 30 ± 15 nM. (B) A Job's plot of the fluorescence changes in NTA–AC and Cu2+, indicative of the formation of the Cu–NTA–AC complex with a ratio of NTA–AC:Cu2+ of 1:1. | ||
We next examined the selectivity of Mn+-TRACER towards amino acids, and increases in fluorescence at 448 nm (ΔF > 0.1, excited at 365 nm) were observed for histidine and cysteine upon incubation with selected Mn+-TRACER (Fig. 2A). More specifically, Ni2+-, Bi3+- and Fe2+-TRACER exhibit a high selectivity towards histidine or cysteine, respectively, whereas the Cu2+- and Co2+-TRACER prefer both histidine and cysteine. In contrast, almost no changes in fluorescence occurred for TRACER alone upon mixing with these amino acids. The binding properties of Ni2+-TRACER towards histidine-containing peptides were also studied. Significant fluorescence changes (ΔF > 0.1) were observed only for peptides containing no less than two histidines, i.e. HGGHGGH, HGHGH and HGGH, but not the single histidine-containing peptide (GHGGG), upon incubation with Ni2+-TRACER (Fig. 2B), indicative of the preference of Ni2+-TRACER towards histidine-rich motifs. Taken together, these data demonstrate that Mn+-TRACER is a “metal-tunable” probe with a selectivity dependent on the metal coordination properties.
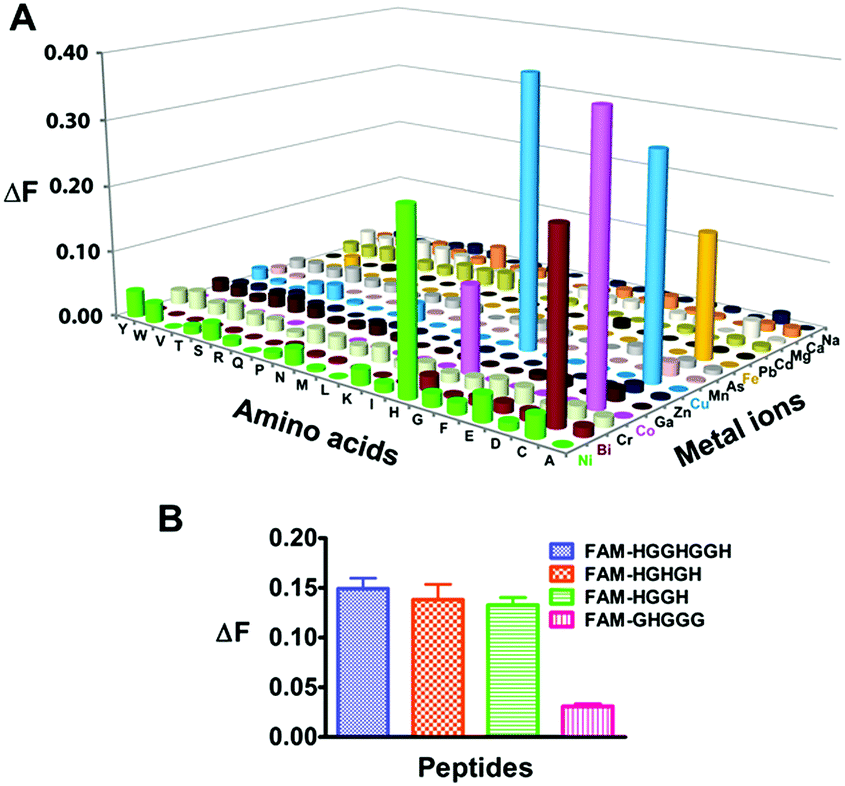 | ||
| Fig. 2 Selectivity of Mn+-TRACER towards amino acids and His-rich peptides. (A) Changes in fluorescence of 20 μM Mn+-TRACER upon incubation with excess amounts of amino acids (100 μM) for 30 min at 25 °C. (B) Fluorescence changes in Ni2+-TRACER upon addition of histidine-containing peptides in 20 mM Tris-buffer, pH 7.2 for 30 min. Note that evident increases in fluorescence were observed for the three histidine-rich peptides. | ||
Fluorescence response of Mn+-TRACER towards proteins in vitro
To examine the response of Mn+-TRACER towards proteins in vitro, we utilized recombinant protein HspA from H. pylori as an example as the protein possesses a unique His-rich C-terminus CCHTGNHDHKHAKEHEACCHDHKKH, and binds a range of transition metal ions.23,24 Titration of HpHspA to Ni2+-TRACER led to a gradual fluorescence increase and levelled off at a molar ratio of [HpHspA]/[Ni2+-TRACER] ca. 0.6 ± 0.1 (Fig. 3A), where the fluorescence was enhanced by about 7.8 fold. Similarly, binding of Cu2+-, Co2+- and Zn2+-TRACER to HpHspA led to a fluorescence increase by 26.4, 40.9 and 4.0 fold, respectively, within 5 min (Fig. S3, ESI†). In contrast, no changes in fluorescence were observed either for Ni2+-TRACER reacting with C-terminus-truncated HpHspA (HpHspAΔC) or for TRACER alone upon addition of HpHspA under identical conditions (Fig. S4, ESI†), confirming that the changes in fluorescence resulted from the selective binding of Ni2+- TRACER to the C-terminus of the protein through Ni2+. The stoichiometry of HpHspA to Ni2+-TRACER was further confirmed using isothermal titration calorimetry, which showed that Ni2+-TRACER binds to HspA with a binding capacity of 2.0 ± 0.1 and a binding constant (Kd) of 9.6 ± 2.2 μM (Fig. 3B). Taken together, each HpHspA binds two Ni2+-TRACER, consistent with the Ni2+-binding stoichiometry of HpHspA.23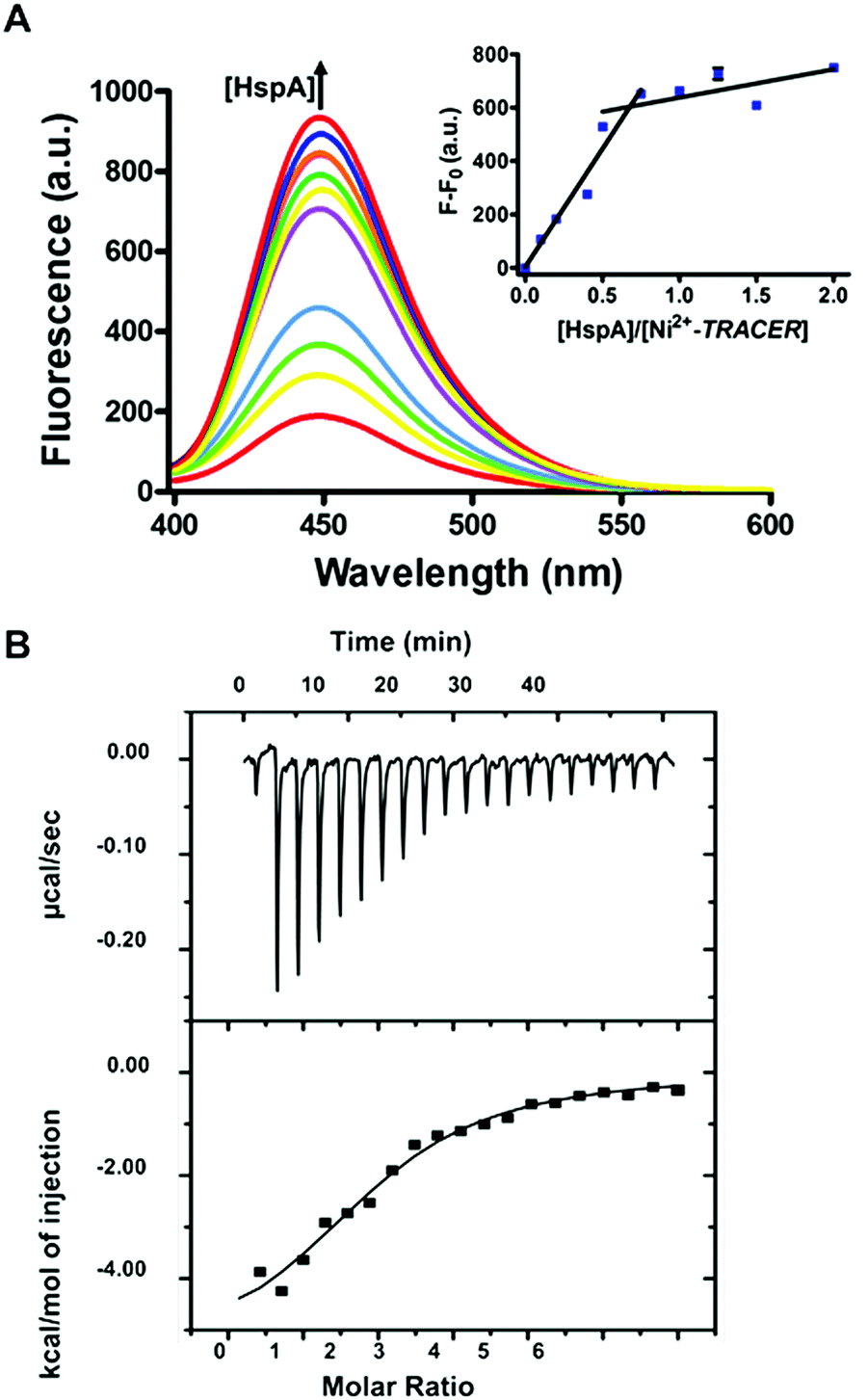 | ||
| Fig. 3 (A) Fluorescence spectra of Ni2+-TRACER (1 μM) with the addition of HpHspA (0, 0.1, 0.2, 0.4, 0.5, 0.75, 1.0, 1.25, 1.5, 2.0 μM, fluorescence signals increase as concentrations increase) upon excitation at λex = 342 nm. Inset: plot of fluorescence at 448 nm versus the molar ratio of HpHspA to Ni2+-TRACER. (B) Binding affinity of Ni2+-TRACER for HpHspA via isothermal titration calorimetry (ITC). The data were fit by a one-set binding model, giving rise to a binding capacity of N = 2.0 ± 0.1 and a dissociation constant of Kd = 9.6 ± 2.2 μM. | ||
Given that various biomolecules such as glutathione (GSH) are present inside cells, we thus examined the potential effect of GSH on the labeling of HpHspA by Ni2+-TRACER. Ni2+-TRACER was incubated with equimolar amounts of HpHspA (10 μM) in the presence of gradient amounts of GSH up to 5 mM, then irradiated with UV prior to gel electrophoresis. Blue fluorescence bands with similar intensities appeared on the gel, attributable to the labeling of HpHspA by Ni2+-TRACER (Fig. 4A), indicating that GSH exerts a negligible effect on protein labeling. This was further confirmed by the observation of only slight changes in the fluorescence of a solution containing Ni2+-TRACER (1 μM), HspA (1 μM) and GSH (5 mM) (Fig. 4B). Metal ions are also unlikely to disrupt the binding of the probe to its target proteins as the cellular concentrations of those metals with a high affinity for NTA are generally at or less than micromolar levels,25 while others with high concentrations (e.g. Na+, K+, Mg2+ and Ca2+) exhibit weak chelation to the NTA moiety of TRACER and are unlikely to enter the cells26 (Fig. S5, ESI†). Therefore, it is feasible to utilize Mn+-TRACER to label metal-associated proteins in living cells.
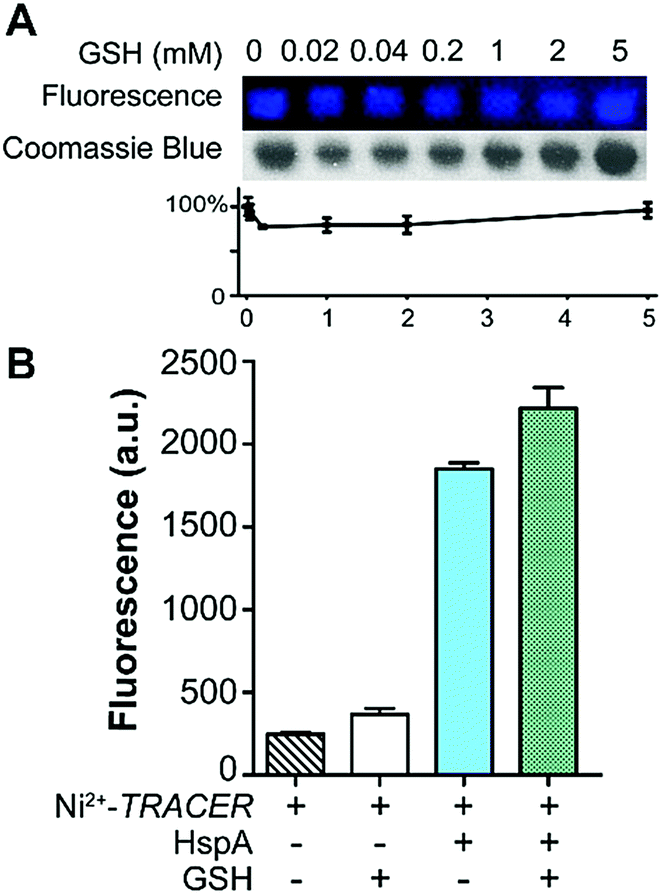 | ||
| Fig. 4 The effect of glutathione (GSH) on the labeling of Mn+-TRACER to proteins as demonstrated by the binding of Ni2+-TRACER and HspA. (A) Fluorescence labeling of HspA (1 μM) by Ni2+-TRACER (1 μM) in the absence and presence of GSH with concentrations up to 5 mM monitored using SDS-PAGE. (B) Fluorescence responses of Ni2+-TRACER (1 μM) incubated with HspA (1 μM) in the absence and presence of GSH (5 mM). | ||
Labeling Ni2+-binding proteins in living E. coli cells
Next we examined the capability of Ni2+-TRACER to “light-up” intracellular HpHspA as a proof of concept. The E. coli cells overexpressing HpHspA, but not HpHspAΔC, gave rise to intense blue fluorescence throughout the cells upon treatment with 10 μM Ni2+-TRACER for 30 min (Fig. 5A), in agreement with the previous in vitro experiments on HspA. No cells could be stained with propidium iodide, indicating that the cells remain viable upon labelling. The cells were subjected to UV irradiation at 365 nm for 10 min using a UVP UVGL-25 Mineralight® UV lamp (720 μW cm−2) prior to lysis to ensure the formation of covalent bonds between HpHspA and the probe, and the cell lysates were subjected to electrophoresis and a fluorescence image was captured. Only one blue fluorescent band corresponding to the molecular weight of HpHspA (ca. 13 kDa) was observable on the SDS-PAGE (Fig. 5B), verifying that Ni2+-TRACER indeed labels HpHspA inside live cells.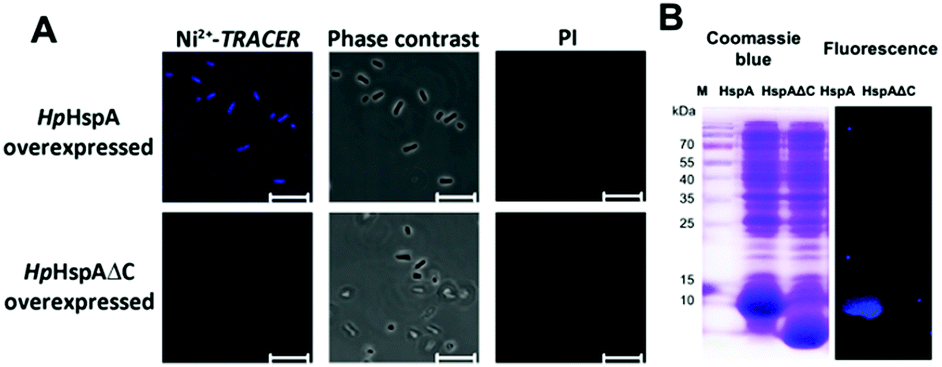 | ||
| Fig. 5 (A) Confocal fluorescence and phase contrast images of live E. coli cells with HpHspA or HpHspAΔC overexpressed after incubation with 10 μM Ni2+-TRACER (n = 5). Scale bar: 5 μm. (B) SDS-PAGE and fluorescence imaging analysis of E. coli cells with overexpression of HpHspA or HpHspAΔC upon incubation with Ni2+-TRACER (10 μM) for 1 h at 30 °C. Note that only the cells with HpHspA but not HpHspAΔC overexpressed exhibit intense fluorescence. | ||
Tracking Ni2+-proteome in microbes using H. pylori as a showcase
Nickel is essential for many microbes, in particular for the survival and pathogenesis of the gastric pathogen H. pylori, which infects nearly half of the world population and is responsible for chronic gastritis, peptic ulcers and even stomach cancers.27 Nickel homeostasis is thus tightly regulated by a battery of metalloproteins or metallochaperones.28–30 We utilized Ni2+-TRACER to track the nickel proteome in H. pylori given that many Ni2+-binding proteins have been extensively characterized in vitro.23,31–37 Upon supplementation of a culture of H. pylori with 50 μM Ni2+-TRACER for 30 min, we observed intense blue fluorescence throughout the pathogen (Fig. 6A), indicating that Ni2+-TRACER had entered the cells to label endogenously expressed nickel-binding proteins in the living bacterial cells. Nevertheless, we could not differentiate the location and abundance of the labeled proteins owing to the small size of each H. pylori cell (∼3 μm × 0.5 μm).38 Cells were subjected to ultraviolet irradiation at 365 nm for 10 min prior to lysis, enabling the formation of a covalent linkage between the arylazide and those lit-up proteins. Subsequently, Ni2+-associated proteins were separated via 2-dimensional gel electrophoresis and the fluorescence was captured. As expected, a quantity of protein spots retained blue fluorescence on the gel (Fig. 6B and Fig. S6, ESI†). These spots were excised and subjected to protein identification using MALDI-TOF-MS. A series of horizontal spots with similar molecular masses for each of these proteins were noted on the gel, likely attributable to the different isoforms resulting from post-translational modifications, leading to a slight shift in their electrophoretic mobility.39,40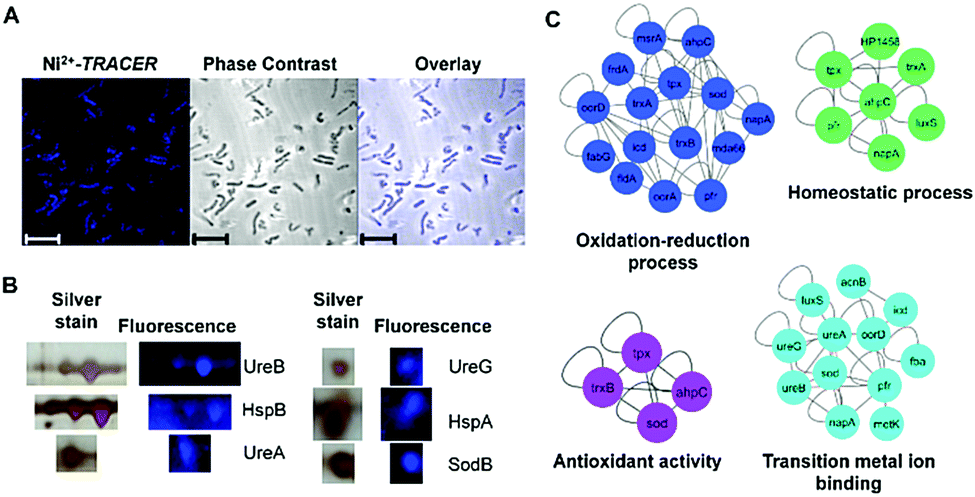 | ||
| Fig. 6 (A) Microscopic imaging of live H. pylori labeled with 50 μM Ni2+-TRACER (n = 5). Scale bar: 5 μm. (B) Selected spots of Ni2+-associated proteins in H. pylori identified in 2-DE by fluorescence and silver staining. Full 2-DE is shown in Fig. S6 (ESI†). (C) Protein–protein interaction networks demonstrating the functional categories of identified Ni2+-binding proteins. Proteins are represented by gene names and proteins without connections are not shown in the network. | ||
Using this strategy, we have identified 44 proteins from the cytosolic fractions of H. pylori (Table S1, ESI†), among which 11 proteins were also previously identified using immobilized-metal affinity chromatography (IMAC).39,40 Further examination of the identified proteins revealed that many proteins were demonstrated previously to be Ni2+-binding proteins based on in vitro characterization including HpUreB, HpHspA and HpUreG. These proteins have been demonstrated to play critical roles in nickel homeostasis.23,24,41 For example, HpHspA, a GroES homologue as a co-chaperonin in H. pylori, plays an essential role in Ni2+ transport as a specialized nickel chaperone protein, and may also serve as a drug target.42HpUreB, one of the urease subunits, has a partially buried dinuclear nickel site,43 consisting of five histidines (His136, His138, His221, His248 and His274) directly involved in the formation of the dinuclear center with three of them located on the flap (Fig. S7, ESI†).43 The ability of Ni2+-TRACER to label UreB suggests that the probe, given its small size, can approach the cavity of the protein and binds to the histidine-rich motif in the native states of UreB. HpUreG is a specific nickel-dependent GTPase and plays an important role in urease maturation by interactions with other metallochaperones such as UreE and HypA.32,44 Surprisingly, HpHspB, a protein that does not bind Ni2+in vitro, was also identified (Table S1, ESI†). This protein was also identified using a conventional IMAC approach.40 To further investigate this unexpected phenomenon, recombinant HpHspB was overexpressed and purified, and subjected to SDS-PAGE analysis after incubation with Ni2+-TRACER, but no blue fluorescence bands corresponding to the protein spots were observed on the gel (Fig. S8, ESI†). HpHspA and HpHspB at a molar ratio of 1:2 were incubated with equimolar amounts of Ni2+-TRACER (12 μM) with respect to HpHspA for 1 h and were then subjected to SDS-PAGE analysis upon UV irradiation for 10 min. To our surprise, we observed two blue bands in the gel corresponding to the molecular masses of both HspA and HspB. In contrast, no blue band corresponding to HspB was found in the gel without prior UV irradiation (Fig. S8, ESI†). We therefore conclude that the UV irradiation accounted for the formation of an HspA–HspB complex. The probe which bound to HspA is in close proximity to HspB within the complex, thus subsequent photo-activation of arylazide leads to the generation of a strong covalent linkage between the probe and HspB, which prevents the protein–probe complex from dissociation even under denaturing conditions. Other proteins such as superoxide dismutase B (SodB) were identified, which have also been reported as Ni2+-binding in other bacteria.28
The identified Ni2+-binding proteins were subjected to Gene Ontology (GO) enrichment analysis to characterize the GO categories that are significantly enriched (cutoff p-value < 0.05). In total, 7 biological processes, 13 molecular functions and 3 cellular component GO terms were associated with the identified proteins (Tables S2–S4, ESI†). As shown in Fig. 6C, the majority of proteins fall into 4 functional categories, involving the oxidation–reduction process (p = 1.70 × 10−5), transition metal ion binding (p = 3.67 × 10−3), the homeostatic process (p = 3.80 × 10−5) and antioxidant activity (p = 1.26 × 10−2), and thus revealing the importance of Ni2+ and Ni2+-associated proteins in the biological processes of these microbes.30,45
Visualization and identification of Cu2+-binding proteins in mammalian cells
We then examined the feasibility of Mn+-TRACER in labeling metal-binding proteins in mammalian cells. We first investigated the membrane permeability of the probe using Cu2+-TRACER as an example. Incubation of HeLa cells with 25 μM of Cu2+-TRACER led to an intense blue fluorescence within 10 min (Fig. 7A), and the intensity further increased almost 5-fold in 20 min (Fig. S9, ESI†), clearly demonstrating that Cu2+-TRACER enters cells rapidly to label intracellular copper-binding proteins in live cells. The potential toxicity of various Mn+-TRACER probes was examined using an MTT assay. According to Fig. S10 (ESI†), the cytotoxicity of the probes remained negligible (except Zn2+-TRACER with ca. 12% loss of cell viability), implying that the probes exhibit no toxicity to mammalian cells at the concentration used.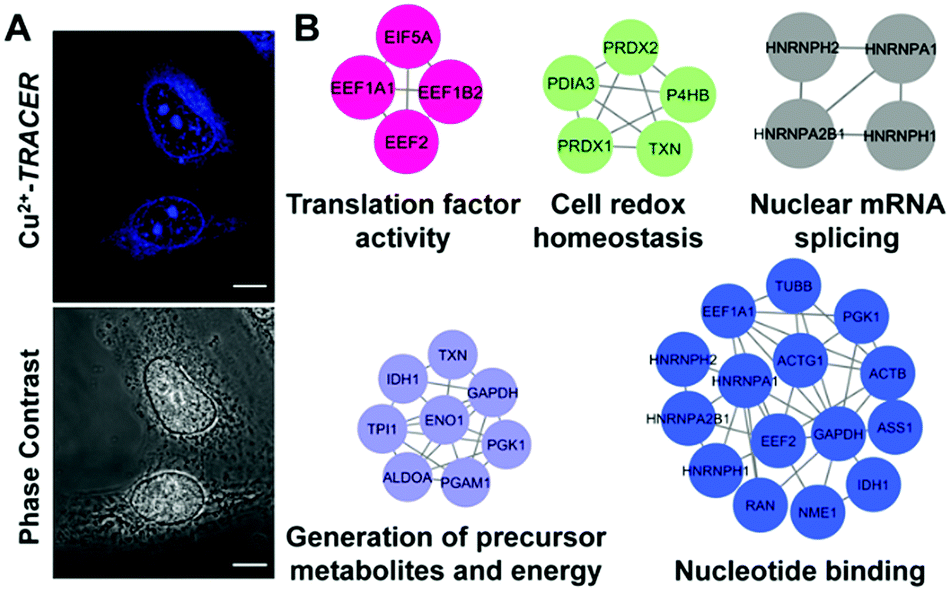 | ||
| Fig. 7 (A) Confocal imaging of HeLa cells incubated with 25 μM Cu2+-TRACER (n = 3). Scale bar: 10 μm. (B) Protein–protein interaction (PPI) networks illustrating the functional categories of Cu2+-binding proteins identified in HeLa. Proteins are represented by gene names and proteins without connections are not shown in the network. | ||
Copper is essential for most living organisms and serves as a cofactor for many enzymes that are critical for various biological functions. Therefore, copper ions are tightly regulated through complex systems to fulfill the cellular copper requirements and in the meantime to minimize the potential toxicity of free copper ions.46–48 To further examine the feasibility of our probe in tracking metallo-proteomes in mammalian cells, Cu2+-TRACER was selected as a showcase. We incubated 50 μM of Cu2+-TRACER with HeLa cells and intense blue fluorescence was observed mainly in the nucleus (Fig. 7A). The cells were UV irradiated prior to lysis and soluble fractions were subjected to 2D gel electrophoresis and peptide mass fingerprinting analysis to allow the labelled proteins to be identified (Fig. S11 and Table S5, ESI†). In total, 54 Cu2+-binding proteins were identified, among which 17 proteins have also been previously reported using an IMAC approach.49 Those identified proteins were further subjected to GO enrichment analysis. Sixty-four biological processes, 19 molecular functions and 24 cellular component GO terms were associated with the identified proteins (Fig. 7B and Tables S6–S8, ESI†). Many of the proteins are associated with nucleotide binding (p = 5.77 × 10−4) and nuclear mRNA splicing (p = 1.05 × 10−3), which support our observation that blue fluorescence was mostly observed in the nucleus. Furthermore, certain identified proteins are associated with energy generation (p = 3.21 × 10−5), cell redox homeostasis (p = 3.44 × 10−5) and translation factor activity (p = 3.37 × 10−3), suggesting that Cu2+ might play important roles in these processes in the eukaryotic system.
Conclusion
We have reported, to our knowledge, the first strategy to visualize the subcellular localization of endogenous metallo-proteomes in living cells, subsequently identifying these proteins through proteomics approaches using a unique fluorescent probe. The probe can serve as a fluorescent reporter, and rapidly cross cell membranes to target specific proteins, leading to considerable fluorescence enhancement. Importantly, the probe can be further covalently anchored to the labeled proteins through arylazide photoactivation, allowing downstream protein identification. Furthermore, the current approach might also be easily manipulated to achieve high throughput analysis of various endogenous metallo-proteomes in living cells as long as metal ions possessing two to three vacant sites for protein targeting after chelation to the NTA moiety. While for those metal ions binding weakly to NTA (e.g. Mg2+ and Ca2+), the metal-chelated probe could not enter the cells to label proteins because of the highly charged NTA-moiety. However, caution has to be taken during analysis of these metalloproteins. Similar to IMAC, a method that was initially developed for protein purification50 and is now frequently utilized to enrich metalloproteins from the total proteomes,39,51 metal-binding proteins identified using the current method might not be intrinsic. Nevertheless, our approach has the advantage that it can track endogenous metalloproteins which bind metal ions transiently or weakly, that cannot be identified using IMAC. Therefore, to fully characterize a metallo-proteome in cells, an integration of this approach with others such as continuous-flow gel electrophoresis with ICP-MS (GE-ICP-MS) and other metal-based approaches is necessary.15,18 Moreover, our strategy will open up an informative approach to mine proteome-wide potential targets of metal-based drugs, a class of drugs which has been widely used for the treatment and diagnosis of various diseases,10,11 and also has the potential to serve as an adjuvant of antibiotics to cope with current antibiotic resistance,52 and in-turn to facilitate the rational design of better metallo-drugs.2Experimental section
Probe synthesis
The fluorescent ligand TRACER was synthesized according to our recent report21 and the probe Mn+-TRACER was generated by reacting metal ions (as chloride salts) with equimolar amounts of TRACER in 20 mM Tris–HCl buffer at pH 7.2.Peptide synthesis
All peptides used were synthesized using standard 9-fluorenylmethoxycarbonyl (Fmoc) based on solid phase synthesis modified from a previous procedure.53 While still attached to the resin, the peptides were labelled with 5(6)-carboxyfluorescein (FAM) at their N-termini in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N,N-diisopropylethylamine (DIPEA) in DMF overnight. Afterwards, the peptides were cleaved from the resin via a regular TFMSA cleavage. The crude peptides were purified using reverse-phase HPLC and lyophilized.Fluorescence spectroscopic measurements
Inorganic compounds and amino acids were received from Sigma-Aldrich and USB Chemicals, and used directly by dissolving in MilliQ water for further use. Bovine serum albumin (BSA) was purchased from USB Chemicals and used as received. All fluorescence measurements were performed on a Hitachi F-7000 fluorescence spectrophotometer with a 1000 W xenon lamp using a 1 cm × 1 cm quartz cuvette (1.5 mL sample volume). Fluorescence responses of Mn+-TRACER towards amino acids and peptides were determined by incubating 20 μM of the probe with 100 μM of the amino acids, and were determined using a Beckman Coulter DTX 880 Multimode Reader with the excitation and emission filters set at λex = 360 nm and λem = 465 nm. Fluorescence responses of Ni2+-TRACER towards His-containing peptides were determined when the probe was incubated with equimolar amounts of the peptides. Fluorescence signals of the proteins were determined by reacting 5 μM Mn+-TRACER with different molar equivalents of the proteins at 25 °C. The binding stoichiometry of TRACER to metal ions was measured using Cu2+ as an example as follows: a series of 10 μM (total concentration) of TRACER and Cu2+ solutions containing different amounts of TRACER and Cu2+ were prepared, and incubated at 25 °C for 30 min. Fluorescence spectra were then recorded to yield a Job's plot. The dissociation kinetics of Cu2+-TRACER was performed by measuring the fluorescence changes in the displacement of Cu2+ from Cu2+-TRACER (5 μM) upon the addition of nitrlinotriacetic acid (NTA) (500 μM) against time using a Thermo Varioskan LUX Microplate Reader at 25 °C. The effect of glutathione on HpHspA labeling was determined by monitoring the fluorescence responses of Ni2+-TRACER (1 μM) incubated with or without equimolar amounts of HpHspA and GSH (5 mM) simultaneously for 30 min in 25 °C.Isothermal titration calorimetry (ITC)
HspA protein was freshly prepared in 20 mM HEPES, 100 mM NaCl, pH 7.4, while Ni–NTA–AC was pre-incubated in the same buffer overnight at 4 °C before use. For the probe–protein interaction study, Ni–NTA–AC was titrated into HspA. All ITC experiments were performed at 25 °C on an ITC200 isothermal titration microcalorimeter (GE Micro).Expression and purification of HpHspA and HpHspB
H. pylori proteins, HpHspA and HpHspAΔC (with histidine-rich C-terminus deletion), were overexpressed and purified as described previously.23,24 For HpHspB, the wild-type hspB gene was amplified via PCR from genomic DNA of H. pylori 26695 using the primer pair HspB-up/HspB-down, and the PCR product was digested using NdeI and EcoRI and inserted into a pET32a (+) plasmid vector, generating pET32-HspB. The plasmid was transformed into E. coli Rosetta (DE3) (Novagen), and HspB was overexpressed at 30 °C for 5 h with the induction by 0.2 mM isopropyl β-D-thiogalactoside (IPTG). HspB was purified into homogeneity using a two-column purification procedure. The HspB-overexpressed E. coli cells were lysed in Buffer A (20 mM Tris–HCl, pH 8.0 containing 50 mM NaCl, 1 mM EDTA and 1 mM DTT) and the lysates were loaded onto a Resource Q column (GE Healthcare). HspB was then eluted out with the linear gradient from 50 mM to 500 mM NaCl on an ÄKTAFPLC system (GE Healthcare). The fractions containing HspB were pooled together, concentrated, and then applied to a HiPrep Saphacryl S-300 HR (GE Healthcare) equilibrated with buffer (20 mM Tris–HCl, pH 8.0, 250 mM NaCl, 1 mM EDTA, 1 mM DTT).Fluorescence imaging of E. coli
pET32a-HpHspA and pET32a-HpHspAΔC transformed BL21(DE3) cells were grown in Luria–Bertani (LB) containing 100 μg mL−1 ampicillin overnight at 37 °C, and then subcultured in a 1:100 dilution. When OD600 reached 0.6, IPTG (0.2 mM) was added to induce protein expression at 30 °C for 3 h. Ni2+-TRACER (10 μM) was added and further incubated in darkness. The HpHspA and HpHspAΔC overexpressed E. coli cells were then washed with 20 mM Tris–HCl, 100 mM NaCl, pH 7.2 at 4 °C. The densities of the cells were adjusted to 0.3 (OD600) with buffer, and propidium iodide (PI) (1 μg mL−1) was added to the samples prior to imaging to examine cell viability. Fluorescence microscopic images were captured using a Carl Zeiss LSM700 Inverted Confocal Microscope with a Plan-Apochromat 63× 1.40 NA oil-immersion objective, utilizing a 405 nm laser (for TRACER excitation) and a 555 nm laser (for PI excitation). HpHspA and HpHspAΔC expression were visualized and confirmed using SDS-PAGE electrophoresis of the cell lysates. The covalent linkage of Ni2+-TRACER to the bound proteins was achieved via photo-activation by ultraviolet irradiation at 365 nm for 10 min using a UVP UVGL-25 Mineralight® UV lamp (720 μW cm−2) at room temperature. SDS-PAGE analysis was performed using 15% resolving gel, and the fluorescence gel images were captured using ImageQuant 350 of GE Healthcare (λex = 365 nm, λem = 460–500 nm), and compared with the images of Coomassie Blue staining.
H. pylori cell culture
H. pylori 26695 was purchased from American Type Culture Collection (ATCC). The bacteria glycerol stock was recovered from −80 °C, and cultured on Columbia agar plates supplemented with 7% (v/v) laked horse blood and selective antibiotics all from Oxoid. The plates were incubated in a microaerophilic atmosphere generated using an Oxoid CampyGen Kit at 37 °C for 3 days. After that, the cells on the plates were transferred to autoclaved Brucella broth (BD) with 0.2% β-cyclodextrin (Sigma), and cultured in an anaerobic jar using an Oxoid CampyGen Kit at 37 °C overnight with 100 rpm agitation. Cell pellets were resuspended and washed with Hank's Balanced Salt Solution (HBSS) for cell imaging and 2D gel.Mammalian cell culture
HeLa cell lines were purchased from American Type Culture Collection (ATCC). All chemicals related to cell culturing were purchased from Life Technologies (Thermo Fisher Scientific) unless otherwise stated. HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM) with the supplementation of 10% fetal bovine serum (FBS) and 1% antibiotics (Pen Strep), and the cells were all cultured in a 5% CO2 incubator at 37 °C. HBSS was used to replace the culture medium for probe-related experiments prior to imaging.Fluorescence imaging of H. pylori
OD600 was measured to monitor the growth of H. pylori 26695 in Brucella broth. At the mid-log phase, Ni2+-TRACER (50 μM) was added to the culture medium for incubation in the dark at 37 °C for 30 min with 100 rpm agitation. The cells were collected and washed with cold PBS buffer (pH 7.4) three times. The density of the cells was adjusted to 0.3 (OD600) prior to confocal imaging.Identification of Ni2+-binding proteins in H. pylori
Different concentrations of Ni2+-TRACER (25, 50, 100 μM) were applied to H. pylori for 2-DE analysis and 50 μM of Ni2+-TRACER was chosen as the optimum for better gel imaging with less nonspecific binding. H. pylori was grown in 100 mL of liquid medium to the mid-log phase. Cells were collected and washed with PBS three times. Then, 10 mL of culture were supplied with 50 μM of Ni2+-TRACER and incubated at 37 °C in darkness for 30 min (sample A), and another 10 mL of culture was used for comparison (sample B). Cells were then centrifuged at 3500g for 5 min at 4 °C and washed with PBS buffer (pH 7.4) three times. Pellets of sample A were then resuspended in buffer and exposed to ultraviolet radiation at 365 nm for 15 min. Both samples were resuspended in 1 mL of lysis buffer and the proteins were recovered by centrifugation at 14000g for 30 min at 4 °C. Protein concentration was determined using a BCA protein assay kit (Thermo Scientific) and 150 μg of total proteins for each sample were used. The samples were desalted using a 2-D Clean-Up Kit (GE Healthcare) and rehydrated, then isoelectric focusing (IEF) was performed using a precast 13 cm Immobiline DryStrip (GE Healthcare) with a nonlinear pH gradient at 3–10, followed by second dimensional SDS-PAGE (13.5% acrylamide gel) with both samples A and B being used in parallel. After electrophoresis, the gels were imaged using a MY ECL Imager (Thermo scientific), and stained with silver staining for visualization of protein spots. Only intense blue fluorescent protein spots could be observed from the gel of sample A, and they were consistent with the location of the silver protein spots of interest on the gel. The selected fluorescent protein spots were subjected to trypsin digestion and then identified using MALDI-TOF-MS.
Microscopic imaging of Cu2+-TRACER
HeLa cells cultured in confocal dishes were grown to 70% confluency, then washed with HBSS three times to remove culture medium for confocal imaging using a Carl Zeiss LSM700 Inverted Confocal Microscope (405 nm laser) under a Plan-Apochromat 63× 1.40 NA oil-immersion objective, while the measuring range of emission (430–500 nm) was fixed. Imaging was initiated every 30 s after Cu2+-TRACER (25 μM) was supplemented to the cells, the time required to label intracellular targets by the probe was quantified. Fluorescence signals of the probe observed in HeLa cells were quantified using Zen software (Carl Zeiss) and plotted accordingly.Determination of cell viability (MTT)
MTT experiments were conducted according to the MTT Cell Proliferation Assay Kit Protocol provided by Thermo Fisher. HeLa cells were seeded into 96-well plates (10000 cells per well) and incubated in the respective medium overnight at 37 °C with 5% CO2. The medium was replaced and the cells were incubated with different concentrations (0, 25 and 50 μM) of Ni2+, Cu2+, Co2+, and Zn2+-TRACER in medium for 30 min at 37 °C and protected from light. The medium was then removed and replaced with fresh medium, 10 μL of (3-4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT, 5 mg mL−1 in sterile PBS) were added and further incubated for 4 h. After removing all but 25 μL of medium, 50 μL of DMSO were applied and the samples were incubated for 10 min at 37 °C. The absorbance of each well was recorded at 540 nm using a microplate reader (BIO-RAD, iMark™), and the cell viability is reported relative to those of the untreated cells.
Identification of Cu2+-binding proteins in HeLa cells
HeLa cells cultured in dishes were washed with HBSS three times. After incubation with 50 μM Cu2+-TRACER for 30 minutes, the cells were washed with HBSS three times and subjected to ultraviolet irradiation at 365 nm for 10 min using a UVP UVGL-25 Mineralight® UV lamp (720 μW cm−2). The treated cells were washed twice with cold PBS and collected via centrifugation. Cells were then resuspended in 20 mM HEPES, 100 mM NaCl, pH 7.2 with the supplementation of a Protease inhibitor cocktail (Sigma-Aldrich) and 1 mM TCEP, and subsequently lysed via sonication to separate the supernatant (cytosolic fraction) and insoluble debris (the fraction including the membrane and unlysed organelles). Protein concentration was determined using a BCA protein assay kit (Thermo Scientific) and 150 μg of the soluble protein samples were desalted using a 2-D Clean-Up Kit (GE Healthcare) and rehydrated, then isoelectric focusing (IEF) was performed using a precast 13 cm Immobiline DryStrip (GE Healthcare) with a nonlinear pH gradient of 3–10, followed by second dimensional SDS-PAGE (13.5% acrylamide gel) with both labeled and unlabeled samples being run in parallel. After electrophoresis, the gels were imaged using a MY ECL Imager (Thermo scientific), and stained with silver staining for visualization of the protein spots. The selected blue fluorescent protein spots were subjected to trypsin digestion and then identified using MALDI-TOF-MS.GO enrichment analysis
DAVID v6.7 was used to perform gene ontology (GO) enrichment analysis to identify statistically enriched GO terms and functionally categorize for the selected Ni-binding proteins in H. pylori 26695, and the Cu-binding proteins in HeLa. The Uniprot accession number of each protein was obtained as input data. The most significant GO terms with a p-value <0.05 were selected and reported.PPI network analysis
The protein–protein interaction (PPI) network of H. pylori 26695 was built using the String database and a series of literature reported PPIs in H. pylori 26695.54,55 The PPI network of HeLa was built using the String database. The final PPI networks were all exported and visualized using Cytoscape v3.4.0.Acknowledgements
This work is supported by the Research Grants Council of Hong Kong (HKU704612, HKU703913, HKU17304614, HKU17305415) and by the University of Hong Kong for an e-SRT on Integrative Biology, a fellowship for Y. Yang. We acknowledge the Li Ka Shing Faculty of Medicine Faculty Core Facility (the University of Hong Kong) for the support on confocal imaging. We thank the Center for Genomic Sciences, Li Ka Shing Faculty of Medicine for the mass spectrometry facilities.Notes and references
- I. Bertini, H. Gray, E. Stiefel and J. Valentine, Biological inorganic chemistry: structure and reactivity, University Science Books, 1st edn, 2007 Search PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef CAS PubMed.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342–355 CrossRef CAS PubMed.
- M. Farina, D. S. Avila, J. B. T. da Rocha and M. Aschner, Neurochem. Int., 2013, 62, 575–594 CrossRef CAS PubMed.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 7, 25–35 CrossRef CAS PubMed.
- Z. Ma, F. E. Jacobsen and D. P. Giedroc, Chem. Rev., 2009, 109, 4644–4681 CrossRef CAS PubMed.
- T. W. Hambley, Science, 2007, 318, 1392–1393 CrossRef CAS PubMed.
- S. J. Lippard, Nat. Chem. Biol., 2006, 2, 504–507 CrossRef CAS PubMed.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- H. Sun and Z. F. Chai, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2010, 106, 20–38 RSC.
- S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119–1138 RSC.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole Ii, F. E. Jenney Jr, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779–782 CrossRef CAS PubMed.
- L. Banci, L. Barbieri, I. Bertini, F. Cantini and E. Luchinat, PLoS One, 2011, 6, e23561 CAS.
- L. Banci, L. Barbieri, I. Bertini, E. Luchinat, E. Secci, Y. Zhao and A. R. Aricescu, Nat. Chem. Biol., 2013, 9, 297–299 CrossRef CAS PubMed.
- L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y.-T. Lai, G. Jiang and H. Sun, Angew. Chem., Int. Ed., 2013, 52, 4916–4920 CrossRef CAS PubMed.
- H. Li and H. Sun, Metallomics, 2014, 6, 69–76 RSC.
- E. Luchinat, L. Barbieri, J. T. Rubino, T. Kozyreva, F. Cantini and L. Banci, Nat. Commun., 2014, 5, 5502 CrossRef CAS PubMed.
- Y.-T. Lai, Y.-Y. Chang, L. Hu, Y. Yang, A. Chao, Z.-Y. Du, J. A. Tanner, M.-L. Chye, C. Qian, K.-M. Ng, H. Li and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2948–2953 CrossRef CAS PubMed.
- D. K. Ryan and J. H. Weber, Anal. Chem., 1982, 54, 986–990 CrossRef CAS.
- S. Cun, H. Li, R. Ge, M. C. M. Lin and H. Sun, J. Biol. Chem., 2008, 283, 15142–15151 CrossRef CAS PubMed.
- S. J. Cun and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4943–4948 CrossRef CAS PubMed.
- R. J. Williams, Dalton Trans., 2007, 991–1001 RSC.
- G. Anderegg, Pure Appl. Chem., 1982, 54, 2693–2758 CrossRef CAS.
- A. Covacci, J. L. Telford, G. Del Giudice, J. Parsonnet and R. Rappuoli, Science, 1999, 284, 1328–1333 CrossRef CAS PubMed.
- T. Cheng, H. Li, W. Xia, L. Jin and H. Sun, Coord. Chem. Rev., 2016, 311, 24–37 CrossRef CAS.
- H. de Reuse, D. Vinella and C. Cavazza, Front. Cell. Infect. Microbiol, 2013, 3, 152–157 Search PubMed.
- A. M. Sydor and D. B. Zamble, Met. Ions Life Sci., 2013, 12, 375–416 Search PubMed.
- Y. B. Zeng, N. Yang and H. Sun, Chem. – Eur. J., 2011, 17, 5852–5860 CrossRef CAS PubMed.
- X. Yang, H. Li, T. Cheng, W. Xia, Y.-T. Lai and H. Sun, Metallomics, 2014, 6, 1731–1736 RSC.
- K. A. Higgins, C. E. Carr and M. J. Maroney, Biochemistry, 2012, 51, 7816–7832 CrossRef CAS PubMed.
- W. Xia, H. Li, K.-H. Sze and H. Sun, J. Am. Chem. Soc., 2009, 131, 10031–10040 CrossRef CAS PubMed.
- M. Bellucci, B. Zambelli, F. Musiani, P. Turano and S. Ciurli, Biochem. J., 2009, 422, 91–100 CrossRef CAS PubMed.
- B. Zambelli, M. Bellucci, A. Danielli, V. Scarlato and S. Ciurli, Chem. Commun., 2007, 3649–3651 RSC.
- A. M. Sydor, H. Lebrette, R. Ariyakumaran, C. Cavazza and D. B. Zamble, J. Biol. Chem., 2014, 289, 3828–3841 CrossRef CAS PubMed.
- C. S. Goodwin, R. K. Mcculloch, J. A. Armstrong and S. H. Wee, J. Med. Microbiol., 1985, 19, 257–267 CrossRef CAS PubMed.
- R. Ge, X. Sun, Q. Gu, R. M. Watt, J. A. Tanner, B. C. Wong, H. H. Xia, J. D. Huang, Q. Y. He and H. Sun, J. Biol. Inorg. Chem., 2007, 12, 831–842 CrossRef CAS PubMed.
- X. Sun, R. Ge, J. F. Chiu, H. Sun and Q. Y. He, Met.-Based Drugs, 2008, 2008, 289490 Search PubMed.
- N.-C. Ha, S.-T. Oh, J. Y. Sung, K. A. Cha, M. H. Lee and B.-H. Oh, Nat. Struct. Mol. Biol., 2001, 8, 505–509 CAS.
- H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef CAS PubMed.
- N.-C. Ha, S.-T. Oh, J. Y. Sung, K. A. Cha, M. H. Lee and B.-H. Oh, Nat. Struct. Mol. Biol., 2001, 8, 505–509 CAS.
- X. M. Yang, H. Y. Li, T. P. Lai and H. Z. Sun, J. Biol. Chem., 2015, 290, 12474–12485 CrossRef CAS PubMed.
- Y. Li and D. B. Zamble, Chem. Rev., 2009, 109, 4617–4643 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Nature, 2010, 465, 645–648 CrossRef CAS PubMed.
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CrossRef CAS PubMed.
- T. V. O'Halloran and V. C. Culotta, J. Biol. Chem., 2000, 275, 25057–25060 CrossRef PubMed.
- S. D. Smith, Y. M. She, E. A. Roberts and B. Sarkar, J. Proteome Res., 2004, 3, 834–840 CrossRef CAS PubMed.
- J. Porath, J. A. N. Carlsson, I. Olsson and G. Belfrage, Nature, 1975, 258, 598–599 CrossRef CAS PubMed.
- Y. M. She, S. Narindrasorasak, S. Yang, N. Spitale, E. A. Roberts and B. Sarkar, Mol. Cell. Proteomics, 2003, 2, 1306–1318 CAS.
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190ra181 Search PubMed.
- J. Wang, X. Jin, J. Liu, C. Khosla and J. Xia, Chem. – Asian J., 2012, 7, 992–999 CrossRef CAS PubMed.
- R. Häuser, A. Ceol, S. V. Rajagopala, R. Mosca, G. Siszler, N. Wermke, P. Sikorski, F. Schwarz, M. Schick, S. Wuchty, P. Aloy and P. Uetz, Mol. Cell. Proteomics, 2014, 13, 1318–1329 Search PubMed.
- J. C. Rain, L. Selig, H. De Reuse, V. Battaglia, C. Reverdy, S. Simon, G. Lenzen, F. Petel, J. Wojcik, V. Schachter, Y. Chemama, A. Labigne and P. Legrain, Nature, 2001, 409, 211–215 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mt00169f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |