
An overview of the structural, textural and morphological modulations of g-C3N4 towards photocatalytic hydrogen production
Sulagna Patnaik
,
Satyabadi Martha
and
K. M. Parida
*
Centre for Nano Science and Nano Technology, Siksha ‘O’ Anusandhan University, Bhubaneswar-751030, India. E-mail: kulamaniparida@soauniversity.ac.in; satyabadimartha@soauniversity.ac.in; Fax: +91-674-2350642; Tel: +91-674-2351777
First published on 25th April 2016
Abstract
Graphitic carbon nitride (g-C3N4) is gaining more and more importance as a photocatalytic material due to its promising electronic band structure and high thermal and chemical stability. Very recently, a variety of nanostructured g-C3N4 photocatalysts with varying shapes, sizes, morphologies and electronic band structures have been reported for application in photocatalytic research. This critical review represents an extensive overview of the synthesis of a variety of g-C3N4 nanostructured materials with a controllable structure, morphology and surface modification for superior electronic properties. This article highlights the design of efficient photocatalysts for the splitting of water into hydrogen gas using solar energy. Finally, in the summary and outlook, this article highlights the ongoing challenges and opportunities of g-C3N4. It is also hoped that this review will stimulate further investigation and will open up new possibilities to develop new hybrid g-C3N4 materials with new and exciting applications.
 Sulagna Patnaik | Sulagna Patnaik completed her master's degree at the Regional Engineering College, Rourkela, in 1989 in chemistry. Then, she became a lecturer in chemistry at Rayagada Autonomous College, Odisha, in 1991. She is presently working at Nimapara Autonomous College and pursuing research work at the Centre for Nanoscience and Nanotechnology, SOA University, Bhubaneswar. She has published one research article in an international journal. Her research work is focused on the development of modified C3N4-based photocatalysts for hydrogen energy production and pollution abatement. |
 Satyabadi Martha | Dr Satyabadi Martha completed his Master of Science (M.Sc.) in 2008 from Utkal University, Bhubaneswar. Then, he completed his PhD degree from North Orissa University under the guidance of Prof. Kulamani Parida. His research work is focused on the development of visible light responsive photocatalysts for hydrogen production, CO2 reduction and pollution abatement. He also spent 1 year at the School of Energy and Chemical Engineering, UNIST, South Korea, as a postdoc researcher with Prof. Jae Sung Lee. He has published 26 research articles in international journals and three national patents and presented his works in several national and international symposiums and conferences. Currently, he is working as assistant professor at the Centre for Nanoscience and Nanotechnology, ITER, SOA University. Currently, his research work is focused on powder- and film-based semiconductor material for solar fuel production. |
 K. M. Parida | Prof. K. M. Parida is currently working as professor in Chemistry and as Director at the Centre for Nanoscience and Nanotechnology, ITER, Siksha ‘O’ Anusandhan University, Bhubaneswar. Before coming to ITER, he worked as a Chief Scientist and head of Colloids and Materials Chemistry Department at the CSIR-Institute of Minerals and Materials Technology, Bhubaneswar, Odisha, India, and Professor at the Academy of Scientific and Innovative Research (AcSIR), New Delhi, India. He is the author of more than 290 international journals and 18 national and international patents as well as three book chapters. His research interest is focused on the design and development of a wide cross section of materials, such as metal oxides, metal phosphates, metal sulfates, cationic and anionic clays, perovskites, zeolites, graphene, C3N4 and nano-metal/metal oxide/complex promoted mesoporous materials and naturally occurring materials, such as manganese nodules, manganese nodules leach residue and manganese oxides of natural origin for energy and environmental applications. |
1. Introduction
Photocatalytic water splitting for the production of H2 energy has become more important in recent decades. The hydrogen evolved from this process is green, environmental friendly and a carbon neutral fuel.1–3 Since the report of Fujishima and Honda about photoelectrochemical hydrogen evolution, many photocatalytic and photoelectrochemical systems have been developed. However, their practical applications are still hindered because of the relatively high cost of photocatalytic H2 production methods. Therefore, the development of various sustainable semiconducting materials, especially those consisting of earth-abundant elements, has attracted growing attention. Among these, recently developed polymeric g-C3N4 is a novel metal-free visible-light-induced organic semiconductor photocatalyst and is considered especially attractive to researchers. It offers a promising high performance due to its hardness, light weight, preparation from easily available starting material, excellent stability at ambient conditions and as it has suitable band gap energy of 2.7 eV and can absorb blue light up to 450 nm.1 This novel nanostructured material was found to have many potential applications, such as energy conversion, the purification of contaminated water, good performance in the photo-oxidation of dyes,1 as a base metal-free catalyst for NO decomposition,2 as a reference material in differentiating oxygen activation sites for oxidation reactions, as a functional material to synthesise nanosized metal particles and as a stable photocatalyst for H2 evolution from water under visible light irradiation and also in fuel cells.3–7This polymeric derivative carbon nitride (C3N4) was first synthesised by Berzelius, and then named by Liebig in 1834![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 as melon, which is regarded as one of the oldest structures of synthetic polymers. The structure of this compound was first described by Franklin in 1922.9 C3N4 exists in five different allotropic forms: α-C3N4, β-C3N4, graphitic-C3N4, cubic C3N4, and pseudo-cubic-C3N4. g-C3N4 possesses a stacked 2-dimensional (2D) graphite-like planar structure, with N-heteroatoms substituted in the graphite framework containing π-conjugated systems maintaining a distance of 0.326 nm between two layers. g-C3N4 is made up of only carbon and nitrogen and is stable in both acidic and basic media due to the presence of strong covalent bonds between the carbon and nitrogen atoms. Bonding consists of a N-atom (N1) in a planar sp2 configuration and a 3-coordinated C-atom, which is sp2 hybridised and is bonded to 2-coordinated N-atom (N2) in a 1,3,5-triazine ring. Both the nitrogen atoms (labelled as N1 and N2) are sp2 hybridised (Fig. 1). In the molecular crystal, strong covalent bonds exist, but inside the molecular building blocks, there exists weak interactions, such as hydrogen bonding and van der Waals forces.10 As a semiconductor material with a suitable bandgap, g-C3N4 absorbs visible light to cause photoexcitation, leading to a spatial charge separation between the electron in the CB and hole in the VB, which can take part in subsequent redox reactions with the surface adsorbed molecules to yield the ultimate products. The N-atoms are the preferred oxidation sites and the C-atoms provide the reduction sites.
8 as melon, which is regarded as one of the oldest structures of synthetic polymers. The structure of this compound was first described by Franklin in 1922.9 C3N4 exists in five different allotropic forms: α-C3N4, β-C3N4, graphitic-C3N4, cubic C3N4, and pseudo-cubic-C3N4. g-C3N4 possesses a stacked 2-dimensional (2D) graphite-like planar structure, with N-heteroatoms substituted in the graphite framework containing π-conjugated systems maintaining a distance of 0.326 nm between two layers. g-C3N4 is made up of only carbon and nitrogen and is stable in both acidic and basic media due to the presence of strong covalent bonds between the carbon and nitrogen atoms. Bonding consists of a N-atom (N1) in a planar sp2 configuration and a 3-coordinated C-atom, which is sp2 hybridised and is bonded to 2-coordinated N-atom (N2) in a 1,3,5-triazine ring. Both the nitrogen atoms (labelled as N1 and N2) are sp2 hybridised (Fig. 1). In the molecular crystal, strong covalent bonds exist, but inside the molecular building blocks, there exists weak interactions, such as hydrogen bonding and van der Waals forces.10 As a semiconductor material with a suitable bandgap, g-C3N4 absorbs visible light to cause photoexcitation, leading to a spatial charge separation between the electron in the CB and hole in the VB, which can take part in subsequent redox reactions with the surface adsorbed molecules to yield the ultimate products. The N-atoms are the preferred oxidation sites and the C-atoms provide the reduction sites.
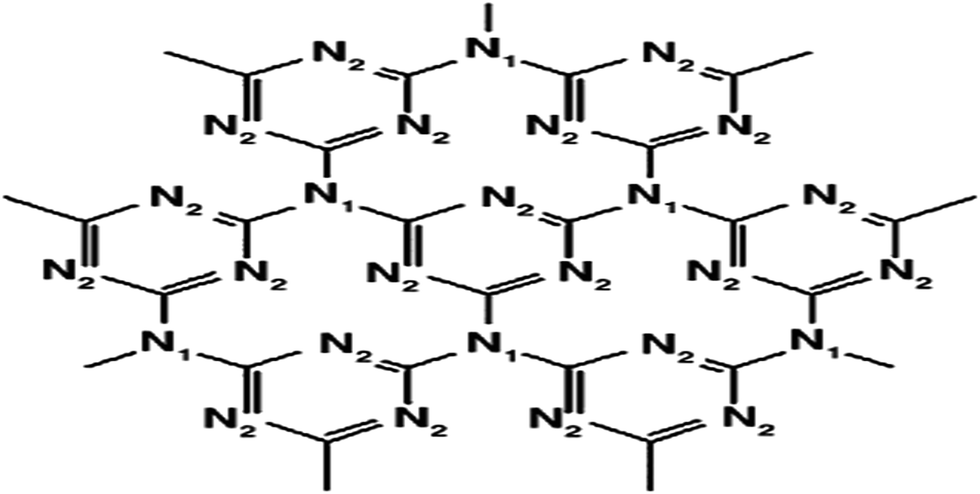 | ||
| Fig. 1 Structure of planar g-C3N4. Nitrogen occupies two positions in the layer, labelled N1 and N2 (reproduced from ref. 8). | ||
Due to the different degree of condensation, the polymerized structure of g-C3N4 develops optimization in its packing, which makes the material behave as a multifunctional catalyst. The presence of a π-conjugated system, as shown in Fig. 2, modifies its bulk electronic structure and surface properties to allow it to exibit:11
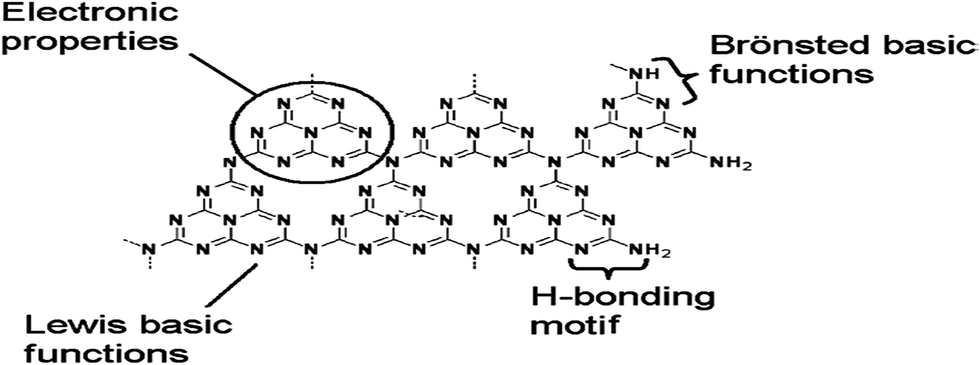 | ||
| Fig. 2 Structure of planar g-C3N4 (reproduced from ref. 11). | ||
• Electronic properties;
• Nucleophilic properties;
• The ability to form H-bonds;
• Photocatalytic activity.
Through its special electronic properties, it can activate the Friedel–Craft reaction, Diels–Alder reactions and the timerisation of alkynes.12 Due to its nucleophillic properties, it helps in the activation of CO2.13 Its ability to form H-bonds favours the trimerization of nitrides.
2. g-C3N4 as a photocatalyst
g-C3N4 has been identified as an efficient visible-light-active polymeric semiconductor photocatalyst for photochemical reactions due to its unique electronic band structure for both water reduction and oxidation. When g-C3N4 receives photon energy ≥ Eg (bandgap = 2.7 eV) from a light source, electrons are excited from the valence band (VB) to the conduction band (CB), leaving a corresponding number of holes in the valence band. It is said to be in its photoexcited state when this results in electron (e−)–hole (h+) pairs. These excited charge carriers (electrons and holes) act as highly reactive species with robust reducing and oxidizing capacities.10,11 They may recombine or get trapped in metastable surface states or can react with suitable electron acceptors/donors adsorbed on the surface of the catalyst. In the case of common photocatalysts in the bandgap region, no energy levels are available. This allows a delay in the recombination of electron–hole pairs because of the void energy region (bandgap). Efficient electron–hole separation and fast charge transport restricts the bulk and surface charge recombination, which is essentially required for the photogenerated charge carries to migrate to the surface reaction sites. Due to the migration of the photoexcited electrons and holes to the active sites on the surface, they can split water into hydrogen and oxygen, depending on the sacrificial agent used. The surface properties of g-C3N4 intrinsically favour the separation and transfer of charge carriers by generating surface states where electrons and holes are spatially trapped and transferred for subsequent redox reactions, as shown in Scheme 1.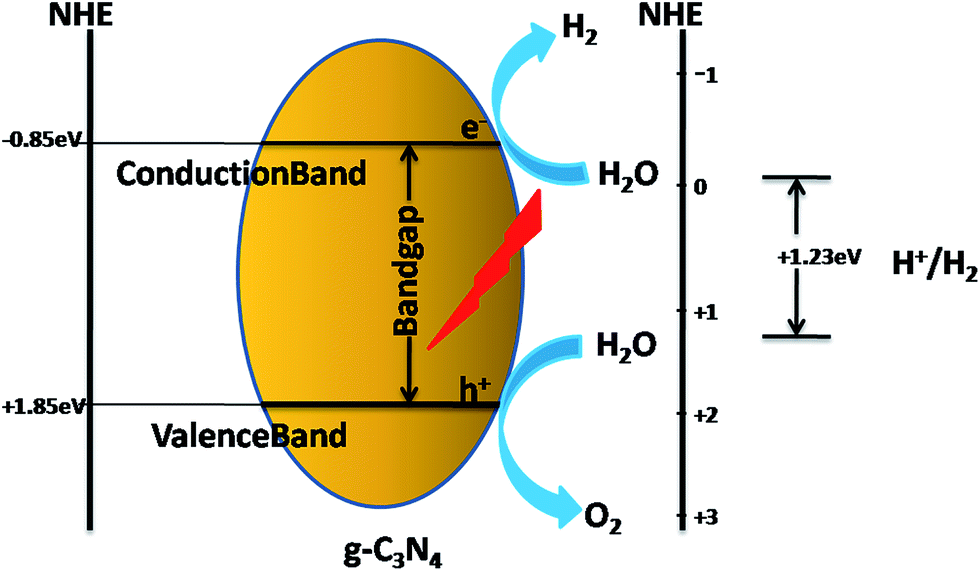 | ||
| Scheme 1 Charge transfer mechanism of neat g-C3N4 as a photocatalyst. | ||
2.1. Disadvantages of g-C3N4 as a photocatalyst14–17
Various theoretical techniques such as molecular dynamics methods, first principle pseudopotentials, routines based on Hartree–Fock and local density approximations, etc. have been applied to study different forms of carbon nitrides. g-C3N4 is the most stable allotrope among various carbon nitride compounds and has attracted much attention due to its potential application in splitting water and decomposing organic pollutants under visible light. However, its photocatalytic activity is non-satisfactory due to:18–21(1) Its low quantum efficiency, due to fast recombination of the photogenerated the electron–hole pairs;
(2) Pure g-C3N4 can absorb only blue light in the solar spectrum (450 nm), which limits the utilization of a broad spectrum of solar light;
(3) During synthesis, the high degree of condensation of the monomers renders the materials with a low surface area (∼10 m2 g−1) and without forming textured pores;
(4) The grain boundary effect, which disrupts the delocalization of electrons from the surface of a photocatalyst through the interface.
2.2. Steps to overcome the limitations
To take better advantage of g-C3N4, it is important to optimize the material to explore more efficient photocatalytic reactivity. The design of high-performance g-C3N4 is highly dependent on the size, morphology, surface area, abundant surface active sites and even extended light harvesting capacity. Some of the important strategies to improve the photocatalytic hydrogen production of g-C3N4 are via:(i) Preparation of mesoporous and ordered mesoporous g-C3N4;22–38
(ii) Preparing texturally and morphologically controlled g-C3N4;39–45
(iii) Doping with non-metals, such as B, S, P, F, I, etc.;46–58
(iv) Loading co-catalysts (especially noble metals, such as Ag, Pt, Au, etc.);59–79
(v) Preparing heterojunction/composites with transitional metal-/metal oxide-based materials.80–96
In order to absorb light effectively in the whole visible spectrum, the bandgap of the photocatalyst should correspond well to the wavelength of the irradiated light. Here, in this review, we discuss some of the methods, giving special emphasis to heteroatom doping, loading a co-catalyst, designing composites/heterojunctions, etc., which can effectively alter the electronic band structure as well as the redox potential of g-C3N4 in order to extend the light absorption and to promote photocatalytic hydrogen generation in the visible region.
Heteroatom doping mainly involves the incorporation of atoms or ions into a crystalline lattice, and modifies the bulk structure of crystallites. Doping with non-metals reduces the energy gap to enhance the visible light absorption of g-C3N4. The increased dispersion of the contour distribution of the HOMO and LUMO brought about by doping facilitates the charge carrier mobility and favours the charge separation. The wider the VB, the higher the mobility of the generated holes, and this thus improves the photo-oxidation efficiency of the holes. To increase the VB width by non-metal doping, the dopant atom must have a lower electronegativity than the substituted atom and there must be a homogeneous distribution of the dopant. The mechanism behind the enhanced activity is explained in the photocatalytic Scheme 2, taking the example of S-doped g-C3N4. Liu et al.55 reported S-doped mpg-C3N4 with a unique electronic structure that showed an increased VB width along with an elevated CB minimum and a slightly reduced absorbance, and its photoreactivity for H2 evolution increased 7.2 to 8.0 fold. Whereas, Hong et al.56 reported an upliftment of VB and a decrease in the bandgap to 2.61 eV by in situ S-doping for increased activity (H2 evolution increased 30-fold).
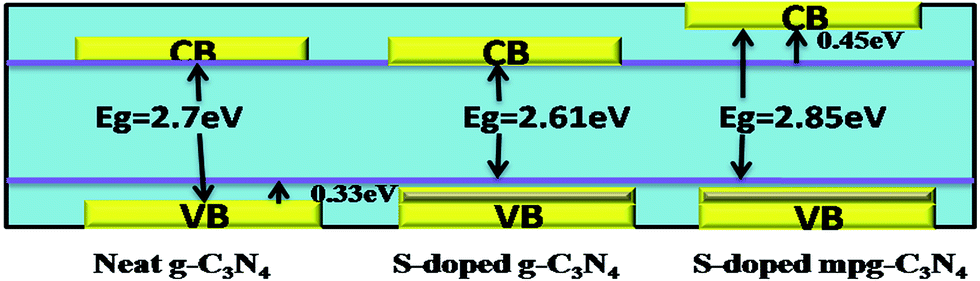 | ||
| Scheme 2 Upliftment of the band edge in S-doped g-C3N4. | ||
Semiconductor hybridization and the creation of heterojunctions/composites with an appropriate semiconducting material is another effective strategy to broaden the utilization of g-C3N4 for visible light photocatalytic hydrogen generation. This is based on the band alignment between g-C3N4 and the other semiconductors to achieve a better efficiency for photogenerated charge separation and to promote photocatalytic activity. This promotion effect was explained by creating heterojunctions by using three different mechanisms, as described below.
(1) Though sensitization, in which electrons generated by visible light irradiation in g-C3N4 migrate to a wider bandgap semiconductor with a VB and CB at a higher potential (such as in ZnO, TiO2, etc., which is explained further in the later part of this review), while the photogenerated electrons from the CB of g-C3N4 are transferred to the CB of wider bandgap semiconductors, as shown in Scheme 3, and help in photoreduction and the holes are transferred to the CB of g-C3N4, thus resulting in effective charge separation.
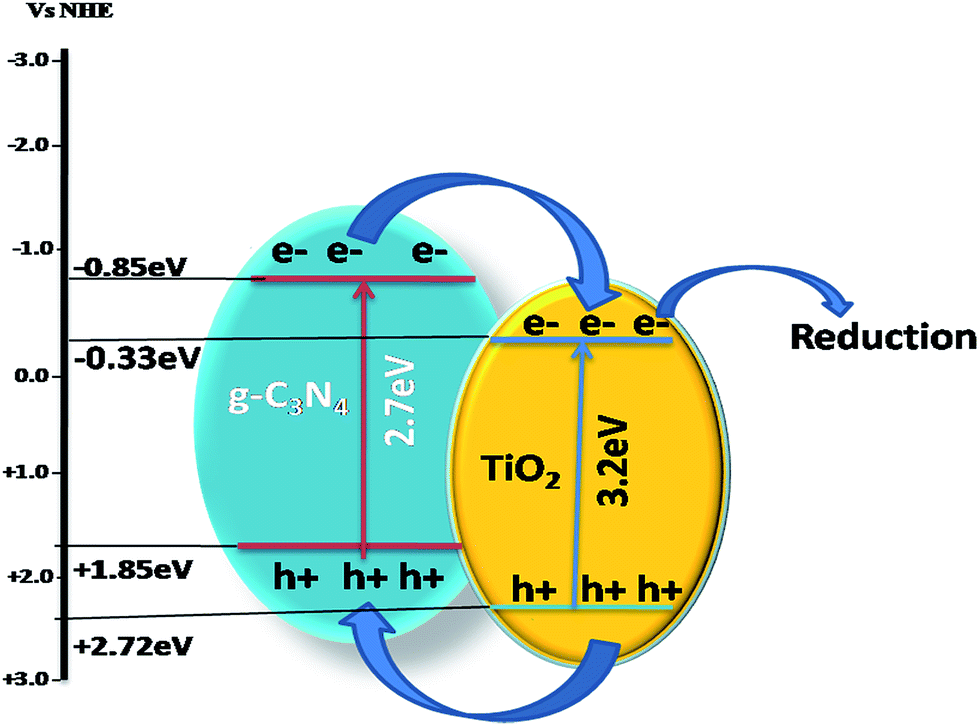 | ||
| Scheme 3 Charge transfer mechanism in a heterojunction of g-C3N4 with a wider bandgap semiconductor with both VB and CB at a higher potential. | ||
(2) Via a heterojunction/composite photocatalyst, in which both semiconductors are visible light active and can be excited to form electron–hole pairs. The photogenerated electrons migrate from Cu2O with a higher CB to another semiconductor with a lower CB, while the photogenerated holes migrate from g-C3N4 with a lower VB to another semiconductor (Cu2O) with a higher VB, as shown in Scheme 4. The double charge transfer may also lead to the separation of electrons and holes and enhance the photocatalytic efficiency.
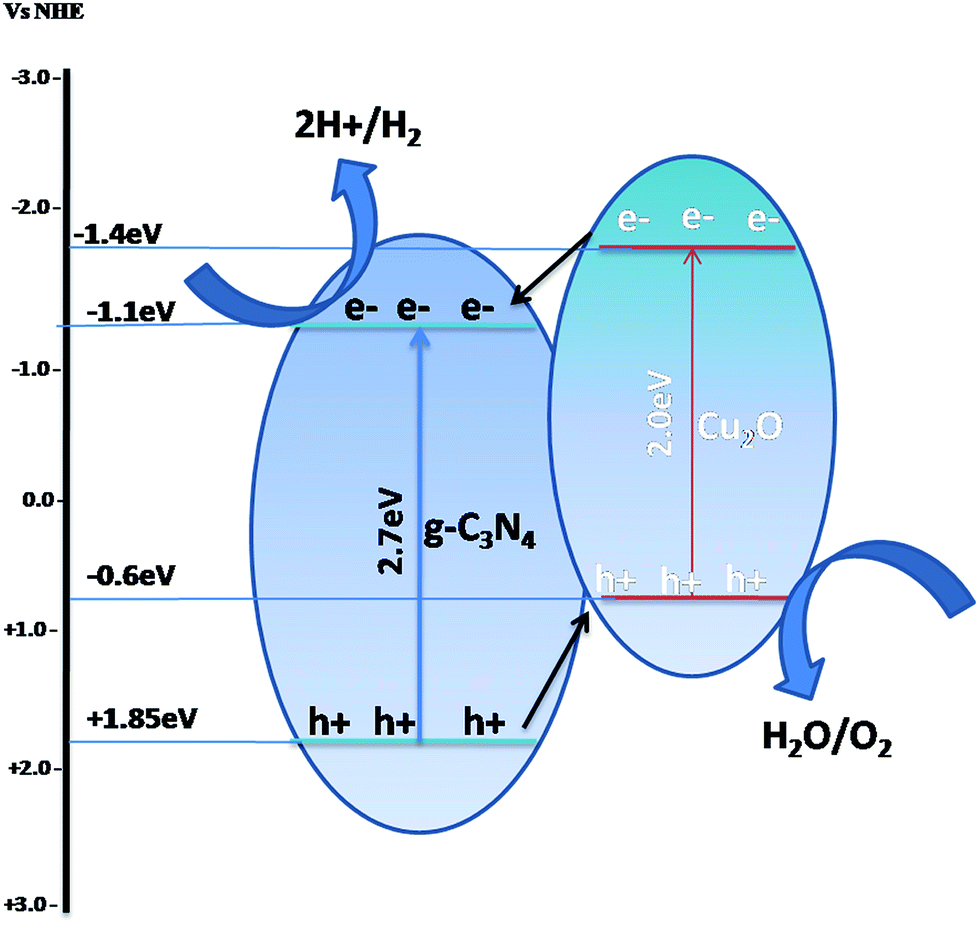 | ||
| Scheme 4 Charge transfer mechanism in a heterojunction of g-C3N4 with narrow-bandgap semiconductors with a VB and CB at lower potentials. | ||
(3) Through a direct Z-scheme type mechanism, in which the photogenerated electrons migrate in a Z-scheme route and greatly affect the band edge potentials of the semiconductors. The photogenerated electrons from a semiconductor with a lower CB recombine with photogenerated holes from the g-C3N4 with a higher VB. Meanwhile, the photogenerated electrons with strong reducibility were left on the semiconductor with higher CB, and the holes with strong oxidizability were left on the semiconductor with a lower VB, as shown in Scheme 5. These photogenerated electrons and holes could participate in the photocatalytic process.
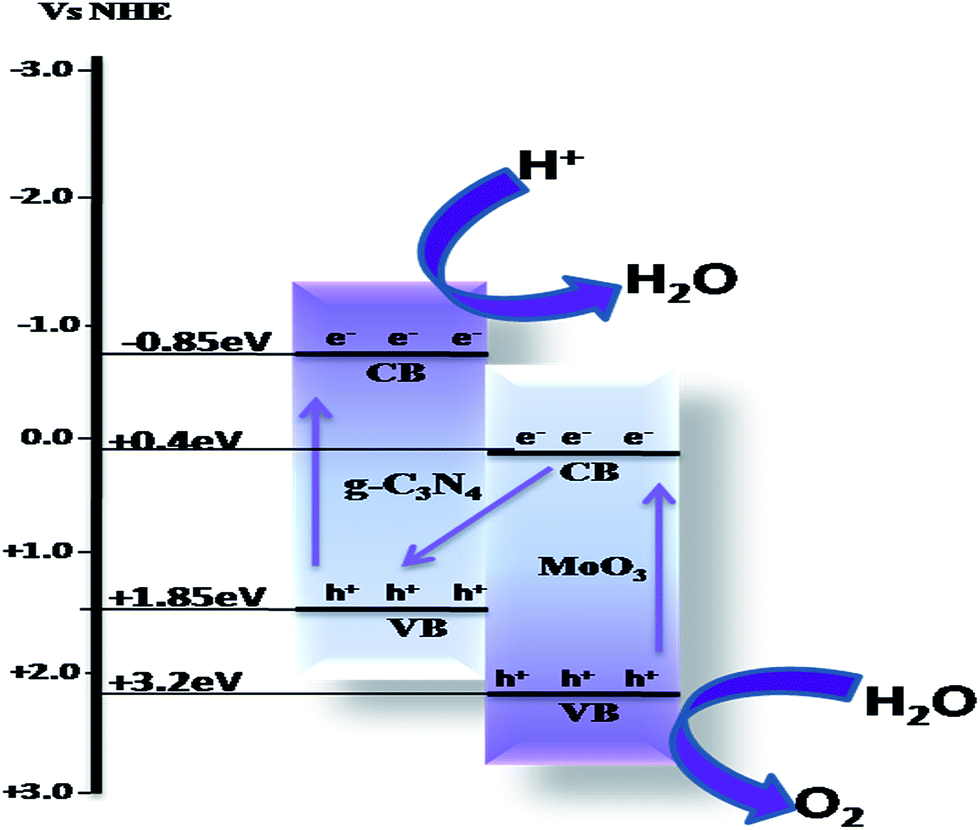 | ||
| Scheme 5 Photocatalytic charge transfer by a Z-scheme mechanism. | ||
Although all the three mechanisms have been widely used by different groups of scientists to explain their works, the band edge potential of the semiconductor is generally considered as the key factor to choose the mechanism most suitable for the specific research.
g-C3N4/co-catalyst hybrids with noble metal co-catalysts, such as Pt, Au, Ag, etc., have improved interfacial contact, which helps to capture the conduction-band electrons for photocatalytic hydrogen generation. Nanoparticles of noble metals can strongly absorb visible light due to their surface plasmon resonance, which can be tuned by varying their shape, size and surroundings. When the surface of the photocatalyst is loaded with the noble metals, the photogenerated electrons migrate to the surface of the photocatalyst and are transferred easily to the noble metal co-catalyst, as in Scheme 6, because the Fermi levels of noble metals lie at a lower potential than that of the semiconductor photocatalyst. The performance of g-C3N4 can be significantly improved by the use of co-catalysts, such as Pt, Ru, Rh and Ni, which are often employed as co-catalysts in energy storage processes. In the case of photocatalytic hydrogen evolution, the co-catalysts are usually considered as electron traps, thus delaying the charge recombination process and making electrons and holes readily available for the photocatalytic process and acting as thermal redox active centres. Several other factors, such as the choice of the sacrificial agent, the intensity of the light source, the pH of the solution and the loading procedure also affect the performance of the co-catalyst.
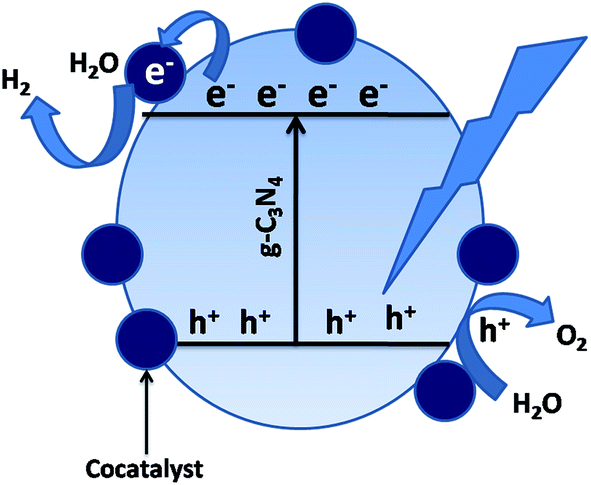 | ||
| Scheme 6 Photocatalytic charge transfer by co-catalyst loading. | ||
3. Synthesis of neat g-C3N4
Different groups of scientist have reported various methods to modify the structure, morphology and texture of neat g-C3N4 material by synthesizing nanopowders, nanosheets, nanowires (or nanorods), nanobelts, nanotubes and nanospheres.3.1. g-C3N4 nanopowder
This material is of great interest because of the fact that it can be synthesized from easily available and cost-effective starting materials, such as cyanamide, dicyandiamide, melamine, urea, cyanurchloride, 2-amino-4,6-dichlorotriazine, etc., by a facile pyrolysis method.12,13 The graphitic-C3N4 synthesized from various amine precursors develops different degrees of photocatalytic efficiency. Zhang et al.22 prepared g-C3N4 by heating cyanamide, dicyanamide and melamina in an alumina crucible with a cover to maintain a semi-closed atmosphere to prevent sublimation. The obtained yellow coloured polymer was g-C3N4 nanopowder. The optical and electronic properties of g-C3N4 are dependent on the C/N ratio and the degree of condensation. The degree of condensation is also dependent on the temperature and starting material. In the case of melamine, the morphology of the products was changed from spherical nanoparticles into layered carbon nitride and graphitic-C3N4 with the increase in temperature. The products were maintained in the crystalline melamine phase when the heating temperature was lower than 300 °C.22 When the temperature increased gradually, it was transformed into crystalline or amorphous graphite-like C3N4 compounds, and the morphology of the product also changed (Table 1).| Temperature | Morphology | Particle size | Structure |
|---|---|---|---|
| 300 °C | Granular | 50–80 nm | Crystalline |
| 350–400 °C | Layered | 0.5–0.8 mμ | Crystalline or amorphous |
| 500 °C | Sheet-like | 0.5–1.0 mμ | Completely amorphous |
| 600 °C | Flakes | 0.5–1.5 mμ | Amorphous |
The major limitation in the synthesis of g-C3N4 is its easy sublimation at elevated temperatures. Its easy sublimation can be suppressed to a large extent if the melamine co-exists with others species where H-bridges are present. The synthesis of g-C3N4 from melamine mainly involves a combination of a polyaddition and polycondensation. Polycondensation proceeds via the elimination of NH3 molecules (Fig. 3).23 During polymerization, melamine first forms a C3N4 sheet structure based on triazine units, which on subsequent polymerization forms a C6H8 sheet structure based on tri-s-triazine units.
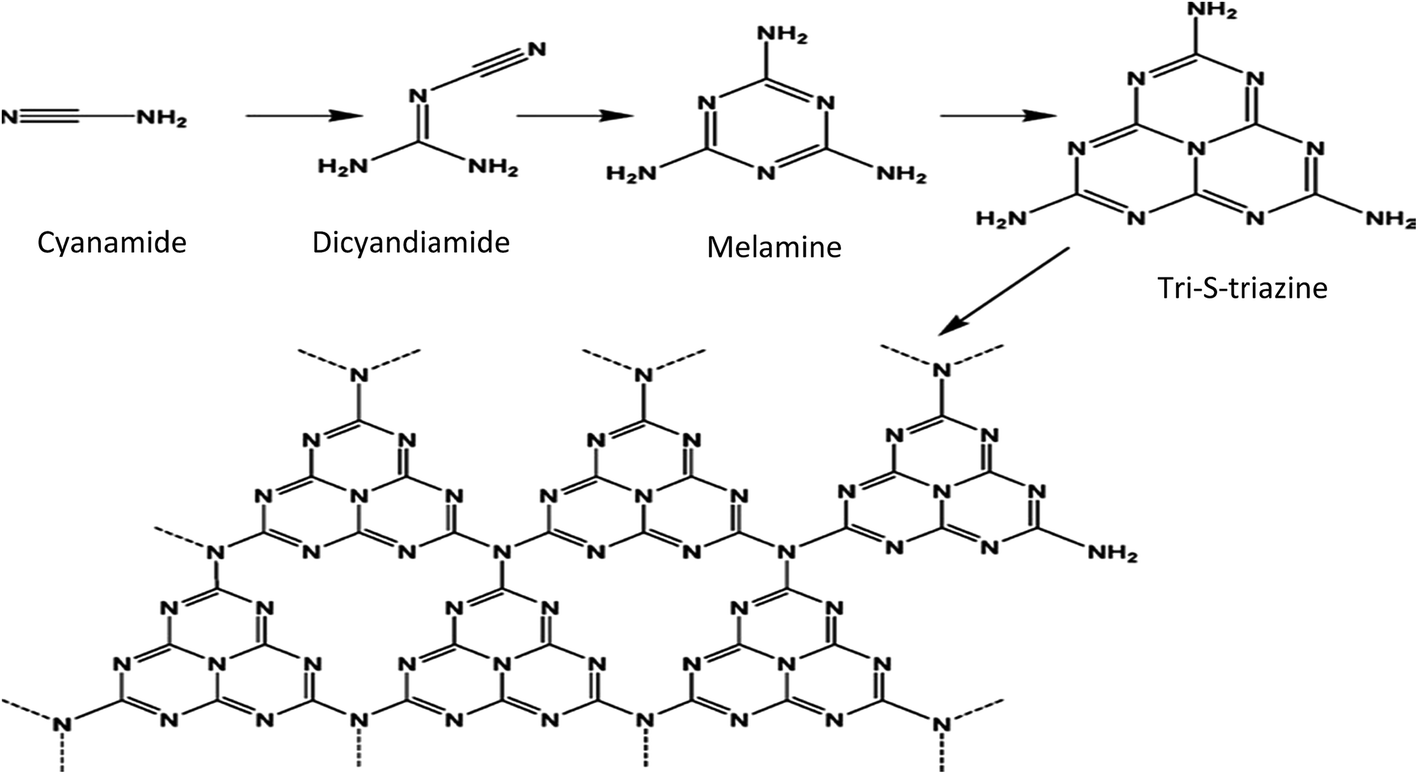 | ||
| Fig. 3 Mechanism of the synthesis of g-C3N4 (reproduced from ref. 11). | ||
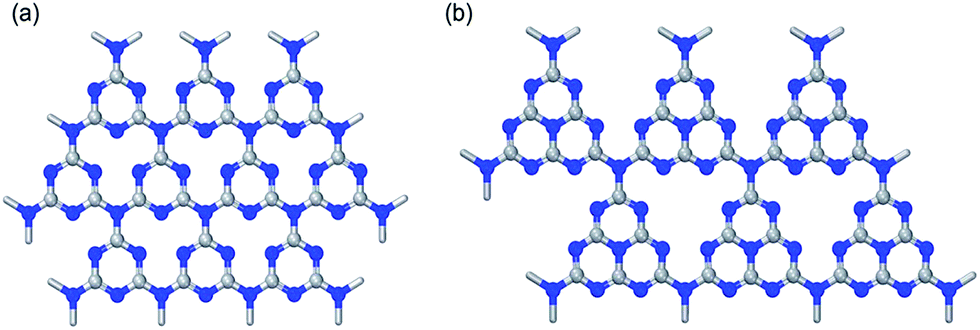 | ||
| Fig. 4 Representation of (a) triazine and (b) tri-s-triazine (reproduced from ref. 27). | ||
3.2. g-C3N4 mesoporous
Nanomaterials should have a large specific surface area as the most important requirement to be effective in photocatalytic applications. It has been found that an additional important feature of nanomaterials for photocatalytic applications is their porous morphology.28–38 Recently, various nanostructured g-C3N4 materials with a mesoporous and highly ordered porous network with a large specific surface area and optimum pore size have been developed. g-C3N4 nanopowder obtained by the thermal polycondensation of various nitrogen-rich starting materials results in bulk materials with a low surface area (∼10 m2 g−1). Low surface area has become one of the main disadvantage hindering the wide potential application of this new emerging material. This is because surface area plays an important role in photochemical reactions by offering more active sites. Porous materials have drawn much attention as heterogeneous catalysts for energy conversion due to their porosity and large surface area.26–31 Mesoporous g-C3N4 shows unique semiconductor properties, along with a large surface area and accessible crystalline pore walls, for mass transfer as a promising metal-free photocatalyst. As compared to the bulk g-C3N4, mpg-C3N4 material exhibits a higher specific surface area of up to 830 m2 g−1 and a larger porosity of up to 1.25 cm2 g−1.32 The most important pathways for the preparation of meso-g-C3N4 are based on templating methods, such as soft templating (self-assembly) and hard templating (nanocasting) methods. In the soft templating method, the porosity is introduced by the co-operative assembly often being carried out under hydrothermal conditions. The most commonly used soft templates are Triton-X-100, P123, F127, Brij30, Brij58 and Brij76, to obtain different pore structures and specific surface areas.33 The preparation of self-porous materials by soft templates is still very difficult. In the case of g-C3N4, the practical problem is that the condensation to form polymeric CN-structures takes place around or above the decomposition temperature of the commonly used soft templates. However, by using primary nanopores in the silica template,34,35 nanostructured g-C3N4 in the form of nanowires/nanospheres can be replicated to form stable replica arrays. After removal of the template, the desired nanoporous g-C3N4 is obtained, which is an inverse replica of the silica template. Groenewolt et al.38 prepared mpg-C3N4 nanoparticles using mesoporous silica as the hard template. The most commonly used hard templates are silica nanoparticles, 2D hexagonal SBA-15 and 3D cubic KIT-6, IBN-4, etc. Mesoporous-C3N4, when synthesized by using silica nanoparticles as the hard template, has an increased specific surface area but there is a decrease in crystallinity, which acts against effective excited charge separation. Therefore, enhancement of the crystallinity of the g-C3N4 polymer and, at the same time, the surface area is still a challenge. Also, the silica nanoparticles can be removed by using aqueous NH4HF2 or HF, which is hazardous and not environmentally friendly. Synthesizing ordered mesoporous g-C3N4 results in a large external surface and improves the activity of the photocatalyst.31 By using ethylenediamine and CCl4 as the precursor and SBA-15 as a template, ordered mesoporous carbon nitride can be prepared. The ompg-C3N4 maintains the rod-like morphology of the SBA-15 template. Also, the surface area increases to 239 m2 g−1.32,33 Recently various template-free methods have been explored to overcome the difficulties in the removal of the template.3.3. g-C3N4 nanorods
Most researchers found that g-C3N4 has a nanoporous or nanosphere morphology. It has been reported that the synthesis of g-C3N4 nanorods involves the transformation process of g-C3N4 from nanoplates to nanorods. The dimensionality and size of the material have been regarded as critical factors to enhance the photocatalytic activity. g-C3N4 nanorods can be synthesized with well-controlled dimensionality by removing the surface defects and producing more active lattice faces to enhance the photocatalytic activity significantly. Bai et al.39 prepared g-C3N4 nanorods by a simple reflux method, which increased the number of active lattice faces by eliminating surface defects. The morphology and the crystal growth of the products were controlled by changing the reaction time and reactant solvent concentration.3.4. g-C3N4 nanosheets
g-C3N4 nanosheets are obtained by the exfoliation of bulk g-C3N4. For the synthesis of nanosheets, bulk g-C3N4 was suspended in different types of solvents, such as IPA, N-methyl pyrrolidone (NMP), water, ethanol and acetone. Then, the dispersion was subjected to sonication at room temperature. There followed a gradual exfoliation of the material to form g-C3N4 nanosheets with the increase in sonication time. Yang et al.40 exfoliated g-C3N4 by using isopropanol (IPA) to form g-C3N4 nanosheets (Fig. 5) of ∼2 nm thickness. However, the use of inorganic solvents or water for the sonochemical exfoliation method suffers from a long ultrasonic treatment required and results in a low exfoliation efficiency. Cheng et al.41 reported the formation of a stable colloid of g-C3N4 nanosheets of ∼9 nm thickness on a large scale following a H2SO4 exfoliation route.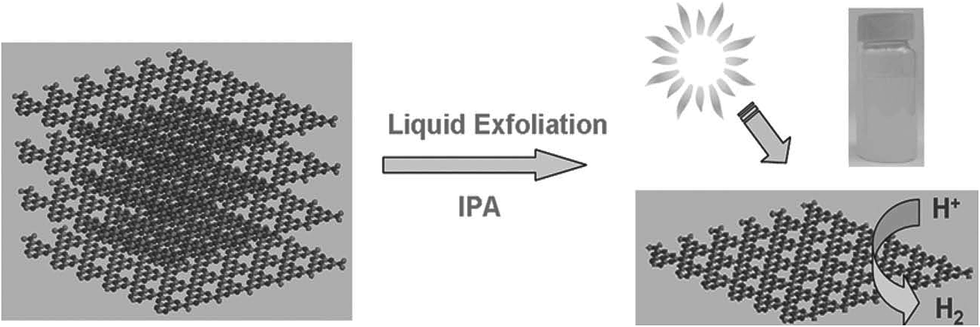 | ||
| Fig. 5 Fabrication of nanosheets of g-C3N4 by the liquid exfoliation method from g-C3N4 powder (reproduced from ref. 40, licence number 3767461234354). | ||
3.5. Structure-distorted g-C3N4 nanosheets
Chen et al.42 reported the formation of structure-distorted g-C3N4 nanosheets (Fig. 6) by thermal etching, and the structure showed extended optical absorption in the visible region. Due to the distortion, the g-C3N4 nanosheets were formed. These distorted nanosheets reduce the repulsive interactions between the lone pairs of electrons present in the N-atoms. The distortion is possible when the condensation temperature is gradually increased higher than 550 °C (550, 600, 625, 650, 675 and 700 °C) and maintained for 2 h at the final temperature.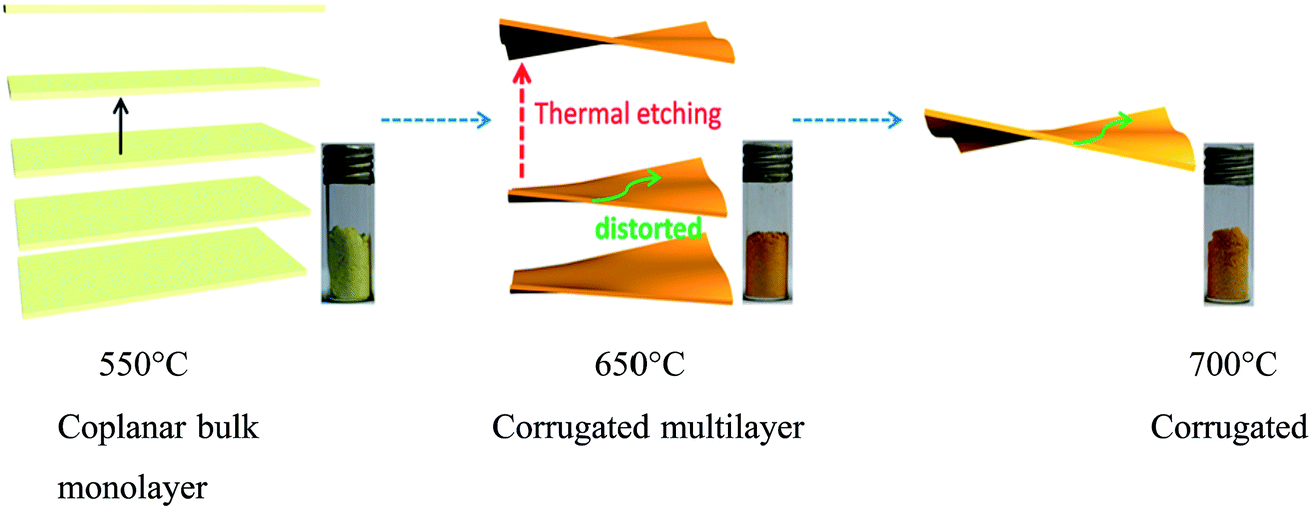 | ||
| Fig. 6 Fabrication structure of distorted carbon nitride nanosheets (reproduced with permission from ref. 42, Copyright American chemical society, 2014). | ||
3.6. g-C3N4 hollow nanospheres
Zheng et al.43,44 designed a hollow carbon nitride nanosphere by grafting an organic group into the structure of g-C3N4 to extend the pi-conjugation and to enhance its photocatalytic activity for hydrogen evolution. During polymerization, ATCN (2-aminothiophene-3-carbonitrile) was added to cyanamide as the precursor. In the product, the thiophene groups are grafted in place of amino groups by nucleophillic reaction. A similar carbon nitride nanospheres network was also obtained by using some other organic compounds, such as barbituric acid and 2-aminobenzonitrile. Zheng et al.,44 designed hollow carbon nitride nanospheres (HCNS) by using silica spheres as the template. After removal of the template by using NH4HF2, a highly stable polymeric semiconductor material was obtained. Bai et al.45 also designed hollow g-C3N4 nanospheres by using silica spheres as the template in a solvothermal technique.3.7. Synthesis of non-metal-doped g-C3N4 (C, B, F, S, P, I, etc.)
Chemical doping is an effective strategy to modify the electronic structure of g-C3N4. Heteroatoms have been used to shift the optical absorption in the visible region by creating localized/delocalized states in the bandgap. Both the composition and the properties of g-C3N4 can be further engineered by introducing some non-metallic elements, such as N, C, B, F, S, P, I, etc.46–58 into the matrix in order to modify the molecular structure.Wang et al.47 synthesized fluorinated polymeric g-C3N4-solids (CNF) by directly incorporating NH4F as the doping reagent during the condensation process of classical g-C3N4. By incorporating different amounts of NH4F, the fluorine concentration in the resulting CNFs can be controlled. Lin et al.48 reported fluorine-doped g-C3N4 by using BimmBF4 and urea. Wang et al.49 synthesized B-doped g-C3N4 (CNB) by using BH3NH3 as a molecular doping source during the condensation process rather using oxygen-containing precursors or the generation of HF. The resulting50,51 CNB materials were found to have a potential oxidation strength. B-Doped g-C3N4 can also be prepared by substituting the H-atom at the terminal of the melon structure with boron, forming C-NB and C-NB2 functional groups. Yan et al.51 synthesized B-doped g-C3N4 by heating a mixture of melamine and boron oxide. Zhang et al.52 reported the synthesis of P-heteroatom doped polymeric g-C3N4 by co-condensation between dicyandiamide and a phosphorous containing ionic liquid. 1-Butyl-3-methylimidazolium hexaflouro phosphate (BmimPF6) is the most common ionic liquid used as a mild P-source for doping polymeric g-C3N4 as they are thermally stable during the polycondensation. With an increase in temperature, PF6− is expected to react with the amine groups to join the C–N framework. The P-atom is found to be homogenously distributed on the surface of the doped g-C3N4 solid. Dong et al.53 synthesized carbon self-doped g-C3N4 by using melamine pre-treated with absolute ethanol as the precursor to provide more carbon, whereas Zhang et al.54 reported the synthesis of carbon self-doped g-C3N4 by the thermal condensation of dicyanamide with barbituric acid. Liu et al.55 synthesized sulphur-doped g-C3N4 by the post-treatment of pristine g-C3N4 at 450 °C in a gaseous H2S atmosphere. Besides this, sulphur can also be doped into the g-C3N4 matrix56 by using sulphur-enriched trithiocyanaric acid as the precursor via a polycondensation process using post-treatment, where the terminal –SH groups act as the leaving group. Hong et al.56 synthesized mesoporous-S–g-C3N4 from thiourea using SiO2 nanoparticles as the hard template. During the co-polymerisation, the C–N network is converted into a S–N bond. Zhang et al.57 synthesized I–g-C3N4 by using dicyandiamine and iodide ion as the carbon nitride source and dopant (a facile in situ method). Wang et al.58 synthesized B- and F-enriched mesoporous g-C3N4 by using a simple soft template, such as ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate (BmimBF4). Due to its special solvent structure, BmimBF4 is an interesting solvent in the synthesis of nanoparticles derived from the ion–ion interaction and tendency to form hydrogen bonding.
3.8. Synthesis of noble-metal-loaded g-C3N4
Although g-C3N4 is a good photocatalyst, its application is limited due to electron–hole recombination. To make it chemically productive, different precious noble metal species, such as Ag,59–64 Pt,65,66 Rh,67–69 Au,70–74 Pd75–79 and RuO2, must be used as co-catalysts to increase the separation of photoinduced charge carriers from the bulk to the surface, in order to enhance the production of H2 gas by the splitting of water. These co-catalysts usually act as electron traps to prevent charge recombination and to enhance the rate of the photocatalytic process.Zhang et al.59 synthesized Ag3PO4–g-C3N4 bulk heterojunctions by mixing g-C3N4, Na2HPO4 and AgNO3 solution following an ion-impregnating method. Jiang et al.60 reported the formation of Ag2S–g-C3N4 composite photocatalysts via a simple precipitation method by using g-C3N4, AgNO3 solution in ethanol and thioacetamide (TAA). Platinum can be loaded on the surface of the catalyst either by the impregnation method or by photodeposition or by reduction. Chen et al.31 reported the synthesis of Pt-doped g-C3N4 by using H2PtCl4 and g-C3N4 as the precursor by a photodeposition technique. Pt catalysts supported on carbon nitride nanotubes were prepared by Zeng et al.65 with a borohydride reduction method. Zhang et al.67 synthesized a Rh–g-C3N4 composite by a photodeposition method using PVP (polyvinyl pyrrolidone), with the Rh nanoparticles in the range of 4–9 nm prepared by the polyol reduction method. To control the particle size, PVP (polyvinyl pyrrolidone) was used as a capping agent. For the preparation of Rh nanoparticles, RhCl3·nH2O salt is preferably used, but to get comparatively bigger nanoparticles Rh(III), acetylacetonate Rh(acac)3 is used in THF (tetrahydrofuran). Then, these nanoparticles are incorporated in g-C3N4 by photodeposition by adding a suspension of Rh nanoparticles in a dropwise manner to a suspension of g-C3N4 in glycerol. Samanta et al.70 prepared Au–g-C3N4 by the deposition–precipitation method by using HAuCl4, urea solution and g-C3N4 powder as the precursor. In another way, Au–g-C3N4 nanocomposites were prepared by Chang et al.71 by a facile citrate-reduction method using HAuCl4, sodium citrate solution and g-C3N4 as the precursor. Chang et al.75 fabricated palladium-modified mesoporous graphitic carbon nitride polymer (Pd/mpg-C3N4) by mixing an equal volume of PdCl2 solution to mpg-C3N4 under vigorous magnetic stirring, followed by the addition of KBH4.
3.9. Synthesis of transitional metal- and metal oxide-promoted g-C3N4
Noble metals, such as Au, Pt, Pd, Rh and Ag, are very effective for trapping the photogenerated electrons, thus helping to improve the separation of charge carriers. However, the high cost of noble metals restricts their practical applications. The optical and electronic properties of g-C3N4 could also be modified by incorporating a transition metal into the network of g-C3N4. Transition metal loading is helpful for modification of the electronic properties of g-C3N4 and can extend its optical absorption in the visible region, which enhances the photocatalytic performance.80–97 Transition metal-based heterojunctions of g-C3N4 can be prepared by mixing transitional metal salts of Fe(III), Cu(II), Zn(II), etc. with the precursor of g-C3N4 by a co-thermal condensation.3.10. Synthesis of composites of g-C3N4 with wide-bandgap semiconductors
Various composites and heterojunctions of g-C3N4 with transitional metal ions and transition metal oxides have been reported with good photocatalytic activity. However, most of the transition metal oxides (with wide-bandgap energy) are only ultraviolet (UV) light active. To make them visible light active (420 nm < λ < 800 nm) with a bandgap less than 3 eV and practically useful for photocatalysis, the wide-bandgap transition metal oxides are tailored towards the visible region by coupling them with graphitic carbon nitride. Herein, we present a systematic overview on the synthesis of some such wide-bandgap transitional metal oxide semiconductors.Sun et al.80 prepared g-C3N4–ZnO composite from melamine and Zn(CH3COO)2 by direct calcinations at 520 °C. In the case of g-C3N4–ZnO composite, the strong interaction is due to the condensation between the amino group of triazine and the surface hydroxyl groups of ZnO to form Zn–N bonds. Zn can also be implanted into the network of g-C3N4 by a post-annealing method by using Zn(NO3)2·6H2O solution. The highly active g-C3N4–TiO2 composite was fabricated by Yan et al.81 by mixing an appropriate amount of g-C3N4 and TiO2 and by applying a simple impregnation method. Yin et al.82 synthesized SnO2–g-C3N4 nanocomposites by an ultrasonic-assisted deposition method by using melamine and SnCl4·5H2O as the precursors at room temperature. It was found that the sphere-like SnO2 nanoparticles with sizes of 2–3 nm were dispersed on the surface of g-C3N4 evenly in the SnO2–g-C3N4 nanocomposites. SnO2–g-C3N4 composite photocatalysts were synthesized by using SnCl4·5H2O solution in methanol by an ultrasonic-assisted deposition method at room temperature. Wang et al.83 synthesized CeO2–g-C3N4 nanocomposite by mixing CeO2 with a suitable amount of g-C3N4 following a simple calcination technique. CeO2 nanocrystals were synthesized by a solid-state decomposition reaction of (NH4)2CeO(NO3)6 at 500 °C, followed by mixing with different amounts of g-C3N4 in the solid state and then calcined to obtain the CeO2–g-C3N4 composite. The CeO2/g-C3N4 (13 wt%) composite represents the most optimal activity.
3.11. Synthesis of composites of g-C3N4 with narrow-bandgap transition metal species semiconductors
Many of the transition metal oxides can realize visible light photocatalysis due to their narrow bandgap. Some such transition metal oxides are discussed below.By coupling N,S-TiO2 with g-C3N4, the bandgap energy of TiO2 decreases, making it visible light active and enhancing its light absorption ability and charge separation efficiency. Pany et al.84 synthesized a N,S-TiO2/g-C3N4 nanocomposite by using TiOSO4·xH2O and thiourea as precursor materials through a cost-effective thermal polymerization method. Fe2O3 was used by Hu et al.85 to prepare an Fe3+-doped g-C3N4 composite photocatalyst, where the Fe3+ ions by coordinating to N-atoms were inserted at the interstitial position of g-C3N4. Chen et al.86 prepared a mesoporous Fe–C3N4 nanocomposite by using SiO2 nanoparticles as a hard template and dicyandiamide as the precursor to which FeCl3 solution was added. Cheng et al.87 recently reported a one-step method to incorporate FeOx into the g-C3N4 network by using ferrocene and urea as the starting materials. To improve the photocatalytic efficiency of g-C3N4, it was also coupled with a well-known p-type semiconducting photocatalytic material Cu2O, with a bandgap energy of 2.0–2.4 eV. The advantage of using Cu2O is its low toxicity, low price and good environmental acceptability. Peng et al.88 prepared a g-C3N4/Cu2O composite by using Cu(CH3COO)2·H2O solution by an alcohol-aqueous-based chemical precipitation method. In another method, a g-C3N4/Cu2O composite was obtained by a hydrothermal method using a mixture of g-C3N4 and Cu(NO3)2. Zhou et al.89 prepared a Cu(OH)2/g-C3N4 composite by using mpg-C3N4, CuCl2 and NaOH aqueous by a facile precipitation method. By the preparation of a Cu(OH)2/g-C3N4 nanocomposite, the absorbed wavelength was estimated to be 470 nm, corresponding to a bandgap of 2.63 eV. He et al.90 prepared a MoO3–g-C3N4 composite from MoO3 and melamine as the precursor by a direct mixing and calcination method; whereas Yuming et al.91 synthesized MoS2/g-C3N4 heterojunction photocatalysts via a simple impregnation and heating method. A WO3–g-C3N4 composite was prepared by Doan et al.92 by mixing crystalline WO3 powder and melamine as the raw materials. WO3 and melamine were mixed in 1:3 ratio and then subjected to calcination. The composite was then synthesized by the simple decomposition of melamine in the presence of WO3 at 500 °C. When the sample was prepared by thermal treatment of the mixture at 500 °C, WO3 was attached by a thin layer of g-C3N4 on the surface to form a g-C3N4–WO3 composite. However, when the samples were treated at 600 °C and 700 °C, instead of the disappearance of g-C3N4, the presence of doping N in WO3 was achieved. An In2O3–g-C3N4 hybrid was synthesized by Cao et al.93 in a simple solvothermal method by mixing In(AO)3 and g-C3N4 in the requisite proportions. In the In2O3–g-C3N4 hybrid, small In2O3 nanocrystals generated by the solvothermal process were uniformly distributed on the surface of g-C3N4. To develop different types of photocatalysts for water splitting, cobalt-based compounds play an important role. It has been reported that the coupling of spinel Co3O4, porous cobalt phosphate and cobalt hydroxide with g-C3N4 significantly enhances the water oxidation rate. Zhang et al.94,95 synthesized Co3O4–g-C3N4 from Co(NO3)·6H2O solution and neat g-C3N4 by an ultra-sonication method while maintaining the pH. Among different transition metal oxides, NiO also acts as an efficient co-catalyst for photocatalytic water splitting. Zhang et al.96 synthesized Ni- and NiO-loaded g-C3N4 by using Ni(NO3)2·6H2O solution by a direct reduction method at 350 °C for 2 h or by oxidation in the presence of air at 200 °C for 1 h.
4. Anatomy of the structural and morphological characterizations of g-C3N4
Various research groups have worked a lot to optimize graphitic-C3N4 by structural, textural and morphological modifications. After the optimisation, different types of instrumental analysis, such as XRD, SEM, and TEM, were used to identify the crystal phase of the materials, the interlayer stacking, size of the nanoparticles and nature of the distribution of the nanoparticles in the composites. It was observed that in almost all the modified forms, g-C3N4 maintains its crystallinity and aromatic motif, showing the two typical characteristic peaks of the g-C3N4 polymer at 13.1° and at 27.4° in the XRD analysis. This indicates that the graphitic-like layered stacking of tri-s-triazine units in g-C3N4 is chemically robust towards textural modification. Zhang et al.22 reported that when the temperature increases gradually, g-C3N4 is gradually transformed into a crystalline or amorphous state as the degree of condensation depends on the starting material as well as on the temperature, which was confirmed by the slight broadening of the peaks. The morphology of the product also changes from spherical nanoparticles to layered g-C3N4 with an increase in temperature. The SEM and XRD analysis of the g-C3N4 obtained by the thermal polycondensation of melamine by varying the temperatures is shown in Fig. 7 and 8, respectively. | ||
| Fig. 7 SEM images of the g-C3N4 synthesized at different temperatures (a) 300 °C, (b) 400 °C, (c) 500 °C and (d) 600 °C (reproduced from ref. 22, licence number 3767470249553). | ||
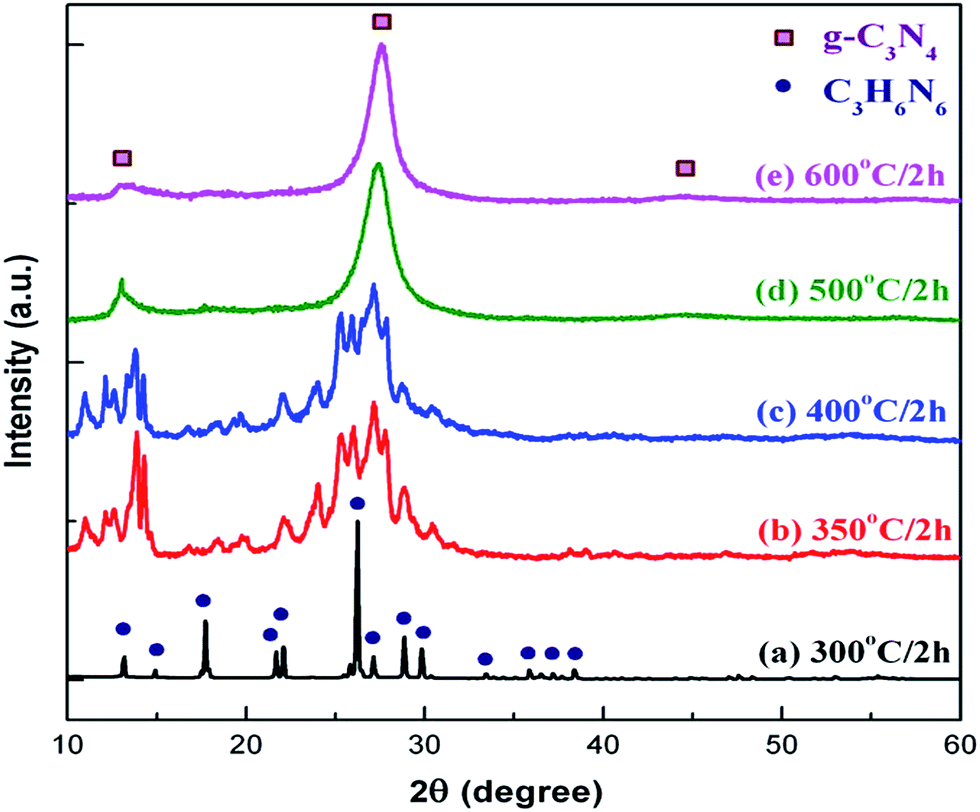 | ||
| Fig. 8 XRD patterns of g-C3N4 synthesized at different temperatures for 2 h (reproduced from ref. 22, licence number 3767470249553). | ||
In the case of a mesoporous structure, there is a gradual decrease of the peak intensity in the XRD analysis with increasing the condensation temperature because of the enlarged surface area and evident decrease in crystallinity due to the presence of mesopores in the polymeric structure. When there were texture changes, there was a considerable broadening of the XRD peaks, with a decreased intensity, thus indicating the size- and shape-dependent properties of the nanomaterials. According to Yang et al.,40 after exfoliation, the intensity of the (002) peak in Fig. 9 is significantly decreased, clearly demonstrating the formation of g-C3N4 nanosheets.
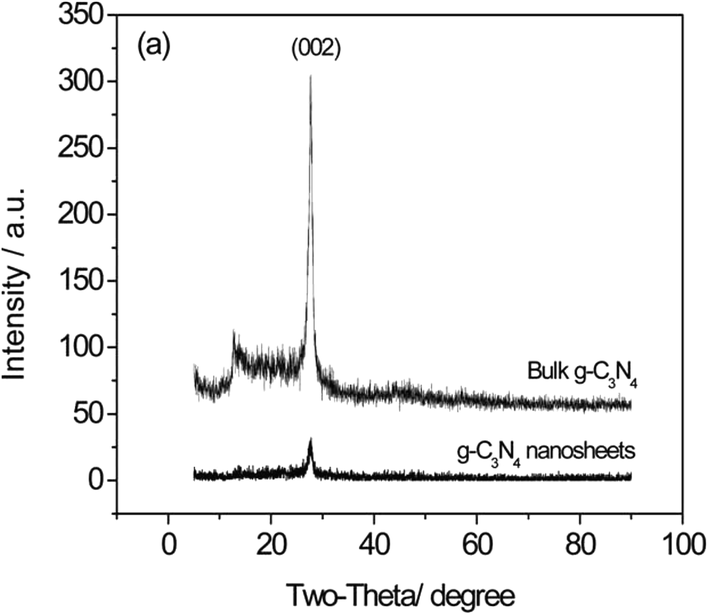 | ||
| Fig. 9 XRD patterns of g-C3N4 nanosheets after exfoliation (reproduced from ref. 40, licence number 3767461234354). | ||
The surface morphology of different nanostructured g-C3N4 were characterized by SEM and TEM images. The surface study confirmed the nanoarchitecture and geometrical features of various textural modifications. The TEM image in Fig. 10 reveals the textural information about the nanoarchitecture of the material.
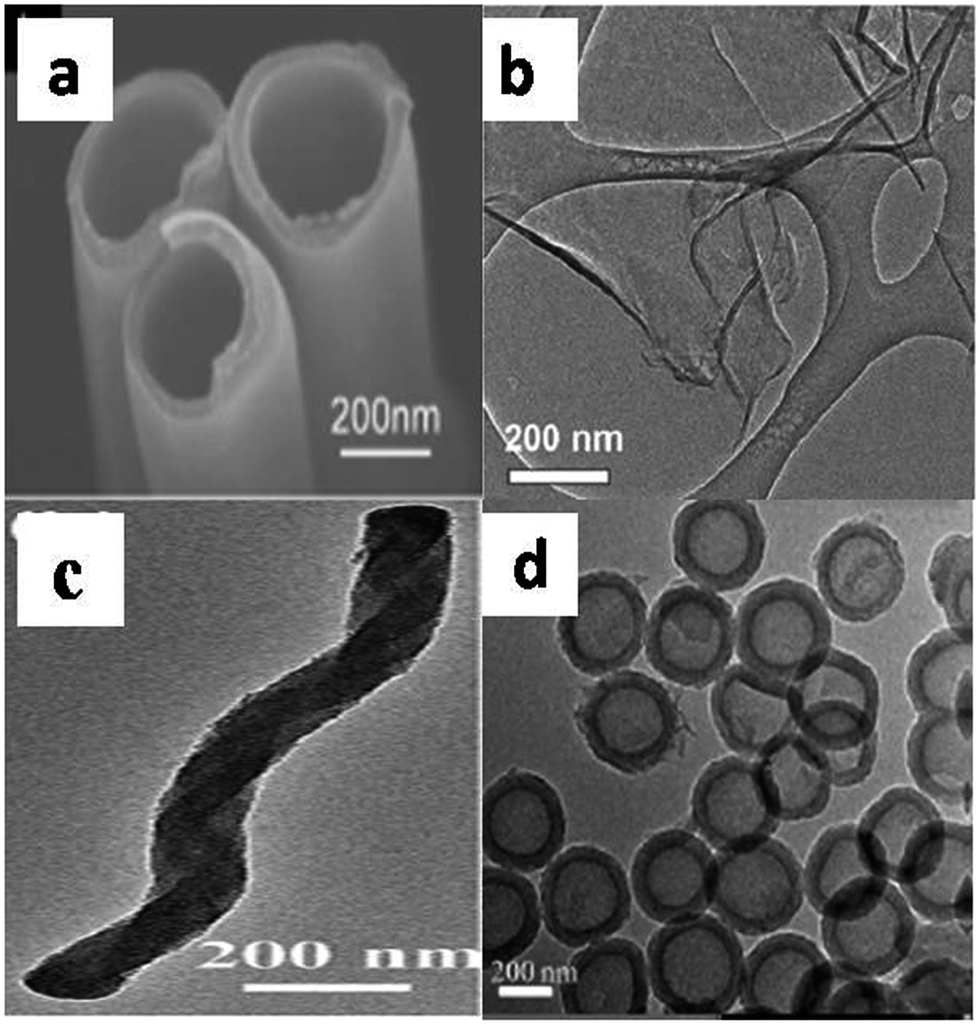 | ||
| Fig. 10 TEM image of g-C3N4 nanotubes (a), nanosheets (b), nanorods (c) and hollow nanospheres (d) (reproduced with permission from ref. 20, Copyright American chemical society, 2009, ref. 40 (licence number 3767461234354), ref. 19 (licence number 3767470642681), ref. 44). | ||
Modifications by different heteroatoms cause a narrowing of the bandgaps by lowering the HOMO levels. When the heteroatoms occupy the substitutional and interstitial positions, they alter their electronic properties to increase the efficiency. To modify the properties of g-C3N4 materials, non-metal doping has been extensively used. In F-, B- and P-modified g-C3N4 frameworks, the N-atom is substituted by the heteroatoms, resulting in a partial conversion of C-sp2 to C-sp3. The incorporation of different amounts of doping reagents results in disturbance of the graphitic structure, which could be seen in the increased intensity of the (100) peak in the XRD analysis.
In I-doped g-C3N4, the sp2 bonded N-atom is substituted by I atoms.57 The electrons in I1− were transferred into the carbon nitride network, and the iodine atom acted as the electron donor. The resultant I+ and I7+ were stabilized by the available lone pair of electrons in the carbon nitride matrix. The π-conjugated system enhances the mobility of the photogenerated carriers by an interaction between iodine and g-C3N4. In the process of doping, the apparent colour of the CN–Ix sample changes with the increasing content of iodine from pale yellow to deep brown. In the CN–Ix sample, many micropores on the surface of the g-C3N4 layers were found, which resulted in an increased surface area. The surface area increased from 12 m2 g−1 (g-C3N4) to 23 m2 g−1 for I–g-C3N4.58 In B- and F-co-doped materials, it was found that the B atoms enter C sites in the g-C3N4 network, while the F atoms saturate the residual bonds. The BN covalent bond also makes the material extremely stable. Boron- and fluorine-doped polymeric graphitic carbon nitride solid with a narrow pore size shows a spongy “morel-like” mesoporous structure of approximately 9.5 nm size, which shows its improved visible-light-induced photocatalytic activity.
Nanoparticles of noble metals, such as Ag, Au, Pt, Pd and Rh, can readily absorb visible light due to their surface plasmon resonance, which can be tuned by changing their size, shape and surrounding. The optical and electronic properties of metal nanoparticles, which are size and shape dependent, enhance the photocatalytic activity by promoting interfacial charge transfer in semiconductor–metal nanocomposites. The bandgap energy of the noble metal incorporated into the g-C3N4 nanoparticles can be lowered on the basis of surface plasmon effects62–75 and can show low photoluminescence intensity and excellent visible light absorption and superior photocurrent generation. When the photodeposition method is used as the loading process, Pt nanoparticles with an average diameter of 0.6 to 1.3 nm are obtained. However, when the impregnated method is used, the average diameter increases to 1.2 to 2.3 nm, with a uniform distribution of Pt nanoparticles on the surface. The oxidation state of Pt nanoparticles and the amount of the platinum species mainly depends on the different loading methods. The Pt nanoparticles are present on the surface of the catalyst in the form of Pt2+ or Pt4+ ions, which trap the photogenerated electrons to retard the recombination process; whereas in the Rh–g-C3N4 nanocomposite, the Rh NPs deposited on g-C3N4 surface are spherical and have approximately the same size as those of the neat one.67 Between the Rh nanoparticles and the carbon nitride surface, a direct interaction exists.70 In Au–g-C3N4, most of the Au NPs were dispersed in the range of 12–16 nm on the g-C3N4 support with different densities. The small Au NPs can be found on the layer of g-C3N4, which ultimately results in a composite photocatalyst. In the Au/g-C3N4 nanocomposite, g-C3N4 maintains a sheet-like layered structure consisting of graphitic planes stacked together with a conjugated aromatic system of triazine units. The Au NPs were found to be present linked to the surface and edges of the g-C3N4 sheet. TEM images of Au/g-C3N4 samples with different Au loading are presented in Fig. 11.
 | ||
| Fig. 11 TEM images of Au/g-C3N4 samples with different Au loading: (A–D) 20, 60, 80 and 90 wt% (reproduced from ref. 71, licence number 3767470882063). | ||
Li et al.73 reported that the XRD patterns of Au-, Pt- and Pd-co-doped g-C3N4 show the same reflections as for mesoporous g-C3N4 along with the characteristic peaks for the noble metals, as shown in the Fig. 12.
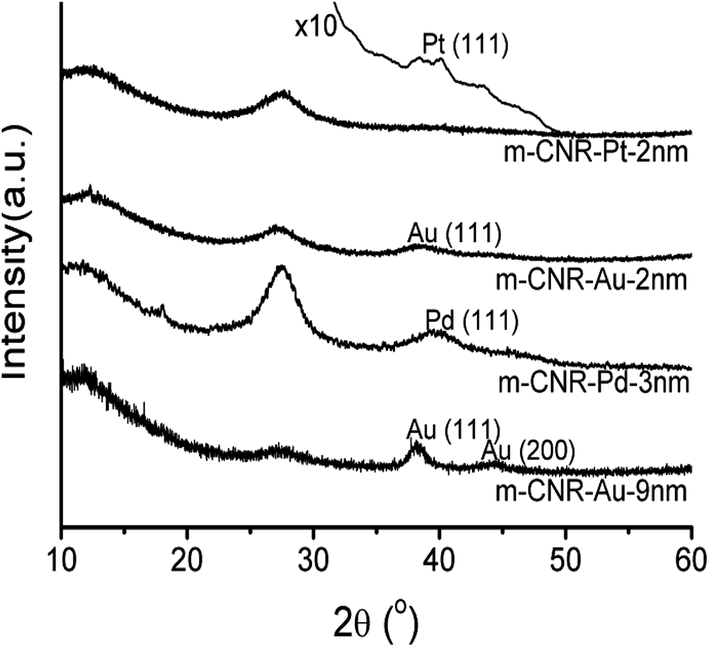 | ||
| Fig. 12 XRD patterns of m-CNR (mesoporous carbon nitride)-supported noble metal NPs, (reproduced from ref. 73). | ||
Transition metal ion or oxide nanoparticles were incorporated into g-C3N4, and the XRD patterns remained almost unchanged, typically showing two diffraction peaks corresponding to the 002 and 100 planes, thus maintaining the same crystallinity in the composite. In few cases, a slightly shifted peak from 27.4° towards a higher angle represented a certain decrease in the interplanar stacking distance. The interaction between the transition metal ions or oxides nanoparticles and g-C3N4 are strong and were dispersed evenly on the surface of g-C3N4. Wang et al.83 reported that in CeO2/g-C3N4 (13 wt%), CeO2 nanoparticles with diameters of about 5–20 nm are attached to the surface and to the edge of the g-C3N4. The agglomeration of CeO2 particles can be observed with an increase in the CeO2 wt% in the CeO2–g-C3N4 nanocomposite. The interaction between the CeO2 nanoparticles and the g-C3N4 layered materials was so strong that even after a long ultrasonication treatment at room temperature, the composite could not be destroyed. Fig. 13 shows the TEM images of the CeO2/g-C3N4 samples, showing the formation of the composite.
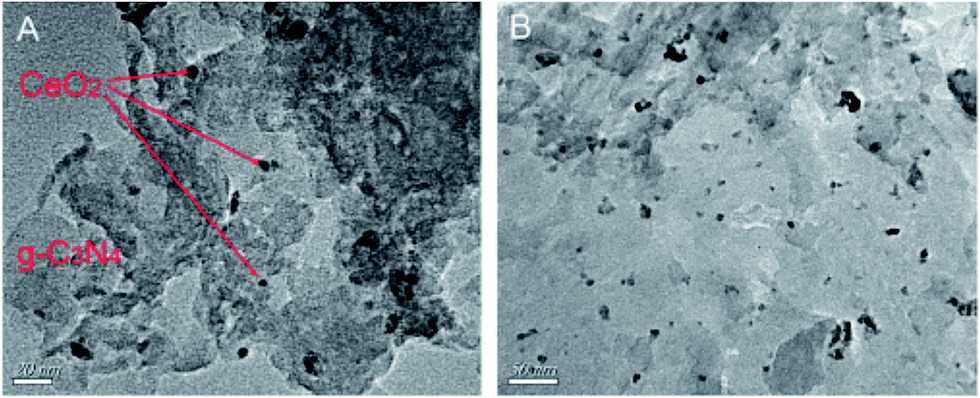 | ||
| Fig. 13 TEM images of (A) CeO2/g-C3N4 (5.9%) composite and (B) CeO2/g-C3N4 (13.0%) composite (reproduced from ref. 83). | ||
The morphologies and the particle sizes of the synthesized composites were studied by SEM (Fig. 14). In the case of neat g-C3N4, a large number of irregular particles were found because of the evolution of gases during the calcinations. However, the SEM images of the composites reveal that the nanocrystals were well spread out over the surface of g-C3N4. However, overcoverage of the nanocrystals may reduce the number of reactive sites on the surface of g-C3N4, thus resulting in a decreased photocatalytic activity.
 | ||
| Fig. 14 SEM images of: (a) g-C3N4, (b) g-C3N4–FeO composite (c) g-C3N4–ZnO (8.4), and (d) g-C3N4–ZnO (15.6) (reproduced from ref. 80 and 85). | ||
5. Study of the optical and electronic properties of g-C3N4-based materials
The main purpose of the modification of g-C3N4 is to modulate its electronic and optical properties. Bandgap narrowing can be effectively used to modify the absorption edge of a semiconductor photocatalyst to extend its absorption in the visible region. The modulation of g-C3N4 by doping with non-metals or by the formation of composites with other narrow-bandgap photocatalysts could effectively enhance the light absorption property of g-C3N4 in the wide range of the visible region, which was confirmed by UV-Vis DRS. Many researchers have reported that morphological changes and the inclusion of various metal-ion species in the g-C3N4 network alter its bandgap energy and enhance its light absorption significantly.An insight into the optical property of g-C3N4 indicates that neat g-C3N4 shows absorption up to 450 nm, which then changes significantly by morphological variation. Yan et al.30 reported a remarkable red-shift in optical absorption from 450 nm to 800 nm by synthesising mesoporous g-C3N4 using the P123 surfactant. The following figures (Fig. 15a and b) show the UV-Vis DRS of neat g-C3N4 prepared from various precursors and mesoporous g-C3N4 prepared with different amounts of P123, in which the arrow indicates the increasing amount of P123.
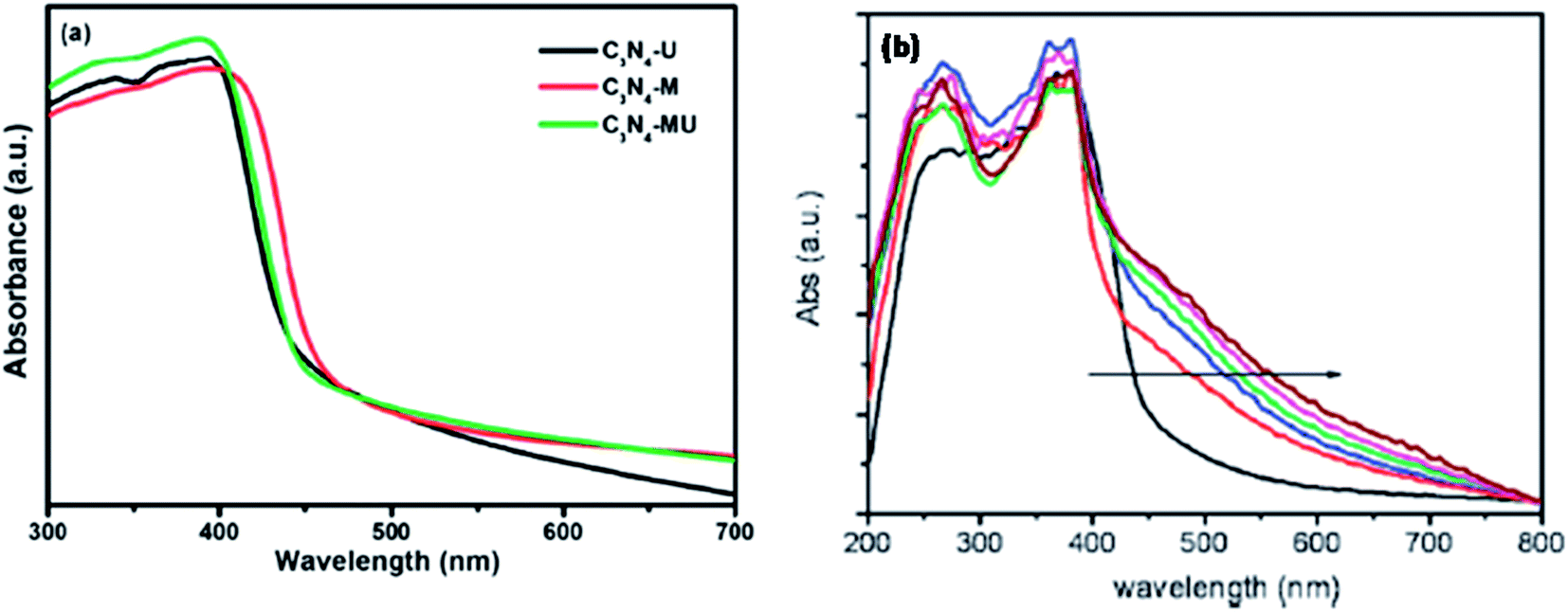 | ||
| Fig. 15 (a) UV-Vis DRS of neat g-C3N4 and (b) mesoporous g-C3N4 prepared by using different amounts of P123 (reproduced from ref. 3 and 30). | ||
By preparing ompg-C3N4, the surface area increased to 239 m2 g−1 and it exhibited better optical absorption than bulk g-C3N4, especially in the 430–550 nm region. According to Yang et al.,40 after exfoliation, the surface area of the material was as high as 384 m2 g−1 and it contained an ample number of nitrogen-containing active sites, which are favourable to enhance its photocatalytic activity. Bai et al.45 reported that while preparing hollow carbon nitride nanospheres, with the increase in the amount of added organic compound, the sample showed a red-shift from 420 nm to 700 nm.
Photoluminescence is another useful technique to study the migration, transfer and separation efficiency of semiconducting materials. The separation of charge carriers in g-C3N4 can be effectively minimized by doping with cations, by loading of noble metals or by the formation of composites with other metal oxides etc. to improve its photocatalytic efficiency towards hydrogen production. Martha et al.3 reported the synthesis of a highly active g-C3N4 photocatalyst from a mixture of urea and melamine, which showed a reduced electron–hole recombination rate and enhanced photocatalytic activity, as shown in Fig. 16.
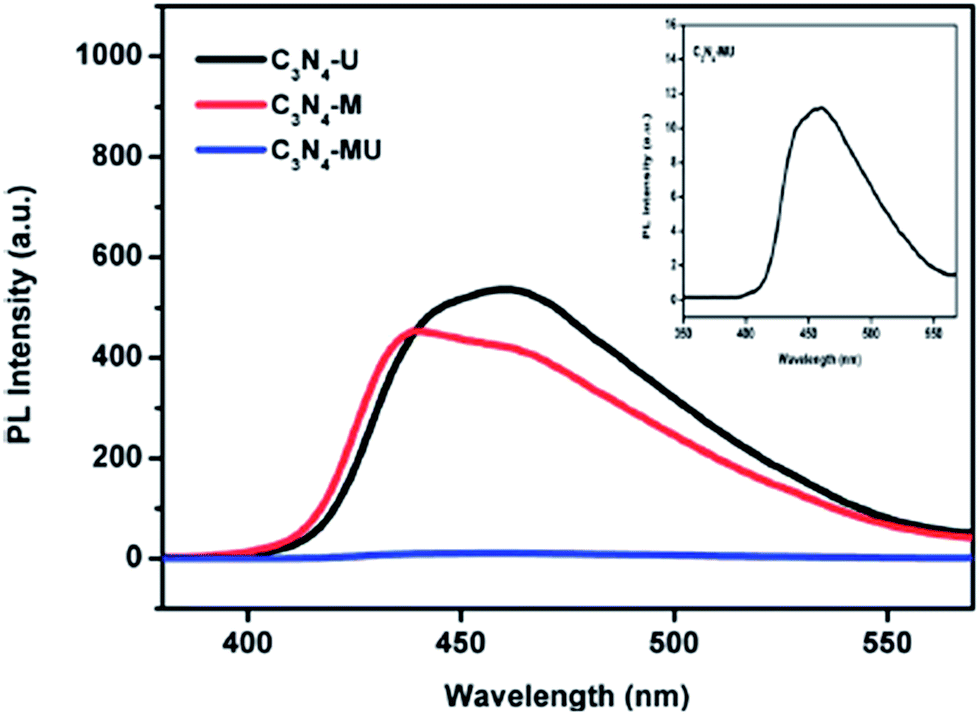 | ||
| Fig. 16 PL spectra of g-C3N4 prepared from a mixture of urea and melamine (reproduced from ref. 3). | ||
Heteroatoms such as N, C, B, F, S, and P are most commonly used to shift the optical absorption in the visible region by creating localized/delocalized states in the bandgap, providing improved activities in the samples for photocatalytic hydrogen production. Non-metal doping decreases the energy gap to enhance the visible light absorption of g-C3N4, which is at a maximum when the doping occurs in the interstitial sites and is due to a significant change in the distribution pattern of the g-C3N4 sheets. The bandgap of CNF decreases from 2.69 eV for g-C3N4 to 2.63 eV by47 fluorine doping. With the decrease in bandgap energy, the semiconductor properties of the F-doped g-C3N4 have slightly been changed, with a shift of the absorption in the visible region. Similarly B-doped g-C3N4 also shows a slightly reduced bandgap of 2.66 eV as compared to pure g-C3N4 (2.7 eV).49 In the case of52 P-doped g-C3N4, it was also found that with the increasing P percentage, the optical bandgap energy gradually changes to lower energy. Compared to undoped g-C3N4, the P-doped g-C3N4 showed a significantly enhanced electrical conductivity by up to 4 orders of magnitude. It was also found that the photocurrent generation was increased 5-fold. Both of these are important steps towards the photovoltaic application of g-C3N4. When the percentage of P-atoms is moderate, the bandgap changes in such a manner that, UV-Vis absorption indicates the presence of some intermediate states between VB and CB. In the case of C–g-C3N4, the bandgap energy value was found to be 2.56 eV.53 As photoexcitation strongly depends on the bandgap of the semiconductor and the wavelength of the light used, C–g-C3N4 should absorb more visible light than g-C3N4. In S-doped g-C3N4, it was found that the nitrogen atoms of the melon ring were replaced by sulphur atoms, resulting in a unique electronic structure.55 There was an increase in the bandgap, which showed a slightly decreased absorbance in the visible region. The bandgap of g-C3N4–xSx was shifted from 2.73 eV to 2.85 eV.56 The most interesting synergistic phenomenon of sulphur involved a widening and upshifting of the VB, which was achieved through the homogenous distribution of the S dopant and a reduction in particle size after doping. Enhanced light harvesting was evidenced from the UV-Vis diffuse reflectance spectra (DRS) by the upshift of the absorbance of S–g-C3N4 compared to porous and non-porous g-C3N4 samples. For mpg-CNS, the bandgap energy decreases to 2.61 eV, which results in a downshift of the conducting band, as shown in Fig. 17.
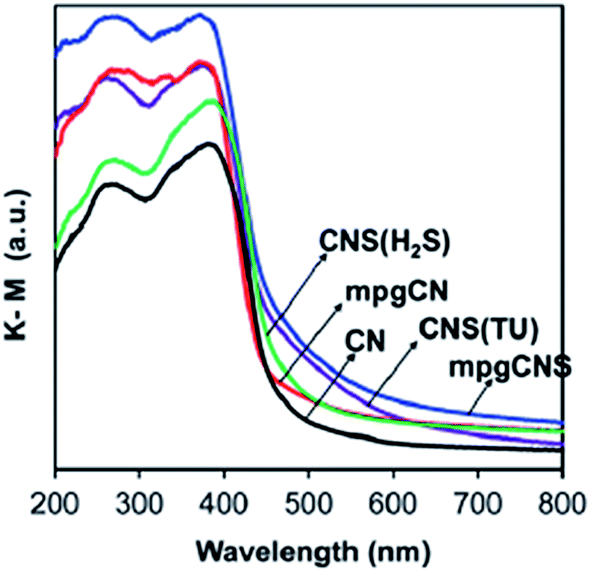 | ||
| Fig. 17 UV-Vis DRS of mpg-CNS and mgp-CN (reproduced from ref. 56). | ||
The absorption band edge extends from 420 nm to 600 nm in57 I-doped g-C3N4 causing a red-shift. The red-shift is due to the effective extension of the aromatic carbon nitride heterocycle by iodide ions and due to the presence of iodine atoms as an impurity in the energy levels above the valence band edge. The band gap energy decreased from 2.75 eV to 2.69 eV (∼0.06 eV). The self-doping strategy of g-C3N4 tunes the surface property, electronic structure and, hence, its photoreactivity. In carbon self-doped g-C3N4, it was found that the bridging N-atoms are substituted by the C-atoms. This increases the delocalized π-bonds between the substituted carbons, which increases the electrical conductivity of g-C3N4 as delocalized π-bonds favour the electron transfer. The bandgap of g-C3N4 decreases with carbon self-doping and enhances its visible light absorption and photocatalytic performance. According to Dong et al., when preparing C–g-C3N4, the particle size decreases in comparison to g-C3N4. Therefore, carbon self-doping induces electrical conductivity increases due to the reduction in particle size. The photocurrent generation of C–g-C3N4 is 1.39 times higher than that of g-C3N4. Besides this post-treatment doping, when sulphur is doped into the g-C3N4 matrix by the polycondensation of sulphur-enriched trithiocyanaric acid, –SH groups present at the terminal of the melon ring are the leading group. The absorption in the visible region increases due to the presence of residual S in the resulting S-doped g-C3N4 as compared to undoped g-C3N4.
Nanoparticles of noble metals, such as Ag, Au, Pt, Pd, Rh, can strongly absorb visible light due to their surface plasmon resonance, which can be tuned by changing their size, shape and surroundings. The optical and electronic properties of metal nanoparticles that are size and shape dependent, enhances the photocatalytic activity by promoting interfacial charge transfer in semiconductor–metal nanocomposites. The bandgap energy of the noble metal nanoparticles can be lowered on the basis of the surface plasmon effects and show low photoluminescence intensity and excellent visible light absorption as well as superior photocurrent generation. In Pt–g-C3N4 nanospheres (NS-g-C3N4), the surface area increases to 160 m2 g−1 and the pore volume was found to be 0.4 cm3 g−1. NS-g-C3N4 shows a hypsochromic shift of the absorption edge from 465 nm to 430 nm and a bandgap energy increase from 2.67 eV to 2.86 eV. Due to the multiple reflections of the sharp edges in the defect sites, the absorption region extends from 430 nm to 590 nm. The 3D interconnected nanosheets shorten the distance for charge migration and help the electron delocalization.70–74 Au and Ag nanoparticles show good photocatalytic performances by lowering the bandgap energy due to the surface plasmon effects. The photocatalytic activity of g-C3N4 can be improved by coupling it with simple silver salts, like Ag3PO4, or by preparing Ag/AgCl/g-C3N4 composites or Ag/AgBr/g-C3N4 nanocomposites. Zhang et al.59 reported that silver orthophosphate (Ag3PO4) is a novel silver salt that shows an extremely high quantum yield of about 80% at wavelengths less than 480 nm for O2 evolution. When Ag3PO4 is coupled with highly stable metal-free polymeric g-C3N4, the photocatalytic activity of both g-C3N4 and Ag3PO4 increases. The insoluble g-C3N4 protects Ag3PO4 from dissolution and enhances its stability. The CB and VB potentials of g-C3N4 are more negative than that of Ag3PO4, which enhances the charge transfer separation process through their interface. The efficient charge separation process was further confirmed by photocurrent measurements (Fig. 18). The pure g-C3N4 and Ag3PO4 exhibited a photocurrent density of 4.7 and 3.1 μA cm−2, respectively. However, due to the synergistic effect between g-C3N4 and Ag3PO4, 5:1 g-C3N4/Ag3PO4 bulk heterojunction showed a photocurrent density of 11.2 μA cm−2, which was even more than the sum of the photocurrent density of pure g-C3N4 and pure Ag3PO4.
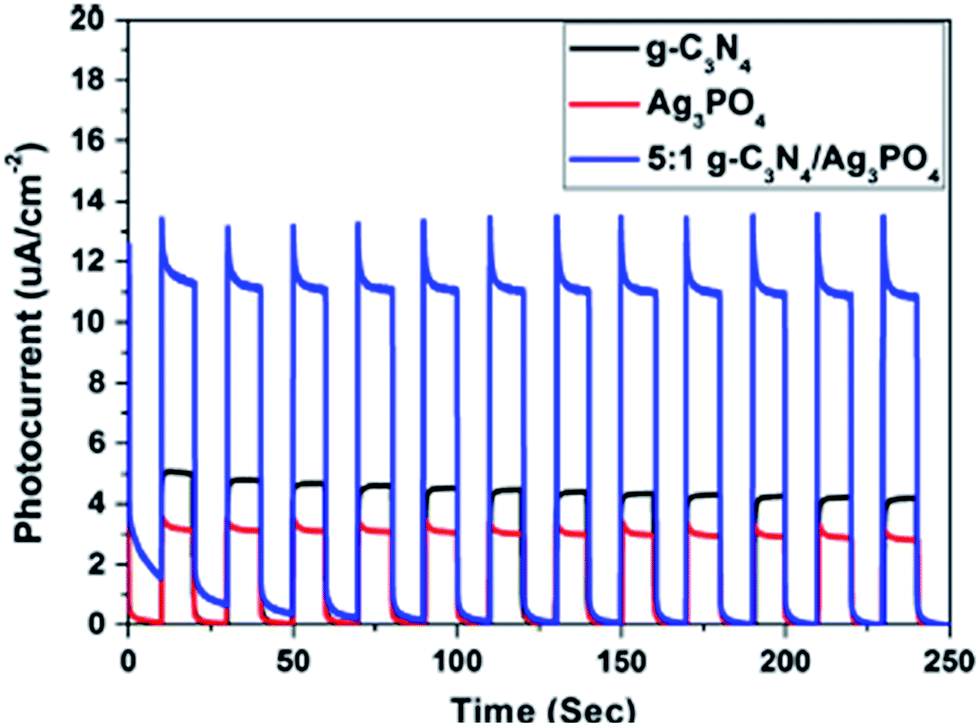 | ||
| Fig. 18 Photocurrent density of 5:1 g-C3N4/Ag3PO4 bulk heterojunction (reproduced from ref. 59). | ||
As a direct semiconductor, a narrow bandgap of Ag2S (1.1 eV) increases its absorption and extends its application in photovoltaic cells and photocatalytic applications. Au nanoparticles show good absorption in visible light due to the surface plasmon resonance, which is necessary for the enhancement of photocatalytic activity, causing a red-shift compared to that of neat g-C3N4. The bandgap energies also change from 2.71 eV for neat g-C3N4 to 2.63 eV in the case of Au/g-C3N4. This reduction in bandgap energy of 1 wt% Au/g-C3N4 is due to the plasmonic effect of AuNPs, resulting in an enhancement of photocatalytic H2 production. The incorporation of AuNPs onto the layer of g-C3N4 restricts recombination between the opposite charge carriers and enhances the photocatalytic activity. It was observed that with the deposition of AuNPs onto g-C3N4, the current density was significantly increased. 1 wt% Au/g-C3N4 produced a photocurrent density of 49.5 mA cm−2, whereas in neat g-C3N4, it was only 15 μA cm−2, which is more than 3000 times higher, which was simply due to the enhanced visible light absorption. Also the enhancement of photocatalytic activity of the nanocomposite photocatalysts can be explained on the basis of their PL spectra, which further confirm the migration of photogenerated charge carriers and the delayed recombination process. As compared to neat g-C3N4, the decreased PL intensity of all the Au/g-C3N4 nanocomposites in Fig. 19 indicates the improved electron–hole separation.
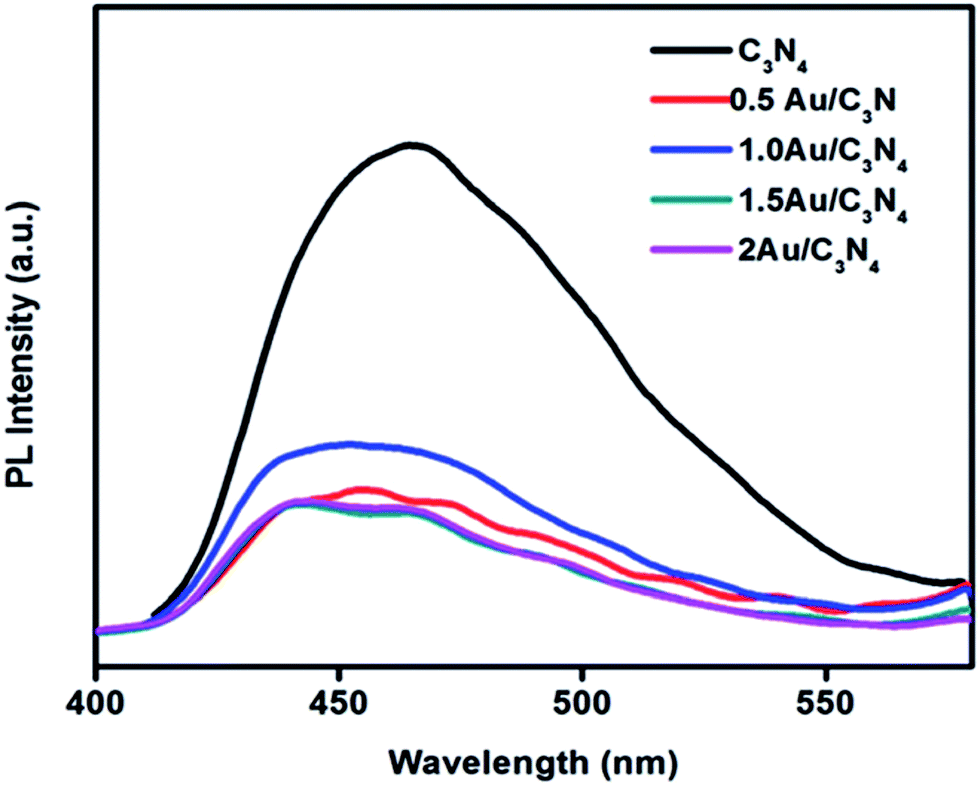 | ||
| Fig. 19 PL spectra of neat g-C3N4 and Au/g-C3N4 nanocomposites (reproduced from ref. 70). | ||
Chang et al.75 reported that Pd/mpg-C3N4 shows a significant increase in light absorption in the visible region. On increasing the Pd content, the colour of the catalysts changed from yellowish to light grey, and their light absorbance increased gradually from 200 nm to 700 nm. When the Pd content was more than 1.5%, the absorbance even shifted to the near infrared. Pd/mpg-C3N4 with 4.50% Pd is the optimum loading and shows a wide absorbance in the UV-Vis region. In Pd/mpg-C3N4, Pd is present as Pd0 and acts as an electron trapping agent, and the separation of photogenerated electrons and holes increases, and consequently the photocatalytic performance is improved.
The electronic and optical functions of g-C3N4 can be easily modified after transition metal-ion doping. The transition metal species, by lowering the bandgap, extends the light absorption into the visible region of the electromagnetic spectrum. In determining electrical conductivity in a material, the bandgap is a major factor and it directly determines the upper wavelength limit of light absorption, and hence affects the photocatalytic activity. Tetragonal tin dioxide (SnO2) is a multifunctional material has been widely used in the fields of solar cells, gas sensing, photovoltaic conversion and photocatalysis. However, the bandgap of SnO2 is so wide (3.7 eV) that it can absorb only UV light for the degradation of pollutants in water and air. However, SnO2 is a better electron acceptor than TiO2 and ZnO. So by preparing a SnO2–g-C3N4 composite, the electron–hole separation tendency of g-C3N4 can be increased and hence so can its photocatalytic activity. The rare earth oxide CeO2 finds application as a photoactive material in solar cells and also as a photocatalyst in the degradation of organic pollutants, as well as in the production of hydrogen.83 But it mainly absorbs in the near UV region. However, when it is coupled with g-C3N4, the CeO2–g-C3N4 composite has an extended application as a photocatalyst in the visible region. In another example, the absorption wavelength of TiO2 nanofibres was under 400 nm, but after coupling g-C3N4 with TiO2 nanofibres, the hybrid showed more efficient light absorption from 400 nm to 800 nm in comparison with the pure g-C3N4 and TiO2.81 Although, the surface area of the g-C3N4–TiO2 composite is less, the notable increase in the photocatalytic activity might be related with the participation of g-C3N4 in the charge separation process. Thus, a higher efficiency in the electronic step leads to higher photocatalytic activity, with an improvement up to 70%. Similarly, the absorption edge of TiO2 corresponds to a bandgap of 3.2 eV, which is in the UV region. However, according to Pany et al. in N,S-TiO2/g-C3N4 nanocomposites, compared to TiO2, the absorption edge is shifted towards the shorter wavelength region, making it visible light active. Moreover, in N,S-TiO2/g-C3N4 photocatalysts, the gradual increase in wt% of thiourea plays a key role in the absorption shift towards the red end with respect to Ti. Cu2O is a well-known p-type semiconducting photocatalytic material with a bandgap energy of 2.0–2.4 eV. To improve the photocatalytic efficiency of g-C3N4, it was also coupled with Cu2O. Peng et al.88 reported that when Cu2O was coupled with g-C3N4, it exhibited enhanced photocatalytic activity, because of the well-matched overlapping band structures and resulting highly efficient separation of photogenerated charges. Pure g-C3N4 possesses an absorption wavelength of ∼450 nm, whereas Cu2O possess a broad absorption in the visible region from 400 to 600 nm. So after combining these two semiconductors, the absorption of g-C3N4–Cu2O in the visible light range remarkably increases. Also, the absorption intensity of g-C3N4–Cu2O increases with the increase in Cu2O content. It was found that when93 In2O3 and g-C3N4 with appropriate band alignments were coupled, there was an interfacial change transfer, which increases the separation efficiency of the photogenerated electrons and holes. The well-dispersed In2O3 nanocrystals on sheet-like g-C3N4 surfaces form an intimate interaction. Since the conduction band (CB) and the valence band (VB) positions of In2O3 lie at ∼−0.6 eV and +2.2 eV, respectively, and as both are lower than those of g-C3N4, i.e. ∼−1.1 eV and +1.6 eV, respectively, this allows effective interfacial charge transfer across the In2O3 and g-C3N4 hybrid.
6. Photocatalytic hydrogen production
After the breakthrough report by Fujishima and Honda in 1972, research has been going on to develop photocatalysts with high efficiency and stability for water splitting to produce hydrogen gas. Studies by different groups of scientists have found that neat g-C3N4 can undergo two cycles of water splitting under visible light successfully. The semiconductor photocatalyst g-C3N4 is considered to be suitable for water splitting as it has a bandgap value greater than 1.23 eV (redox potential of water) and also proper positioning of the CB and VB levels. For water splitting, the bottom level of the conduction band of the semiconductor has to be more negative than the redox potential of H+/H2 (0 V vs. the normal hydrogen electrode (NHE)), whereas the valence band of the semiconductor has to be more positive than the redox potential of O2/H2O (1.23 eV). One half of the reaction is a water-reduction half-cycle, which produces H2, and the other half is a water-oxidation half-cycle to form O2. Although the band potentials are appropriate, g-C3N4's photocatalytic efficiency is still low due to the fast recombination of charge carriers and the mismatch between its bandgap and solar spectra. Because of this, the material has been optimized by morphological and textural variation, non-metal doping, noble metal incorporation and transition metal- and metal oxide modification. In this review, we have tried to summarize the key modifications of g-C3N4 to increase the quantum yield. Doping and various modifications are expected to increase the carrier mobility, while the non-coplanar HOMO and LUMO improve the separation of photogenerated e−/h+ pairs to enhance the photocatalytic activity.6.1. Hydrogen production by neat g-C3N4
For the first time, Wang et al. predicted that g-C3N4 is thermodynamically stable under light irradiation and is capable of water splitting in the presence of a sacrificial agent. The proper bandgap of g-C3N4 in particular makes it suitable for applications in photochemistry and photocatalysis. g-C3N4 can be prepared from different precursors (550 °C, 5 °C min−1 ramp rate), and when tested for HER in an aqueous sacrificial solution containing triethanolamine (TEOA) at room temperature and atmospheric pressure, it was found that urea-derived g-C3N4 exhibited the highest H2 evolution in comparison to DCDA or thiourea-derived g-C3N4 under visible light irradiation. The HER of urea-derived g-C3N4 is 20000 μmol h−1 g−1, which is 15 times more than that of DCDA-derived g-C3N4 and 8 times more than that of thiourea-derived g-C3N4. However, the g-C3N4 obtained from dicyanamide exhibits the highest activity.4
g-C3N4 is a better semiconductor photocatalyst than the other developed traditional photocatalysts owing to its stability, visible light absorption property and appropriate band edge potentials for water splitting reactions. Herein, we have briefly presented a comparative study of various class of photocatalysts, including oxides, sulphides, nitrides, oxynitrides, etc., which have been widely used for H2 production. The details regarding materials, the light source, reaction solution and H2 evolution are presented in Table 2.
| Photocatalyst | Incident light (λ) | Reaction solution | H2 evolution (μmol h−1 g−1) | Reference |
|---|---|---|---|---|
| TiO2 | 250–400 nm | CH3OH | Pt/∼3300 | 97 |
| TiO2 nanosheets | 250–400 nm | CH3OH | 117.6 | 98 |
| ZnO | ≥420 nm | Na2S | 44000 |
99 |
| CeO2 | ≥300 nm | Ce4+ | — | 100 |
| LaTiO3 | 250–400 nm | Pure water | NiOX/137 | 101 |
| SrTiO3 | 250–400 nm | NaOH | NiOX/∼70 | 102 |
| NaTiO3:La |
250–400 nm | Pure water | NiO/19800 |
103 and 104 |
| PbWO4 | 290–600 nm | Pure water | RuO2/96 | 105 and 106 |
| BiVO4 | ≥420 nm | AgNO3 | — | 107 |
| CdS | ≥420 nm | Na2S + Na2SO3 | Pt/27333 |
108–110 |
| In2S3 | ≥400 nm | Na2S + Na2SO3 | Pd/960.2 | 111 |
| ZnIn2S4 | ≥420 nm | Na2S + Na2SO3 | Pt/231 | 112 |
| AgGaS2 | ≥420 nm | Na2S + Na2SO3 | Pt/2960 | 113 |
| CuGa3S2 | ≥420 nm | Na2S + Na2SO3 | NiS/∼2800 | 114 |
| ZnS | ≥420 nm | Na2S + Na2SO3 | 232.7 | 115 |
| Cd In2S4 | ≥420 nm | H2S + KOH | 6960 | 116 |
| CuInS2 | ≥420 nm | Na2S + Na2SO3 | Pt/84 | 117 |
| TiN | ≥400 nm | Na2S | Pt/150 | 118 |
| Ta3N5 | ≥420 nm | CH3OH/AgNO3 | Pt/∼125 | 119 |
| Ge3N4 | ≥200 nm | H2SO4 | RuO2/3600 | 120 |
| GaN | ≥300 nm | H2SO4 | Rh2−xCrxO3/63.3 | 121 |
| TaON | ≥420 nm | C2H5OH/AgNO3 | Ru/300 | 122 |
| La TiO2N | ≥420 nm | CH3OH/AgNO3 | Pt/∼15 | 123 |
| Y2Ta2O5N2 | ≥420 nm | C2H5OH/AgNO3 | Pt–Ru/833 | 124 |
| Zr2ON2 | ≥420 nm | CH3OH/AgNO3 | Pt/7 | 125 |
| Ga–Zn-in mixed oxy nitride | ≥400 nm | CH3OH/AgNO3 | Rh/50 | 126 |
| TaOxNy | ≥400 nm | CH3OH | 3.12 mmol g−1 h−1 | 127 |
| Zn–Ga–Ge–N–O | ≥400 nm | H2C2O4 | (∼62 μmol h−1) | 128 |
g-C3N4 is further modified in various ways for enhancement of its H2 production ability. Groenewolt et al.38 reported the preparation of mpg-C3N4 nanoparticles, where the particle size of g-C3N4 could be easily replicated, ranging from 5 to 7 nm, by using a mesoporous silica template, and managed to increase the specific surface area to 400 m2 g−1, which would result in the photocatalytic H2 evolution being enhanced nearly 10-fold.
According to Yang et al.,40 after exfoliation, the photocatalytic HER of g-C3N4 nanosheets was evaluated from water/triethanolamine mixtures in visible light and with Pt (3 wt%) as the co-catalyst, and it was found to be 93 μmol h−1, which is much higher than the value for bulk g-C3N4, which is 10 μmol h−1.
Zheng et al.42,43,45 extensively studied the photocatalytic H2 evolution of hollow carbon nitride nanospheres (HCNS) and found that the H2 evolution was increases 3-fold (278 μmol h−1) by using Pt (3 wt%) as a co-catalyst. Also, by changing the condensation temperature, the photocatalytic H2 evolution varies. The HCNS-550 °C exhibited a HER of 275 μmol h−1, which is about 2 times higher than that of HCNS. The HER of HCNS-300 °C was found to be 202 μmol h−1, which is much higher than the HCNS-400 °C and HCNS-500 °C samples. The comparative study of H2 evolution rates are presented in Table 3.
| Texture | Morphology | Surface area (m2 g−1) | Incident light (λ) | Reaction solution | H2 evolution (μmol h−1) | Reference |
|---|---|---|---|---|---|---|
| Bulk g-C3N4 | Nanopowder | 13.4 | ≥420 nm | Triethanolamine | 12.5 | 18 |
| g-C3N4 | Nanosheets | 84.2 | ≥420 nm | Methanol | 93 | 40 |
| Structure-distorted nanosheets | 99 | ≥420 nm | Triethanolamine | 177 | 42 | |
| Hollow nanospheres | 221 | ≥420 nm | Triethanolamine | 275 | 44 | |
| mpg-C3N4 using silica nanoparticles | 400 | ≥420 nm | Triethanolamine | 125 | 38 | |
| mpg-C3N4 using soft template (10 wt% P123) | 90 | ≥420 nm | Triethanolamine | 148.2 | 30 | |
| ompg-C3N4 | o-Cylindrical pores | 142 | ≥420 nm | Triethanolamine | 26.5 | 31 |
6.2. Hydrogen production by non-metal-doped g-C3N4
Wang et al.47 studied the photocatalytic activity of F-doped g-C3N4 towards HER from water under visible light (λ > 420) and in the presence of Pt (3 wt%) as a co-catalyst, as shown in Fig. 20, and found that CNFx samples (x = 0.5 g) showed higher H2 evolution activity over g-C3N4, which was about 2.7 times more than that of neat polymeric g-C3N4. These catalysts are highly stable and can be easily separated from the reaction solution by simple filtration. Furthermore, they can be reused for several cycles without losing activity. Hence, the CNF samples provide a modified texture with a decreased optical bandgap and improved activities for the photocatalytic hydrogen production from water/TEA.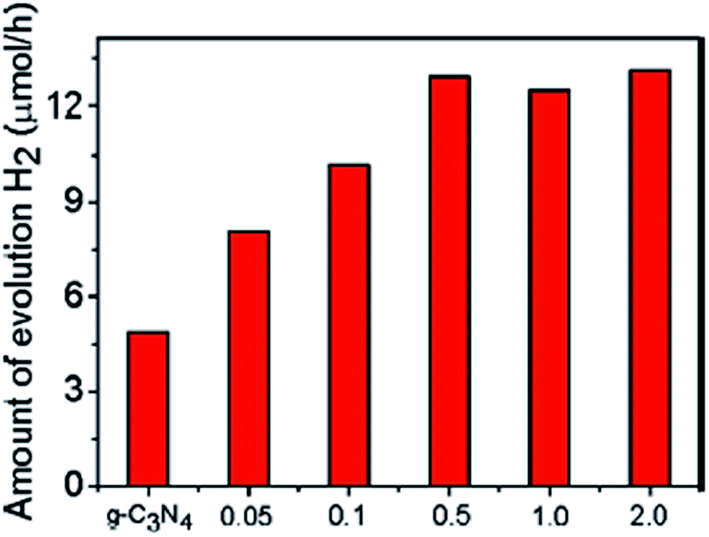 | ||
| Fig. 20 Photocatalytic activity of F-doped g-C3N4 towards HER from water/TEA under visible light (λ > 420) (reproduced from ref. 47, licence number 3767471256893). | ||
The HER of C–g-C3N4 was 1.42 times greater than that of neat g-C3N4. Carbon self-doped g-C3N4 synthesised by Zhang et al. by the copolymerization of dicyandiamide with barbituric acid increased the HER by nearly 4-fold that of neat g-C3N4. All these enhanced photocatalytic activities were due to the separation and transfer of photoinduced charge carriers. The polymeric CNS sample undergoes the water oxidation reaction without the need for the addition of co-catalysts, which is not possible for the undoped g-C3N4. Similarly, S-doped g-C3N4, due to its unique electronic structure, had a special photocatalytic activity towards HER, which was 8 times54 higher than that of neat g-C3N4 under visible light. However, according to Hong et al.,56 by doping a small percentage of S in mesoporous g-C3N4, the photocatalytic HER was remarkably improved. The HER of mpg-CNS was about 136 μmol h−1, which was 214% higher than that of CNS and 36% higher than that of the mpg-CN samples. Due to both the mesoporous structure and sulphur doping, the activity of mpg-CNS towards hydrogen evolution from photocatalytic water splitting was enhanced almost 30-fold56 compared to that of pure g-C3N4, although the surface increase was only 10-fold, as shown in Fig. 21. The quantum efficiency was found to be as high as 5.8% at 440 nm in an aqueous solution of triethanolamine. But ex situ sulphur doping was found to be less effective towards photocatalytic performance.
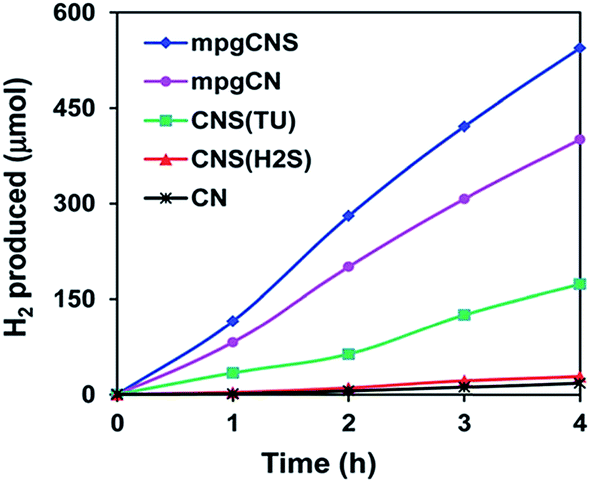 | ||
| Fig. 21 Hydrogen evolution of S-doped g-C3N4 from an aqueous solution of TEA under visible light (λ > 400) (reproduced from ref. 56, licence number 3767480308846). | ||
Zhang et al.57 reported that iodine modification is considered to be a good dopant to enhance the photocatalytic performance based on the optical characterization and hydrogen evolution activity. Also, the photocatalytic activities of halide-ions-doped g-C3N4 were greater in comparison to pure g-C3N4. An I–g-C3N4 sample showed the fastest HER (about 38 μmol h−1), approximately 2 times more in comparison to pure g-C3N4 (14 μmol h−1). The photocurrent generation for I-doped g-C3N4 was also nearly 3 times higher than that of pure g-C3N4, which explains the increased mobility of the photogenerated charge carriers. As compared to undoped g-C3N4, the P-doped g-C3N4 showed a significantly enhanced electrical conductivity of up to 4 orders of magnitude higher. It was also found that the photocurrent generation increased 5-fold.
6.3. Hydrogen production by noble metal-loaded g-C3N4
Among all the noble metals, platinum was found to be most efficient, with HER varying from 20 to 200 mmol h−1 g−1 depending on several factors, like the pH of the solution, the intensity of the light source, the selection of the sacrificial agent and the loading procedure of the co-catalyst. The HER increases with increasing Pt content to around 2–4%, and then it decreases. The deposition of Pt nanoparticles enhances the H2 production, and the HER was about 172 μmol h−1, which is 7 times more than that of neat g-C3N4 when the catalyst is synthesized from an organo sulphur compound and calcined at 550 °C, which indicates that the presence of S during calcination controls the polymerization and hence enhances its photocatalytic activity. Photocatalytic H2 evolution using65 Pt as a co-catalyst showed a value of 574 μmol h−1 for 3 wt% Pt/NS-g-C3N4, whereas for 3 wt% Pt/bulk g-C3N4 it was only 12.5 μmol h−1 and for 3 wt% Pt/mpg-C3N4 with a surface area of 182 m2 g−1, the H2 evolution rate was found to be 123 μmol h−1, as shown in Table 4.| Catalysts | Surface area (m2 g−1) | Incident light (λ) | Reaction solution | H2 evolution (μmol h−1) |
|---|---|---|---|---|
| 3 wt% Pt/NS-g-C3N4 | 160 | ≥420 nm | Triethanolamine | 574 |
| 3 wt% Pt/bulk g-C3N4 | 10 | ≥420 nm | Triethanolamine | 12.5 |
| 3 wt% Pt/mpg-C3N4 | 182 | ≥420 nm | Triethanolamine | 123 |
When hollow carbon nitride nanospheres were synthesized by grafting the organic group ATCN (2-aminothiophene-3-carbonitrile) into the structure of g-C3N4, the photocatalytic activity towards H2 evolution increased 3-fold (278 μmol h−1) by using Pt (3 wt%).44 With the increase in the amount of the added organic compound, the sample also showed a red-shift from 420 nm to 700 nm. When Pt was doped on exfoliated g-C3N4 nanosheets,40 the surface area of the material was as high as 384 m2 g−1, and it contained an ample number of nitrogen-containing active sites, which are favourable for enhancing its photocatalytic activity. The photocatalytic HER of Pt-doped g-C3N4 nanosheets was found to be 93 μmol h−1, which is much higher than that of neat g-C3N4 (10 μmol h−1). When 3 wt% of Pt was loaded onto the surface of the catalyst by an in situ photodeposition method, the HER of the sample was found to be dependent on the processing temperature. When the processing temperature was below 650 °C, the absorption edge was blue-shifted, but when it was above 650 °C, the band edge was slightly red-shifted (Table 5).
| Catalyst | Surface area (m2 g−1) | Incident light (λ) | Reaction solution | H2 evolution (μmol h−1) |
|---|---|---|---|---|
| g-C3N4 | 9 | ≥420 nm | Triethanolamine | 10 |
| CN-550 | 4 | ≥420 nm | Triethanolamine | 5 |
| CN-600 | 9 | ≥420 nm | Triethanolamine | 14 |
| CN-625 | 15 | ≥420 nm | Triethanolamine | 30 |
| CN-650 | 20 | ≥420 nm | Triethanolamine | 63 |
| CN-675 | 68 | ≥420 nm | Triethanolamine | 147 |
| CN-700 | 99 | ≥420 nm | Triethanolamine | 177 |
Pt-doped hollow mpg-C3N4 nanospheres (with a decreased shell thickness of the nanospheres of 10–30 nm) showed enhanced photocatalytic hydrogen evolution due to increased light absorption in the visible region. The hollow CNS samples with 3 wt% of Pt as a co-catalyst showed a HER of 224 μmol h−1, which is 25 times higher than that of the bulk. The photocatalytic activity was enhanced towards water splitting under visible light irradiation by incorporating Rh metal into the g-C3N4 network with the decrease of the particle size. When the size of the metallic Rh nanoparticles was around 9 nm, the hydrogen evolution rate increased, whereas when the size of the metallic Rh nanoparticles was around 25–50 nm, Rh particles were preferred as catalysts for the oxidation reaction (CH4 + 2H2O → CO2 + 8H).67 The added Rh nanoparticles with particle sizes ranging from 1 to 5 nm behave as efficient photocatalysts for water splitting to produce hydrogen in the presence of methanol as the sacrificial agent. The H2 production rate of the most active catalyst, Rh(PVP)/C3N4, was 60 μmol during a reaction time of 4 h. When the Rh particles become larger, the H2 production rate is significantly decreased. Hence, the photodeposition method is the preferred method to deposit nanoparticles on the surface of g-C3N4. Lei et al.61 reported that with an increase in Ag loading, the H2 production rate was found to increase. The neat g-C3N4 sample showed a HER of 0.862 μmol h−1 g−1 under visible light, whereas after Ag loading H2 evolution rate was 10.105 μmol h−1 g−1, which was about 11.7 times more than that of neat g-C3N4. An optimum (1.0 wt%) Ag loading results in good dispersion on the surface of g-C3N4, which enhances the photocatalytic activity of the composite significantly, as shown in Fig. 22.
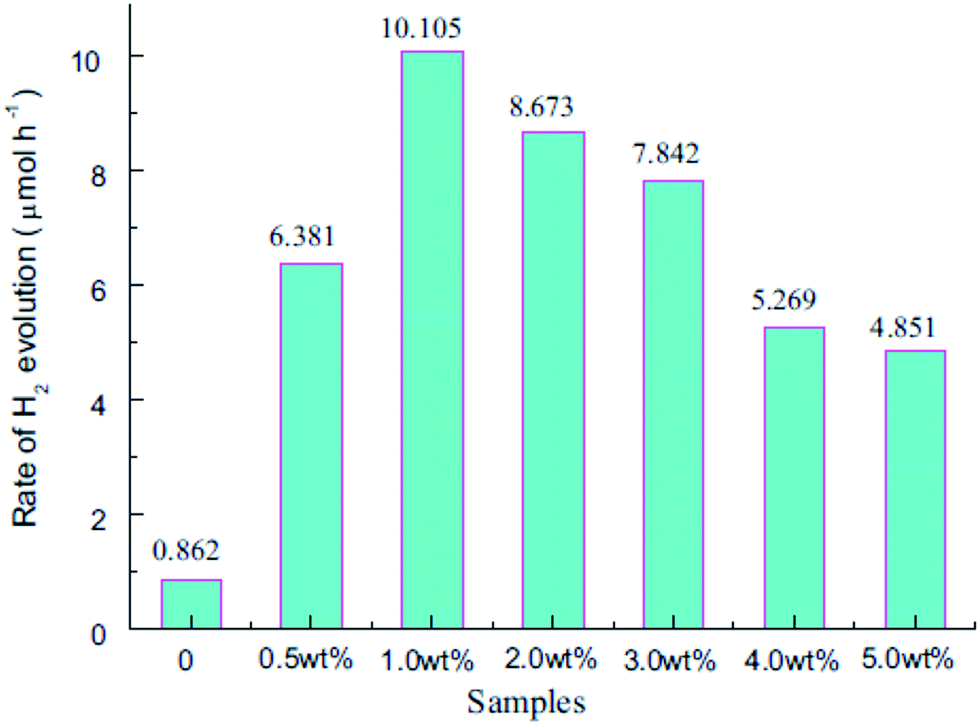 | ||
| Fig. 22 Photocatalytic activity of Ag/g-C3N4 composites for H2 evolution from methanol solution under visible light (reproduced from ref. 61). | ||
However, in Ag2S-modified g-C3N4 composite, the photocatalyst shows a significantly improved H2 production compared to that of pristine g-C3N4 in the range of 460 to 490 nm, which is around 100 times than that of pure g-C3N4.61 The maximum activity was observed with 5 wt% Ag2S–g-C3N4 when the size of the Ag2S nanoparticles were 8 to 16 nm. Also the H2 evolution was about 532.2 μmol when the reaction was performed under visible light illumination (λ > 400 nm). The optimum 1 wt% AuNPs-loaded g-C3N4 produced 23 times71 more H2 gas than neat g-C3N4. However, when doubling the concentration of the AuNPs, the amount of H2 gas production decreased to nearly half that value. The mechanism of photocatalytic hydrogen production and the surface plasmon resonance of Au in the Au/C3N4 nanocomposite is presented in Scheme 7. As the Fermi level of the Au NPs exists between the conduction band maximum and the valence band minimum of the polar semiconducting material, the surface wave can propagate easily at the interface between the g-C3N4 and Au NPs, resulting in the formation of plasmonic photocatalysts, and hence the photocatalytic performance of g-C3N4 can be further improved.
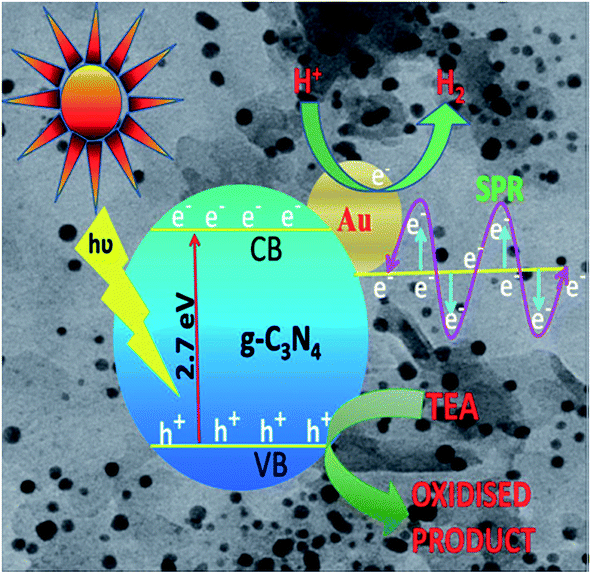 | ||
| Scheme 7 Mechanism of photocatalytic hydrogen production and surface plasmon resonance of Au in an Au/C3N4 nanocomposite (reproduced from ref. 70). | ||
A comparative study of the synthesis methods and the activity over noble metal-doped g-C3N4 is presented in Table 6.
| Nobel metal | Reagent used | Synthesis method | Catalyst | Incident light (λ) | Reaction solution | H2 evolution (μmol h−1 g−1) | Reference |
|---|---|---|---|---|---|---|---|
| Ag | AgNO3 | Facile heating method | 1 wt% Ag/g-C3N4 | ≥420 nm | Methanol | 10.105 | 61 |
| Au | HAuCl4, AuCl3 | Deposition–precipitation method | 1 wt% Au/g-C3N4 | ≥400 nm | Triethanolamine | 532 | 70 |
| Pt | H2PtCl4 | Deposition-method | 1 wt% Pt/g-C3N4 | ≥420 nm | Triethanolamine | 732 | 44 |
| Rh | RhCl3·nH2O | Polyol reduction method | Rh(PVP)/g-C3N4-4:1 |
420–630 nm | Methanol | 14.9 | 67 |
6.4. Hydrogen production by transition metal- and metal oxide-based composites and heterojunctions with g-C3N4
Sun et al.80 reported that Zn/g-C3N4 shows a red-shift in the absorption edge, and the new organic metal hybrid material shows a 10 times higher rate of photocatalytic H2 evolution under visible light. The synergistic combination of the enhanced specific surface area, the crystalline anatase phase, the small crystallite size, the effective charge separation and enhanced visible light absorption ability makes the N,S-TiO2/g-C3N4 photocatalyst effective for photocatalytic H2 evolution under visible light irradiation. Because of the faster recombination of photogenerated charge carriers, the amount of hydrogen evolution (125 μmol h−1) in neat g-C3N4 is limited. However, in the84 N,S-TiO2/g-C3N4 photocatalyst comparison to neat g-C3N4, there could be seen an enhanced activity towards H2 evolution. As the wt% of thiourea (i.e. from 1 to 10) is increased, gradually the H2 evolution rate increases (i.e. from 161 to 317 μmol h−1). According to Zhou et al.,89 by preparing a Cu(OH)2/g-C3N4 composite, the photocatalyst showed a higher H2-production rate (48.7 μmol h−1 g−1), which was 16.5 times greater than that of pure g-C3N4. In2O3–g-C3N4 hybrids are used as efficient photocatalysts for H2 generation and CO2 reduction, rather than pure In2O3 and g-C3N4, as the pure In2O3 shows no noticeable H2 evolution,93 while the pure g-C3N4 shows a H2 generation rate of only 0.19 μmol h−1; whereas In2O3–g-C3N4 (10 wt%) hybrid exhibits a 5 times higher H2 generation rate (0.99 μmol h−1) than pure g-C3N4. However, an increase in the concentration of In2O3 nanocrystals (>10 wt%) on the g-C3N4 surfaces results in a decrease in photocatalytic activity for H2 generation. The photocatalytic CO2 reduction into hydrocarbon fuels is also a promising application of the In2O3–g-C3N4 hybrid photocatalyst. The In2O3–g-C3N4 hybrid photocatalyst has exhibited a CH4 production more than 3 times higher than that of neat g-C3N4 and more than 4 times higher than that of neat In2O3, with an optimum concentration of 10 wt% In2O3. The mechanism of charge transfer in the In2O3–g-C3N4 hybrid photocatalyst is presented in Scheme 8.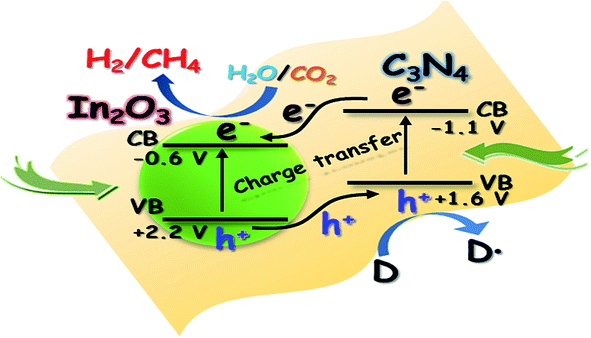 | ||
| Scheme 8 Schematic of charge transfer in In2O3–g-C3N4 (reproduced from ref. 93, licence number 3767471444007). | ||
Zhang et al.96 also studied the photocatalytic activity of Ni/NiO–g-C3N4 composites and found that Ni/NiO nanoparticles were properly dispersed on the surface of g-C3N4, indicating a strong interaction between Ni/NiO nanoparticles and the soft interface of the polymeric C3N4, thus promoting the surface kinetics by the channelization of the photoelectrons and hindering the recombination rate. The generated photoelectrons are first transferred to the Ni-core surface and then to the NiO shell and therefore speeding up the catalytic reaction, leading to a much higher HER compared to neat g-C3N4. In Ni/NiO–g-C3N4 (1 wt%), the HER is enhanced to 5 μmol h−1. But after 4 h of visible light irradiation, the HER increases to 40 μmol h−1. However, further increases in the Ni/NiO wt% decreases the photocatalytic activity and HER, although the long-term H2 evolution shows no decrease in the activity of 2 wt% Ni/NiO modified g-C3N4 as shown in Fig. 23. The hydrogen evolution data of various metallic elements and metal oxides heterojunctions with g-C3N4 is represented in Table 7.
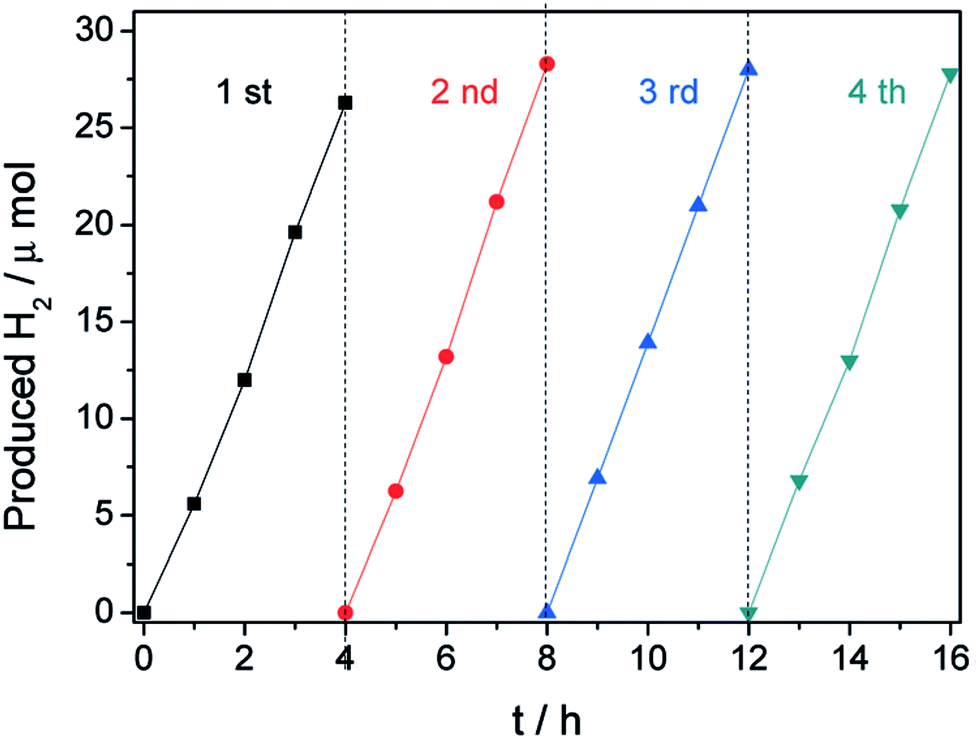 | ||
| Fig. 23 Long-term H2 evolution by a Ni/NiO/C3N4 nanocomposite under visible light (reproduced from ref. 96, licence number 3767480083704). | ||
| Metal/metal oxide | Precursor | Synthesis method | Photocatalysts | Incident light (λ) | Reaction solution | H2 evolution (μmol g−1 h−1) | Reference |
|---|---|---|---|---|---|---|---|
| Ti | TiO2 and g-C3N4 | Solid-state pyrolysis | 50 wt%TiO2/g-C3N4 | ≥436 nm | Methanol | Pt/22.4 | 81 |
| Zn | (1) Zn(NO3)2·6H2O (2) Zn(CH3COO)2 | Post-annealing solid-state pyrolysis | 10%-Zn/g-C3N4 | ≥420 nm | Methanol | 59.5 | 80 |
| CeO2 | (NH4)2Ce(NO3)6 | Solid-state reaction | 8% CeO2/g-C3N4 | ≥420 nm | Lactate solution | 73.12 | 128 |
| Sn | (i) SnCl4·5H2O in methanol | Ultrasonic deposition | 47.5% SnO2–g-C3N4/SnO2–Pt | ≥420 nm | Triethanol-amine | 900 | 82 |
| (ii) K2SnO3 in ethanol | 129 | ||||||
| N,S-TiO2 | TiOSO4·xH2O and thiourea | Thermal polymerization | 10% N,S-TiO2/g-C3N4 | ≥400 nm | Methanol | 317 | 84 |
| FeOx modified g-C3N4 | Ferrocene and urea | One-step calcination | 100 FeOx/g-C3N4 (100 Fe–CN) | ≥420 nm | Triethanol-amine | 108 | 87 |
| MoS2 | MoS2 | Solid-state reaction | 3% MoS2/g-C3N4 | ≥420 nm | 0.1 M Na2SO4 solution | 50 | 91 |
| Cu | (1) Cu(CH3COO)2·H2O | Chemical precipitation | 0.34 mol%, Cu(OH)2/g-C3N4 | ≥400 nm | Methanol | 48.7 | 89 |
| (2) Cu(NO3)2 | Hydrothermal precipitation | ||||||
| (3) CuCl2 and NaOH | |||||||
| In2O3 | In(AO)3 + g-C3N4 | Simple solvothermal | 10 wt% In2O3/g-C3N4 | ≥420 nm | 0.1 M L-ascorbic acid | 0.99 | 93 |
| NiO | Ni(NO3)2·6H2O | Direct reduction | 2 wt% Ni/g-C3N4 | ≥420 nm | Triethanol-amine | 10 | 96 |
7. Summary and outlook
The present review summarized the recent development on g-C3N4-based photocatalysts for efficient hydrogen generation under visible light irradiation. It is concluded that the visible light photocatalytic performance of g-C3N4 can be significantly improved by careful design of the nanostructure and nanoporous structure, by bandgap engineering, by surface functionalization with noble metals, non-metals and transition metals and by heterojunction construction. Although energy band engineering has been widely studied to modify the optical property of g-C3N4, further efforts are still being made to achieve the desired efficiency. More research is still required to modify the band structure, porous network and crystallinity of g-C3N4 to achieve a significant quantum yield for hydrogen production. The main drawback in g-C3N4 research lies with the high electron–hole recombination at the surface of g-C3N4, which limits its potential application. Therefore, the combination of energy band engineering with other modification strategies, such as noble metal loading/heterojunction formation, is highly encouraged to find superior ways to improve the performance of g-C3N4. Also further research on the surface activation of g-C3N4 for the purpose of the specific binding of functional groups to modify the light absorbing property in higher wavelengths is highly necessary. The potential applications of g-C3N4 can also be further extended to a wide range of heterogeneous catalysis for various reactions, like artificial photosynthesis for significant CO2 reduction to generate hydrocarbon fuels, as a reactive template for metal nitride synthesis, in nanocasting to obtain a special nanostructure, as electrocatalysts for fuel cells, in lithium ion batteries, and in sensors and solar cell applications.Acknowledgements
The authors are highly obliged to the president of ITER, S‘O’A University for the financial support for carrying out the work.References
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen and J. D. Epping, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- J. J. Zhu, Y. C. Wei, W. K. Chen, Z. Zhao and A. Thomas, Chem. Commun., 2010, 46, 6965 RSC.
- S. Martha, A. Nasim and K. M. Parida, J. Mater. Chem. A, 2013, 1, 7816–7824 CAS.
- P. W. J. Wang, J. Zhao, L. Guo and F. E. Osterloh, J. Mater. Chem. A, 2014, 2, 20338–20344 Search PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- B. Long, J. Lin and X. Wang, J. Mater. Chem. A, 2014, 2, 2942 CAS.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 118 CrossRef PubMed.
- J. Liebig, Ann. Pharm., 1834, 10, 10 Search PubMed.
- E. C. Franklin, J. Am. Chem. Soc., 1922, 44, 486 CrossRef CAS.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schloglb and J. M. Carlssonc, J. Mater. Chem., 2008, 18, 4893 RSC.
- C. Li, X. Yang, B. Yang, Y. Yan and Y. Qian, Mater. Chem. Phys., 2007, 103, 427 CrossRef CAS.
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717 CrossRef CAS PubMed.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- R. M. N. Yerga, M. C. A. Galvan, F. del Valle, J. A. V. de la Mano and J. L. Fierro, ChemSusChem, 2009, 2, 471 CrossRef PubMed.
- Y. Zheng, L. Lin, X. Ye, F. Guo and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 11926–11930 CrossRef CAS PubMed.
- S. W. Bian, Z. Ma and W. G. Song, J. Phys. Chem. C, 2009, 113, 8668–8672 CAS.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083 RSC.
- X. X. Zou, G. D. Li, Y. N. Wang, J. Zhao, C. Yan, M. Y. Guo, L. Li and J. S. Chen, Chem. Commun., 2011, 47, 1066 RSC.
- Y. Guo, S. Chu, S. Yan, Y. Wang and Z. Zou, Chem. Commun., 2010, 7, 325 Search PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang and J. Qiu, Sci. Rep., 2013, 3, 1943 Search PubMed.
- M. Mattesini, S. F. Matar, A. Snis, J. Etourneau and A. Mavromaras, J. Mater. Chem., 1999, 9, 3151 RSC.
- M. Mattesini, S. F. Matar and J. Etourneau, J. Mater. Chem., 2000, 10, 709 RSC.
- Q. X. Guo, Y. Xie, X. J. Wang, S. C. Lv, T. Hou and X. M. Liu, Chem. Phys. Lett., 2003, 380, 84 CrossRef CAS.
- X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe, K. Domen, Y. D. Hou, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniecc and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- A. Taguchi and F. Sch€uth, Microporous Mesoporous Mater., 2005, 77, 1 CrossRef CAS.
- J. Wang, C. Zhang, Y. Shen, Z. Zhou, J. Yu, Y. Li, W. Wei, S. Liu and Y. Zhang, J. Mater. Chem. A., 2015, 3, 5126–5131 CAS.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- X. Chen, Y. S. Jun, K. Takanabe, M. Kazuhiko, K. Domen, F. Xianzhi, A. Markus and X. Wang, Chem. Mater., 2009, 21, 4093–4095 CrossRef CAS.
- Z. Wang, W. Guan, Y. Sun, F. Dong, Y. Zhouc and W. K. Ho, Nanoscale, 2015, 7, 2471–2479 RSC.
- S. Kumar, T. Surendar, B. Kumar, A. Baruah and V. Shanker, RSC Adv., 2014, 4, 8132–8137 RSC.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- Z. Yang, Y. Zhang and Z. Schnepp, J. Mater. Chem. A, 2015, 3, 14081–14092 CAS.
- X. F. Chen, J. S. Zhang, X. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- Y. S. Jun, W. H. Hong, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 4270–4274 CrossRef CAS.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117, 9952–9961 CAS.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- F. Cheng, H. Wang and X. Dong, Chem. Commun., 2015, 51, 7176 RSC.
- Y. Chen, B. Wang, S. Lin, Y. Zhang and X. Wang, J. Phys. Chem. C, 2014, 118, 29981–29989 CAS.
- D. Zheng, C. Pang, Y. Liu and X. Wang, Chem. Commun., 2015, 51, 9706–9709 RSC.
- D. Zheng, C. Huang and X. Wang, Nanoscale, 2015, 7, 465 RSC.
- X. Bai, J. Lee, C. Cao and S. Hussain, Mater. Lett., 2011, 65, 1101 CrossRef CAS.
- Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244 RSC.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS.
- Z. Lin and X. Wang, ChemSusChem, 2014, 6, 1547 CrossRef PubMed.
- Y. Wang, H. Li, J. Yao, X. Wanga and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC.
- X. Ma, L. Yanhui, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Qing (Max) Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006–15012 RSC.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 48, 451 CrossRef.
- F. J. Zhang, F. Z. Xie, S. F. Zhu, J. Liu, J. Zhang, S. F. Mei and W. Zhao, Chem. Eng. J., 2013, 228, 435–441 CrossRef CAS.
- D. Jiang, L. Chen, J. Xie and M. Chen, Dalton Trans., 2014, 43, 4878–4885 RSC.
- L. Ge, C. Han, J. Liu and Y. Li, Appl. Catal., A, 2011, 409–410, 215–222 CrossRef CAS.
- L. Shi, L. Liang, J. Ma, F. Wang and J. Sun, Catal. Sci. Technol., 2014, 4, 758–765 CAS.
- S. Meng, X. Ning, T. Zhang, S. F. Chen and X. Fu, Phys. Chem. Chem. Phys., 2015, 17, 11577–11585 RSC.
- P. He, L. Song, S. Zhang, X. Wu and Q. Wei, Mater. Res. Bull., 2014, 51, 432–437 CrossRef CAS.
- J. H. Zeng and J. Y. Lee, J. Power Sources, 2005, 140, 268–273 CrossRef CAS.
- G. Zhang, Z. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Y. Zhang, D. A. J. Michel Ligthart, X. Y. Quek, L. Gao and E. J. M. Hensen, Int. J. Hydrogen Energy, 2014, 39, 11537–11546 CrossRef CAS.
- X. Y. Quek, Y. Guan and E. Hensen, Catal. Today, 2012, 183, 72–78 CrossRef CAS.
- T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467–2473 CAS.
- S. Samanta, S. Martha and K. M. Parida, ChemCatChem, 2014, 6, 1453–1462 CAS.
- S. Chang, A. Xie, S. Chen and J. Xiang, J. Electroanal. Chem., 2014, 719, 86–91 CrossRef CAS.
- J. Zhuang, W. Lai, M. Xu, Q. Zhou and D. Tang, ACS Appl. Mater. Interfaces, 2015, 7(15), 8330–8338 CAS.
- X. H. Li, X. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170 RSC.
- N. Chang, J. Tian, Q. Liu, C. Ge, A. H. Qusti, A. M. Asiri, A. O. A. Yousi and X. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6815–6819 Search PubMed.
- C. Chang, Y. Fu, M. Hu, C. Wang, G. Shan and L. Zhu, Appl. Catal., B, 2013, 142, 553–560 CrossRef.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- H. Dai, X. Gao, E. Liu, Y. H. Yang, W. Q. Hou, L. M. Kang, J. Fan and X. Hu, Diamond Relat. Mater., 2013, 38, 109–117 CrossRef CAS.
- C. Langhammer, Z. Yuan, I. Zorić and B. Kasemo, Nano Lett., 2006, 6, 833–838 CrossRef CAS PubMed.
- Y. Xiong, B. Wiley, J. Chen, Z. Y. Li, Y. Yin and Y. Xia, Angew. Chem., Int. Ed., 2005, 44, 7913–7917 CrossRef CAS PubMed.
- J. K. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shena and J. F. Zhub, Dalton Trans., 2012, 41, 6756–6763 RSC.
- H. Yan and H. Yang, J. Alloys Compd., 2011, 509, 26–29 CrossRef.
- R. Yin, Q. Luo, D. Wang, H. Sun, Y. Li, X. Li and J. An, J. Mater. Sci., 2014, 49, 6067–6073 CrossRef CAS.
- Y. Wang, B. Li, C. Zhang, L. Cui, S. Kang, X. Li and L. Zhou, RSC Adv., 2013, 3, 22269–22279 RSC.
- S. Pany and K. M. Parida, Phys. Chem. Chem. Phys., 2015, 17, 8070–8077 RSC.
- S. Hu, R. Jin, G. Lu, D. Liub and J. Gu, RSC Adv., 2014, 4, 24863–24869 RSC.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- R. Cheng, L. Zhang, X. Fan, M. Wang, M. Li and J. Shi, Carbon, 2016, 101, 62–70 CrossRef CAS.
- B. Peng, S. Zhang, S. Yang, H. Wang, H. Yu, S. Zhang and F. Peng, Mater. Res. Bull., 2014, 56, 19–24 CrossRef CAS.
- X. Zhou, L. Z. Zhihui, P. Tao, B. Jin, Z. Wu and Y. Huang, Mater. Chem. Phys., 2014, 143, 1462–1468 CrossRef CAS.
- Y. He, L. Zhang, X. Wang, Y. Wu, H. Lin, L. Zhao, W. Weng, H. Wan and M. Fan, RSC Adv., 2014, 4, 13610–13619 RSC.
- T. Yuming, G. Lei, W. Kaiyue and C. Yueshenga, Mater. Charact., 2014, 87, 70–73 CrossRef.
- A. T. Doan, D. X. Nguyen, T. P. H. Nguyen, T. V. N. Nguyen, S. J. Kim and V. Vo, Bull. Korean Chem. Soc., 2014, 35, 1794–1798 CrossRef CAS.
- S. W. Cao, X. F. Liu, Y. P. Yuan, Z. Y. Zhang, Y. S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef CAS.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS.
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215 CrossRef CAS PubMed.
- G. Zhang, G. Li and X. Wang, ChemCatChem, 2015, 7, 2864–2870 CrossRef CAS.
- J. Shi, J. Chen, Z. Feng, T. Chen, Y. Lian, X. Wang and C. Li, J. Phys. Chem. C, 2007, 111, 693 CAS.
- J. Jitputti, T. Rattanavoravipa, S. Chuangchote, S. Pavasupree, Y. Suzuki and S. Yoshikawa, Catal. Commun., 2009, 10, 378 CrossRef CAS.
- X. Lu, G. Wang, S. Xie, J. Shi, W. Li, Y. Tong and Y. Li, Chem. Commun., 2012, 48, 7717–7719 RSC.
- G. R. Bamwenda, T. Uesigi, Y. Abe, K. Sayama and H. Arakawa, Appl. Catal., A, 2001, 205, 117 CrossRef CAS.
- J. Kim, D. Hwang, H. G. Kim, S. W. Bae, J. S. Lee, W. Li and S. H. Oh, Top. Catal., 2005, 35, 295 CrossRef CAS.
- K. Domen, S. Naito, W. M. Som, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS PubMed.
- A. Kudo and H. Kato, Chem. Phys. Lett., 2000, 331, 373 CrossRef CAS.
- H. Kadowaki, N. Saito, H. Nishiyama, H. Kobayashi, Y. Shimodaira and Y. Inoue, J. Phys. Chem. C, 2007, 111, 439 CAS.
- N. Saito, H. Kadowaki, H. Kobayashi, K. Ikarashi, H. Nishiyama and Y. Inoue, Chem. Lett., 2004, 33, 1452 CrossRef CAS.
- J. Yu, Y. Zhang and A. Kudo, J. Solid State Chem., 2009, 182, 223 CrossRef CAS.
- J. S. Jang, U. A. Joshi and J. S. Lee, J. Phys. Chem. C, 2007, 111, 13280 CAS.
- N. Bao, L. Shen, T. Takata, D. Lu and K. Domen, Chem. Lett., 2006, 35, 318 CrossRef CAS.
- N. Bao, L. Shen and T. Takata, Chem. Mater., 2008, 20, 110 CrossRef CAS.
- X. Fu, X. Wang, Z. Chen, Z. Zhang, Z. Li, D. Y. C. Leung, L. Wu and X. Fu, Appl. Catal., B, 2010, 95, 393 CrossRef CAS.
- Z. Lei, W. You, M. Liu, G. Zhou, T. Takata, M. Hara, K. Domen and C. Li, Chem. Commun., 2003, 17, 2142 RSC.
- J. S. Jang, S. H. Choi, N. Shin, C. Yu and J. S. Lee, J. Solid State Chem., 2007, 180, 1110 CrossRef CAS.
- M. Tabata, K. Maeda, T. Ishihara, T. Minegishi, T. Takata and K. Domen, J. Phys. Chem. C, 2010, 114, 11215–11219 CAS.
- G. Wang, B. Huang, Z. Li, Z. Lou, Z. Wang, Y. Dai and M. H. Whangbo, Sci. Rep., 2015, 5, 8544 CrossRef CAS PubMed.
- B. B. Kale, J. O. Baeg, S. M. Lee, H. Chang, S. J. Moon and C. W. Lee, Adv. Funct. Mater., 2006, 16, 1349 CrossRef CAS.
- L. Zheng, Y. Xu, Y. Song, C. Wu, M. Zhang and Y. Xie, Inorg. Chem., 2009, 48, 4003 CrossRef CAS PubMed.
- W. Xiao, J. Yuan, Y. Zhang and W. Shangguan, Mater. Chem. Phys., 2007, 105, 6 CrossRef CAS.
- M. Hara, G. Hitoki, T. Takata, J. N. Kondo, H. Kobayashi and K. Domen, Catal. Today, 2003, 78, 555 CrossRef CAS.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, Bull. Chem. Soc. Jpn., 2007, 80, 1004 CrossRef CAS.
- N. Arai, N. Saito, H. Nishiyama, Y. Inoue, K. Domen and K. Sato, Chem. Lett., 2006, 35, 796 CrossRef CAS.
- M. Hara, T. Takata, J. N. Kondo and K. Domen, Catal. Today, 2004, 90, 313 CrossRef CAS.
- A. Kasahara, K. Nukumizu, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2003, 107, 791 CrossRef CAS.
- M. Liu, W. You, Z. Lei, G. Zhou, J. Yang, G. Wu, G. Ma, G. Luan, T. Takata, M. Hara, K. Domen and C. Li, Chem. Commun., 2004, 19, 2192 RSC.
- T. Mishima, M. Matsuda and M. Miyake, Appl. Catal., A, 2007, 324, 77 CrossRef CAS.
- K. Kamata, K. Maeda, D. Lu, Y. Kako and K. Domen, Chem. Phys. Lett., 2009, 470, 90 CrossRef CAS.
- Y. Xie, F. Wu, X. Sun, H. Chen, M. Lv, S. Ni, G. Liu and X. Xu, Sci. Rep., 2016, 6, 19060 CrossRef CAS PubMed.
- N. Tian, H. Huang, C. Liu, F. Dong, T. Zhang, X. Du, S. Yua and Y. Zhang, J. Mater. Chem. A, 2015, 3, 17120–17129 CAS.
- Y. Zang, L. Li, X. Li, R. Lin and G. Li, Chem. Eng. J., 2014, 246, 277–286 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |