
MoS2 with an intercalation reaction as a long-life anode material for lithium ion batteries†
Zhe
Hu
a,
Qiannan
Liu
a,
Weiyi
Sun
b,
Weijie
Li
a,
Zhanliang
Tao
*ab,
Shu-Lei
Chou
*a,
Jun
Chen
bc and
Shi-Xue
Dou
a
aInstitute for Superconducting and Electronic Materials, Australian Institute for Innovative Materials, University of Wollongong, Innovation Campus, Squires Way, North Wollongong, NSW 2522, Australia. E-mail: shulei@uow.edu.au
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin, 300071, People's Republic of China. E-mail: taozhl@nankai.edu.cn
cCollaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin, 300071, People's Republic of China
First published on 11th January 2016
Abstract
MoS2 with expanded layers was synthesized and characterized as an anode material for lithium ion batteries in an ether-based electrolyte by cutting off the terminal discharge voltage at 1.0 V to prevent a MoS2 conversion reaction. The as-prepared MoS2 achieved 96% capacity retention even after 1400 cycles and showed good performance in a full cell with LiCoO2 as the counter electrode.
Recently rechargeable batteries have attracted a large amount of attention, mainly because of their cyclability, as a sustainable power supply.1–3 Especially for rechargeable lithium ion batteries (LIBs), practical applications mostly facilitate social development.4,5 Among different kinds of electrode materials, MoS2 has become one of the most popular materials owing to its layered graphite-like structure.6–8 The weak van der Waals force between adjacent layers is easy to break by lithium ion insertion and a full conversion reaction provides a high specific capacity of 670 mA h g−1 (four-electron reaction).9–11 In order to achieve optimized electrochemical performances, the reaction mechanism of MoS2 cycling at 0.1–3.0 V has been widely discussed.12,13 In the first cycle, there is an intercalation process of MoS2 reacting with Li+ to form LiMoS2, which accompanies a phase change from trigonal prismatic MoS2 (2H-MoS2) to octahedral MoS2 (1T-MoS2).10 As the interlayer spacing is much larger than that of graphite, this will introduce less of a volume change regarding the intercalation process.14 Then with continuous Li+ intercalation, the structure of the layered MoS2 decomposes to Mo metal and Li2S. This step possesses large volume expansion (∼103%) leading to electrode pulverization.15–17 Because the charge products can never return back to MoS2 again, the reaction mechanism of the following cycles is the reversible reaction between Li2S and S, just the same as that for Li/S batteries. This presents the problem that MoS2 should also come across the difficulties encountered in Li/S batteries, such as the severe capacity loss owing to the active material dissolution, the polysulfide shuttling effect, and the side reaction between polysulfides and the electrolyte.18–20 So it is urgent to find an appropriate method to solve the problems mentioned above.
The most effective method is the nano-size design together with carbon modification.21 The nano-size design would provide a short ion diffusion path, which will enhance the reaction kinetics.22 By coating with carbon, the active materials can be protected from the negative effect of the volume expansion and accelerate the surface electron transportation. Qiao and coworkers synthesized mesoporous MoS2 with expanded interlayers. The as-prepared product showed an initial capacity of 1052 mA h g−1 and lasted for 100 cycles at 0.1 A g−1.18 Although carbon coating leads to great improvement of MoS2/Li batteries, the cycling performance is still hard to match to the need for commercialization, and the broadened voltage region (0.1–3.0 V) still suffers from safety issues like electrolyte decomposition, large volume change (203% after change), and the precipitation of lithium metal on the anode surface of LIBs. Thus to further improve the electrochemical performance of MoS2, there should be more modification besides carbon coating and a nano-size design.
According to previous work on MoS2, FeS2 and FeSe2, setting an appropriate cut-off voltage to prevent a conversion type reaction happening is an effective way to improve the cycling life.23–25 Py et al. excluded the possibility for lithium/electrolyte co-intercalation.26 After intercalating lithium there is only 0.14 Å expansion of the MoS2 interlayer. The intercalation reaction can also confine the charge and discharge platform mainly locating in the voltage range of 1.5–2.0 V, so this is a prominent improvement in the volume and voltage control of the intercalation reaction over the conversion type reaction. This inspires us to fabricate a full cell using MoS2 as the anode material mainly because of the low volume change and high terminal discharge voltage just like Li4Ti5O12, etc.16,27 As is known, high-voltage cathode materials (e.g. LiCoO2 and LiNi0.5Mn1.5O4) always suffer from the risk of electrolyte decomposition when charging over 4.0 V in practical use. Fig. 1 shows the typical charge and discharge curves of the cathode material LiCoO2, and anode materials MoS2, Li4Ti5O12, and graphite. Graphite is the most popular commercial anode material owing to its cheap price, relatively stable cyclability, and competitive specific capacity. However the charge and discharge curves are almost around 0 V, which probably leads to the deposition of lithium metal on the surface of the anode materials and then causes severe safety issues like short circuiting. However, materials such as MoS2 and Li4Ti5O12, which have much safer voltage regions from 1.0–3.0 V (half cell), can not only avoid safety issues like short circuiting and large volume changes during cycling (full cell) but also lower the risk of electrolyte decomposition when serving as the counter electrode of high voltage cathode materials.14,28 Nevertheless, until now there are only a few papers focusing on MoS2/Li batteries with intercalation reactions.26,29–31
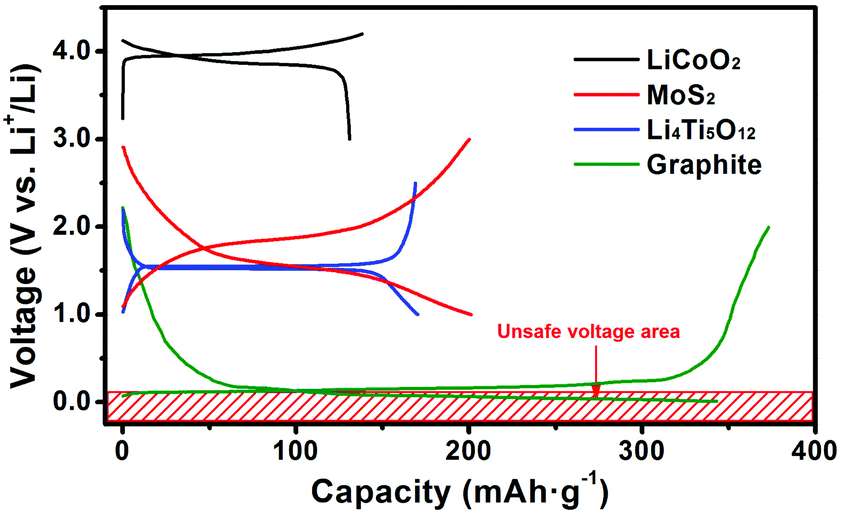 | ||
| Fig. 1 The typical charge and discharge curves of LiCoO2, MoS2, Li4Ti5O12, and graphite. | ||
Herein we have synthesized MoS2 with expanded layers (H-MoS2) through a hydrothermal process and freeze-drying method by modifying the experimental method of our previous work.23 To preserve the layer structure, obtain a relatively high specific capacity and protect the electrolyte from decomposition, the terminal discharge voltage was set to 1.0 V. The stable charge and discharge platform was ∼1.8 V and ∼1.6 V, which ensured possible applications in commercial rechargeable lithium batteries and as anodes in rechargeable LIBs (full cell with LiCoO2 as the counter electrode).
H-MoS2 represents the product procured by hydrothermal treatment, and B-MoS2 represents the bulk MoS2 purchased from Alfa Aesar (10–20 μm). Fig. S1a† illustrates the X-ray diffraction (XRD) patterns of H-MoS2 and B-MoS2. The results show that the H-MoS2 shows broadening characteristic peaks, and lower peak intensities. On the contrary, B-MoS2 retains sharp and strong XRD peaks. Meanwhile the peak shift of the (002) crystal plane indicates that the layers in H-MoS2 slightly expand. Fig. S1b and S1c† depict the high resolution transmission electron microscopy (HRTEM) images of H-MoS2 and B-MoS2. The d-spacing calibrated from the crystal fringes is in accordance with the XRD analysis: the H-MoS2 layers (layer distance of 0.69 nm) are in a disordered arrangement and are defect-rich, however B-MoS2 possesses neatly restacked MoS2 layers with a d-spacing of 0.62 nm. Through a freeze-drying process, which is also used as the most effective way to fabricate 2D/3D graphene, the ice plays an important role in supporting the morphology.32 When the ice is removed from the powders, the MoS2 layers with expanded space could be preserved.
Then the electrochemical performances were tested using the ether-based electrolyte (1.0 M lithium bis(trifluoromethanesulfonyl)imide in tetraethylene glycol dimethyl ether). Fig. 2a and S2a† exhibit the galvanostatic charge and discharge curves of H-MoS2 and B-MoS2 at 0.2 A g−1. Referring to the initial cycle, H-MoS2 has a higher discharge platform (∼1.5 V) and more specific discharge capacity (260 mA h g−1) than B-MoS2 (∼1.1 V and 181 mA h g−1) because of the different d-spacing of (002). A larger layer distance facilitates the kinetics of Li+ intercalation leading to a smaller energy barrier and more stable thermodynamics expressed as a stronger capacity for accommodating more Li+.33,34 From analysis of the cyclic data (Fig. 2b and S2b†), the capacities of the 2nd cycle reveal a slight decrease (the specific values are 195 and 108 mA h g−1 for H-MoS2 and B-MoS2, respectively), which should be ascribed to the partial side reaction and the possibility of traces of residual Li+ inside the layers. Then, the discharge capacity became a little higher (5–10 mA h g−1), which is a result of the activation of the electrode materials.10 What's more, after the 1st cycle the charge and discharge curves change a bit. The mechanism of the 1st discharge process is well investigated. The phase conversion from MoS2(2H) to MoS2(1T) should be the main reason for the curve changes.26Fig. 2b reveals the cyclic performance of H-MoS2. After the 1st cycle, the specific charge/discharge capacities are around 190 mA h g−1. Then with gradual activation, the specific capacity remains stable at 205 mA h g−1 and after 1400 cycles the capacity retention is 96% (compared to the capacity of the second cycle). The coulombic efficiency has a low value in the 1st cycle (75%), and then gradually increases to near 100% and is stable for 1400 cycles. Fig. 2c shows the rate property of H-MoS2. The discharge capacities at 0.2, 1, 2, and 3 A g−1 are 200, 115, 70, 50 mA h g−1, respectively. H-MoS2 displays a high capacity at a low current density (0.2 A g−1), and considerable capacity at 1 A g−1. The H-MoS2 battery can show a good recovery capability in high current density treatments: the discharge capacity can return back to 200 mA hg−1 at 0.2 A g−1 after treating with 3 A g−1.
 | ||
| Fig. 2 Electrochemical performances of the as-prepared MoS2 and the full cell assembled by LiCoO2 with a cathode and MoS2 as an anode. (a) Galvanostatic charge and discharge curves of MoS2 at the 1st and 200th cycles at a current density of 0.2 A g−1. (b) Cyclic performance and (c) rate property of MoS2. (d) Galvanostatic charge and discharge curves of the full cell at the voltage range from 1.5–3.5 V (inset is the cyclic performance). | ||
The half cell performance at 0.2 A g−1 in the voltage range of 0.1–3 V was also tested (Fig. S2c and S2d†). As expected, the large voltage polarization between the charge and discharge process proves the above analysis that it is not suitable for full cell use. Although the discharge capacity can reach 670 mA h g−1 (4 electrons/Li+ ions reaction), the large volume change generated from the formation of the conversion products Mo and Li2S leads capacity fade and worse reaction kinetics, which is proved by GITT characterization (Fig. S3 and S4†). It is clear that after the first discharge process, the H-MoS2 battery cycling between 0.1–3.0 V shows sluggish ion diffusion (the lithium diffusion coefficient decreases by almost 1–2 orders of magnitude) leading to a large voltage polarization (Fig. S3b†). However the H-MoS2 battery cycling from 1.0 to 3.0 V possesses a fast lithium migration (∼10−9 cm2 s−1) ensuring the stable electrochemical performances mentioned above. An carbonate-based electrolyte (1M LiPF6 ethylene carbonate and diethyl carbonate) is used to investigate the influence of the electrolyte. As shown in Fig. S5,† the Li/MoS2 cell also performs well with good cycling stability. According to the above analysis, this means that the cut-off voltage is the most important factor for achieving long life.
To further investigate the possibility of using this material as an anode material, we have fabricated a full cell using LiCoO2 as the cathode material and H-MoS2 as the anode material. The electrochemical performances of the assembled full cell are estimated using the active mass of the cathode material and are tested under 0.1 C (14 mA g−1). The galvanostatic charge and discharge curves are shown in Fig. 2d. The average charge platform is ∼2.80 V and average discharge platform is ∼2.35 V. The slope of the discharge platform is convenient and accurate for the residual capacity indication. The cyclic data inserted in Fig. 2d shows that the 1st discharge capacity is 120 mA h g−1 with a coulombic efficiency of 82%. Then the coulombic efficiency improves to near 99% along with a capacity loss from 120 to 90.5 mA h g−1. Analysis of a graphite/LiCoO2 full cell is also performed (Fig. S6†). The charge and discharge platforms are at 4.0 and 3.6 V, respectively and the discharge capacity is 132 mA h g−1. After cycling 30 times, the capacity retention is 91.6%. Thus the performance of the MoS2/LiCoO2 battery is comparable with that of a commercial type graphite/LiCoO2 battery, and moreover MoS2 possesses a higher safety factor because of its high charge and discharge voltage region (Fig. 1). The full cell technology should be improved in further investigations such as by designing high tapping density MoS2 with high electrochemical performances. However this result shows the possibility of using MoS2 as an anode material for LIBs.
The electrochemical impedance spectroscopy (EIS) measurements of H-MoS2 at different voltage ranges were also characterized (Fig. S7†). Both EIS data from 1.0–3.0 V and 0.1–3.0 V exhibit one circle at high frequency and a line at low frequency. By comparing the different voltage ranges, the charge transfer resistance of the intercalation reaction is much smaller than that of the conversion reaction. Moreover H-MoS2 shows smaller charge transfer resistance than B-MoS2, resulting from the better nano-design that facilitates the electrochemical reaction kinetics.
Fig. 3 shows further investigations of the electrode material after cycling. Transmission electron microscopy (TEM) and HRTEM images of the MoS2 electrode after cycling 100 times are shown in Fig. 3a and b. It is noticed that the graphite (conductive additive, KS-6) served as the carrier for the MoS2 particles and as the conductive substrate between the collector and MoS2 particles. We also find the MoS2 layers preserved after cycling, which means that the conversion reaction does not happen when the terminal discharge voltage is set to 1.0 V. Energy dispersive X-ray spectroscopy (EDX) measurement was also employed to detect the elemental content of Mo and S. The results show that the atomic ratio of Mo versus S is about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, however there is a loss of S of about 2%, which should be ascribed to systematic error and a partial over-discharge effect. Fig. 3d shows the electrode material after cycling 10 times in the voltage range of 0.1–3.0 V. Apparently nano-sized Mo particles are detected and found to be a little aggregated, which would cause separation of Mo and Li2S leading to a severe capacity loss.16
:2, however there is a loss of S of about 2%, which should be ascribed to systematic error and a partial over-discharge effect. Fig. 3d shows the electrode material after cycling 10 times in the voltage range of 0.1–3.0 V. Apparently nano-sized Mo particles are detected and found to be a little aggregated, which would cause separation of Mo and Li2S leading to a severe capacity loss.16
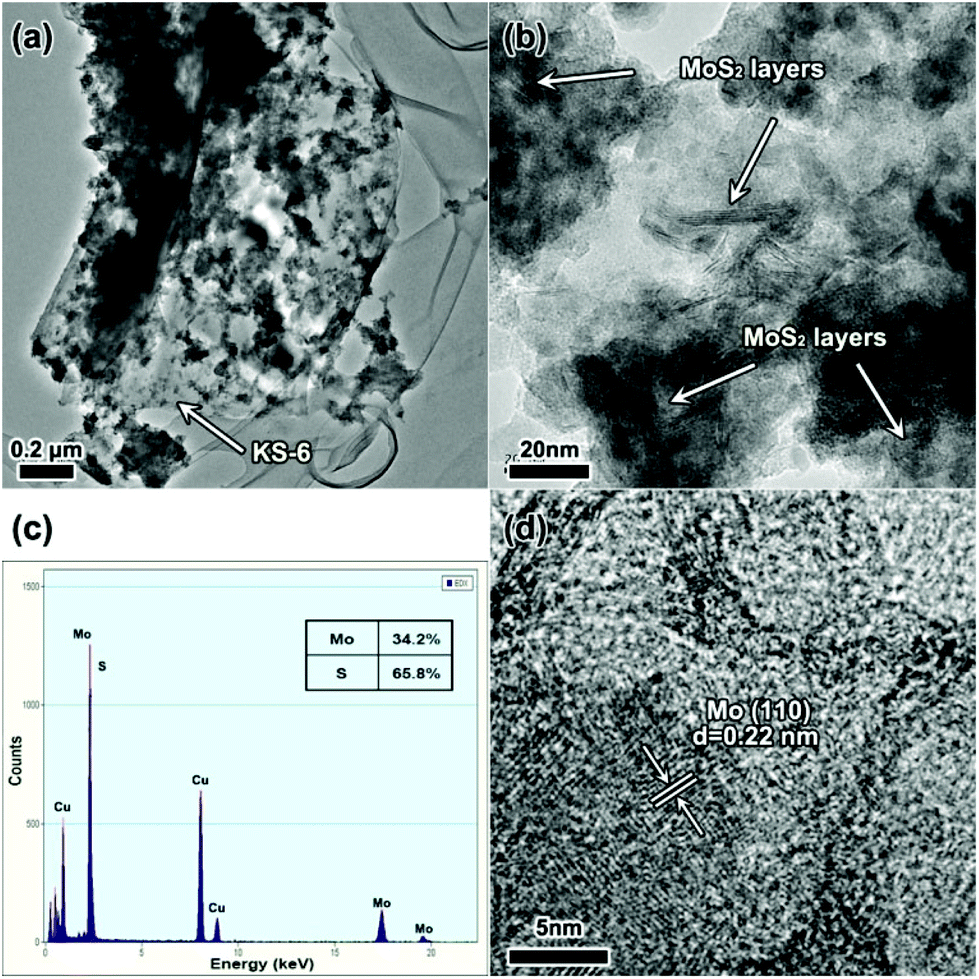 | ||
| Fig. 3 TEM characterization of the electrode material (half cell) after cycling. (a) The TEM image, (b) HRTEM image, and (c) EDX of the MoS2 electrode at 1.0 V after cycling between 1.0 V and 3.0 V 100 times. (d) HRTEM image of the MoS2 electrode at 0.1 V after cycling at 0.1–3.0 V 10 times. | ||
The overall characterization and discussion connect the proof of the excellent electrochemical properties of H-MoS2 cycling in 1.0–3.0 V. Expanded layers provide better thermodynamics and kinetics, expressed as a higher discharge voltage and fast ionic conductivity. By eliminating the conversion reaction, the wholly preserved layer-structured MoS2 ensures a rechargeable ability for MoS2/Li and LiCoO2/MoS2 batteries. A smaller charge transfer resistance reveals improved kinetics leading to a smaller voltage polarization.
Conclusions
The as-prepared MoS2 was synthesized through a hydrothermal process. By cutting off the terminal discharge voltage to 1.0 V in an ether-based electrolyte, H-MoS2 exhibits a high discharge capacity of 200 mA h g−1 at 0.2 A g−1 with a stable charge and discharge platform of ∼1.8 V and ∼1.6 V, respectively. Regarding the cyclability, it can cycle 1400 times with almost no capacity fade. Thus controlling the terminal discharge voltage should be an effective way to improve conversion type materials with two key factors; an intercalation process that occurs before the conversion reaction happens, and easy-control voltage management. More attention should focus on the intercalation reaction so that MoS2 can finally find promising applications as an anode material for rechargeable lithium ion batteries.Acknowledgements
This work is financially supported by the Australian Research Council (ARC) through a Linkage Project (LP120200432) and the Commonwealth of Australia through the Automotive Australia 2020 Cooperative Research Centre (Auto CRC). The authors acknowledge use of the facilities at the UOW Electron Microscopy Centre funded by ARC grants (LE0882813 and LE0237478). The authors would also like to thank Dr Tania Silver for the critical reading of the manuscript.Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- H. J. Yu, Y. Ren, D. D. Xiao, S. H. Guo, Y. B. Zhu, Y. M. Qian, L. Gu and H. S. Zhou, Angew. Chem., Int. Ed., 2014, 53, 8963–8969 CrossRef CAS PubMed.
- Y. Sun, J. Zhu, L. Bai, Q. Li, X. Zhang, W. Tong and Y. Xie, Inorg. Chem. Front., 2014, 1, 58–64 RSC.
- S. Xu, S. Lau and L. A. Archer, Inorg. Chem. Front., 2015, 2, 1070–1079 RSC.
- J. Zhou, J. Qin, X. Zhang, C. Shi, E. Liu, J. Li, N. Zhao and C. He, ACS Nano, 2015, 9, 3837–3848 CrossRef CAS PubMed.
- R. Tenne, Nat. Nanotechnol., 2006, 1, 103–111 CrossRef CAS PubMed.
- X. Huang, Z. Y. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433–3438 CrossRef CAS PubMed.
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS PubMed.
- K. Chang and W. X. Chen, J. Mater. Chem., 2011, 21, 17175–17184 RSC.
- J. Xiao, X. J. Wang, X. Q. Yang, S. D. Xun, G. Liu, P. K. Koech, J. Liu and J. P. Lemmon, Adv. Funct. Mater., 2011, 21, 2840–2846 CrossRef CAS.
- P. Sun, W. Zhang, X. Hu, L. Yuan and Y. Huang, J. Mater. Chem. A, 2014, 2, 3498–3504 CAS.
- Z. Wu, B. Li, Y. Xue, J. Li, Y. Zhang and F. Gao, J. Mater. Chem. A, 2015, 3, 19445–19454 CAS.
- Y. Gong, S. Yang, Z. Liu, L. Ma, R. Vajtai and P. M. Ajayan, Adv. Mater., 2013, 25, 3979–3984 CrossRef CAS PubMed.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 CAS.
- Y. X. Wang, S. L. Chou, D. Wexler, H. K. Liu and S. X. Dou, Chem. – Eur. J., 2014, 20, 9607–9612 CrossRef CAS PubMed.
- H. Liu, D. Su, R. Zhou, B. Sun, G. Wang and S. Z. Qiao, Adv. Energy Mater., 2012, 2, 970–975 CrossRef CAS.
- J. Gao, M. A. Lowe, Y. Kiya and H. D. Abruña, J. Phys. Chem. C, 2011, 115, 25132–25137 CAS.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- C. B. Zhu, X. K. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152–2156 CrossRef CAS PubMed.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309–1316 CAS.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS PubMed.
- M. A. Py and R. R. Haering, Can. J. Phys., 1983, 61, 76–84 CrossRef CAS.
- Z. Hu, K. Zhang, Z. Zhu, Z. Tao and J. Chen, J. Mater. Chem. A, 2015, 3, 12898–12904 CAS.
- Z. Zhu, F. Cheng and J. Chen, J. Mater. Chem. A, 2013, 1, 9484–9490 CAS.
- N. Imanishi, K. Kanamura and Z. i. Takehara, J. Electrochem. Soc., 1992, 139, 2082–2087 CrossRef CAS.
- C. Julien, S. I. Saikh and G. A. Nazri, Mater. Sci. Eng., B, 1992, 15, 73–77 CrossRef.
- Y. Miki, D. Nakazato, H. Ikuta, T. Uchida and M. Wakihara, J. Power Sources, 1995, 54, 508–510 CrossRef CAS.
- L. Qian and H. Zhang, J. Chem. Technol. Biotechnol., 2011, 86, 172–184 CrossRef CAS.
- K. Chang, W. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J.-Y. Lee, J. Mater. Chem., 2011, 21, 6251–6257 RSC.
- S. Q. Yang, D. X. Li, T. R. Zhang, Z. L. Tao and J. Chen, J. Phys. Chem. C, 2011, 116, 1307–1312 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, XRD, TEM, and electrochemical characterization. See DOI: 10.1039/c5qi00237k |
| This journal is © the Partner Organisations 2016 |