
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable and highly efficient light-harvesting antenna systems based on 1,7-perylene-3,4,9,10-tetracarboxylic acid derivatives†
Rajeev K.
Dubey
ab,
Damla
Inan
b,
Sanchita
Sengupta‡
ab,
Ernst J. R.
Sudhölter
a,
Ferdinand C.
Grozema
b and
Wolter F.
Jager
*a
aLaboratory of Organic Materials & Interfaces, Department of Chemical Engineering, Delft University of Technology, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: W.F.Jager@tudelft.nl
bLaboratory of Optoelectronic Materials, Department of Chemical Engineering, Delft University of Technology, Julianalaan 136, 2628BL Delft, The Netherlands
First published on 15th March 2016
Abstract
We report the synthesis and excited-state dynamics of a series of five bichromophoric light-harvesting antenna systems, which are capable of efficient harvesting of solar energy in the spectral range of 350–580 nm. These antenna systems have been synthesized in a modular fashion by the covalent attachment of blue light absorbing naphthalene monoimide energy donors (D1, D2, and D3) to green light absorbing perylene-3,4,9,10-tetracarboxylic acid derived energy acceptors, 1,7-perylene-3,4,9,10-tetracarboxylic tetrabutylester (A1), 1,7-perylene-3,4,9,10-tetracarboxylic monoimide dibutylester (A2), and 1,7-perylene-3,4,9,10-tetracarboxylic bisimide (A3). The energy donors have been linked at the 1,7-bay-positions of the perylene derivatives, thus leaving the peri positions free for further functionalization and device construction. A highly stable and rigid structure, with no electronic communication between the donor and acceptor components, has been realized via an all-aromatic non-conjugated phenoxy spacer between the constituent chromophores. The selection of donor naphthalene derivatives for attachment with perylene derivatives was based on the effective matching of their respective optical properties to achieve efficient excitation energy transfer (EET) by the Förster mechanism. A comprehensive study of the excited-state dynamics, in toluene, revealed quantitative and ultrafast (ca. 1 ps) intramolecular EET from donor naphthalene chromophores to the acceptor perylenes in all the studied systems. Electron transfer from the donor naphthalene chromophores to the acceptor perylenes has not been observed, not even for antenna systems in which this process is thermodynamically allowed. Due to the combination of an efficient and fast energy transfer along with broad absorption in the visible region, these antenna systems are promising materials for solar-to-electric and solar-to-fuel devices.
Introduction
Conversion of sun-light into more useful forms of energy is the most sustainable and promising endeavour to tackle the growing global concerns on energy supply and environmental issues. Solar energy can either be turned directly into electricity using solar cells or converted to high-energy compounds that can be used as fuel. The latter approach is reminiscent of natural photosynthesis, which is often used as a source of inspiration to achieve the so-called artificial photosynthesis.1 The success of both approaches heavily relies on the ability of artificial devices to harvest the maximum amount of incident light energy. Therefore, current research efforts largely focus on the efficient harvesting of solar irradiation, especially between 400 to 920 nm.2In the past, a wide range of organic and organometallic chromophores have been employed as sensitizers in solar-to-electric and solar-to-fuel devices, owing to their high molar absorptivities in the visible region.3 However, the intense transitions of individual chromophores are often narrow, which results in poor overlap of their absorption with the incident solar spectrum. Light-harvesting antenna systems, utilized in natural photosynthesis, contain large numbers of nearly identical chromophores which are precisely positioned in a protein matrix. This approach ensures a high optical density, even for thin layers, but the spectral coverage from essentially one chromophore is generally poor.4 Energy transfer between the individual dye molecules in these systems is extremely fast, in the order of 0.1 ps, and the energy transfer mechanism is generally referred to as quantum coherence.1b
Synthetic light-harvesting antenna systems generally consist of multiple chromophores with distinct chemical structures and complementary absorption spectra. These chromophores absorb light below a threshold wavelength and funnel the excitation energy unidirectionally towards a single chromophore within the array. In these systems, the excitation energy transfer is generally accomplished by dipole–dipole interactions, commonly referred to as Förster Resonance Energy Transfer (FRET).5 This approach ensures a large coverage of the visible spectrum using a less demanding and more straightforward molecular design.6 Obtaining large optical densities on relatively thin layers is an issue that is generally tackled by absorbing dye molecules on a structured interface like TiO2, as is the case for well-known dye-sensitized solar cells (DSCs).7
Artificial light-harvesting (LH) antenna systems are thus an essential building block for the realization of efficient solar energy conversion.2a,b,8 In addition to the careful tuning of the optical properties of the individual chromophores to accomplish fast and efficient energy transfer, a simple and robust antenna design, synthetic accessibility of the components, and elimination of competing photoinduced processes are essential elements for the development of suitable light-harvesting antenna systems that can ultimately be incorporated into devices.
The choice of the acceptor chromophore in light-harvesting antenna systems determines the properties of the antenna to a large extent for several reasons. First of all, the excitation energy of the acceptor determines the upper limit of the spectral range of the antenna system and thereby the energy content of the photons that are harvested. Secondly, the stability of the excited state of the acceptor determines, to a large extent, the stability of the antenna system itself. That is because the energy transfer between the chromophores is generally faster than the electron transfer that follows, and because of that the excitation energy resides at the acceptor chromophore for most of the time.9 Antenna systems based on various energy acceptor chromophores, which are often attached to dendritic scaffolds, have been reported.10 Recently, the use of bio-based binding platforms,8a like DNA11 and sugars,12 and the use of hybrid organic–inorganic materials,13 like metal organic frameworks (MOFs) and periodic mesoporous organosilicas (PMOs), for the construction of light-harvesting assemblies has been demonstrated. A wide variety of organic chromophores, such as BODIPY dyes,2c,10g perylene bisimides,14,15 and aromatic hydrocarbons, have been employed for constructing light-harvesting antenna molecules. A particularly attractive chromophore for the design of such light-harvesting arrays is perylene bisimide (PBI), a molecule that is well-known for its exceptional photo-chemical stability, strong and broad absorption in the visible region, and synthetic versatility.16 Perylene-based antenna molecules carrying various donors have been reported,14 along with larger antenna systems in which perylenebisimides are intermediate energy donors.15 In addition, the strong absorption of PBIs has been successfully utilized to improve the spectral coverage of C60.17 An unfavorable feature of PBI dyes is their high electron deficiency,16a and therefore PBI assemblies tend to undergo facile charge-separation when PBIs are coupled with even moderately electron-rich chromophores.18 In addition, the strong tendency of PBIs to form π–π aggregates is a severe limitation that needs to be accounted for in the design of molecular PBI based arrays.19 As a result, the construction of PBI based light-harvesting arrays requires careful design, with regard to matching the electrochemical properties of the donor and acceptor chromophores, the selection of the spacers that connect these units, and the prevention of undesirable aggregation. Otherwise, electron-transfer processes which are undesirable within the antenna system will compete with the energy-transfer.14e,h
In this work, we report on the design, synthesis and characterization of a series of functional, robust, and highly efficient light-harvesting antenna molecules based on perylene-3,4,9,10-tetracarboxylic acid derivatives. In the design of these molecules we have used a modular synthetic approach, employing three naphthalene monoimide-derived energy donor molecules, D1–D3, along with three perylene-3,4,9,10-tetracarboxylic acid derived energy acceptors; perylene-3,4,9,10-tetrabutylester (A1), perylene-3,4,9,10-monoimide dibutylester (A2),20 and perylene-3,4,9,10-bisimide A3, see Fig. 1. The recently developed acceptors A1 and A2 have significantly reduced electron deficiencies and increased solubilities compared to PBI (A3). The donor fragments 4-(isopentylthio)naphthalene monoimide (D1), 4-(n-butylamino)naphthalene monoimide (D2), and 4-(dimethylamino)naphthalene monoimide (D3) were selected because these molecules are stable, readily accessible, and have appropriate redox properties to limit electron transfer.16a,21 Also, these compounds are highly fluorescent, with emission spectra that strongly overlap with the absorption spectra of the perylene derivatives, which facilitates efficient excitation energy transfer by the Förster mechanism. From the nine possible donor–acceptor combinations depicted in the matrix in Fig. 1, we have synthesized the antenna molecules D1A1, D1A2, D2A2, D2A3 and D3A3. In particular, for the “diagonal compounds” D1A1, D2A2 and D3A3, the donor and acceptor absorptions are expected to be complementary and a broad continuous absorption is anticipated for these compounds. Also, the best overlap between donor emission and acceptor absorption, the main prerequisite for efficient Förster energy transfer, is expected for these “diagonal” compounds. For the other compounds, D1A2 and D2A3, donor absorptions are blue-shifted and a “hole” in the absorption spectrum is anticipated. For the remaining compounds, the donor and acceptor chromophores absorb more or less in the same wavelength region, and for that reason these compounds have not been synthesised.
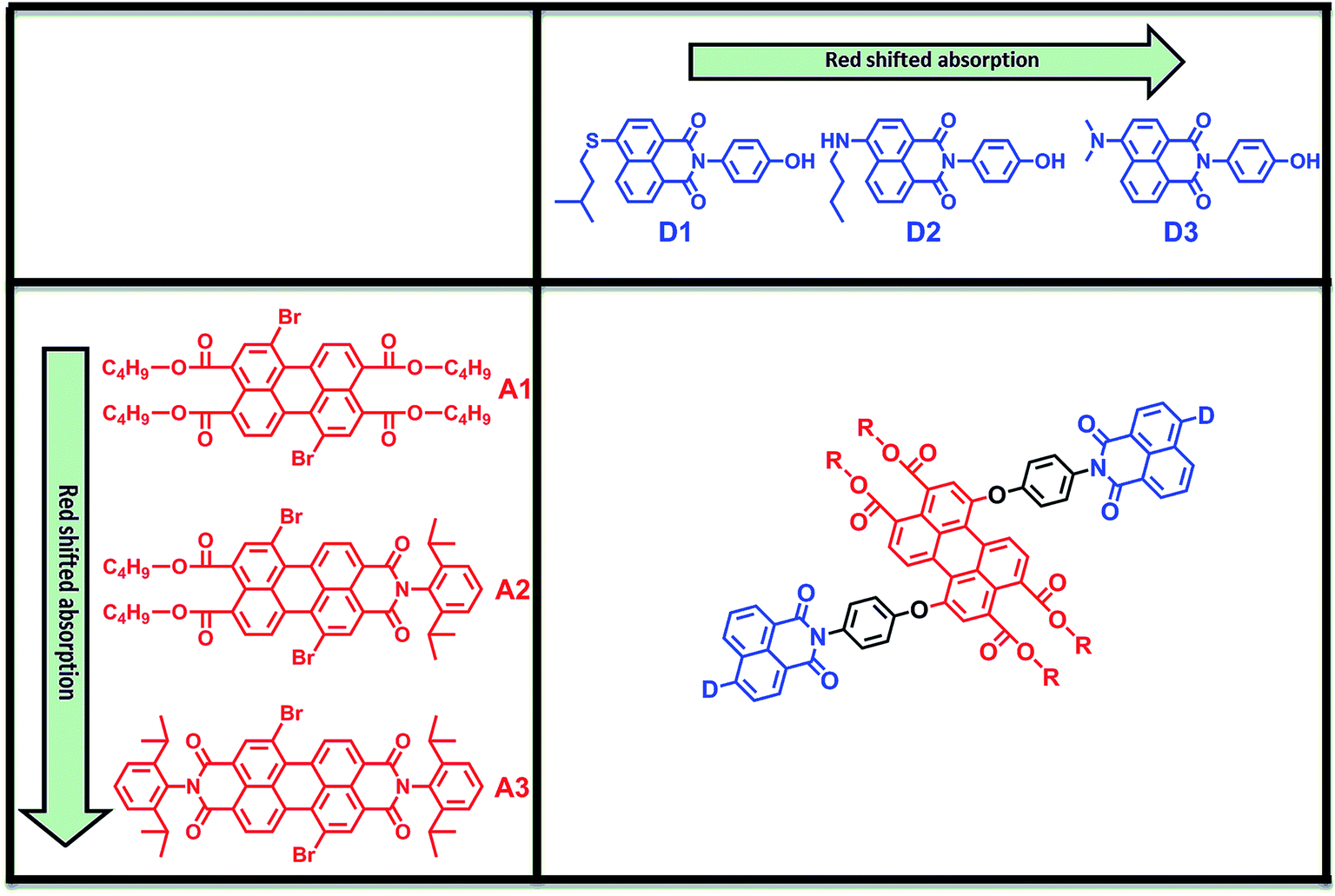 | ||
| Fig. 1 The modular design of the light-harvesting antennae based on perylene tetracarboxylic acid derivatives (A1, A2, and A3) and naphthalene monoimides (D1, D2, D3). | ||
For the covalent attachment of donors, we have chosen the phenoxy substitution approach to achieve a high chemical stability and a rigid well-defined conformation of the antenna molecules. Also, the phenoxy coupling approach ensured that the donor and acceptor chromophores are electronically decoupled, which implies that the absorption spectra and the electrochemical properties of the donor and acceptor fragments are unaffected by the coupling. This makes the physical properties of these compounds highly predictable and enables the developments of these antenna molecules by a truly modular approach. A detailed photophysical characterization of all the antenna systems was carried out using steady-state and time-resolved spectroscopy, along with cyclic voltammetry, in order to gain a good understanding of the energy transfer process and the effect of perylene-structure modification on the excited-state dynamics.
Results and discussion
Synthesis and characterization
The energy donor naphthalimide derivatives (3–5), outlined in Scheme 1, were obtained from commercially available 4-bromo-1,8-naphthalic anhydride 1. The compounds were synthesized in two steps in high yields. First, imidization of 1 with 4-aminophenol yielded N-(4′-hydroxyphenyl)-4-bromonaphthalene monoimide 2.22 Subsequently, the bromine was substituted by isopentylthio, n-butylamino, and dimethylamino groups to obtain compounds 3, 4, and 5, respectively.23 | ||
| Scheme 1 Synthesis of 4-substituted naphthalene monoimide derivatives D1, D2, and D3 (3–5). | ||
The synthesis of light-harvesting antenna systems was carried out from the regioisomerically pure 1,7-dibromoperylene-3,4,9,10-tetracarboxylic acid derivatives 1,7-dibromoperylene monoimide dibutylester 6, 1,7-dibromoperylene bisimide 9, and 1,7-dibromoperylene monoanhydride diester 12. These perylene derivatives were obtained according to a previously reported procedure.20 For the synthesis of antenna systems D1A2 and D2A2 (Scheme 2), the naphthalene derivatives 3 and 4 were separately reacted with regioisomerically pure 1,7-dibromoperylene monoimide diester 6, (A2). This phenoxy substitution reaction was carried out in dry toluene in the presence of K2CO3 and 18-crown-6 at 95 and 105 °C to obtain D1A2 and D2A2, respectively, in good yields. The same procedure was followed to synthesize antenna systems D2A3 and D3A3 using the naphthalene derivatives 4 and 5, respectively, and 1,7-dibromoperylene bisimide 9 (A3) as shown in Scheme 3.
 | ||
| Scheme 2 Synthesis of the antenna systems D1A2 (7) and D2A2 (8) from the perylene derivative 6 (A2). | ||
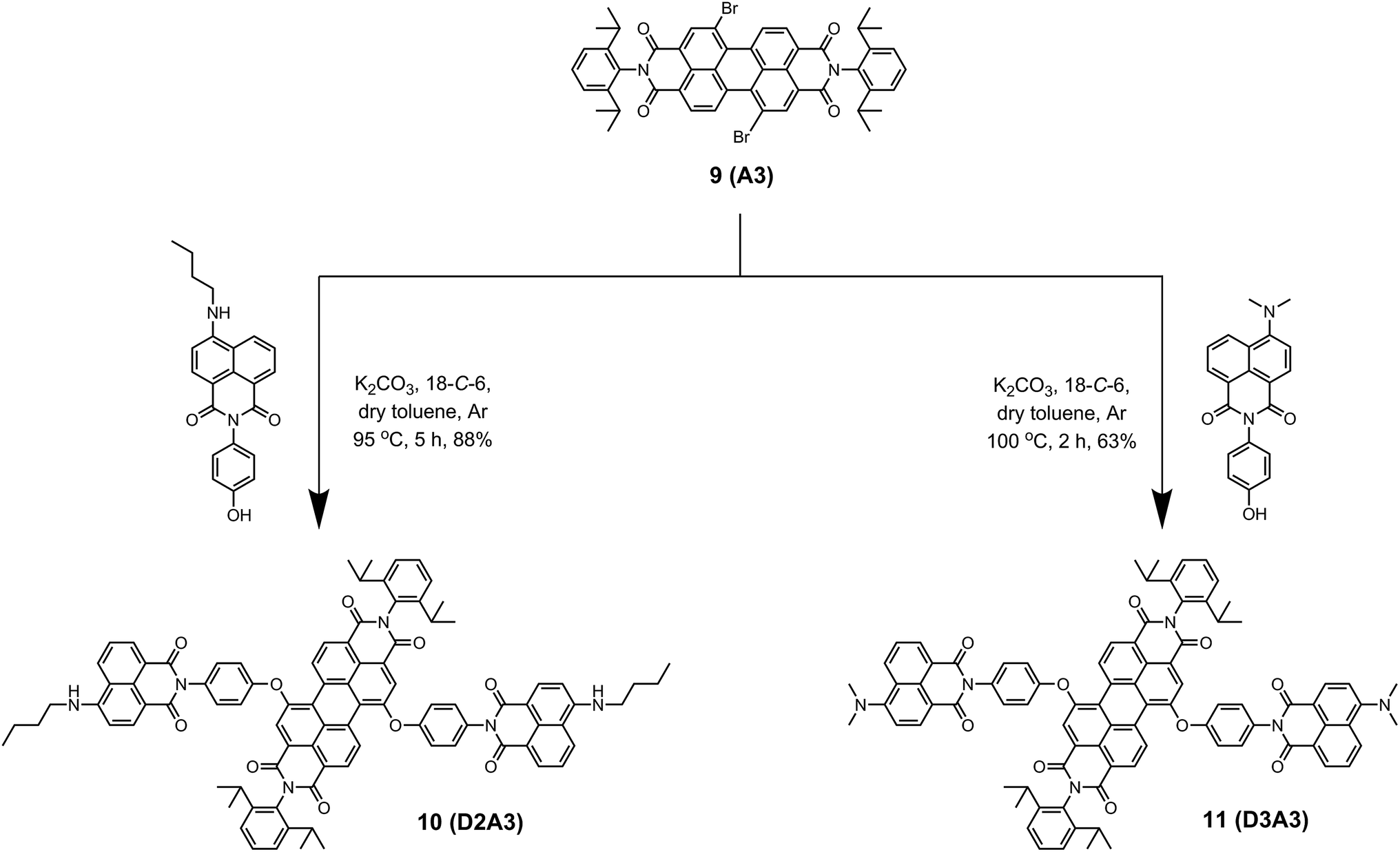 | ||
| Scheme 3 Synthesis of antenna systems D2A3 (10) and D3A3 (11) from 1,7-dibromoperylene bisimide 9 (A3). | ||
The synthesis of the antenna system D1A1 was found to be more challenging (Scheme 4). The coupling reaction was first attempted by reacting 1,7-dibromoperylene tetrabutylester 15, (A1) with naphthalene derivative 3. However, compound D1A1 was not obtained, not even after employing the harshest possible reaction conditions; refluxing DMF in the presence of Cs2CO3. This result can be rationalized based on the fact that perylene tetrabutylester 15 is significantly less electron deficient compared to perylene monoimides and bisimides, due to the absence of strong electron withdrawing imide groups.20 This decreased electron deficiency of the perylene core in 15 reduces its reactivity towards the nucleophilic substitution reaction. Therefore, the synthesis of D1A1 was carried out using the perylene derivative 12, which is more reactive than 15 owing to the presence of an electron withdrawing anhydride group. Following this approach, D1A1 was obtained in two steps. The first step involved the substitution of bromines with 3 in anhydrous NMP at 120 °C to obtain compound 13 in 39% yield. Subsequently, the esterification of the anhydride group yielded the antenna system D1A1 in 76% yield. The low yield in the first step of this reaction was most likely due to the reaction of the nucleophilic phenol with the anhydride group.
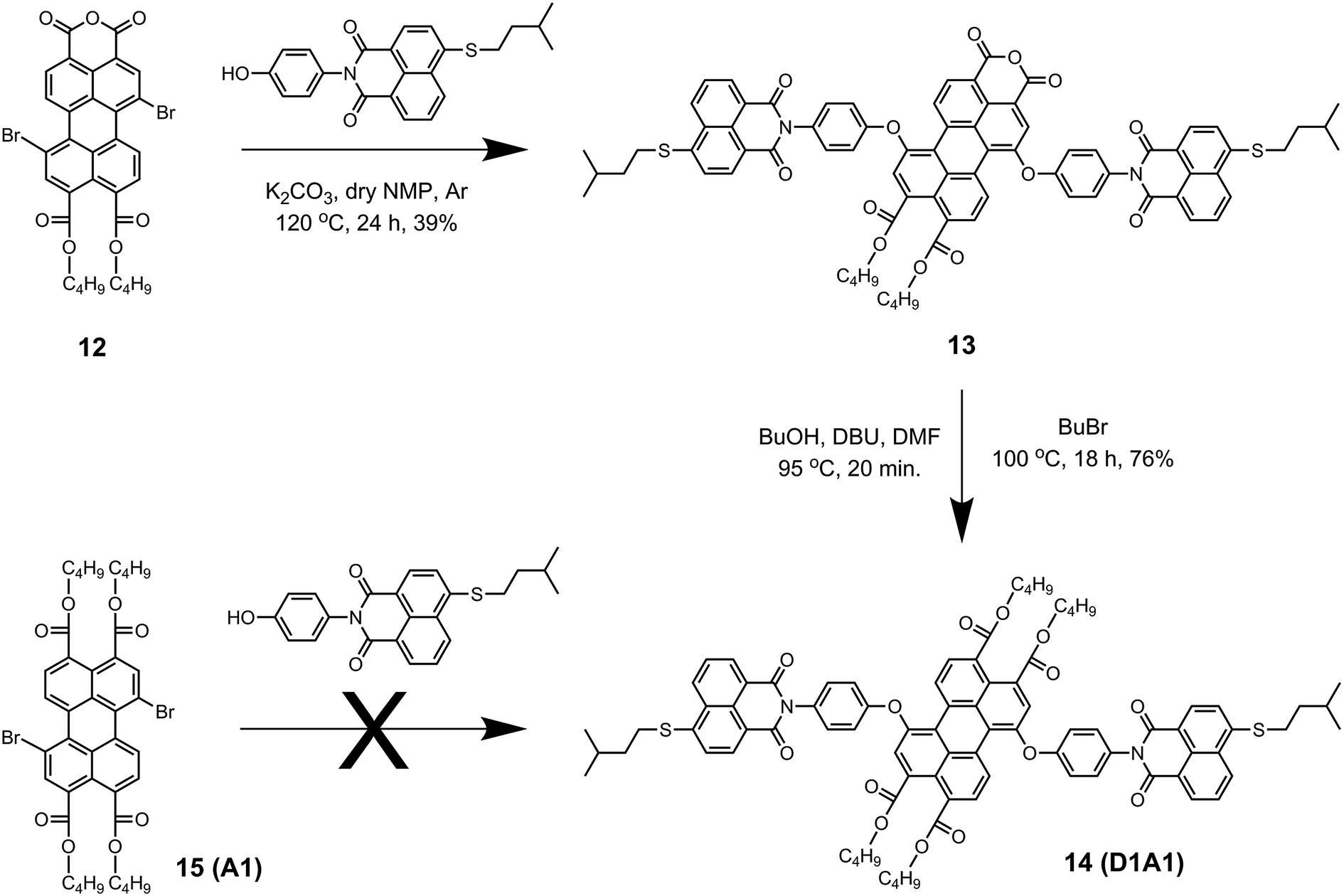 | ||
| Scheme 4 Synthesis of the antenna D1A1. | ||
For spectroscopic and electrochemical characterization, reference energy-donor compounds ref-D1, ref-D2, and ref-D3, and reference acceptor compounds ref-A1, ref-A2, and ref-A3, were synthesized (Scheme 5). The reference energy-donors were prepared according to the similar procedure as followed for D1, D2, and D3.24 The synthesis of the reference perylene compounds was achieved according to previously reported procedures.20,25
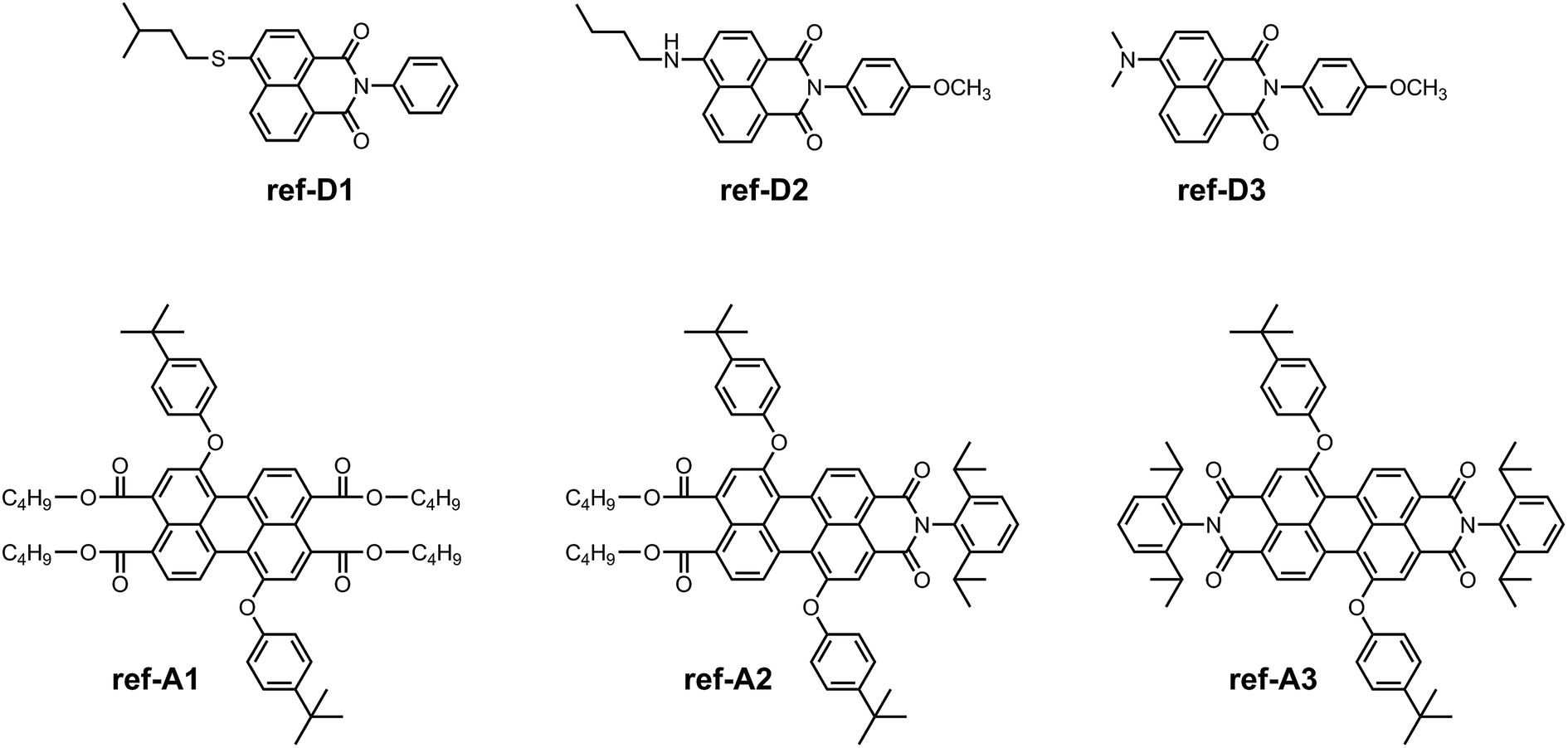 | ||
| Scheme 5 Structure of model energy-donors and acceptors used in the spectroscopic and electrochemical studies. | ||
1H NMR analysis of antenna system D2A2
The aromatic region of the 1H NMR spectrum of D2A2 was compared with the spectrum of ref-D2 to obtain information regarding the mutual orientation of the two moieties in the antenna systems (Fig. 2). The assignment of the various signals to the naphthalene ring protons was performed with the help of 1H–1H COSY measurements (Fig. S1, ESI†). The perylene core protons are indicated by asterisks. In case of the close-proximity of the two chromophores, the aromatic ring current generated from the perylene moiety will affect the chemical shifts of the proton signals of the naphthalene rings and vice versa.26 In the antenna system D2A2, however, all the naphthalene core protons (H2, H3, H5, H6, and H7) have retained their chemical shift values compared to ref-D2. This observation leads to the conclusion that the two chromophores are oriented away from each other and do not experience any through-space interaction. This observation is also indicative of the absence of ground-state interactions between the two chromophores, i.e. the absence of conjugation between them. The same phenomenon was observed for all other antenna systems that we synthesized. Another noteworthy aspect of the NMR spectrum of D2A2 is the small ∼0.07 ppm difference in the chemical shift that is observed for all the naphthalene proton resonances. This chemical shift difference reveals that both naphthalene units in compound D2A2 are inequivalent. For all antenna molecules based on the centrosymmetric acceptors A1 and A3, the proton resonances of both naphthalene units are identical and chemical shift differences have not been observed. Finally, it should be noted that the absence of additional resonances in the 1H NMR spectrum of D2A2 reflects the high purity of this compound.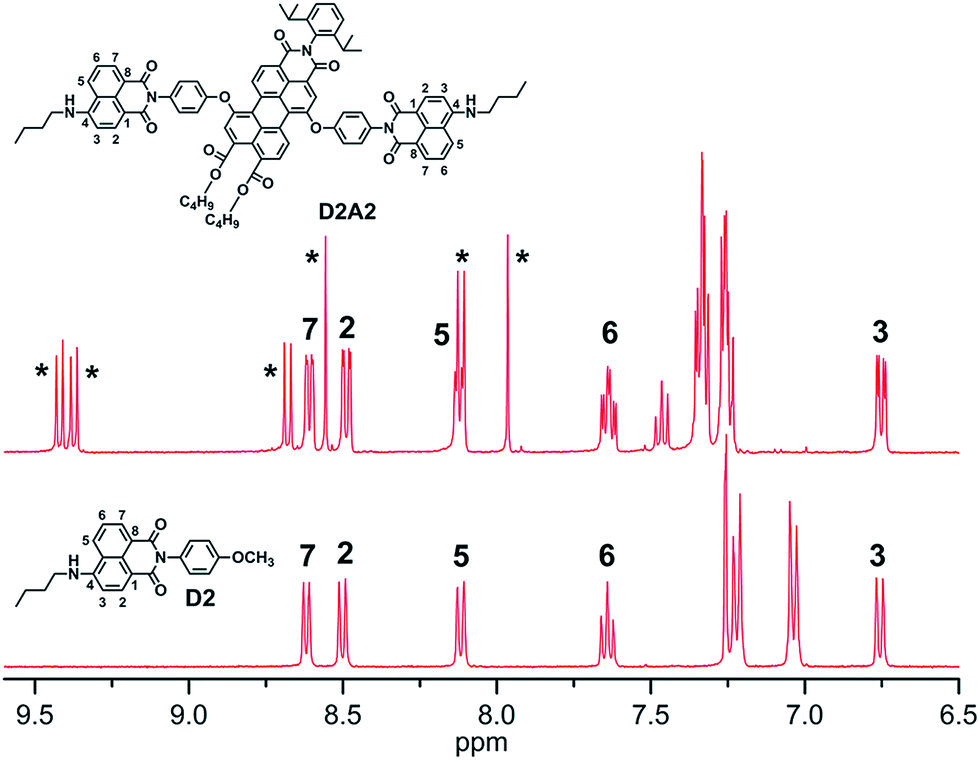 | ||
| Fig. 2 Comparison of the 1H NMR spectra (aromatic regions) of compounds D2A2 and ref-D2 in CDCl3. The perylene core protons are indicated by asterisks. | ||
Electrochemical studies
The redox properties of the antenna systems and model compounds were investigated using cyclic voltammetry in dichloromethane. The obtained redox potentials (V vs. Fc/Fc+) for the antenna systems are reported in Table 1. For the reference donors ref-D2 and ref-D3, first oxidation potentials were measured at 0.81 and 0.75 V, respectively (Table S1, ESI†). For ref-D1, no oxidation was observed in the measured potential window. The oxidation potential of ref-D1 is expected to be higher than that of ref-D2, based on the weaker electron-donating nature of the isopentylthio group compared to the butylamino group.27 For all antenna molecules, the obtained values for the oxidation of D2 and D3 units were similar to those of the reference compounds ref-D2 and ref-D3 (Table 1). Once more, this indicates the lack of conjugation between the two types of chromophores in the antenna molecules. For the perylene moieties in the antenna molecules (A1, A2, and A3), first reduction potentials were obtained at ca. −1.55, −1.33, and −1.08 V, respectively. These values, which are virtually identical to those measured for the reference compounds ref-A1, ref-A2 and ref-A3, shown in Table S1,† indicate that the electron-deficiency significantly increases upon moving from A1 to A3.| Compound | Naphthalimide | Perylene | E CS (D˙+–A˙−)d | ΔGCSe | ||
|---|---|---|---|---|---|---|
| E 1/2ox | E S1 (eV) | E 1/2red | E S1 (eV) | In DCM (eV) | In DCM (eV) | |
| a Scan rate 0.10 V s−1. b Energy of the first singlet excited state of the naphthalimide unit calculated from the absorption and emission measurements. c Energy of the first singlet excited state of the perylene moiety obtained electrochemically. d Energy of the lowest charge-separated state. e Driving force for charge separation with respect to perylene singlet excited state, ES1 − ECS (D˙+–A˙−). f No oxidation signal was detected in the measured potential window. g Value taken from compound 15. | ||||||
| D1A1 | —f | 2.80 | −1.55g | 2.41 | >2.13 | — |
| D1A2 | —f | 2.80 | −1.34 | 2.37 | >2.13 | — |
| D2A2 | +0.80 | 2.60 | −1.33 | 2.35 | 2.13 | 0.22 |
| D2A3 | +0.79 | 2.58 | −1.09 | 2.15 | 1.88 | 0.27 |
| D3A3 | +0.74 | 2.58 | −1.07 | 2.15 | 1.81 | 0.34 |
For the antenna molecules, the energies of the charge-separated (CS) states, in DCM, were estimated by calculating the difference between the first oxidation potential of the donor and the first reduction potential of the acceptor [ECS = E1ox(D) − E1red(A)]. The energies of the charge-separated states (D˙+–A˙−) for antenna systems D2A2, D2A3, and D3A3 are estimated at ca. 2.13, 1.88, and 1.81 eV, respectively. Among these three systems, D2A2 has the highest energy of the CS states, which is due to the higher reduction potential of A2 compared to A3. The energies of the CS states could not be estimated for systems D1A1 and D1A2 because no redox activity for D1 has been observed. However, in view of the weaker electron-donating capability of D1, the CS states of D1A1 and D1A2 should have values that are significantly higher than the 2.13 eV estimated for D2A2. The data in Table 1 clearly indicate that the energies of the charge separated states of D2A2, D2A3 and D3A3 are below those of the singlet excited acceptor state. Therefore photo-induced electron transfer is a thermodynamically allowed process for these compounds in dichloromethane. In apolar toluene, the CS states of all antenna systems should be around 0.20–0.30 eV higher than the corresponding values estimated for DCM.28,14e This implies that in toluene, the CS states in most of the antenna systems will be of approximately the same value (or even higher) than that of the first singlet excited state of the respective perylene component, and no photoinduced electron transfer is expected to occur. However, for antenna molecules containing A3, in particular antenna molecule D3A3, photo-induced electron transfer from the singlet acceptor excited state of the acceptor is an energetically favorable process.
Steady-state absorption studies
The absorption spectra of all the reference compounds and antenna systems in toluene are shown in Fig. 3.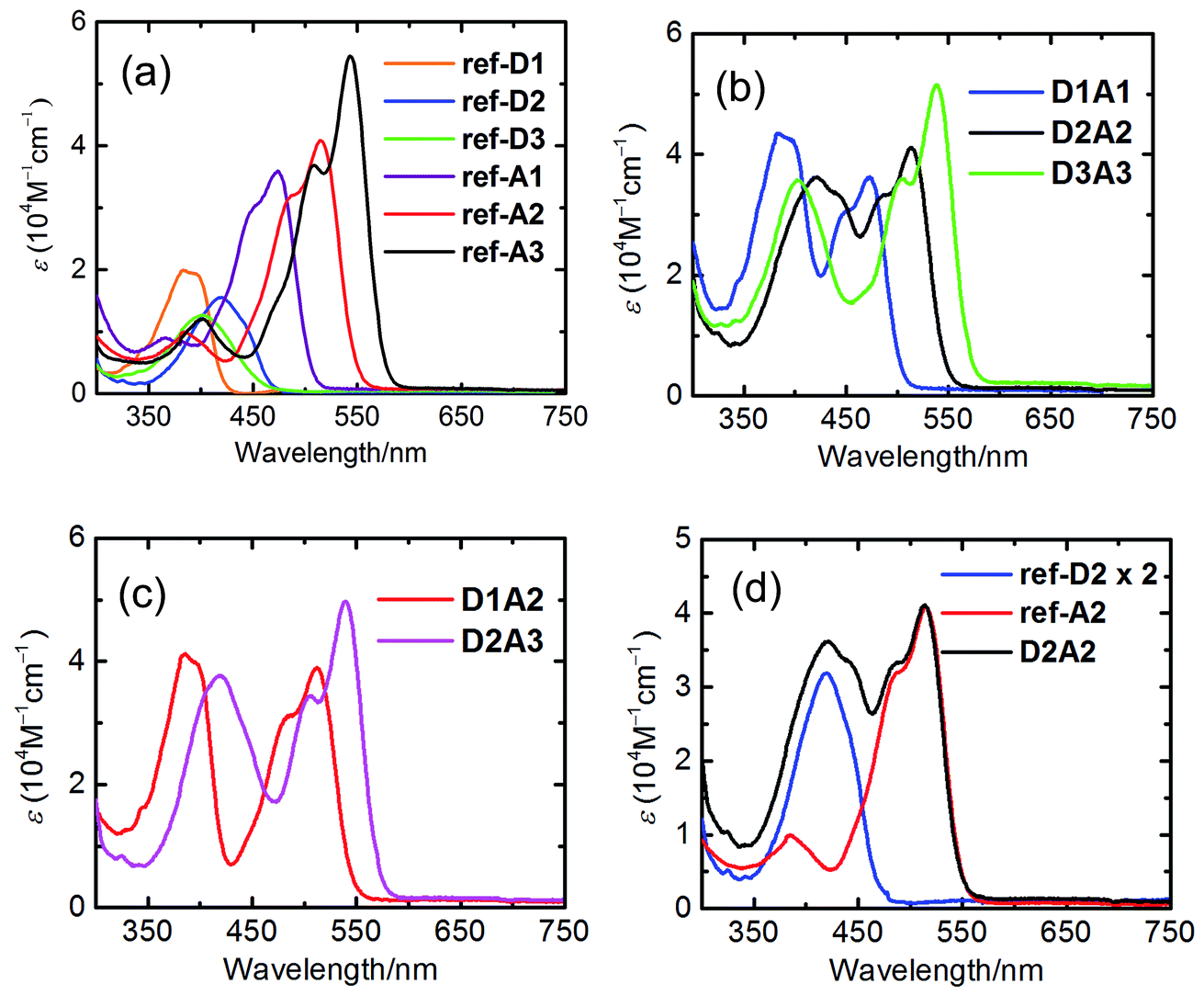 | ||
| Fig. 3 UV/Vis absorption spectra in toluene: (a) reference donor and acceptor compounds; (b) antenna systems D1A1, D2A2 and D3A3; (c) antenna systems D1A2 and D2A3; (d) antenna system D2A2 with ref-D2 and ref-A2 (note: the spectrum of ref-D2 is multiplied by two). | ||
The absorption spectra of the reference acceptors (ref-A1, ref-A2, and ref-A3) exhibit strong absorptions at wavelengths longer than ca. 430 nm. The absorption band shifts to longer wavelengths by 30–40 nm and the molar absorptivity increases upon going from ref-A1 to ref-A2 to ref-A3 (Table 2). The reference donors (ref-D1, ref-D2, and ref-D3) exhibit absorption at shorter wavelengths in the range of 350–450 nm. Among them, ref-D1 has the most blue-shifted absorption with λmax at 383 nm. The absorption of compounds ref-D2 and ref-D3 are red-shifted by 20–40 nm relative to ref-D1. Surprisingly, the absorption of D3 is not red-shifted compared to that of D2.27,29 The molar absorptivity of donor molecules is significantly lower compared to those of the acceptors. For that reason, the antenna systems are designed with two donors linked to a single acceptor.
| Compound | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ f | τ f | τ EET |
|---|---|---|---|---|---|---|
| a Fluorescence quantum yield. b Fluorescence lifetime (λex = 400 nm). c Lifetime data for energy transfer obtained from femtosecond transient absorption experiments. d Obtained after selective excitation of the perylene moiety. e Obtained after predominant excitation of the naphthalene moiety. | ||||||
| ref-D1 | 383 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
447 | 0.77 | 5.50 ns | — |
| ref-D2 | 420 | 15500 |
489 | 0.81 | 8.84 ns | — |
| ref-D3 | 402 | 12600 |
494 | 0.82 | 7.86 ns | — |
| ref-A1 | 474 | 35900 |
515 | 0.92 | 4.56 ns | — |
| ref-A2 | 515 | 40800 |
554 | 0.95 | 4.67 ns | — |
| ref-A3 | 543 | 54500 |
576 | 0.96 | 4.52 ns | — |
| D1A1 | 382 | 43400 |
512 | 0.93d | 4.50e ns | 0.99 ps |
| 473 | 36300 |
0.92e | ||||
| D1A2 | 386 | 41200 |
551 | 0.94d | 4.65e ns | 1.31 ps |
| 512 | 39000 |
0.92e | ||||
| D2A2 | 421 | 38000 |
553 | 0.92e | 4.60e ns | 1.16 ps |
| 514 | 41100 |
0.95e | ||||
| D2A3 | 421 | 38300 |
572 | 0.96d | 4.54e ns | 0.92 ps |
| 539 | 50300 |
0.97e | ||||
| D3A3 | 402 | 35100 |
571 | 0.96d | 4.51e ns | 0.87 ps |
| 538 | 50900 |
0.94e | ||||
The absorption spectra of the antenna systems clearly revealed the characteristic features of both donor and acceptor moieties (Fig. 3). At shorter wavelengths, absorption is dominated by the naphthalene chromophores, whereas the absorption at longer wavelengths originates exclusively from the perylene chromophores. Moreover, the spectra of all the antenna systems correspond very closely to the sum of the spectra of constituent chromophores, as can be seen in Fig. 3d. These results reflect the absence of any ground-state interaction between the two chromophores, which can be explained by the absence of conjugation between them. Notable is the excellent spectral coverage of most antenna molecules. Most antenna molecules have a high and uninterrupted absorption over a 200 nm wavelength span. Only in the case of compound D1A2, a distinct hole in the absorption spectrum around 430 nm is observed.
Steady-state and time-resolved fluorescence studies
Efficient transfer of excitation energy from the outer naphthalene chromophores to the inner perylene moiety is a prerequisite for a good light-harvesting antenna system. Therefore, we first examined the overlap between the donor's emission and acceptor's absorption, which is an important condition for efficient fluorescence resonance based energy transfer. In all antenna systems, the donor's emission overlapped strongly with the absorption of perylene derivatives. For example, the donor compound ref-D2 emits strongly in the range of 450–575 nm and ref-A2 absorbs strongly in the same range as is shown in Fig. 4.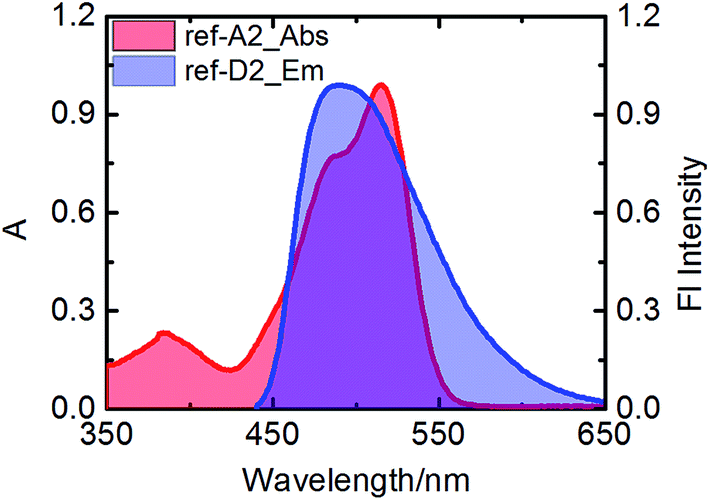 | ||
| Fig. 4 Overlap of the emission of ref-D2 with the absorption of ref-A2 in toluene. | ||
All the reference acceptor compounds are highly emissive with fluorescence quantum yields of ca. 0.95 and singlet-state life-times around 4.6 ns (Table 2). Their emission bands are red-shifted compared to the reference donors as depicted in Fig. 5a. The reference donors also exhibit strong emissions (Φf > 0.75) with singlet life-times in the range of 5.5–8.8 ns.
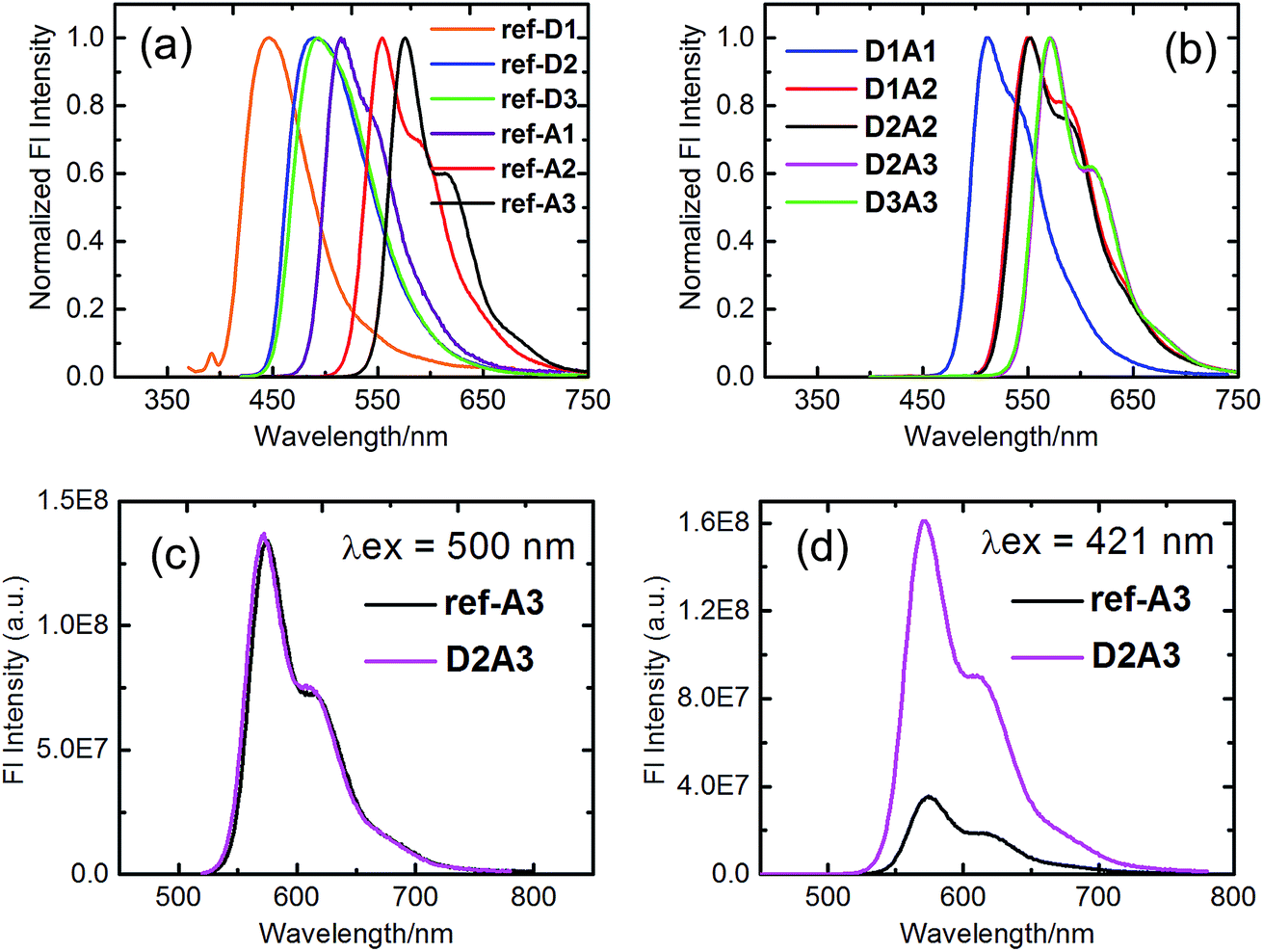 | ||
| Fig. 5 Normalized steady-state emission spectra in toluene: (a) the reference compounds; (b) the antenna systems obtained after excitation at the absorption maxima of the naphthalene moieties. (c) Excitation at 500 nm; ref-A3 (A = 0.16) and D2A3 (A = 0.16). (d) Excitation at 421 nm; ref-A3 (A = 0.04) and D2A3 (A = 0.18). | ||
The fluorescence emission studies of the antenna molecules were performed at two separate wavelengths to achieve selective excitation of only one chromophore. First, the perylene moieties were selectively excited at wavelengths higher than 460 nm and their emission, in terms of intensity and lifetime, was found to be identical to those of reference acceptors (Fig. 5c and Table 2). This indicates that the perylene excited state decays to the ground-state exclusively via emission in all antenna molecules. Subsequently, the antenna molecules were excited at the absorption maxima of the naphthalene chromophores in the range of 380–420 nm, to achieve predominant excitation of the donor chromophores. In these measurements, the emission of the naphthalene chromophores was found to be completely quenched in all the antennae (Fig. 5b). At the same time, all the antenna molecules produced the characteristic sensitized emission of perylene moieties with fluorescence quantum yields and life-times identical to those of the reference acceptors (Fig. 5d and Table 2). These results not only indicate a quantitative photo-induced energy transfer from the naphthalene moieties to the perylene chromophores, but also show an absence of other photo-induced processes in these antenna systems, such as photo-induced electron transfer from the naphthalene to the perylene unit. This conclusion was further verified by the excitation spectra of the antenna molecules measured at 600 nm (i.e. at the emission band of the perylene component), which resemble their absorption spectra within experimental error (Fig. 6).
 | ||
| Fig. 6 Excitation spectrum of D2A3 (dashed-line) measured at λem = 600 nm along with the absorption spectrum (solid-line) in toluene. | ||
Transient absorption studies
Finally, the excited state dynamics of all antenna molecules in toluene was examined on the femtosecond time-scale to gain insight into the kinetics of the intramolecular photo-induced energy transfer process. For these measurements, the antenna systems were excited at two different excitation wavelengths. First at the absorption maxima of the perylene chromophores to achieve an exclusive excitation of the perylene component. And secondly, at 400 nm, which resulted in the predominant excitation of the donor naphthalene moieties.Excitation of all the antenna molecules at the perylene absorption maximum resulted in the instant formation of the first singlet excited state of the perylene acceptor, as illustrated for antenna D1A2 in Fig. 7. These perylene singlet excited states are characterized by their typical features, i.e. strong absorption between 650–850 nm and a bleach of the ground state absorption at ca. 500 nm,14e,h,26a as has been reported for perylene bisimides. The absorption spectra of the singlet excited state of the perylene tetraester A1 and the perylene monoimide diester A2, which have not been reported previously, are slightly blue shifted compared to the excited state absorption spectrum of A3 (Fig. S5, ESI†). No changes in the absorption spectra of the perylene excited state at picosecond delay times were observed. At nanosecond delay times, the magnitude of the time resolved absorption spectra gradually decreases. This leads to the conclusion that for all antenna molecules, the perylene excited state decays directly to the ground-state via emission from the singlet excited state.
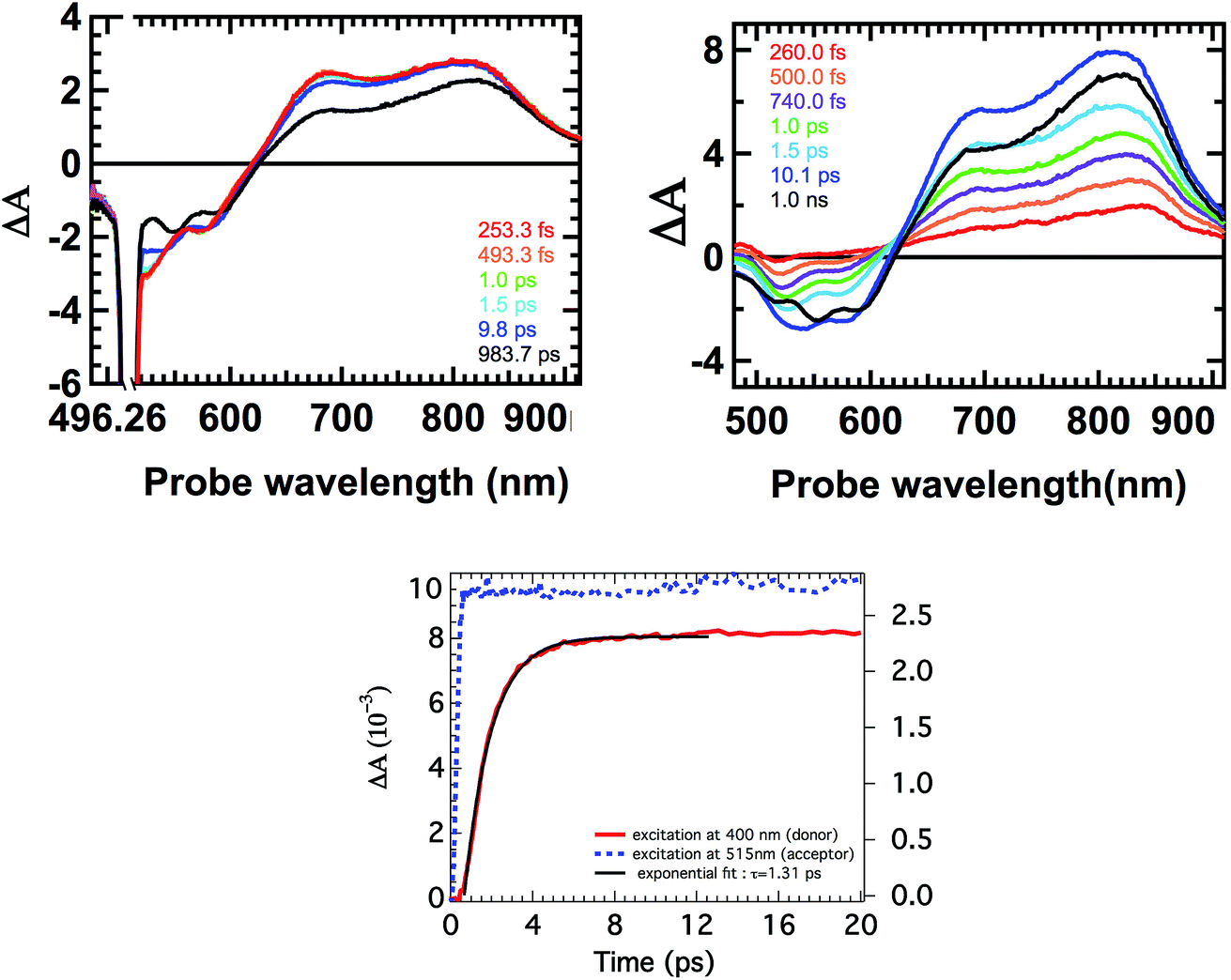 | ||
| Fig. 7 Time evolution of the femtosecond transient absorption spectra of the antenna system D1A2 after selective excitation of the perylene chromophore at 515 nm (top left) and after predominant excitation of the naphthalene chromophore at 400 nm (top right). Kinetics of D1A2 at 820 nm obtained after excitation at 400 and 515 nm (bottom). | ||
Excitation of the antenna molecules at the donor absorption maxima is illustrated for antenna D1A2 in Fig. 7. Immediately after the laser excitation of antenna D1A2 at 400 nm, absorption due to the singlet excited state of the perylene unit was observed. The immediate appearance of the perylene singlet excited state can be rationalized based on the fact that the perylene chromophore also has a limited absorption at the excitation wavelength (i.e. 400 nm). With the increase in delay times in the range of 0–10 ps, the perylene singlet excited state absorption increases drastically. At nanosecond delay times the magnitude of the time resolved absorption spectra gradually decreases, due to fluorescence from the acceptor. Similar results were observed for the other antenna systems as well (Fig. S5, ESI†).
The kinetics of the formation of the perylene singlet excited state absorption that was obtained after the excitation of the antenna molecules at 400 nm,30 was compared with the one obtained after the selective excitation of the perylene chromophore at 515 nm, and is represented in Fig. 7. These kinetic profiles are consistent with a fast excitation energy transfer from the naphthalene chromophore to the perylene component and subsequent slow decay of the perylene singlet excited state to the ground-state via emission. The time constants τEET for the energy transfer processes were determined by fitting the curves in Fig. 7 and S5,† and are compiled in Table 2. All antenna molecules show ultrafast energy transfer with time constants ranging from 0.9 to 1.3 ps. This ultrafast energy transfer is explained by the large values of the overlap integrals of donor emission and acceptor absorption, and the short distances between the donor and acceptor moieties in the antenna molecules. For synthetic light-harvesting antenna molecules containing perylene energy acceptors, energy transfer with significantly longer time constants, typically in the order of 5–50 ps, have been reported.10,14 This is most likely due to the larger D–A distances along with a decreased overlap between the donor emission and acceptor absorption spectra in these systems. The modest increase in energy transfer rates, upon going from antenna molecules based on A2 to A3, may be explained by a gradual increase of the acceptor extinction coefficients in this series.
As far as the occurrence of electron transfer processes are concerned, no spectral evidence for the formation of the perylene radical anions, which in the case of perylenebisimides can be easily recognized by their characteristic strong and relatively narrow absorption at ca. 725 nm,14h,26a,31 has been detected. This observation as such does not fully exclude the occurrence of photo-induced electron transfer within the antenna molecules. This is because very small quantities of CT species (<1%) may not be detected by transient absorption. Also, accumulation of CT states will not occur if the back electron transfer (BET) to the ground state is faster than the forward photo-induced electron transfer. Nevertheless, the lack of spectral evidence for photo-induced charge transfer is fully in line with the results obtained by steady state fluorescence, notably the unaltered lifetimes and fluorescence quantum yields of the singlet states of the perylene acceptors moieties upon incorporation in light-harvesting antenna molecules.
Discussion
We have designed and synthesized a series of five light-harvesting antenna molecules that exhibit 150 to 200 nm wide strong absorptions in the 350–580 nm wavelength region. For these compounds the absorption wavelength region is tunable in steps of 30–40 nm. The high absorption throughout a large part of the visible wavelength region is in contrast to most other light-harvesting molecules that generally exhibit large differences in the extinction coefficient within their absorption range. All antenna molecules exhibit the desired photo-physics for light-harvesting antenna systems, depicted in Fig. 8; quantitative and ultrafast energy transfer from the donor excited state towards the acceptor, D*–A ® D–A*, no charge transfer from the acceptor excited state, D–A* ® D+˙–A, and a long lifetime of the acceptor excited state.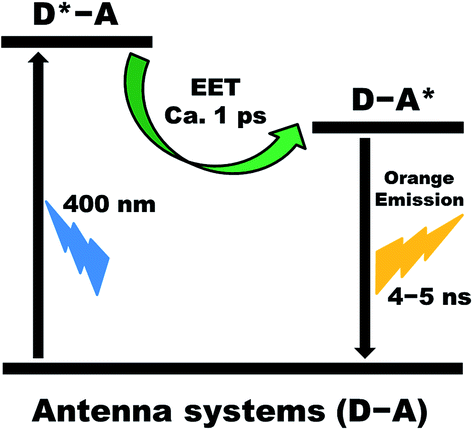 | ||
| Fig. 8 Summary of processes that take place in our antenna systems upon excitation of the naphthalene chromophore in toluene. | ||
The absence of electron transfer in all antenna molecules that we report here is remarkable. For example, a structurally similar antenna molecule, in which four 4-dimethylamino-naphthalimide moieties (similar to D3) were covalently linked to the bay positions of perylene bisimide using a long and flexible linker, was earlier reported by Flamigni et al.14e The photophysics of this molecule were investigated in toluene. Energy transfer from the peripheral naphthalimide donors to the PBI acceptor is relatively slow with τET = 54 ps and 90% efficient due to competitive electron transfer from the excited naphthalene unit to the perylene, a process with τCT = 0.6 ns. From the perylene excited state, charge-transfer to the naphthalene moieties is slower (τCT = 2.1 ns) and in competition with perylene fluorescence (τF = 6.0 ns). Interestingly, we did not observe any charge-separation in our PBI based antenna D3A3, which has the same donor and a slightly more electronegative acceptor chromophore compared to the above mentioned system. This difference in photo-induced behavior can only be explained by the different design of the linkers. In our antenna systems, the rigid structure of the spacer keeps the two acting moieties away from each other, which reduces the rate of electron transfer. This finding underlines the importance of employing rigid linkers in the construction of perylene based antenna systems.
Finally, it is worthwhile to evaluate the utility of the reported light-harvesting antenna molecules for constructing practical devices for photovoltaics or solar fuel generation. In such devices, fast and efficient energy transfer towards the acceptor should be followed by an efficient electron transfer from the acceptor excited state towards other components in the device, to produce either a potential difference for photovoltaics or charged catalysts that produce a solar fuel. Thus, the basic device requirements for such devices are that the energy transfer from the donor to the acceptor within the antenna molecule outcompetes other photo-physical processes from the donor excited state, notably electron transfer from the donor to the acceptor within the antenna system. In addition, the desired electron transfer processes starting from the acceptor excited state must outcompete other photo physical processes, notably electron transfer from the donor towards the acceptor and acceptor fluorescence. In this perspective it is important to realize that electron transfer processes are strongly enhanced in polar media, whereas energy transfer for rigid molecules does not depend on solvent polarity. For our antenna molecules, the energy transfer process from the naphthalene donor to the perylene acceptor, D*–A ® D–A*, occurs in approximately 1 ps, which is relatively fast for a synthetic antenna system, and will be fast enough to outcompete electron transfer processes. From the acceptor excited state thus obtained, electron transfer should be the dominant process. Time constants for the electron transfer from perylene excited states to good electron donors,18,32 are typically 2 orders of magnitude below the 4–5 ns fluorescence lifetimes of our perylene acceptors. In solvents of high polarity, electron transfer from the naphthalene donor towards the perylene excited state, D–A* ® D+˙–A−˙, will be thermodynamically allowed, in particular from the antenna systems based on the electron deficient PDI acceptor A3. However, it should be a rather straightforward exercise to design the antenna systems, such that the desired electron transfer process outcompetes electron transfer within the antenna molecules. Choosing appropriate linkers to limit electron transfer within the antenna molecules, like the phenoxy linker described herein, and applying acceptors with a decreased electron affinity, like D1 or D2, are the most obvious design criteria for limiting intermolecular energy transfer within the antenna.
Conclusions
A series of light-harvesting antenna molecules have been synthesized in a modular fashion, by a highly efficient covalent attachment of two identical naphthalene chromophores to the 1,7-bay positions of perylene-3,4,9,10-tetracarboxy derivatives. All the antenna molecules showed an efficient light absorption over a 200 nm wavelength span that is tunable in the 350–580 nm wavelength region. All antenna molecules exhibit good overlap between the donor's emission and the acceptor's absorption, which is a prerequisite for an efficient excited energy transfer (EET) by the Förster mechanism. In toluene, the light harvested by the naphthalene unit is transferred quantitatively to the perylene chromophore in picosecond time-scale in all antenna systems. Subsequently, the perylene singlet excited state decays to the ground state by the exclusive emission of light on a nanosecond time-scale (Fig. 8).No signs of photoinduced electron transfer were observed in our antenna molecules, despite the fact that the estimated energies of the charge-separated states obtained by cyclic voltammetry indicate that excited state deactivation by electron transfer is thermodynamically allowed and may already compete with the energy transfer process in apolar solvents like toluene. Therefore, we conclude that the rigid phenol linker group that is used in our antenna molecule systems prevents electron transfer in toluene.
In solvents of higher polarity, electron transfer from the energy donor to the energy acceptor gets increasingly favourable. Therefore, the antenna molecules containing the less electron deficient energy acceptors A1 and A2, are the most promising candidates for the construction of efficient light-harvesting components in light-driven devices. That is the case because the lowest electron transfer rates within the antenna molecules is expected for these antenna molecules. Further photophysical characterization of antenna systems in more polar solvents and their functionalization for the attachment to solid surfaces and molecular catalysts are currently undertaken in our research group.
Experimental section
Materials
All the reagents utilized in the synthesis were purchased from commercial suppliers and used as received unless otherwise stated. Toluene was dried over sodium under an argon atmosphere prior to use. NMP used for the reaction was of anhydrous grade. Purification of the products was performed using column chromatography (silica gel 60, mesh size 0.063–0.200 mm). TLC plates and the sorbent for the column chromatography were purchased from commercial suppliers.Instrumentation and characterization
The NMR spectra were recorded with a 400 MHz pulsed Fourier transform NMR spectrometer in either CDCl3 or DMSO-d6 at room temperature. The chemical shift values are given in ppm and J values in Hz. High-resolution mass spectra were collected on an AccuTOF GCv 4G JMS-T100GCV Mass Spectrometer (JEOL, Japan). The FD/FI probe (FD/FI) was equipped with an FD Emitter, Carbotec (Germany), FD 10 μm. Typical measurement conditions were as follows: current rate 51.2 mA min−1 over 1.2 min; counter electrode −10 kV; ion source 37 V. The samples were prepared in dichloromethane.Electrochemical behavior of the compounds was studied using cyclic voltammetry (CHI 600D electrochemical analyzer) in a three-electrode single-compartment cell consisting of a platinum sheet as the working electrode, Ag wire as the reference electrode, and a Pt wire as the counter electrode (scan-rate = 0.10 V s−1). The cell was connected to a computer-controlled potentiostat (CH Instruments Inc. 600D). Pre-dried CH2Cl2 containing 0.1 M tetrabutylammonium hexafluorophosphate was used as the solvent. The measurements were done under continuous flow of nitrogen. The concentration of the prepared samples was ca. 0.5 mM. Under these experimental conditions, the ferrocene oxidation was observed at 0.52 V.
All the spectroscopic measurements were carried out at room temperature. The emission spectra were corrected for the wavelength response of the detection system. Fluorescence quantum yields were determined by the comparative method, using perylene-3,4,9,10-tetracarboxylictetramethyl ester (Φf = 0.95 in CH2Cl2) as a reference.33 Optical densities at the excitation wavelengths were maintained at around 0.1 to avoid re-absorption.
Fluorescence lifetime measurements were performed on a Lifespec-ps Fluorescence spectrometer from Edinburgh Instruments. Samples were placed in 1 cm quartz cuvettes and their time correlated fluorescence was analysed by exponential tail fit with F900 Lifespec software. Transient absorption measurements were performed in a Pharos SP YKGBW Light Conversion laser, which is sent from 180 fs pulses at 1028 nm and amplified. This main beam is divided into a highly energetic pump beam and a white light probe beam, which is generated in a sapphire. The delay between the pump and probe beam can be adjusted up to 3 ns by using a time delay stage consisting of moveable mirrors. The desired wavelength of the pump beam can be changed by using an optical parametric amplifier (OPA) and an Orpheus second harmonics module from Light Conversion. The light after the sample (probe beam), was collected by a Helios detector from Ultrafast Systems. The samples were placed in 2 mm quartz cuvettes and were stirred with a magnetic stirrer to prevent aggregation.
Synthesis of N-(4′-hydroxyphenyl)-4-bromonaphthalene-1,8-dicarboxy monoimide (2)
A mixture of 4-bromo-1,8-naphthalic anhydride 1 (4.00 g, 14.44 mmol) and 4-aminophenol (1.89 g, 17.24 mmol), in ethanol (120 mL), was refluxed for 18 h. The reaction mixture was filtered after being cooled to room temperature. Thereafter, the residue was washed with ethanol and dried to obtain compound 2 (4.79 g, 13.00 mmol, 90%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ = 9.68 (s, 1H), 8.55 (d, J = 6.4 Hz, 2H), 8.31 (d, J = 7.6 Hz, 1H), 8.22 (d, J = 7.6 Hz, 1H), 8.00 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 8.0 Hz, 2H), 6.87 ppm (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ = 163.8, 163.7, 157.7, 133.0, 132.0, 131.8, 131.3, 130.3, 130.2, 129.5, 129.2, 129.0, 127.0, 123.8, 123.0, 115.9 ppm.Synthesis of N-(4′-hydroxyphenyl)-4-(isopentylthio)naphthalene-1,8-dicarboxy monoimide (3)
N-(4′-Hydroxyphenyl)-4-bromonaphthalene-1,8-dicarboxy monoimide 2 (1.20 g, 3.26 mmol), 3-methyl-1-butanethiol (1.22 mL, 9.78 mmol), and K2CO3 (2.03 g, 14.67 mmol) were taken in DMF (40 mL). The reaction mixture was stirred at 80 °C for 22 h. Afterwards, it was poured into water (200 mL) to precipitate the crude product overnight. The precipitate was filtered off, washed with several portions of water to remove all the residual DMF and 3-methyl-1-butanethiol, and dried in a vacuum oven. Subsequently, it was dissolved in MeOH and filtered to remove insoluble impurities. MeOH was evaporated to afford compound 3 (1.22 g, 3.12 mmol, 96%). 1H NMR (400 MHz, DMSO-d6): δ = 9.68 (br. s, 1H), 8.55–8.42 (m, 2H), 8.31 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.6 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 3.23 (t, J = 7.2 Hz, 2H), 1.85–1.73 (m, 1H), 1.60 (q, J = 7.2 Hz, 2H), 0.94 ppm (d, 6.5 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ = 164.1, 164.0, 157.6, 145.1, 131.4, 130.9, 130.3, 130.0, 129.2, 128.3, 127.6, 127.2, 123.7, 123.4, 119.3, 115.8, 37.1, 29.7, 27.6, 22.6 ppm.Synthesis of N-(4′-hydroxyphenyl)-4-(n-butylamino)naphthalene-1,8-dicarboxy monoimide (4)
A 50 mL round-bottomed flask was charged with N-(4′-hydroxyphenyl)-4-bromonaphthalene-1,8-dicarboxy monoimide 2 (1.00 g, 2.72 mmol), n-butylamine (4.02 mL, 40.80 mmol), and DMSO (30 mL). The reaction mixture was stirred at 80 °C for 24 h and the resultant solution was poured in water (200 mL) to precipitate the crude product. The precipitate was filtered off, washed with several portions of water, and dried in a vacuum oven to afford compound 4 (0.95 g, 2.64 mmol, 97%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 9.60 (s, 1H), 8.75 (d, J = 8.4 Hz, 1H), 8.41 (d, J = 7.2 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.79 (t, J = 5.2 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.05 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.4 Hz, 1H), 3.39 (q, J = 6.8 Hz, 2H), 1.75–1.66 (m, 2H), 1.49–1.39 (m, 2H), 0.96 ppm (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ = 163.9, 163.1, 156.6, 150.4, 134.0, 130.4, 129.7, 129.6, 128.5, 127.1, 123.9, 122.1, 120.0, 115.1, 115.0, 107.5, 103.4, 42.3, 29.7, 19.6, 13.5 ppm.Synthesis of N-(4′-hydroxyphenyl)-4-(dimethylamino)naphthalene-1,8-dicarboxy monoimide (5)
A mixture of N-(4′-methoxyphenyl)-4-bromonaphthalene-1,8-dicarboxy monoimide 2 (0.50 g, 1.36 mmol), 3-(dimethylamino)propionitrile (0.62 mL, 5.43 mmol), and DMSO (15 mL) was stirred at 132 °C overnight. After cooling down to room temperature, the resultant mixture was poured in water (200 mL) to precipitate the crude product. The precipitate was filtered off, washed with several portions of water, and dried. Subsequently, it was refluxed in MeOH (50 mL), filtered off after cooling, and dried to get compound 5 (0.32 g, 0.96 mmol, 71%) as a dark yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 9.59 (br. s, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 7.2 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.4 Hz, 2H), 3.39 ppm (s, 6H). 13C NMR spectrum could not be measured because of the low solubility of the compound.Synthesis of N-(2′,6′-diisopropylphenyl)-1,7-bis[N′-(p-phenyloxy)-(4′′-isopentylthio-1′′,8′′-dicarboxy naphthalene monoimide)]perylene-3,4,9,10-tetracarboxy monoimide dibutylester (7, D1A2)
A mixture of compound 3 (70 mg, 0.18 mmol), K2CO3 (50 mg, 0.36 mmol), and 18-crown-6 (95 mg, 0.36 mmol), in dry toluene (30 mL), was stirred for 45 minutes at room temperature under an argon atmosphere. Subsequently, 1,7-dibromoperylene monoimide dibutylester (6, A2) (50 mg, 0.06 mmol) was added. Thereafter, the reaction mixture was stirred for 16 h at 95 °C under an argon atmosphere. After being cooled to room temperature, the solvent was removed by rotary evaporation. The residue was washed with ethanol and water, and dried. The crude product was purified using column chromatography (silica-60/CHCl3) to afford the antenna system D1A2 (70 mg, 0.05 mmol, 80%). 1H NMR (400 MHz, CDCl3): δ = 9.41 (d, J = 8.0 Hz, 1H), 9.37 (d, J = 8.0 Hz, 1H), 8.68 (d, J = 8.0 Hz, 1H), 8.67–8.60 (m, 4H), 8.56 (s, 1H), 8.51 (dd, J = 8.0 and 1.2 Hz, 2H), 8.12 (d, J = 8.0 Hz, 1H), 7.97 (s, 1H), 7.80–7.75 (m, 2H), 7.56 (dd, J = 8.0 and 2.4 Hz, 2H), 7.50–7.44 (m, 1H), 7.37–7.31 (m, 6H), 7.29–7.24 (m, 4H), 4.38–4.31 (m, 4H), 3.22–3.14 (m, 4H), 2.80–2.72 (m, 2H), 1.89–1.76 (m, 6H), 1.75–1.67 (m, 4H), 1.54–1.41 (m, 4H), 1.20–1.14 (m, 12H), 1.06–0.85 ppm (m, 18H). 13C NMR (100 MHz, CDCl3): δ = 168.26, 167.45, 164.21, 163.54, 162.82, 155.78, 155.69, 152.49, 152.31, 146.37, 146.29, 145.73, 133.99, 133.19, 131.99, 131.24, 131.07, 130.65, 130.58, 129.71, 129.61, 128.63, 128.57, 128.14, 126.75, 126.69, 126.63, 126.44, 125.59, 125.33, 123.99, 123.27, 123.10, 122.49, 121.59, 118.80, 118.65, 118.56, 65.88, 65.55, 37.09, 30.64, 30.48, 30.29, 29.13, 27.69, 24.06, 22.25, 19.29, 19.21, 13.82 ppm. MS (ESI-TOF): [M]+ calculated for C90H81N3O12S2, 1459.52616; found, 1459.53383.Synthesis of N-(2′,6′-diisopropylphenyl)-1,7-bis[N′-(p-phenyloxy)-(4′′-butylamino-1′′,8′′-dicarboxy napthalene monoimide)]perylene-3,4,9,10-tetracarboxy monoimide dibutylester (8, D2A2)
Compound 4 (70 mg, 0.19 mmol), K2CO3 (50 mg, 0.36 mmol), and 18-crown-6 (126 mg, 0.48 mmol) were taken in dry toluene (85 mL). The resultant mixture was stirred at room temperature for 20 minutes and subsequently at 50 °C for another 20 minutes under an argon atmosphere. Afterwards, 1,7-dibromoperylene monoimide dibutylester (6, A2) (50 mg, 0.06 mmol) was added and the reaction mixture was stirred for 24 h at 105 °C under an argon atmosphere. After being cooled to room temperature, the solvent was removed by rotary evaporation. The residue was washed with methanol and water. The dried residue was refluxed in MeOH (100 mL) and filtered off after cooling down to room temperature. Finally, the crude product was purified using column chromatography (silica-60/DCM, CHCl3) to afford the antenna system D2A2 (55 mg, 0.04 mmol, 65%). 1H NMR (400 MHz, CDCl3): δ = 9.42 (d, J = 8.4 Hz, 1H), 9.37 (d, J = 8.0 Hz, 1H), 8.68 (d, J = 8.0 Hz, 1H), 8.61 (d, J = 7.2 Hz, 2H), 8.56 (s, 1H), 8.48 (d, J = 8.8 Hz, 2H), 8.15–8.09 (m, 3H), 7.97 (s, 1H), 7.67–7.60 (m, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.38–7.24 (m, 7H), 6.75 (dd, J = 8.4 and 2.8 Hz, 2H), 5.35–5.28 (m, 2H), 4.38–4.30 (m, 4H), 3.46–3.38 (m, 4H), 2.80–2.71 (m, 2H), 1.86–1.74 (m, 8H), 1.58–1.40 (m, 8H), 1.20–1.15 (m, 12H), 1.09–0.80 ppm (m, 12H). 13C NMR (100 MHz, CDCl3): δ = 168.31, 167.49, 164.85, 164.19, 163.61, 162.91, 155.52, 155.39, 152.66, 152.49, 149.91, 145.78, 134.99, 133.14, 132.04, 131.78, 131.56, 131.26, 131.15, 130.78, 130.72, 130.56, 130.26, 129.69, 129.48, 129.40, 128.57, 128.12, 126.69, 126.60, 126.40, 126.36, 125.50, 125.26, 124.66, 123.98, 123.17, 123.09, 122.66, 121.51, 120.25, 118.64, 118.53, 109.76, 104.40, 65.90, 65.56, 43.42, 30.93, 30.64, 30.48, 29.13, 24.07, 20.33, 19.30, 19.21, 13.86, 13.83 ppm. MS (ESI-TOF): [M]+ calculated for C88H79N5O12, 1397.63271; found, 1397.61057.Synthesis of N,N′-bis(2′,6′-diisopropylphenyl)-1,7-bis[N′-(p-phenyloxy)-(4′′-butylamino-1′′,8′′-dicarboxy naphthalene monoimide)]perylene-3,4,9,10-tetracarboxy bisimide (10, D2A3)
In a dry 100 mL round-bottom flask, weighed amounts of compound 4 (58 mg, 0.16 mmol), K2CO3 (38 mg, 0.28 mmol), and 18-crown-6 (74 mg, 0.28 mmol) were taken. Subsequently, anhydrous toluene (50 mL) was added. The reaction mixture was stirred for 15 min at room temperature and subsequently at 50 °C for another 15 min under an argon atmosphere. Thereafter, 1,7-dibromoperylene bisimide (9, A3) (40 mg, 0.05 mmol) was added and the reaction mixture was stirred at 95 °C for 5 h. After being cooled to room temperature, the solvent was removed by rotary evaporation and the residue was washed with MeOH and water. The dried residue was refluxed in ethanol (200 mL) and filtered off to remove any unreacted compound 4. Finally, column chromatography (silica-60/1:1 DCM–CHCl3) was performed to afford the desired antenna system D2A3 (58 mg, 0.04 mmol, 88%). 1H NMR (400 MHz, CDCl3): δ = 9.65 (d, J = 8.4 Hz, 2H), 8.75 (d, J = 8.0 Hz, 2H), 8.63 (s, 2H), 8.60 (d, J = 7.2 Hz, 2H), 8.48 (d, J = 8.4 Hz, 2H), 8.11 (d, J = 8.4 Hz, 2H), 7.63 (t, J = 7.6 Hz, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 8.8 Hz, 7H), 7.28 (m, 5H), 6.74 (d, J = 8.4 Hz, 2H), 5.33 (s, 2H), 3.46–3.37 (m, 4H), 2.82–2.71 (m, 4H), 1.85–1.75 (m, 4H), 1.55–1.49 (m, 4H), 1.23–1.16 (m, 24H), 1.02 ppm (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ = 164.83, 164.16, 163.34, 162.65, 155.25, 153.82, 149.83, 145.72, 135.03, 133.47, 132.00, 131.61, 131.32, 130.87, 130.44, 130.25, 129.70, 129.61, 129.22, 126.71, 126.56, 126.26, 125.87, 124.72, 124.42, 124.05, 123.12, 122.66, 120.23, 118.45, 109.84, 104.45, 43.43, 30.98, 29.18, 24.08, 20.33, 13.85 ppm. MS (ESI-TOF): [M]+calculated for C92H78N6O10, 1426.57794; found, 1426.58144.
Synthesis of N,N′-bis(2′,6′-diisopropylphenyl)-1,7-bis[N′-(p-phenyloxy)-(4′′-dimethylamino-1′′,8′′-dicarboxy naphthalene monoimide)]perylene-3,4,9,10-tetracarboxy bisimide (11, D3A3)
Weighed amounts of compound 5 (52 mg, 0.16 mmol), K2CO3 (40 mg, 0.29 mmol), and 18-crown-6 (77 mg, 0.29 mmol) were taken in anhydrous toluene (50 mL). The mixture was stirred for 20 min at room temperature and, thereafter, 1,7-dibromoperylene bisimide (9, A3) (40 mg, 0.05 mmol) was added. The reaction mixture was stirred at 100 °C for 2 h. After being cooled to room temperature, the solvent was removed by rotary evaporation and the residue was washed with MeOH and water. The dried residue was refluxed in methanol (150 mL) and filtered off to remove any unreacted compound 5 and other soluble impurities. Finally, column chromatography (silica-60/CHCl3) was performed to afford the desired antenna system D3A3 (40 mg, 0.03 mmol, 63%). 1H NMR (400 MHz, CDCl3): δ = 9.65 (d, J = 8.0 Hz, 2H), 8.75 (d, J = 8.4 Hz, 2H), 8.63 (s, 2H), 8.59 (d, J = 7.2 Hz, 2H), 8.49 (t, J = 7.6 Hz, 4H), 7.68 (t, J = 7.6 Hz, 2H), 7.48 (t, J = 8.0 Hz, 2H), 7.40–7.26 (m, 12H), 7.13 (d, J = 8.0 Hz, 2H), 3.13 (s, 12H), 2.82–2.70 (m, 4H), 1.25–1.14 ppm (m, 24H). 13C NMR (100 MHz, CDCl3): δ = 164.75, 164.14, 163.31, 162.61, 157.32, 155.37, 153.73, 145.70, 133.43, 133.16, 131.71, 131.67, 131.53, 131.33, 130.82, 130.66, 130.42, 129.70, 129.61, 129.21, 126.76, 126.59, 125.91, 125.28, 124.92, 124.46, 124.05, 123.04, 122.70, 118.48, 114.65, 113.31, 44.76, 29.18, 24.07 ppm. MS (ESI-TOF): [M]+ calculated for C88H70N6O10, 1370.51534; found, 1370.51784.Synthesis of 1,7-bis[N′-(p-phenyloxy)-(4′′-isopentylthio-1′′,8′′-dicarboxy naphthalene monoimide)]perylene-3,4,9,10-tetracarboxy tetrabutylester (14, D1A1)
The antenna system D1A1 was synthesized in the following two steps: (i) a mixture of 1,7-dibromoperylene monoanhydride dibutylester 12 (100 mg, 0.15 mmol), compound 3 (200 mg, 0.51 mmol), and K2CO3 (100 mg, 0.72 mmol), in anhydrous NMP (20 mL), was stirred at 120 °C for 24 h under an argon atmosphere. The resultant mixture was poured into water (200 mL) to precipitate the crude product. The precipitate was filtered off and washed with several portions of water. The dried residue was refluxed in ethanol (150 mL) and filtered off to remove any unreacted compound 3 and other soluble impurities. Product 13 (75 mg, 39%) was used as such in the next step without NMR characterization due to its low solubility. (ii) Compound 13 (75 mg, 0.06 mmol), butanol (200 μL, 2.19 mmol), and DBU (200 μL, 1.34 mmol) were stirred in DMF (20 mL) for 20 min at 95 °C. Thereafter, butylbromide (200 μL, 1.85 mmol) was added and the reaction mixture was stirred at 100 °C for 18 h. The resultant mixture was poured into water (200 mL) to precipitate the crude product. The precipitate was filtered off, and washed with several portions of water. The dried residue was refluxed in methanol (100 mL) and filtered off (after cooling) to remove the soluble impurities. The process was repeated two times. Finally, silica gel column chromatography was performed on the crude product. Impurities were removed by eluting with DCM and the pure antenna system D1A1 (63 mg, 0.044 mmol, 76%) was achieved. 1H NMR (400 MHz, CDCl3): δ = 9.07 (d, J = 8.4 Hz, 2H), 8.65 (d, J = 7.2 Hz, 2H), 8.61 (d, J = 8.8 Hz, 2H), 8.50 (d, J = 7.6 Hz, 2H), 8.05 (d, J = 8.4 Hz, 2H), 7.89 (s, 2H), 7.77 (t, J = 8.4 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.8 Hz, 4H), 7.22 (d, J = 8.8 Hz, 4H), 4.37–4.26 (m, 8H), 3.18 (t, J = 7.6 Hz, 4H), 1.87–1.66 (m, 14H), 1.55–1.39 (m, 8H), 1.05–0.94 ppm (m, 24H). 13C NMR (100 MHz, CDCl3): δ = 168.4, 167.6, 164.2, 164.1, 156.0, 150.9, 146.2, 131.9, 131.8, 131.2, 130.9, 130.8, 130.5, 129.9, 129.6, 129.5, 128.6, 127.4, 126.6, 125.3, 125.2, 123.5, 123.2, 122.5, 118.9, 118.6, 65.6, 65.3, 37.1, 30.6, 30.5, 30.3, 27.7, 22.2, 19.3, 19.2, 13.8 ppm. MS (ESI-TOF): [M]+ calculated for C86H82N2O14S2, 1430.52074; found, 1430.53751.Acknowledgements
This project was carried out within the research programme of BioSolar Cells, co-financed by the Dutch Ministry of Economic Affairs, Agriculture and Innovation. This work is also part of the research programme of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organisation for Scientific Research (NWO).References
- (a) R. van Grondelle, J. P. Dekker, T. Gillbro and V. Sundstrom, Biochim. Biophys. Acta., 1994, 1187, 1–65 CrossRef; (b) G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- (a) A. Harriman, Chem. Commun., 2015, 51, 11745–11756 RSC; (b) P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC; (c) R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330–11344 CrossRef CAS PubMed; (d) A. Fermi, P. Ceroni, M. Roy, M. Gingras and G. Bergamini, Chem.–Eur. J., 2014, 20, 10661–10668 CrossRef CAS PubMed.
- (a) M. Grätzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef PubMed; (b) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed; (c) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed; (d) W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed; (e) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed; (f) E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed; (g) F. M. Jradi, D. O'Neil, X. Kang, J. Wong, P. Szymanski, T. C. Parker, H. L. Anderson, M. A. El-Sayed and S. R. Marder, Chem. Mater., 2015, 27, 6305–6313 CrossRef CAS.
- G. Cinque, R. Croce and R. Bassi, Photosynth. Res., 2000, 64, 233–242 CrossRef CAS PubMed.
- J. P. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic and Plenum Publishers, New York, 1999, ch. 9 Search PubMed.
- In case donor and acceptor units exhibit through bond interaction (conjugation), which is generally achieved by using acetylene linkers, energy transfer can take place by an electron exchange mechanism, that is generally referred to as the Dexter mechanism, see ref. 5.
- (a) T. N. Murakami, S. Ito, Q. Wang, M. K. Nazeeruddin, T. Bessho, I. Cesar, P. Liska, R. Humphry-Baket, P. Compte, P. Péchy and M. Grätzel, J. Electrochem. Soc., 2006, 153, A2255–A2261 CrossRef CAS; (b) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- (a) H. Peng, L. Niu, Y. Chen, L. Wu, C. Tung and Q. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS PubMed; (b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- M. A. H. Alamiry, A. Harriman, A. Haefele and R. Ziessel, ChemPhysChem, 2015, 16, 1867–1872 CrossRef CAS PubMed.
- (a) C. Devadoss and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 9635–9644 CrossRef CAS; (b) S. L. Gilat, A. Adronov and J. M. J. Fréchet, Angew. Chem., Int. Ed., 1999, 38, 1422–1427 CAS; (c) A. Andronov, S. L. Gilat, L. M. J. Fréchet, K. Ohta, F. V. R. Neuwahl and G. R. Fleming, J. Am. Chem. Soc., 2000, 122, 1175–1185 CrossRef; (d) T. Weil, E. Reuther and K. Müllen, Angew. Chem., Int. Ed., 2002, 41, 1900–1904 CrossRef CAS; (e) F. C. De Schryver, T. Vosch, M. Cotlet, M. Van der Auweraer, K. Müllen and J. Hofkens, Acc. Chem. Res., 2005, 38, 514–522 CrossRef CAS PubMed; (f) M. Cotlet, T. Vosch, S. Habuchi, T. Weil, K. Müllen, J. Hofkens and F. C. De Schryver, J. Am. Chem. Soc., 2005, 127, 9760–9768 CrossRef CAS PubMed; (g) W. R. Dichtel, S. Hecht and J. M. J. Fréchet, Org. Lett., 2005, 7, 4451–4454 CrossRef CAS PubMed; (h) X. Zhang, Y. Xiao and X. Qian, Org. Lett., 2008, 10, 29–32 CrossRef CAS PubMed; (i) A. Harriman, L. Mallon and R. Ziessel, Chem.–Eur. J., 2008, 14, 11461–11473 CrossRef CAS PubMed; (j) M. Fischer, T. E. Kaiser, F. Würthner and P. Bauerle, J. Mater. Chem., 2009, 19, 1129–1141 RSC; (k) E. Fron, L. Puhl, I. Oesterling, C. Li, K. Müllen, F. C. De Schryver, J. Hofkens and T. Vosch, ChemPhysChem, 2011, 12, 595–608 CrossRef CAS PubMed.
- P. Ensslen and H.-A. Wagenknecht, Acc. Chem. Res., 2015, 48, 2724–2733 CrossRef CAS PubMed.
- P. Bonaccorsi, M. C. Aversa, A. Barattucci, T. Papalia, F. Puntoriero and S. Campagna, Chem. Commun., 2012, 48, 10550–10552 RSC.
- K. V. Rao, K. K. R. Datta, M. Eswarmoorthy and S. J. George, Chem.–Eur. J., 2012, 18, 2184–2194 CrossRef CAS PubMed.
- (a) H. Langhals and S. Saulich, Chem.–Eur. J., 2002, 8, 5630–5643 CrossRef CAS PubMed; (b) J. M. Serin, D. W. Brousmiche and J. M. J. Fréchet, J. Am. Chem. Soc., 2002, 124, 11848–11849 CrossRef CAS PubMed; (c) C. Hippius, F. Schlosser, M. O. Vysotsky, V. Böhmer and F. Würthner, J. Am. Chem. Soc., 2006, 128, 3870–3871 CrossRef CAS PubMed; (d) M. D. Yilmaz, A. Bozdemir and E. U. Akkaya, Org. Lett., 2006, 8, 2871–2873 CrossRef CAS PubMed; (e) L. Flamigni, B. Ventura, C.-C. You, C. Hippius and F. Würthner, J. Phys. Chem. C, 2007, 111, 622–630 CrossRef CAS; (f) J. H. Hurenkamp, W. R. Browne, R. Augulus, A. Pugzlys, P. M. H. van Loosdrecht, J. H. van Esch and B. L. Feringa, Org. Biomol. Chem., 2007, 5, 3354–3362 RSC; (g) M. Konemann, US Patent US2008/0287678 A1, 2008; (h) M. Fulitsuka, K. Harada, A. Sugimoto and T. Majima, J. Phys. Chem. A, 2008, 112, 10193–10199 CrossRef PubMed.
- (a) R. W. Wagner, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1996, 118, 11166 CrossRef CAS; (b) R. F. Kelly, W. S. Shin, B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Am. Chem. Soc., 2007, 129, 3173–3181 CrossRef PubMed; (c) A. J. Jiménez, F. Spänich, S. Rodriguez-Morgade, K. Ohkubu, S. Fukuzumi, D. M. Guldi and T. Torres, Org. Lett., 2007, 9, 2481–2484 CrossRef PubMed; (d) V. M. Blas-Ferrando, J. Ortiz, L. Bouissane, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and A. Sastre-Santos, Chem. Commun., 2012, 48, 6241–6243 RSC.
- (a) C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed; (b) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- (a) R. Gómez, J. L. Segura and N. Martín, Org. Lett., 2005, 7, 717–720 CrossRef PubMed; (b) J. Baffreau, S. Leroy-Lhez, N. V. Anh, R. M. Williams and P. Hudhomme, Chem.–Eur. J., 2008, 14, 4974–4992 CrossRef CAS PubMed; (c) C. C. Hofmann, S. M. Lindner, M. Ruppert, A. Hirch, S. A. Haque, M. Thelakkat and J. Köhler, J. Phys. Chem. B, 2010, 114, 9148–9156 CrossRef CAS PubMed; (d) R. K. Dubey, M. Niemi, K. Kaunisto, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Chem.–Eur. J., 2013, 19, 6791–6806 CrossRef CAS PubMed; (e) S. Pla, L. Martín-Gomis, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and A. Sastre-Santos, Asian J. Org. Chem., 2014, 3, 185–197 CrossRef CAS.
- Commonly undesired electron transfer towards perylenes has been limited by using the perylene monoimides PMIs instead of PBIs, see for example: C. Kirmaier, H. Song, E. Yang, J. K. Schwartz, E. Hindin, J. R. Diers, R. S. Loewe, K. Tomizaki, F. Chevalier, L. Ramos, R. R. Birge, J. S. Lindsey, D. F. Bocian and D. Holten, J. Phys. Chem. B, 2010, 114, 14249–14264 CrossRef CAS PubMed.
- (a) Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed; (b) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- S. Sengupta, R. K. Dubey, R. W. M. Hoek, S. S. P. van Eeden, D. D. Gunbaş, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, J. Org. Chem., 2014, 79, 6655–6662 CrossRef CAS PubMed.
- (a) P. Kucheryavy, G. Li, S. Vyas, C. Hadad and K. D. Glusac, J. Phys. Chem. A, 2009, 113, 6453–6461 CrossRef CAS PubMed; (b) D. Collado, P. Remón, Y. Vida, F. Najera, P. Sen, U. Pischel and E. Perez-Inestrosa, Chem.–Asian J., 2014, 9, 797–804 CrossRef CAS PubMed.
- A. Bamesberger, C. Schwartz, Q. Song, W. Han, Z. Wang and H. Cao, New J. Chem., 2014, 38, 884–888 RSC.
- J. Kollár, P. Hrdlovic, S. Chmela, M. Sarakha and G. Guyot, J. Photochem. Photobiol., A, 2005, 170, 151–159 CrossRef.
- In ref-D1, a phenyl group instead of a methoxyphenyl group was attached to the naphthylimide. This is the case because the electron-rich methoxyphenyl group strongly quenches the excited state of the 4-isopentylthionaphthalene monoimide.
- R. K. Dubey, A. Efimov and H. Lemmetyinen, Chem. Mater., 2011, 23, 778–788 CrossRef CAS.
- (a) R. K. Dubey, M. Niemi, K. Kaunisto, K. Stranius, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Inorg. Chem., 2013, 52, 9761–9773 CrossRef CAS PubMed; (b) R. K. Dubey, T. Kumpulainen, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Eur. J. Org. Chem., 2010, 3428–3436 CrossRef CAS.
- In the energy donation naphthylimides the Hammett substituent constants sp of electron donating groups –S(CH2)2CH(CH3)2, –NH(CH2)3CH3 and –N(CH3)2 are ≈0.05, −0.51, 0.83, respectively, see: C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- A. Weller, Z. Phys. Chem., 1982, 133, 93–98 CrossRef CAS.
- In more polar solvents the absorption and emission spectra of compound D3 are red-shifted compared to D2, due to the strong solvatochromism of this polar compound, see: (a) E. Lippert, Z. Electrochem., 1957, 61, 962–975 CAS; (b) W. F. Jager, A. A. Volkers and D. C. Neckers, Macromolecules, 1995, 28, 8153 CrossRef CAS.
- The kinetic trace in Fig. 7b, resulting from excitation at 400 nm, has been normalized. This has been achieved by subtracting the absorption from the directly excited perylene at t = 0.
- (a) W. E. Ford, H. Hiratsuka and P. V. Kamat, J. Phys. Chem., 1989, 93, 6692–6696 CrossRef CAS; (b) M. S. Rodriguez-Morgade, T. Torres, C. Atienza-Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 15145–15154 CrossRef CAS PubMed.
- (a) A. Prodi, C. Chiorboli, F. Scandola, E. Iengo, E. Alessio, R. Dobrawa and F. Würthner, J. Am. Chem. Soc., 2005, 127, 1454–1462 CrossRef CAS PubMed; (b) L. Flamigni, B. Ventura, M. Tasior, T. Becherer, H. Langhals and D. T. Gryko, Chem.–Eur. J., 2008, 14, 169–183 CrossRef CAS PubMed; (c) L. Flamigni, B. Ventura, A. Barbieri, H. Langhals, F. Wetzel, K. Fuchs and A. Walter, Chem.–Eur. J., 2010, 16, 13406–13416 CrossRef CAS PubMed; (d) R. K. Dubey, G. Knorr, N. Westerveld and W. F. Jager, Org. Biomol. Chem., 2016, 14, 1564–1567 RSC.
- H. Langhals, J. Karolin and L. B.-Å. Johansson, J. Chem. Soc., Faraday Trans., 1998, 94, 2919–2922 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of model donors; 1H–1H COSY spectrum of antenna D2A2 (Fig. S1); cyclic voltammograms of all the ensembles and reference compounds (Fig. S2); fluorescence decay curves (Fig. S3); comparison of absorption and excitation spectra (Fig. S4); transient absorption spectra of antenna systems and their decay kinetics (Fig. S5); 1H and 13C NMR spectra of all synthesized compounds (Fig. S23–S62). See DOI: 10.1039/c6sc00386a |
| ‡ Present address: Interdisciplinary Centre for Energy Research (ICER), Indian Institute of Science (IISc), Bangalore-560012, India. |
| This journal is © The Royal Society of Chemistry 2016 |