
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Superior anion induced shuttling behaviour exhibited by a halogen bonding two station rotaxane†
Timothy A.
Barendt
 ,
Sean W.
Robinson
and
Paul D.
Beer
*
,
Sean W.
Robinson
and
Paul D.
Beer
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk; Fax: +44 (0)1865 272690; Tel: +44 (0)1865 285142
First published on 28th April 2016
Abstract
Two bistable halogen and hydrogen bonding-naphthalene diimide [2]rotaxanes have been prepared and the system incorporating a halogen bond donor anion recognition site is demonstrated to exhibit superior anion induced translational motion of the macrocyclic wheel component relative to the hydrogen bonding analogue. Proton NMR spectroscopy is used to estimate the percentage occupancies of the macrocycle at the respective station and importantly indicates that the halogen bonding rotaxane displays superior positional integrity in competitive protic solvent media as a consequence of strong halogen bond–halide anion binding interactions.
Introduction
With the growing demand for smaller and more efficient computing devices, it has been proposed that the next wave of technology could use individual molecules as a way of rapidly handling vast amounts of data on the nanometre scale.1 As a consequence, the controlling of molecular motion by using dynamic interlocked structures is receiving an increasing amount of interest.2–7 This is motivated by the promise of their potential applications in nanotechnology as machines,7 switches,4,7,8 sensors9,10 or delivery systems11–14 where dynamic behaviour is driven by a variety of light-, chemical- and redox-based stimuli.4,7,15–18 Whilst examples of cation binding-induced co-conformational change4,7 within a rotaxane or catenane are prevalent in the literature, systems that use anions19–27 as an external stimulus to produce molecular motion are relatively rare.According to IUPAC “a halogen bond [XB] occurs when there is evidence of a net attractive interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity”.28 The integration of XB donor motifs into the scaffold of an anion receptor has resulted in contrasting and, in some cases, enhanced anion recognition behaviour in comparison to hydrogen bonding (HB) receptor analogues in protic organic solvent mixtures and recently in pure water.29–32 This has also led to the development of the first interlocked structure utilising XB anion recognition to control translational molecular motion.33
Herein we report the syntheses of two novel cationic XB and HB bistable rotaxanes and demonstrate their capability to perform molecular shuttling upon changing the nature of the counter anion and the solvent. Both rotaxanes comprise of an isophthalamide-containing macrocycle threaded by a potential two station axle. The axle component contains an electron-deficient naphthalene diimide (NDI) motif capable of aromatic donor–acceptor charge-transfer interactions with the electron-rich hydroquinone groups of the macrocycle34,35 and as the second station, an XB iodo- or HB proto-triazolium group capable of anion coordination. Molecular motion of the macrocyclic wheel component from the NDI station to the triazolium group is demonstrated upon addition of a coordinating halide anion as the external stimulus (Fig. 1). Importantly, superior shuttling behaviour is exhibited by the XB rotaxane in competitive CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3OD solvent mixtures as a consequence of strong XB–halide anion binding.
:CD3OD solvent mixtures as a consequence of strong XB–halide anion binding.
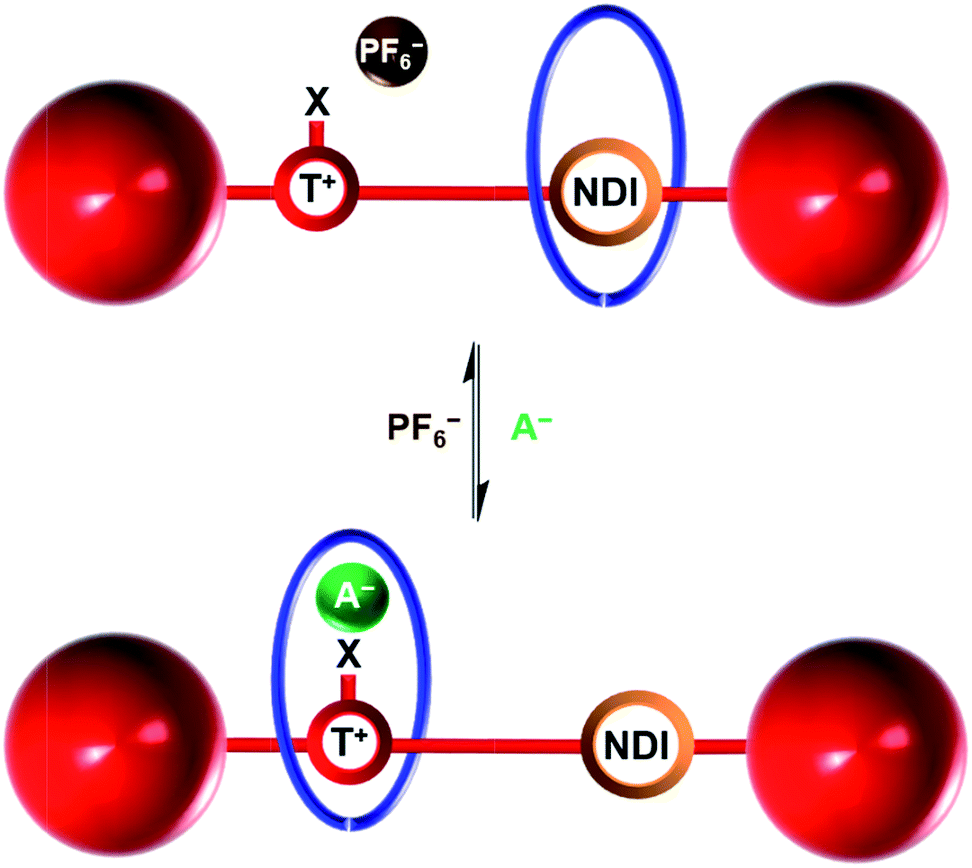 | ||
| Fig. 1 Schematic of the anion induced translational molecular motion within a bistable [2]rotaxane. NDI = naphthalene diimide, T+ = triazolium, X = H (HB donor) or I (XB donor) and A− = Cl− or I−. | ||
Results and discussion
Synthesis of HB and XB two station rotaxanes
The HB and XB two station rotaxanes 4·Cl and 5·Cl were prepared by multistep synthetic pathways as described in the ESI.† Chloride anion-templation was used to facilitate ring closing metathesis (RCM) of a bis-vinyl-functionalised macrocyclic precursor 3 around the appropriate HB or XB asymmetric axle (1·Cl and 2·Cl respectively) using Grubbs' II catalyst in dry dichloromethane. Preparative silica thin-layer chromatography purification afforded the desired rotaxanes (Scheme 1) which were characterised by 1H, 13C and 1H 2D ROESY NMR spectroscopy and by high resolution mass spectrometry (see ESI†).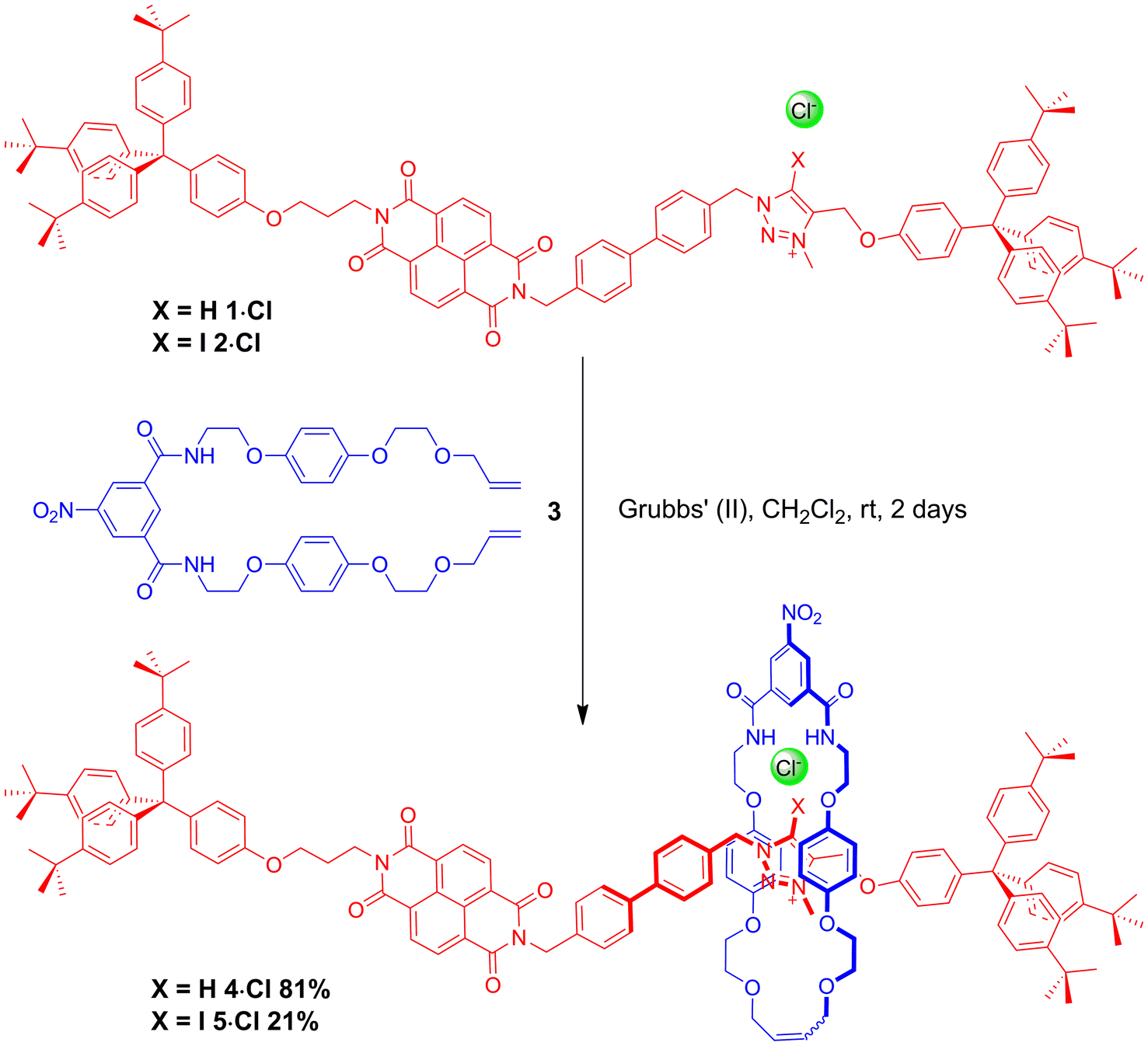 | ||
| Scheme 1 Syntheses of HB and XB rotaxanes 4·Cl and 5·Cl by an anion-templated clipping methodology. | ||
Investigations into anion-induced molecular motion
A comparison of the 1H NMR spectra of the axle components 1·Cl and 2·Cl with their respective rotaxanes 4·Cl and 5·Cl, in CDCl3, was used to indicate the co-conformation of both interlocked structures in the presence of the coordinating chloride anion (Fig. 2). Exclusive upfield shifts of the triazolium proton Hm (in the case of the HB rotaxane 4·Cl), N-methyl triazolium group Hl and the alkyl groups (Hk and Hn) either side of the triazolium station are observed on comparing the 1H NMR spectra of the axle and rotaxane. This is indicative of halide anion coordination by multiple HB and, in the case of 5·Cl, XB, interactions within the unique three-dimensional cavity created between the axle and the macrocycle occupying the triazolium station. In contrast there is no change in the chemical shift of the NDI aromatic protons He,e′ or its proximal alkyl protons Hf and Hd which suggests the macrocycle resides almost exclusively at the triazolium station in both rotaxanes. These co-conformations were confirmed by two-dimensional 1H ROESY NMR experiments in CDCl3 that showed, amongst other evidence, only cross coupling between hydroquinone protons of the macrocycle Hγ,γ′ and triazolium N-methyl protons Hl, with no interaction between Hγ,γ′ and the NDI protons He,e′ (see ESI†).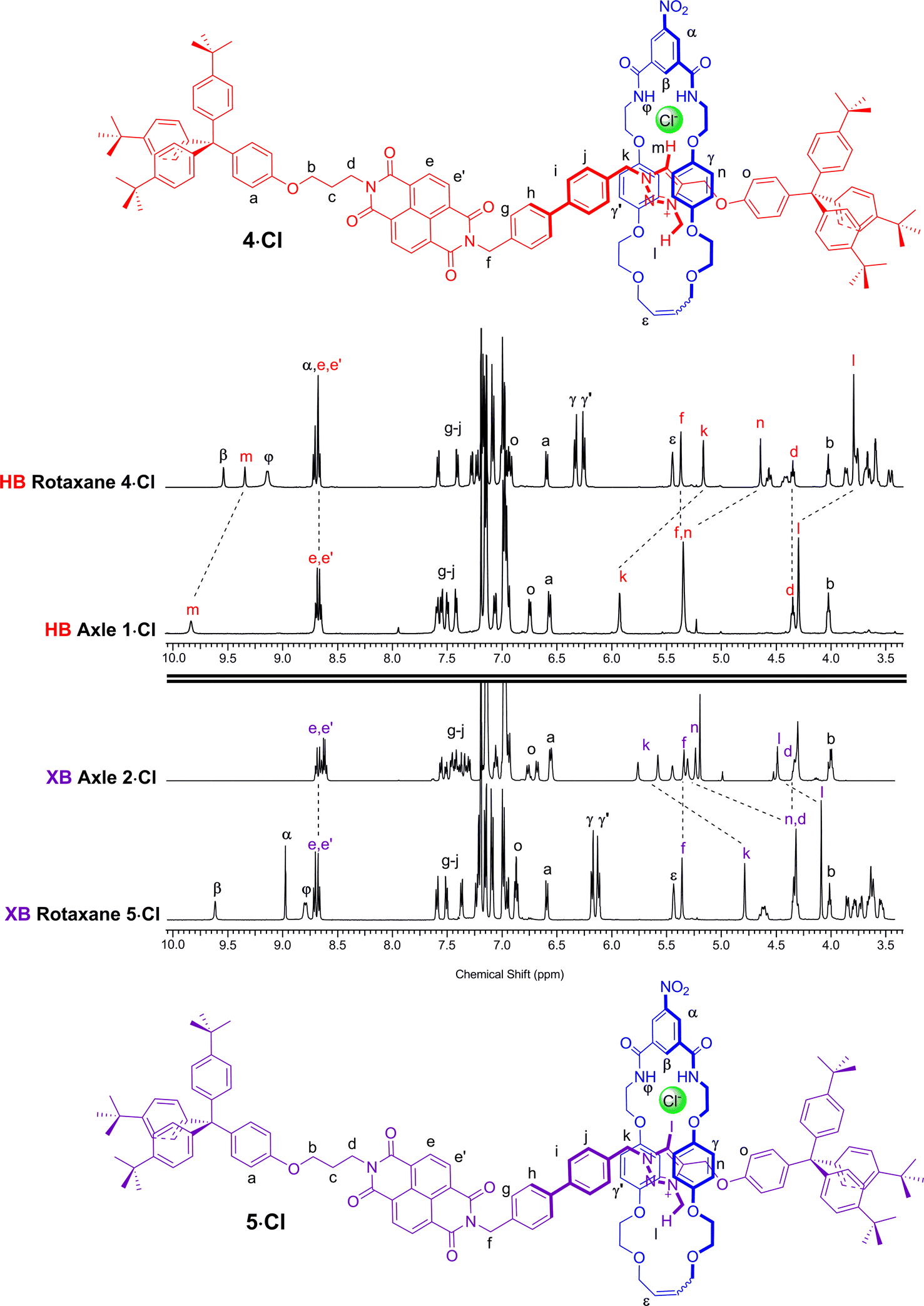 | ||
| Fig. 2 Comparison of the truncated 1H NMR spectra (CDCl3, 298 K, 400 MHz) of axles 1·Cl and 2·Cl and [2]rotaxanes 4·Cl and 5·Cl with their respective expected co-conformations. | ||
Anion exchange to the corresponding hexafluorophosphate salts was achieved by repeatedly passing a solution of the rotaxane through a column containing an Amberlite® IRA-400 resin loaded with PF6−. A comparison of the resulting 1H NMR spectra of rotaxanes 4·PF6 and 5·PF6 with their respective chloride salts 4·Cl and 5·Cl in CDCl3 indicated a change in co-conformation had occurred as a result of shuttling of the macrocycle component to occupy the NDI station of the axle (Fig. 3). This is inferred by upfield shifts of the macrocycle hydroquinone protons Hγ,γ′ which are indicative of stronger aromatic donor–acceptor charge-transfer interactions between the axle's electron-deficient NDI motif and macrocycle hydroquinone groups. In addition, upfield shifts of the NDI station protons He,e′, in contrast to a downfield shift of the N-methyl triazolium protons Hl, also suggests that shuttling of the macrocyclic component to the NDI station had taken place. Interestingly, the largest shifts are observed in the XB two station rotaxane 5·PF6 indicating a greater degree of translocation within this system has occurred than the HB analogue 4·PF6. The changes to the co-conformations are further evidenced by the 1H 2D ROESY NMR spectra of 4·PF6 and 5·PF6, both showing the appearance of new correlations between resonance signals arising from the macrocycle hydroquinone protons Hγ,γ′ and the NDI protons He,e′ (see ESI†).
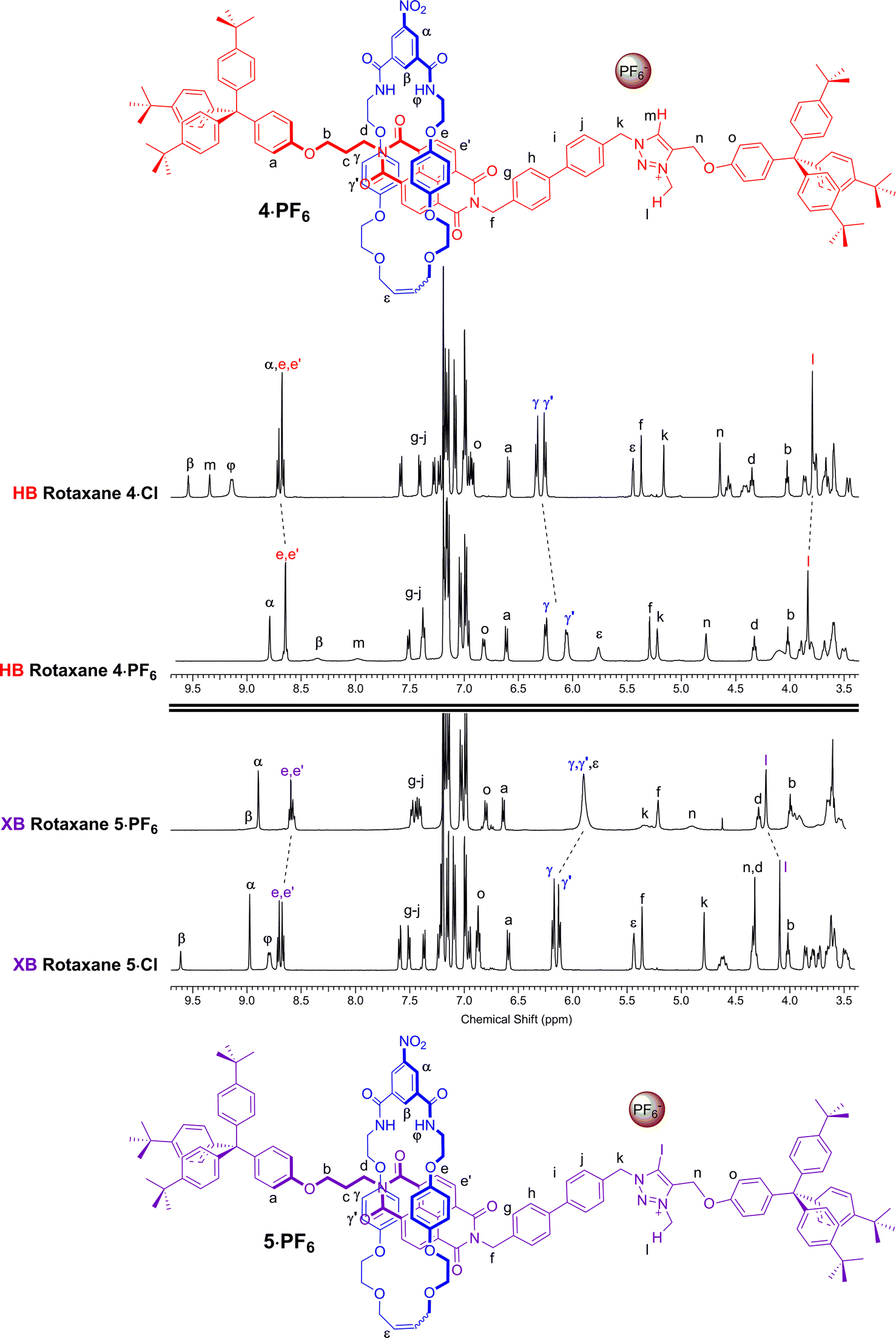 | ||
| Fig. 3 Comparison of the truncated 1H NMR spectra (CDCl3, 298 K, 400 MHz) of [2]rotaxanes 4·Cl, 5·Cl, 4·PF6 and 5·PF6 with their predicted co-conformations in the presence of PF6−. | ||
Originally colourless as chloride salts 4·Cl and 5·Cl, donor–acceptor charge-transfer interactions between the macrocycle hydroquinone motifs and the extended π surface of the NDI station resulting from the shuttling behaviour upon conversion to rotaxanes 4·PF6 and 5·PF6 gave rise to a bright, distinctive orange colour (Fig. 4). Hence molecular motion can be detected by the naked eye through this colorimetric sensing response and was also demonstrated to be reversible using UV-vis spectroscopic studies (see ESI†).
 | ||
| Fig. 4 Schematic of the anion-induced molecular shuttling exhibited by rotaxanes 4·A and 5·A and the colour of their two co-conformations in CDCl3 solution. | ||
X-ray crystallography36
Crystals suitable for X-ray diffraction structural analysis were obtained for the chloride salt of the XB rotaxane 5·Cl by slow evaporation of a solution of the rotaxane in 1:1 CDCl3:CD3OD. The rotaxane crystallises in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) where the asymmetric unit comprises the rotaxane as a 1:1 complex with chloride, as well as one molecule each of chloroform and water. Notably, the solid-state structure of the chloride salt of the XB rotaxane shows the macrocycle situated exclusively at the triazolium station, templated by the presence of the coordinating chloride anion (DI⋯Cl− = 2.987(6) Å) (Fig. 5). This chloride anion⋯iodo-triazolium halogen bond is much shorter than the sum of the van der Waals radii (80%), indicating a very strong interaction. The structure is in agreement with the aforementioned 1H NMR evidence for the favoured co-conformation of the molecular shuttle in the presence of a coordinating anion.
where the asymmetric unit comprises the rotaxane as a 1:1 complex with chloride, as well as one molecule each of chloroform and water. Notably, the solid-state structure of the chloride salt of the XB rotaxane shows the macrocycle situated exclusively at the triazolium station, templated by the presence of the coordinating chloride anion (DI⋯Cl− = 2.987(6) Å) (Fig. 5). This chloride anion⋯iodo-triazolium halogen bond is much shorter than the sum of the van der Waals radii (80%), indicating a very strong interaction. The structure is in agreement with the aforementioned 1H NMR evidence for the favoured co-conformation of the molecular shuttle in the presence of a coordinating anion.
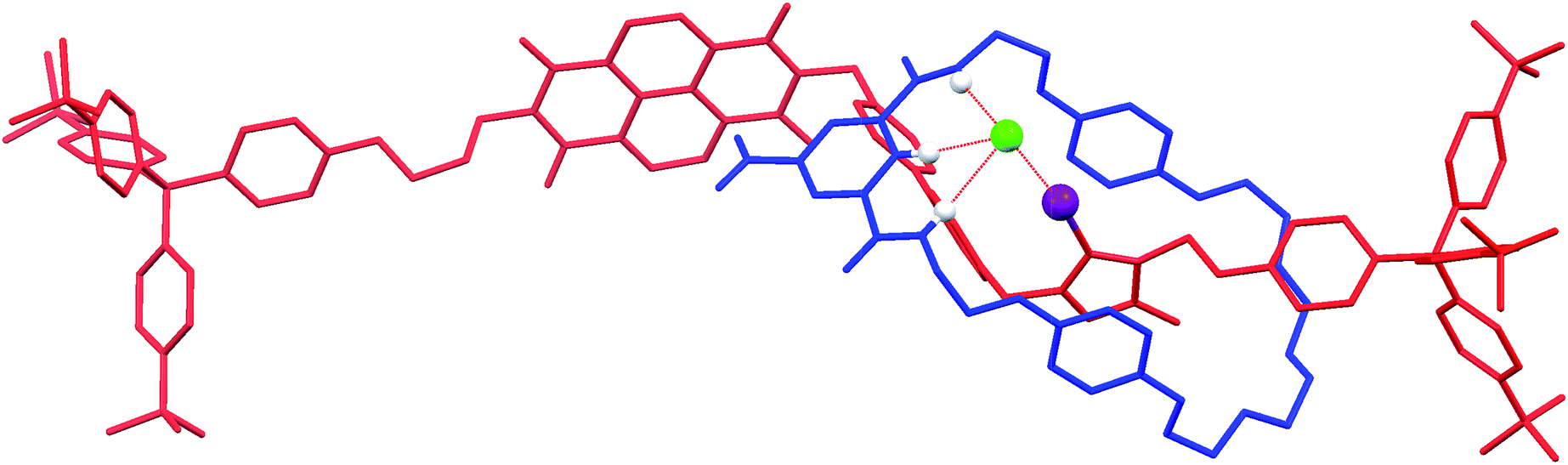 | ||
| Fig. 5 X-ray crystal structure of 5·Cl showing the macrocycle (blue) situated at the iodo-triazolium station on the axle (red), templated by the coordinated chloride anion (green). Notable hydrogen and halogen bonding interactions are shown as dotted red lines with the atoms involved shown as spheres. Non-coordinating hydrogens and the co-crystallised chloroform and water molecules are omitted for clarity. | ||
Effect of anion and solvent variation on percentage occupancies
In order to further investigate how the effect of varying the nature of the counter anion influenced the co-conformation of the XB and HB molecular shuttles, three new one-station rotaxanes containing either a proto-triazolium (6·Cl), iodo-triazolium (7·Cl) or NDI (8) motif were synthesised by multistep synthetic pathways and characterised by 1H and 13C NMR spectroscopy and by high resolution mass spectrometry as described in the ESI† (Fig. 6). These rotaxanes acted as models for the two station shuttles and enabled estimations of the station percentage occupancies by the macrocycle to be determined in the presence of non-coordinating PF6− and chloride and iodide anions.37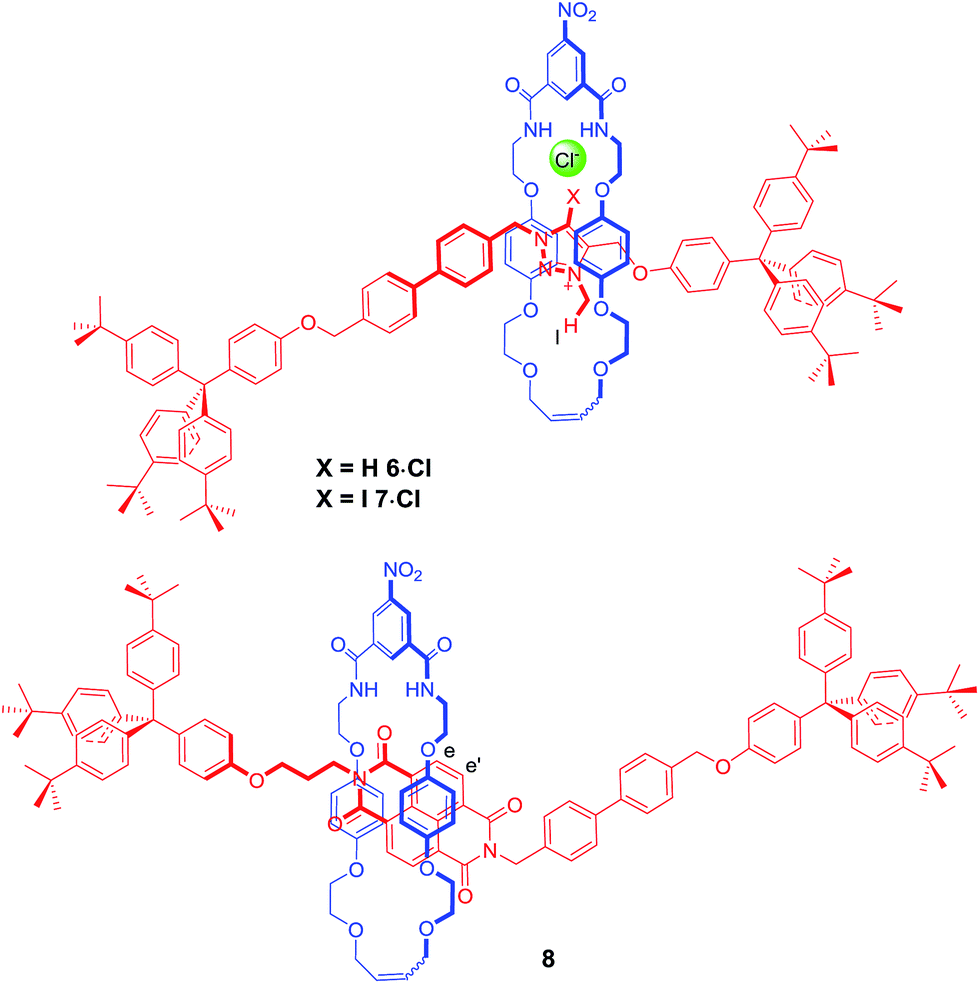 | ||
| Fig. 6 The structures of the three model one station rotaxanes 6·Cl, 7·Cl and 8. | ||
The 1H NMR spectra of these three rotaxanes in CDCl3 gave the proton chemical shift values of each station's diagnostic protons when there was complete occupancy by the macrocycle; specifically the N-methyl protons of the triazolium ring (Hl) and those of the NDI aromatic protons (He,e′). The chemical shift values for the same protons at 0% occupancy were obtained from the 1H NMR spectra in CDCl3 of the two-station axles 1·A and 2·A for a particular anion (A). Using these data, chemical shift scales were determined with respect to each station in the presence of each anion, to characterise the two extremes of macrocycle occupation (see ESI†). This method was necessary because the dynamic behaviour of the bistable rotaxanes was fast on the 1H NMR timescale.
The complete occupation of the triazolium station by the macrocycle in the HB two station rotaxane system in the chloride salt (4·Cl) was confirmed by comparison of the chemical shift of the N-methyl triazolium protons in 4·Cl (δ(Hl) = 3.88 ppm) with the δ HB N-methyl triazolium chloride chemical shift scale. This was supported by an analogous comparison of the chemical shift of the NDI protons, δ(He,e′), in 4·Cl with the NDI chemical shift scale which indicated zero occupancy of the NDI station. This method was used to determine the macrocycle occupancies of both HB and XB rotaxane 4·A and 5·A halide and PF6− salts using the appropriate N-methyl triazolium chemical shift scale (reported in Fig. 7) and concurred using the NDI chemical shift scale (see ESI†).
 | ||
| Fig. 7 The estimated percentage occupancies (based upon Hl protons) of the HB and XB rotaxanes 4·A and 5·A as chloride, iodide and PF6− salts as determined by 1H NMR spectroscopy in CDCl3 solution. A− = Cl− or I− and Trz = triazolium. | ||
From these data it may be seen that both two-station rotaxanes exhibit anion induced molecular motion in CDCl3 solution (Fig. 7). The preference for the triazolium station in the chloride salts (4·Cl and 5·Cl) was evident for both HB (100% occupancy of the triazolium station) and XB (92%) rotaxane switches, giving excellent positional integrity within this state. The same was true for the iodide salts (4·I and 5·I), with the HB and XB rotaxanes estimated to have 100% and 95% occupancies of their triazolium stations respectively. Upon exchange to the non-coordinating PF6− salt, both systems demonstrated an increase in occupancy of the NDI station (Fig. 7), as expected from the one- and two-dimensional 1H NMR spectroscopy data discussed earlier. The largest occupation of the NDI station was found to occur with the XB rotaxane 5·PF6 (62%) relative to the HB rotaxane 4·PF6 (24%).38 This indicated the XB rotaxane behaves as a superior molecular shuttle to the HB analogue on account of the larger swing in percentage occupancy of the stations upon anion exchange (>54% for the XB rotaxane vs. 24% for the HB rotaxane).
The propensity for anion receptors containing XB donor groups to exhibit enhanced halide, in particular iodide, anion recognition properties over their HB analogues in protic organic solvents30,31 provided the impetus for investigating the shuttling behaviour in more competitive solvent mixtures. The percentage occupancies of the HB and XB rotaxane PF6−, chloride and iodide salt systems were estimated in competitive 4:1 CDCl3:CD3OD and 1:1 CDCl3:CD3OD solvent mixtures via analogous 1H NMR spectroscopic experiments monitoring the chemical shifts of the respective rotaxane salt N-methyl triazolium Hl protons. Importantly, the results revealed a significantly enhanced positional integrity, in favour of the triazolium station for the XB rotaxane halide salt systems.
In 4:1 CDCl3:CD3OD the difference in behaviour between XB and HB rotaxanes was most strongly manifested within the iodide salt of the rotaxanes. XB rotaxane 5·I exhibited a 92% occupancy of the triazolium station whereas the HB analogue 4·I was estimated at 70% (Fig. 8). The differences between the chloride salt occupancies of the triazolium stations of the two rotaxanes were, by contrast, smaller: XB rotaxane 5·Cl was 100% vs. 87% for the HB rotaxane 4·Cl (Fig. 8). This is a consequence of superior halide affinity afforded by strong XB formation from the rotaxane's iodo-triazolium anion recognition site in comparison to the weaker proto-triazolium HB donor motif in the competitive solvent medium. Upon exchange to their respective PF6− salts, an enhanced positional integrity in favour of the NDI station was observed within the XB system 5·PF6 (48% occupancy of the NDI station), relative to the HB rotaxane 4·PF6 (33%), the same trend as found in CDCl3 solution.
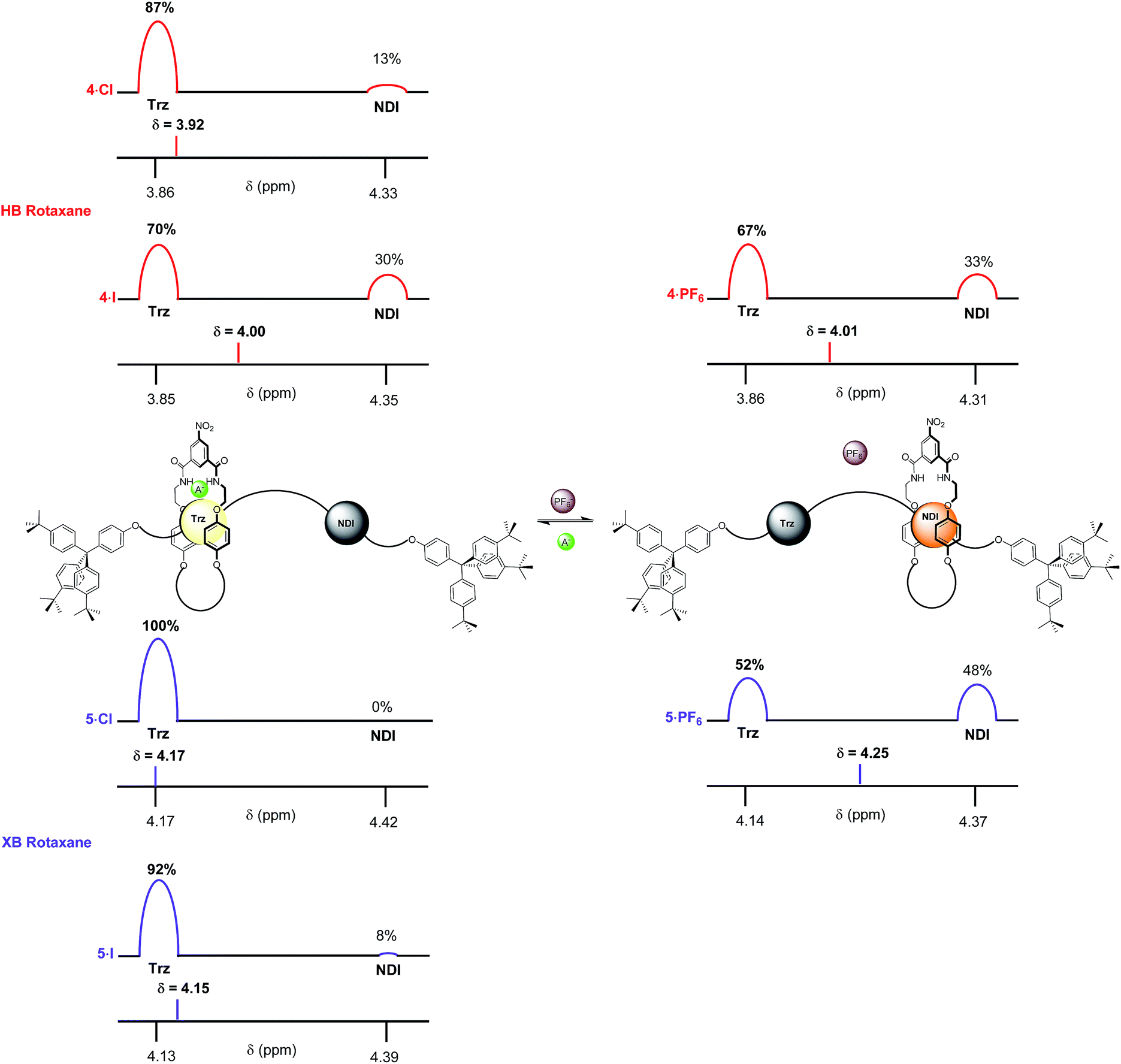 | ||
| Fig. 8 The estimated percentage occupancies (based upon Hl protons) of the HB and XB rotaxanes 4·A and 5·A as chloride, iodide and PF6− salts as determined by 1H NMR spectroscopy in 4:1 CDCl3:CD3OD solution. A− = Cl− or I− and Trz = triazolium. | ||
Finally, the percentage occupancies of both XB and HB rotaxane iodide and PF6− salts were estimated in the more competitive solvent medium of 1:1 CDCl3:CD3OD using 1H NMR spectroscopy experiments based upon the Hl protons (Fig. 9). The HB and XB rotaxanes were estimated to have the same occupancy of the NDI station as their PF6− salts (67%). However, a significantly larger preference for the triazolium station was exhibited in the iodide salt by the XB system 5·I (63% occupancy of the triazolium station), over the HB system 4·I (36%). This enabled the XB shuttle to still perform effectively in 1:1 CDCl3:CD3OD and produce a significant percentage of iodide induced molecular motion whereas the HB system afforded only negligible changes in the occupancies of the stations upon anion exchange. Analogous to previous solvent systems, there were smaller differences between occupancies of the triazolium stations in the chloride salts of HB and XB rotaxanes 4·Cl and 5·Cl (Fig. 9).
 | ||
| Fig. 9 The estimated percentage occupancies (based upon Hl protons) of the HB and XB rotaxanes 4·A and 5·A as iodide and PF6− salts as determined by 1H NMR spectroscopy in 1:1 CDCl3:CD3OD solution. A− = Cl− or I− and Trz = triazolium. | ||
These experiments demonstrate that an enhanced positional integrity for the triazolium station in the halide state is achievable upon increasing the competitive nature of the solvent system, but only when it is facilitated by the XB interaction with iodide. As such, the XB rotaxane switch is demonstrated to be a superior example of a synthetic molecular shuttle over its HB analogue in both CDCl3 and competitive CDCl3:CD3OD solvent mixtures.
Conclusions
In conclusion, the synthesis of five novel rotaxanes including two bistable rotaxanes and three model mono station systems are reported. Both cationic XB/HB two station rotaxanes were demonstrated to undergo halide anion induced molecular switching of their co-conformations in CDCl3 solution, with the added benefit that this shuttling behaviour resulted in a distinct colour change of the solution, giving a naked eye response to the translocation of the macrocyclic component. The XB two station rotaxane 5 demonstrated superior molecular shuttling in CDCl3 solution over the HB analogue rotaxane 4. Upon increasing the competitive nature of the solvent mixture to 4:1 and 1:1 CDCl3:CD3OD, the XB two station rotaxane again demonstrated superior molecular shuttling. This was driven by the XB interaction between iodide and the iodo-triazolium XB donor anion recognition site within the axle component. These results were reflected by determined estimations of the percentage occupancies of the stations using 1H NMR spectroscopy. This is the first example of using XB in concert with donor–acceptor charge-transfer interactions to exert significant control over solution phase molecular shuttling motion within an interlocked supramolecular system. Importantly this highlights the real and exciting potential XB has in the future design of dynamic interlocked mechanically bonded nanotechnological switchable systems.
Acknowledgements
T. A. B. would like to thank the EPSRC for a DTA studentship. S. W. R would like to thank the Clarendon Fund and St John's College, Oxford for funding. The authors would like to thank Dr Nicholas G. White (ANU) and Dr Amber L. Thompson (Oxford) for assistance with the crystallography as well as Diamond Light Source for an award of beamtime (MT9981).Notes and references
- V. Balzani, A. Credi and M. Venturi, in Molecular Devices and Machines – A Journey into the Nano World, Wiley-VCH Verlag GmbH & Co. KGaA, 2003 Search PubMed.
- J. F. Stoddart, Acc. Chem. Res., 2001, 34, 410–411 CrossRef CAS PubMed.
- R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445–455 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2011, 41, 19–30 RSC.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS PubMed.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 CAS.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 CrossRef CAS PubMed.
- S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, J. Am. Chem. Soc., 2015, 137, 7656–7659 CrossRef CAS PubMed.
- F. Vögtle, W. M. Müller, U. Müller, M. Bauer and K. Rissanen, Angew. Chem., Int. Ed. Engl., 1993, 32, 1295–1297 CrossRef.
- V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399–9400 CrossRef CAS.
- J. Berná, C. Franco-Pujante and M. Alajarín, Org. Biomol. Chem., 2013, 12, 474–478 Search PubMed.
- G. T. Spence, M. B. Pitak and P. D. Beer, Chem.–Eur. J., 2012, 18, 7100–7108 CrossRef CAS PubMed.
- N. H. Evans, C. J. Serpell and P. D. Beer, Chem.–Eur. J., 2011, 17, 7734–7738 CrossRef CAS PubMed.
- C. J. Serpell, R. Chall, A. L. Thompson and P. D. Beer, Dalton Trans., 2011, 40, 12052–12055 RSC.
- E. V. Dzyuba, L. Kaufmann, N. L. Löw, A. K. Meyer, H. D. F. Winkler, K. Rissanen and C. A. Schalley, Org. Lett., 2011, 13, 4838–4841 CrossRef CAS PubMed.
- J. J. Gassensmith, S. Matthys, J.-J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem.–Eur. J., 2010, 16, 2916–2921 CrossRef CAS PubMed.
- H. Zheng, W. Zhou, J. Lv, X. Yin, Y. Li, H. Liu and Y. Li, Chem.–Eur. J., 2009, 15, 13253–13262 CrossRef CAS PubMed.
- T.-C. Lin, C.-C. Lai and S.-H. Chiu, Org. Lett., 2009, 11, 613–616 CrossRef CAS PubMed.
- M. J. Barrell, D. A. Leigh, P. J. Lusby and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 8036–8039 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- A. Caballero, L. Swan, F. Zapata and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11854–11858 CrossRef CAS PubMed.
- L. Raehm, D. G. Hamilton and J. K. Sanders, Synlett, 2002, 2002, 1743–1761 CrossRef.
- G. Kaiser, T. Jarrosson, S. Otto, Y.-F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959–1962 CrossRef CAS PubMed.
- Single crystal X-ray diffraction data for the rotaxane structure were collected using synchrotron radiation at Diamond Lightsource ( H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435 CrossRef CAS PubMed ). Raw frame data (including data reduction, inter-frame scaling, unit cell refinement and absorption corrections) were processed using CrysAlis Pro (Agilent Technologies, 2010). Structures were solved using SUPERFLIP ( L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef ) and refined using CRYSTALS ( P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef; R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100 CrossRef; P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef PubMed ). For further details of all crystallography, see the ESI.†.
- The rotaxanes were demonstrated to be excellent models of the two-station rotaxanes because the chemical shifts of the diagnostic protons for each station, that is the N-methyl protons of the triazolium chloride groups Hl and the NDI aromatic protons He,e′, of the model one station axles were identical to those found in the 1H NMR spectra for the HB and XB two-station axles 1·Cl and 2·Cl.
- The difference in behaviour of 4·PF6 and 5·PF6 under these conditions is not clear at present, however, we tentatively ascribe the lower occupancy of the iodo-triazolium station to potential steric repulsion between the macrocycle and the large iodine atom.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1454515. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00783j |
| This journal is © The Royal Society of Chemistry 2016 |