
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of cationic and neutral (allenylidene)(carbene) and bis(allenylidene)gold complexes†
Liqun
Jin
a,
Mohand
Melaimi
 a,
Arseni
Kostenko
b,
Miriam
Karni
b,
Yitzhak
Apeloig
b,
Curtis E.
Moore
a,
Arnold L.
Rheingold
a and
Guy
Bertrand
*a
a,
Arseni
Kostenko
b,
Miriam
Karni
b,
Yitzhak
Apeloig
b,
Curtis E.
Moore
a,
Arnold L.
Rheingold
a and
Guy
Bertrand
*a
aUCSD-CNRS Joint Research Chemistry Laboratory (UMI 3555), Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093-0358, USA. E-mail: guybertrand@ucsd.edu
bSchulich Faculty of Chemistry, Technion - Israel Institute of Technology, Haifa 32000, Israel
First published on 16th November 2015
Abstract
The one-electron reduction of a cationic (allenylidene)[cyclic(alkyl) (amino)carbene]gold(I) complex leads to the corresponding neutral, paramagnetic, formally gold(0) complex. DFT calculations reveal that the spin density of this highly robust coinage metal complex is mainly located on the allenylidene fragment, with only 1.8 and 3.1% on the gold center and the CAAC ligand, respectively. In addition, the first homoleptic bis(allenylidene)gold(I) complex has been prepared and fully characterized.
Introduction
Allenylidene transition metal complexes were first isolated in 1976 by Fischer and Berke.1 The π interactions between the metal ion and the rigid, linear allenylidene framework make these complexes of interest for molecular wires and electronic materials; they have also been postulated as key intermediates in catalysis.2 The chemistry of metal allenylidene complexes is largely dominated by Cr, W, Mn, Ru, Os, Ir, Pd and Pt,3 while studies dealing with coinage metals are limited. Hashmi et al.4 and Che et al.5 prepared the Au(I) and Au(III) complexes A and B, respectively, featuring an (alkoxy) (amino)allenylidene, while we isolated a silver(I) complex C, featuring the (diamino)allenylidene6 (Chart 1). Recently, we and others reported that cyclic (alkyl) (amino)carbene (CAAC)7 ligands allow for the isolation of complexes featuring a metal in the formal zero oxidation state,8,9 including the neutral complex D, the first example of a compound with a gold(0) center.10 The unusual stability of D is due to the π-accepting properties of CAAC ligands.11 Theoretical calculations have clearly established that allenylidenes are likewise σ-donor and π-acceptor ligands with a dominant contribution of the latter component to the bonding, and thus are attractive ligands for stabilizing gold(0) complexes.11,12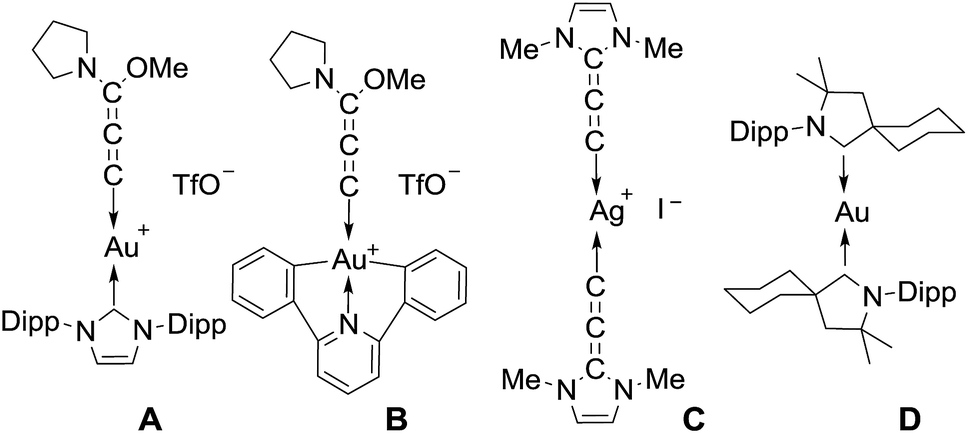 | ||
| Chart 1 | ||
Herein, we report the preparation of several stable allenylidene gold complexes, including the first neutral (allenylidene)(carbene)gold and cationic bis(allenylidene)gold complexes.
Results and discussion
Although aryl- or alkyl-substituted allenylidenes have stronger π-accepting properties, their complexes are usually much less stable than those featuring a π-donating heteroatom.4 Therefore we chose to target allenylidenes featuring one amino substituent. As shown in Scheme 1, alkyne 2 was prepared by formal insertion of a CAAC13,14 into the CH-bond of trimethylsilylacetylene, followed by treatment with tetrabutylammonium fluoride. In the presence of sodium hydroxide, alkyne 2 reacts with Ph3PAuCl in methanol, affording the gold–acetylide complex 3 in 84% yield. Lastly, a hydride abstraction15 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), followed by anion exchange with HBF4, led to the cationic allenylidene gold complex 4 as a white solid, but in only 37% yield. Moreover, this complex appeared to decompose rapidly in solution (vide infra). Thus, we decided to replace the triphenylphosphine ligand with a CAAC. Addition at room temperature of CAAC 1 to the acetylide complex 3 readily afforded complex 5, which was obtained in 78% yield. Using the same procedure as for 3, the desired cationic gold allenylidene complexes 6a,b with BPh4 or BF4 counter-anion were isolated as light yellow, air and water stable solids in good yields. The 13C NMR signal of the C3 and C2 nuclei of the allenylidene fragment are at 185.9 and 97.6 ppm, downfield and highfield shifted by 62.5 and 8.4 ppm, respectively, compared to those of 5. A similar trend has been observed for other late transition metal allenylidenes versus acetylide complexes.16 Single crystals suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into a dichloromethane solution of 6b (Fig. 1A).17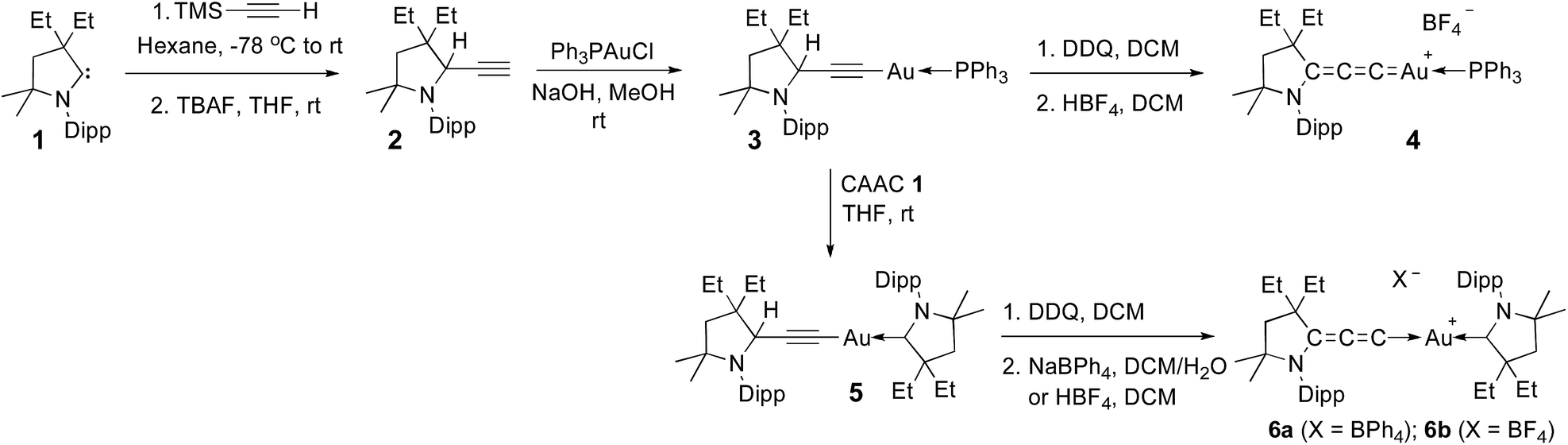 | ||
| Scheme 1 Synthesis of allenylidene gold(I) complexes 4 and 6a,b. Dipp = 2,6-diisopropylphenyl, TBAF = tetrabutylammonium fluoride. | ||
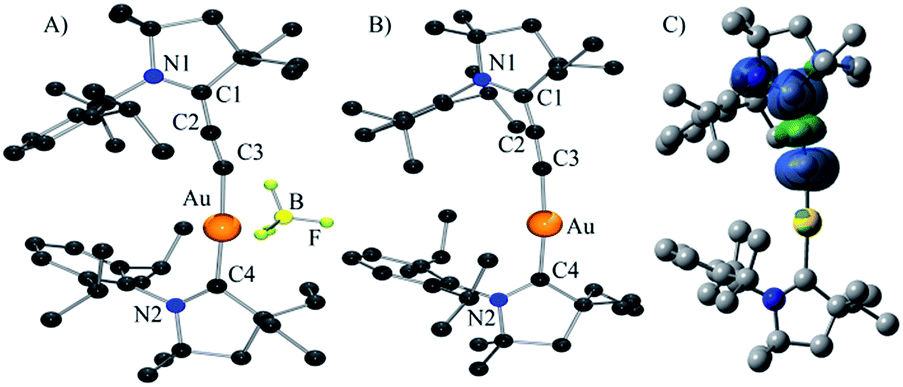 | ||
| Fig. 1 Solid-state structure of complexes 6b (A), 7 (B) (H-atoms omitted for clarity), and calculated spin-density for complex 7 (C). | ||
The cyclic voltammetry of (allenylidene) (CAAC)Au(I) 6b was carried out in THF using 0.1 M nBu4NPF6 as supporting electrolyte. A reversible one electron reduction at E1/2 = −1.73 V versus Fc+/Fc was observed. This reduction potential is shifted towards a more positive value compared to that observed for bis(CAAC)Au(I) complex (E1/2 = −2.24 V),10 demonstrating the superior π-accepting properties of allenylidenes compared to CAACs. Encouraged by these results, the chemical reduction of complex 6b was carried out with one equivalent of KC8 in THF at room temperature. A dark yellow solution was obtained, which was NMR silent and EPR active. The room temperature EPR spectrum of 7 in benzene (see ESI†) shows a broad signal centered at g = 1.983. The highly shifted g factor, compared with free electron g = 2.002, is typical to heavy-element-containing radicals due to strong spin–orbit coupling.
The DFT calculated g factor (at ZORA/B3LYP/TZVP//M05-2X/SDD/def2-SVP level of theory) of 1.985 (for full details, see the “Computational methods” section below) is in excellent agreement with experiment. Yellow single crystals of the neutral complex 7 were grown from a concentrated THF solution at −20 °C (Fig. 1B). Detailed comparison of the geometric parameters of 6b and 7 is not possible because two superimposed molecules are present in the unit cell of 6b. At the M05-2X/SDD/def2-SVP level of theory the C2–C3 and C1–C2 bonds slightly elongates and shortens, respectively, upon reduction [C2–C3: 6b 1.229, 7 1.243; C1–C2: 6b 1.399, 7 1.380]. The calculations also predict a significant elongation of the C1–N1 bond upon reduction [6b: 1.304; 7: 1.386]. These data suggest that the electron is added to the allenylidene ligand. This hypothesis is supported by Mulliken spin density analysis of 7 computed at the M05-2X/SDD/def2-SVP//M05-2X/SDD/def2-SVP level of theory. Indeed, 93.9% of the spin density resides on the CCC fragment, while only 3.8 and 1.8% are located on the CAAC ligand and the Au atom, respectively (Fig. 1C). This is also reflected in the charges. Upon reduction the gold atom and the CAAC moiety gain only 0.02 el. and 0.10 el., respectively, while the calculated charge in the allenylidene moiety increases by 0.88 el. Consequently, 7 has only a very weak gold(0) character, even weaker than complex D, in which 17% of the spin density is located at gold;10 in other words it can be viewed as a gold(I) complex featuring a paramagnetic anionic ligand.
We then turned our attention to the decomposition process of the (allenylidene) (triphenylphosphine)gold(I) complex 4. To our delight, this complex undergoes a dismutation reaction, affording bis(triphenylphosphine)gold(I) along with the bis(allenylidene)gold(I) complex 8, which was isolated in 11% yield as a moisture and air stable solid (Scheme 2). So far, only bis(allenylidene) complexes of Ag,6 Pd,6,18 Pt,18 and Ru16 have been isolated. 1H and 13C NMR spectroscopy of 8 clearly shows a symmetrical structure. Only three different signals ascribed to the cumulenic carbon nuclei were displayed in the 13C NMR spectrum (185.8, 177.7 and 95.9 ppm). Because of the poor yield and rather difficult separation procedure, we looked for a more efficient synthetic route leading to bis(allenylidene) gold complex 8.
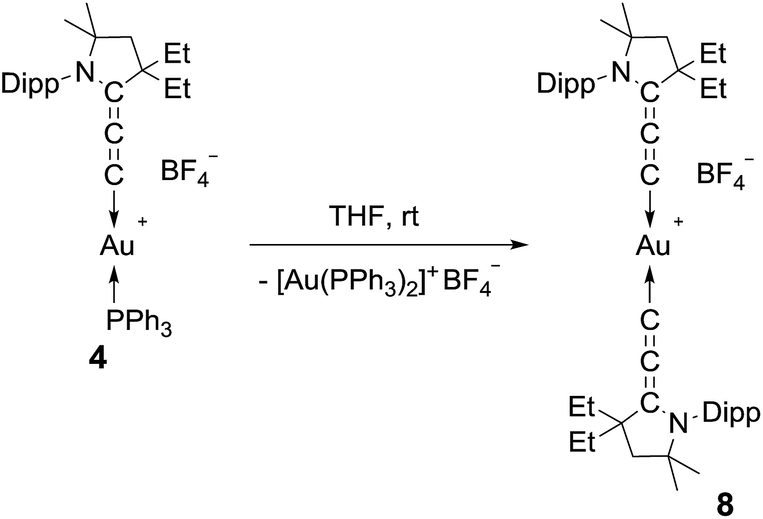 | ||
| Scheme 2 Dismutation of 4 into the homoleptic bis(allenylidene)gold(I) complex 8. | ||
No dismutation was observed with the (allenylidene) (CAAC)gold(I) complex 6a. However, we found that it reacts with one equivalent of (THT)AuCl in the presence of a chloride source to afford (CAAC)AuCl 9 and (allenylidene)AuCl 10 which were isolated in 74 and 82% yields, respectively (Scheme 3). Both compounds were characterized by 1H and 13C NMR spectroscopy, as well as by high-resolution mass spectrometry. By subjecting complex 10 to the same synthetic sequence as for 3, the corresponding gold–acetylide complex 11 was obtained in 76% yield. Subsequent hydride abstraction by DDQ followed by anion exchange led to the bis(allenylidene) gold complex 8 in 76% isolated yield. Single crystals were obtained by slow evaporation of a saturated THF solution, and subjected to an X-ray diffraction study (Fig. 2A). The C1–C2 [1.427(8) Å] and C2–C3 [1.176(8) Å] bond distances are close to standard values for carbon–carbon single and triple bonds, respectively, and therefore the ligands in complex 8 are best described by the resonance form depicted in Fig. 2B.
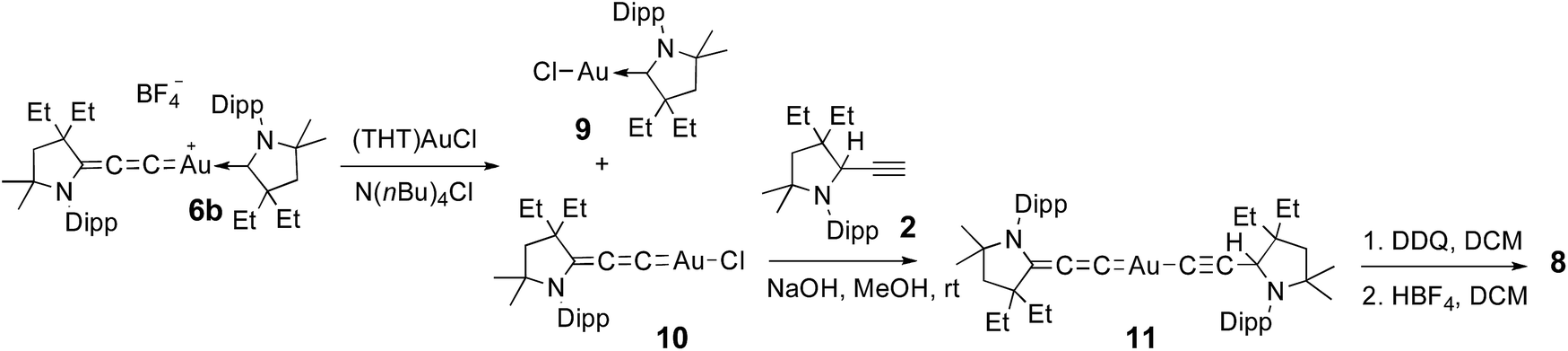 | ||
| Scheme 3 Synthesis of bis(allenylidene)gold(I) complex 8. THT = tetrahydrothiophene. | ||
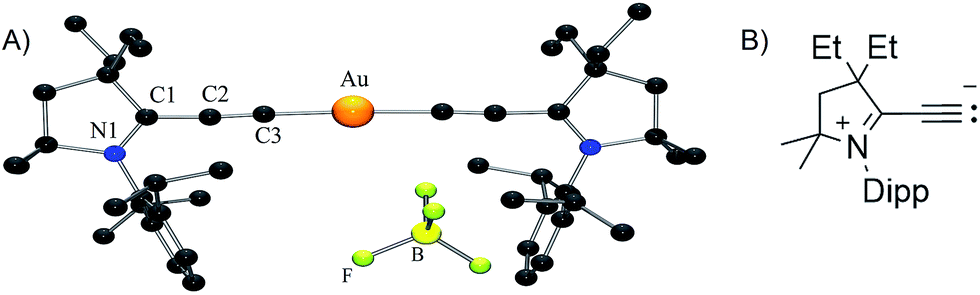 | ||
| Fig. 2 Solid-state structure of complex 8 (H-atoms omitted for clarity). | ||
The cyclic voltammogram of a THF solution of bis(allenylidene)gold(I) complex 8 showed two well defined one-electron reversible reductions at −1.695 and −2.045 V versus Fc+/Fc. The first reduction potential is shifted to an even more positive value than that of the (allenylidene)(CAAC)gold(I) complex 6b, further supporting that the allenylidene behaves as a stronger π-accepting ligand than CAACs. Chemical reduction of 8 with CoCp2* resulted in a dark green solution, which was NMR silent and EPR active, but all attempts to grow single crystals led to decomposition.
Conclusions
Although a variety of cumulene transition metal complexes are known, compounds 6a,b, 7, 8, 10 and 11 are rare examples in the coinage metal series.4–6,19 Because of their π-accepting properties, the allenylidenes, derived from CAACs, proved to be excellent ligands for electron-rich paramagnetic complexes, featuring a formal gold(0) center. The physical properties5 of these novel robust complexes are under active investigation.Experimental section
Synthesis and characterization
Computational methods
Geometry optimizations of 6 and 7 were carried out using the M05-2X method20 (global hybrid functional with 52% HF exchange) with Ahlrichs' def2-SVP basis set.21 The Au relativistic effect was accounted for by the Stuttgart-Dresden ECP.22 This method is referred to as M05-2X/SDD/def2-SVP. NBO analysis was carried out using the NBO 6.0 software.23 The g factor of 7 at its optimized geometry was calculated using the B3LYP (ref. 24) (hybrid functional with 20% HF exchange) functional with the TZVP basis set25 and the zero-order regular approximation (ZORA)26 for the relativistic effect in Au. The method is referred to as ZORA/B3LYP/TZVP. The calculations were performed using the Gaussian 09 (ref. 27) and ORCA 3.0.1 (ref. 28) programs. Full details of the calculations are given in the ESI.†Acknowledgements
We are grateful to the US-Israel Binational Science Foundation (BSF 2012184) and DOE (DE-FG02-13ER16370) for financial support of this work.Notes and references
- (a) E. O. Fischer, H. J. Kalder, A. Frank, F. H. Köhler and G. Huttner, Angew. Chem., Int. Ed. Engl., 1976, 15, 623–624 CrossRef; (b) H. Berke, Angew. Chem., Int. Ed. Engl., 1976, 15, 624–624 CrossRef.
- (a) C. Coletti, A. Marrone and N. Re, Acc. Chem. Res., 2011, 45, 139–149 CrossRef PubMed; (b) P. J. Low, Dalton Trans., 2005, 2821–2824 RSC; (c) C. Bruneau and P. H. Dixneuf, in Metal vinylidenes and allenylidenes in catalysis from reactivity to applications in synthesis, Wiley-VCH: Weinheim, 2008 Search PubMed; (d) J. W. Herndon, Coord. Chem. Rev., 2014, 272, 48–144 CrossRef CAS.
- (a) M. I. Bruce, Chem. Rev., 1998, 98, 2797–2858 CrossRef CAS PubMed; (b) R. Chauvin and Y. Canac, Top. Organomet. Chem., 2010, 30, 1–256 CrossRef.
- M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 4, 1552–1559 RSC.
- X. S. Xiao, W. L. Kwong, X. Guan, C. Yang, W. Lu and C. M. Che, Chem. - Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed.
- M. Asay, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 4796–4799 CrossRef CAS PubMed.
- For reviews on CAACs, see: (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2014, 48, 256–266 CrossRef PubMed.
- For a review, see: C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC.
- (a) G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427–8431 CrossRef CAS PubMed; (b) D. S. Weinberger, N. A. Sk, K. C. Mondal, M. Melaimi, G. Bertrand, A. C. Stuckl, H. W. Roesky, B. Dittrich, S. Demeshko, B. Schwederski, W. Kaim, P. Jerabek and G. Frenking, J. Am. Chem. Soc., 2014, 136, 6235–6238 CrossRef CAS PubMed; (c) P. P. Samuel, K. C. Mondal, H. W. Roesky, M. Hermann, G. Frenking, S. Demeshko, F. Meyer, A. C. Stueckl, J. H. Christian, N. S. Dalal, L. Ungur, L. F. Chibotaru, K. Proepper, A. Meents and B. Dittrich, Angew. Chem., Int. Ed., 2013, 52, 11817–11821 CrossRef CAS PubMed; (d) P. Jerabek, H. W. Roesky, G. Bertrand and G. Frenking, J. Am. Chem. Soc., 2014, 136, 17123–17135 CrossRef CAS PubMed.
- D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold, G. Frenking, P. Jerabek and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 8964–8967 CrossRef CAS PubMed.
- (a) O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS PubMed; (b) R. R. Rodrigues, C. L. Dorsey, C. A. Arceneaux and T. W. Hudnall, Chem. Commun., 2014, 50, 162–164 RSC; (c) A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed.
- For the synthesis of CAACs, see: (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (b) R. Jazzar, R. D. Dewhurst, J. B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899–2902 CrossRef CAS PubMed; (c) R. Jazzar, J. B. Bourg, R. D. Dewhurst, B. Donnadieu and G. Bertrand, J. Org. Chem., 2007, 72, 3492–3499 CrossRef CAS PubMed.
- For an easy access to aza-analogues of CAACs, see: (a) R. Manzano, T. Wurm, F. Rominger and A. S. K. Hashmi, Chem. - Eur. J., 2014, 20, 6844–6848 CrossRef CAS PubMed; (b) A. S. K. Hashmi, D. Riedel, M. Rudolph, F. Rominger and T. Oeser, Chem. - Eur. J., 2012, 18, 3827–3830 CrossRef CAS PubMed; (c) R. Manzano, F. Rominger and A. S. K. Hashmi, Organometallics, 2013, 32, 2199–2203 CrossRef CAS.
- M. Tamm, A. Grzegorzewski, I. Brudgam and H. Hartl, J. Chem. Soc., Dalton Trans., 1998, 3523–3528 RSC.
- S. Rigaut, K. Costuas, D. Touchard, J. Y. Saillard, S. Golhen and P. H. Dixneuf, J. Am. Chem. Soc., 2004, 126, 4072–4073 CrossRef CAS PubMed.
- CCDC 1418631 (6b), 1418632 (7) and 1418633 (8) contain the supplementary crystallographic data for this paper.
- F. Kessler, B. Weibert and H. Fischer, Organometallics, 2010, 29, 5154–5161 CrossRef CAS.
- R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015, 54, 6867–6869 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P. Schwerdtfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker and P. D. W. Boyd, J. Chem. Phys., 1989, 91, 1762–1774 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013, NBO 6.0 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- (a) A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef; (b) A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- D. A. Pantazis, X. Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc.Wallingford CT, 2009 Search PubMed. The full list of authors is given in the ESI†.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1418631–1418633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03654b |
| This journal is © The Royal Society of Chemistry 2016 |