
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequence-regulated vinyl copolymers with acid and base monomer units via atom transfer radical addition and alternating radical copolymerization†
Takamasa
Soejima
a,
Kotaro
Satoh
*ab and
Masami
Kamigaito
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: satoh@apchem.nagoya-u.ac.jp; kamigait@apchem.nagoya-u.ac.jp
bPrecursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 24th June 2016
Abstract
Main- and side-chain sequence-regulated vinyl copolymers with acid and base monomer units were prepared using atom transfer radical addition (ATRA) and alternating radical copolymerization. A series of maleimide-ended sequence-regulated “oligomonomers” were prepared by ATRA of t-butyl acrylate (tBA: Ap) (i.e., a protected acrylic acid (A)) or an amine-functionalized acrylate, (2-(dimethylamino)ethyl acrylate (DMAEA: B)), and styrene (S) followed by an SN2 reaction of furan-protected maleimide (M) anion and its deprotection. The obtained maleimide-ended oligomonomers (M–ApS, M–SAp, M–BS, M–SB) were copolymerized with S in alternating reversible addition–fragmentation chain transfer (RAFT) copolymerizations, resulting in main- and side-chain sequence-regulated copolymers with controlled molecular weights. After deprotection of the t-butyl group, the acid and base interactions between the copolymers were evaluated by dynamic light scattering (DLS) of the polymer solutions. All the mixtures resulted in submicron particles, and the size depended on the sequence of acid monomers in the side chains. The effects of the functionalized monomer sequences on the thermal properties were also examined using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA).
Introduction
Natural functional macromolecules, such as proteins, consist of controlled sequences of “monomers” that possess hydrophobic, hydrophilic, polar, nonpolar, ionic, nonionic, aliphatic, aromatic, acid, base, or other moieties with various properties and functions. The controlled monomer sequences can dictate the shape of macromolecules and the mutual location of each monomer unit in the resulting higher-order structure to endow special properties and functions. However, monomer sequence control in polymer synthesis remains challenging, especially for synthetic vinyl polymers,1–5 because chain-growth polymerizations of comonomers typically proceed in a random or statistical fashion.Many approaches for controlling the monomer sequences in vinyl copolymers have been reported including copolymerization with highly selective propagation, such as alternating copolymerization,6–20 template polymerization,21–31 sequential multi block copolymerization,32–38 iterative single monomer addition reactions,16–18,39–46 and other polymerizations of designed monomers.39,41,46–58 However, the degree of controllability, number of controllable sequence units, and molecular weights varies. There have been only a few studies on the effects of monomer sequences on the properties and functions of these vinyl copolymers.
Recently, we developed a novel method for controlling the monomer sequences in both the main and side chains of vinyl copolymers using a combination of iterative atom transfer radical additions (ATRAs) and alternating radical copolymerization.16–18 In this method, maleimide-ended “oligomonomers” bearing sequence-controlled vinyl monomer units were prepared by iterative ATRAs followed by radical copolymerization with styrene or limonene to afford the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1 alternating copolymers. The monomer sequence in the short side chains can be perfectly controlled by iterative ATRAs, and the alternating radical copolymerization can result in relatively long main chains with alternating monomer sequences that are statistically governed by the comonomer reactivity ratios. This combined approach generates fairly high molecular weight vinyl copolymers in relatively high yield. In addition, the effects of the monomer sequences on the polymer properties (e.g., solubility and thermal properties) of the sequence-regulated vinyl copolymers were studied.
:1 or 2:1 alternating copolymers. The monomer sequence in the short side chains can be perfectly controlled by iterative ATRAs, and the alternating radical copolymerization can result in relatively long main chains with alternating monomer sequences that are statistically governed by the comonomer reactivity ratios. This combined approach generates fairly high molecular weight vinyl copolymers in relatively high yield. In addition, the effects of the monomer sequences on the polymer properties (e.g., solubility and thermal properties) of the sequence-regulated vinyl copolymers were studied.
In this study, we constructed carboxylic acid- and amine-functionalized vinyl monomer units as the monomer sequences of the side chains in the vinyl copolymers and investigated the effects of the functionalized monomer sequences on the acid–base interactions between the copolymers. The acid and base monomer units were chosen to be acrylate monomers (i.e., tert-butyl acrylate (tBA: Ap)), which is a precursor of acrylic acid (A), and 2-(dimethylamino)ethyl acrylate (DMAEA: B), respectively, and a series of maleimide (M)-ended sequence-regulated vinyl dimers with styrene (S) and functionalized acrylates (M–SAp, M–ApS, M–SB, M–BS) were synthesized using ATRAs (Scheme 1). Then, the sequence-regulated vinyl dimers were radically copolymerized with S in a 1:1 alternating fashion, resulting in main- and side-chain sequence-regulated copolymers with sequenced acid or base units in their side chains. Furthermore, the acid–base interactions of the sequence-regulated copolymers with controlled molecular weights, which were prepared by the RAFT copolymerization, were investigated.
 | ||
| Scheme 1 Sequence-regulated functional oligomonomers via atom transfer radical addition and alternating RAFT copolymerization of oligomonomers and styrene for main- and side-chain sequence-regulated vinyl copolymers with acid and base functions. | ||
Results and discussion
Synthesis of sequence-regulated oligomonomers with acid and base monomer units by ATRAs
A series of maleimide-ended sequence-regulated acid- or base-functionalized oligomonomers were prepared by ATRA between tBA, DMAEA, or styrene and their unimer halides followed by SN2 reaction between the resulting dimer halides and furan-masked maleimide anion with a retro Diels–Alder reaction to deprotect the furan group.ATRAs were first conducted between the vinyl monomers (Ap, B, S) and the excess monomeric halides (1-phenylethyl chloride (Cl–S), 1-phenylethyl bromide (Br–S), t-butyl α-bromopropionate (Br–Ap), or 2-(dimethylamino)ethyl α-chloropropionate (Cl–B)) using CuBr/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) or RuCp*Cl(PPh3)2 as the metal catalyst. As previously reported,17,18 we selectively employed RuCp*Cl(PPh3)2 for styrene terminal halides (Cl–S, Br–S) and acrylate terminal chloride (Cl–B), and CuBr/PMDETA was employed for acrylate terminal bromide (Br–Ap). These ATRAs proceeded as those reported for methyl acrylate (MA) with no functional moieties17,18 even though the products gradually decomposed during purification via lactonization of Br–SAp or quaternization between the halide and pendent amine moiety in Cl–SB and Cl–BS. Therefore, after quick purification of the obtained dimer halides, the carbon–halogen terminal was immediately converted into a furan-protected maleimide group via the subsequent SN2 reaction with the furan-masked maleimide anion. Then, the furan moiety was removed via a retro Diels–Alder reaction, resulting in a series of corresponding maleimide-ended sequence-regulated vinyl oligomonomers (M–SAp, M–ApS, M–SB, M–BS) that possess an acid- or base-monomer unit.
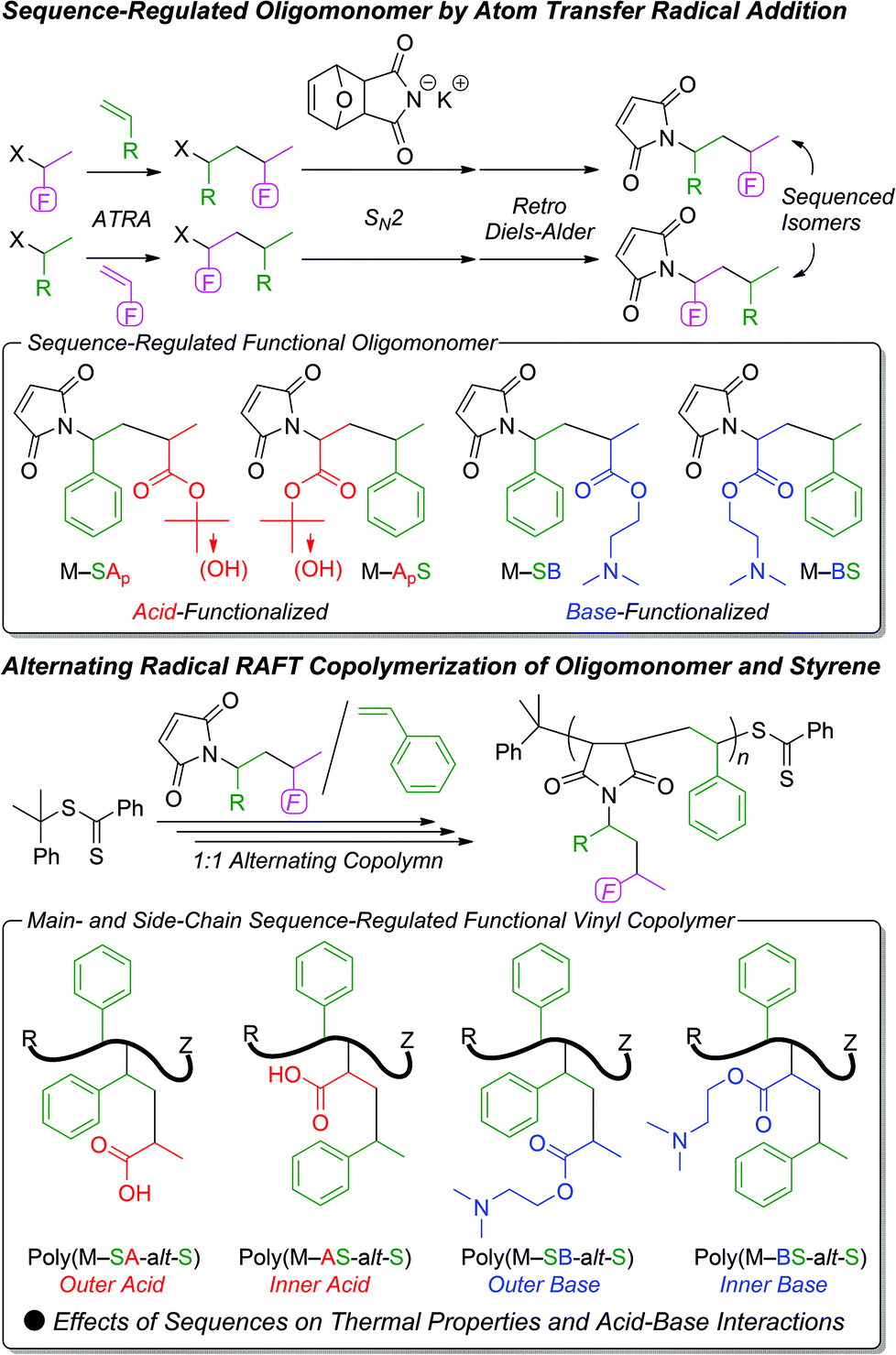
Fig. 1 shows the 1H NMR spectra of the maleimide-ended sequence-regulated oligomonomers. All these oligomonomers exhibited characteristic maleimide olefin protons (a) and dimer methine protons (b) adjacent to the nitrogen atom at 6.3–7.7 ppm and 4.3–5.2 ppm, respectively. In addition, intense peaks corresponding to the methyl protons of the t-butyl ester (f for M–SAp in Fig. 1A and c for M–ApS in Fig. 1B) and dimethylamino group (h for M–SB in Fig. 1C and e for M–BS in Fig. 1D) were observed. The structures were also confirmed by 13C NMR (Fig. S1†). These results indicate the successful synthesis of a series of maleimide-ended sequence-regulated oligomonomers bearing acid- or base-monomer units on the outside or inside of the monomer sequences.
 | ||
| Fig. 1 1H NMR spectra (CDCl3, r.t.) of a series of maleimide-ended sequence-regulated oligomonomers (M–SAp (A), M–ApS (B), M–SB (C), M–BS (D)). | ||
Synthesis of main- and side-chain sequence-regulated vinyl copolymers with acid and base groups via alternating free radical copolymerization with styrene
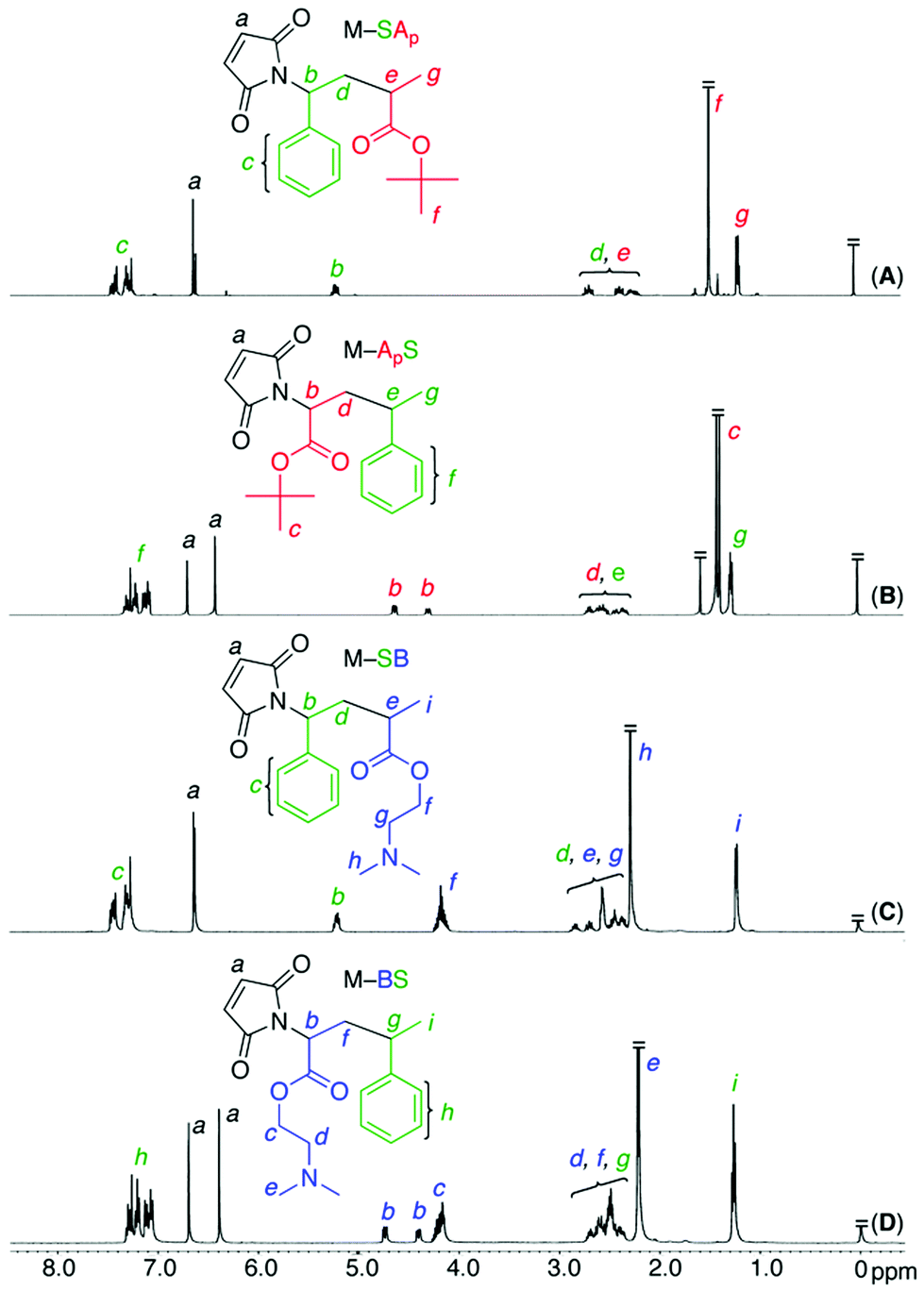
Free radical alternating copolymerization of a series of maleimide-ended sequence-regulated acid- or base-functionalized oligomonomers and styrene was investigated (Table 1 and Fig. 2). For the radical copolymerization of styrene and oligomonomers with a t-butyl acrylate unit (M–SAp, M–ApS) at a 1:1 feed ratio in toluene at 60 °C with AIBN, both styrene and the maleimide group were consumed at nearly the same rate to yield copolymers with relatively high molecular weights (Mn > 180000) similar to the previously reported unfunctionalized maleimide-ended oligomonomers and styrene.17,18 However, radical copolymerization of the amine-functionalized oligomonomers with styrene resulted in only low molecular weight polymers (Mn = 1000–2000) in toluene and DMF at 60 °C due to irreversible chain-transfer reactions caused by the amine moiety despite their consumption at nearly the same rate. However, in a fluorinated alcohol (PhC(CF3)2OH) with weak acidity, the alternating radical copolymerization successfully proceeded at 20 °C with a low temperature radical azo-initiator (V-70), resulting in alternating copolymers with higher molecular weights (Mn = 20000–40000). 1H NMR analysis of the resulting copolymers confirmed a 1:1 incorporation of the styrene and oligomonomers into the copolymers, indicating the formation of sequence-regulated alternating main chains.
 | ||
| Fig. 2 Alternating free radical copolymerization of sequence-regulated functional maleimide-ended oligomonomer (M1) and styrene (S). [M1]0 = [S]0 = 1.0 M. [AIBN]0 = 20 mM in toluene at 60 °C for M–SAp and M–ApS. [V-70]0 = 20 mM in PhC(CF3)2OH at 20 °C for M–SB and M–BS. | ||
| Entry | M1 | Time, h | Conv.c, % |
M
nd |
M
w/Mnd |
Incorp.c, % |
T
ge, °C |
T
d5f,c, °C |
After deprotection | |
|---|---|---|---|---|---|---|---|---|---|---|
| M1/S | M1/S |
T
ge, °C |
T
d5f, °C |
|||||||
| Polymerization conditions:a [M1]0/[S]0/[AIBN]0 = 1000/1000/20 mM in toluene at 60 °C.b [M1]0/[S]0/[V-70]0 = 1000/1000/20 mM in PhC(CF3)2OH at 20 °C.c Determined by 1H NMR.d Determined by SEC.e Determined by DSC.f Determined by TGA. | ||||||||||
| 1a | M–SAp | 5 | 92/91 | 243600 |
2.28 | 49/51 | 129 | 236 | 190 | 348 |
| 2a | M–ApS | 7 | 94/94 | 211400 |
2.32 | 51/49 | 143 | 201 | 194 | 347 |
| 3b | M–SB | 7 | 93/94 | 40800 |
3.01 | 50/50 | 89 | 301 | — | — |
| 4b | M–BS | 7 | 96/98 | 49100 |
2.56 | 49/51 | 98 | 256 | — | — |
Hydrolysis of the t-butyl ester in poly(M–SAp-alt-S) and poly(M–ApS-alt-S) was conducted using CF3COOH in CHCl3 at 20 °C to convert the ester to carboxylic acid groups. After hydrolysis, the obtained products were purified by precipitation in n-hexane and analyzed by 1H NMR. As shown in Fig. S2,† after hydrolysis, the sharp methyl protons of the t-butyl group completely disappeared, which was accompanied by the appearance of broad carboxylic acid signals at approximately 11 ppm. Therefore, main- and side-chain sequence-regulated vinyl copolymers with a carboxylic acid group (i.e., poly(M–SA-alt-S) and poly(M–AS-alt-S)) were successfully synthesized.
Effects of monomer sequences on the properties of sequence-regulated vinyl copolymers with acid and base groups
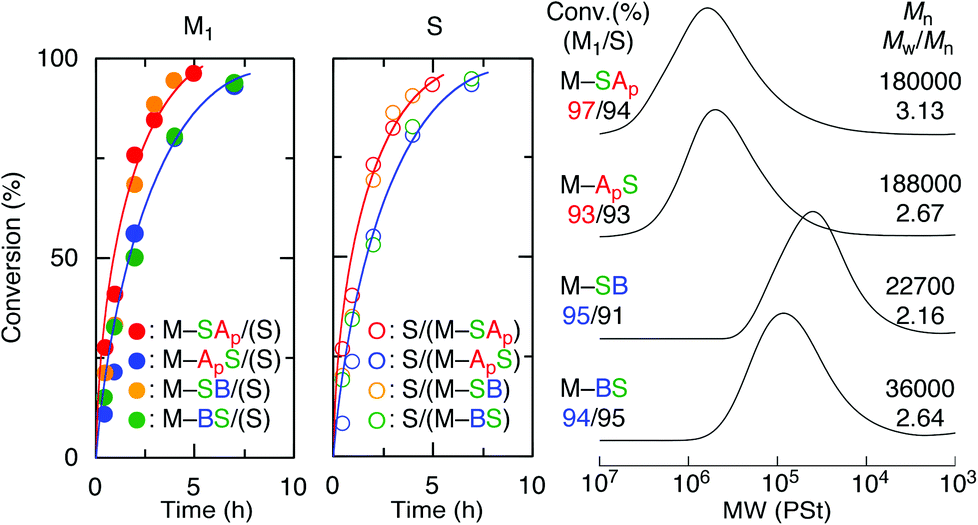
The effects of the monomer sequences and substituents of the acrylate units on the thermal properties of the sequence-regulated vinyl copolymers were evaluated using differential scanning calorimetry (DSC). All the copolymers are amorphous and exhibit glass transition temperatures (Tg) between 90 and 200 °C, and these temperatures primarily depend on the substituents of the acrylate moieties with a minor dependence on the sequences (Fig. 3). The Tg of the polymers with the tBA unit was approximately 130–140 °C (129 °C for poly(M–SAp-alt-S) and 143 °C for poly(M–ApS-alt-S)), and these values increased to 190–195 °C after deprotection of the tert-butyl group to the acid moiety (190 °C for poly(M–SA-alt-S) and 194 °C for poly(M–AS-alt-S)) (Fig. 3A). The higher Tg value of the acid forms may be due to hydrogen bonding between the polymers. However, the polymers with DMAEA exhibited low Tgs of approximately 90–100 °C (89 °C for poly(M–SB-alt-S) and 98 °C for poly(M–BS-alt-S)) (Fig. 3B) due to the amine-substituted alkyl substituents. Comparing the Tgs between a pair of isomeric polymers (i.e., poly(M–SAp-alt-S) vs. (poly(M–ApS-alt-S), poly(M–SA-alt-S) vs. (poly(M–AS-alt-S), and (poly(M–SB-alt-S) vs. poly(M–BS-alt-S)), all the polymers with outer acrylate units exhibited lower Tgs than those with inner acrylate units. As previously reported, for copolymers possessing methyl acrylate (MA) and styrene units in their side chains, the Tgs were not dependent on the sequence for the dimeric units but slightly dependent for the trimeric units.17,18 This dependence of Tg on the monomer sequence may be due to bulkier (tBu) or longer (Me2NCH2CH2) substituents than the methyl group in MA and/or polar functional (CO2H, NMe2) groups. | ||
| Fig. 3 DSC curves and Tg values of a series of main- and side-chain sequence-regulated functional vinyl copolymers (poly(M1-alt-S)) obtained in alternating free radical copolymerization of sequence-regulated maleimide-ended oligomonomers and styrene. (A) Dotted lines: poly(M–SAp-alt-S) and poly(M–ApS-alt-S). Solid lines: poly(M–SA-alt-S) and poly(M–AS-alt-S). (B) poly(M–SB-alt-S) and poly(M–BS-alt-S). | ||
The degradation behavior was also investigated using thermogravimetric analysis (TGA) (Fig. S3†). For the ester polymers, the degradation occurred via two steps. The first weight loss was approximately 12–14%, which was close to the weight% of the ester substituents (i.e., 13.2% for tBu and 16.0% for Me2NCH2CH2) in the copolymers, indicating that the first decomposition corresponds to loss of the substituents in the ester moieties. In addition, the first decomposition occurred at lower temperatures for the polymers with inner acrylate units (poly(M–ApS-alt-S) and poly(M–BS-alt-S)) but the second decomposition was nearly the same and independent of the monomer sequences. The inductive effects of the nitrogen atoms in the adjacent maleimide unit may affect the decomposition of the ester groups. In contrast, the copolymers with acid forms exhibited the same single-step decomposition behaviors, where Td5 (temperature at 5% weight loss) was 347–348 °C and nearly independent of the monomer sequences in the side chains.
Alternating RAFT copolymerization of oligomonomers with styrene for molecular weight control
To precisely determine the monomer sequence effects on the acid–base interactions of the polymers, control of the molecular weight of the sequence-regulated copolymers was investigated by RAFT copolymerization using S-cumyl S′-dithiobenzoate (CDB) as a RAFT agent (Fig. 4 and Table S1†). The RAFT copolymerization proceeded quantitatively at the same consumption rate of oligomonomers and styrene in a fashion similar to the free radical copolymerization, indicating the formation of alternating copolymers. The MWD of the obtained copolymers was narrow but slightly broader for the copolymers with an amine moiety. The molecular weights increased in direct proportion to the monomer conversion even though they were slightly lower than the calculated values based on the assumption that one RAFT agent generates one polymer chain. As we reported previously, the slightly lower molecular weights may be due to the compact hydrodynamic volume of the copolymers with pendent side chains compared to that of polystyrenes as standard samples for the molecular weight calibration.17,18 These results indicate that a series of main- and side-chain sequence-regulated vinyl copolymers with an acid or base function and nearly the same controlled chain lengths (DPn ∼ 100) were successfully synthesized.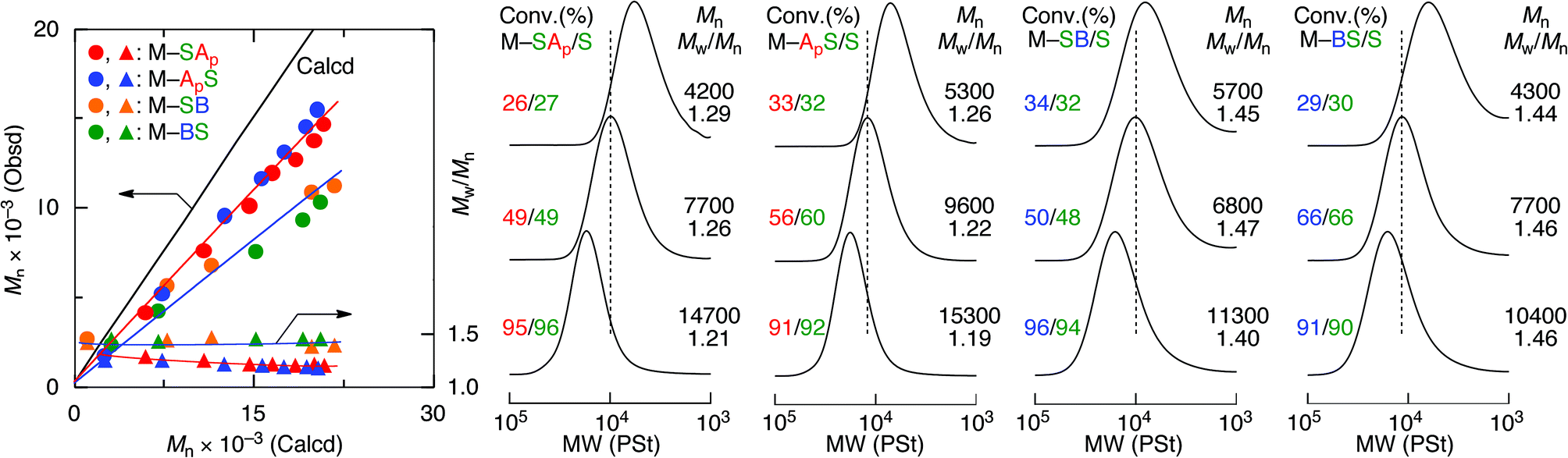 | ||
| Fig. 4 M n, Mw/Mn, and SEC curves for alternating RAFT copolymerization of a series of sequence-regulated functional oligomonomers and styrene in the presence of CDB as a RAFT agent. [M1]0 = [S]0 = 1.0 M, [CDB]0 = 20mM. [AIBN]0 = 20 mM in toluene at 60 °C for M–SAp and M–ApS. [V-70]0 = 20 mM in PhC(CF3)2OH at 20 °C for M–SB and M–BS. | ||
Effects of the monomer sequences on the acid and base interactions of the copolymers
After deprotection of the tert-butyl ester in the copolymers, the acid- and base-functionalized copolymers with controlled molecular weights prepared by RAFT were dissolved in CHCl3. The copolymer solutions were mixed together at the same acid and base concentrations ([CO2H] = [NMe2] = 0.50 mM) and analyzed by dynamic light scattering (DLS) at 20 °C. All the mixtures exhibited the presence of submicron particles, and the size of these particles depended on the functionalized monomer sequences in the side chains (Fig. 5). The mixture containing outer acid units in the side chain contained particles that were ∼330 nm irrespective of the inner or outer base units in the counterpart polymers. However, the inner CO2H copolymers exhibited smaller sizes (i.e., 170–200 nm) that were nearly independent of the inner or outer NMe2 group. Therefore, the copolymers with outer acrylic acid units resulted in larger particles with amine-functionalized copolymers, and the positions of the amino groups barely affected the size of the particles.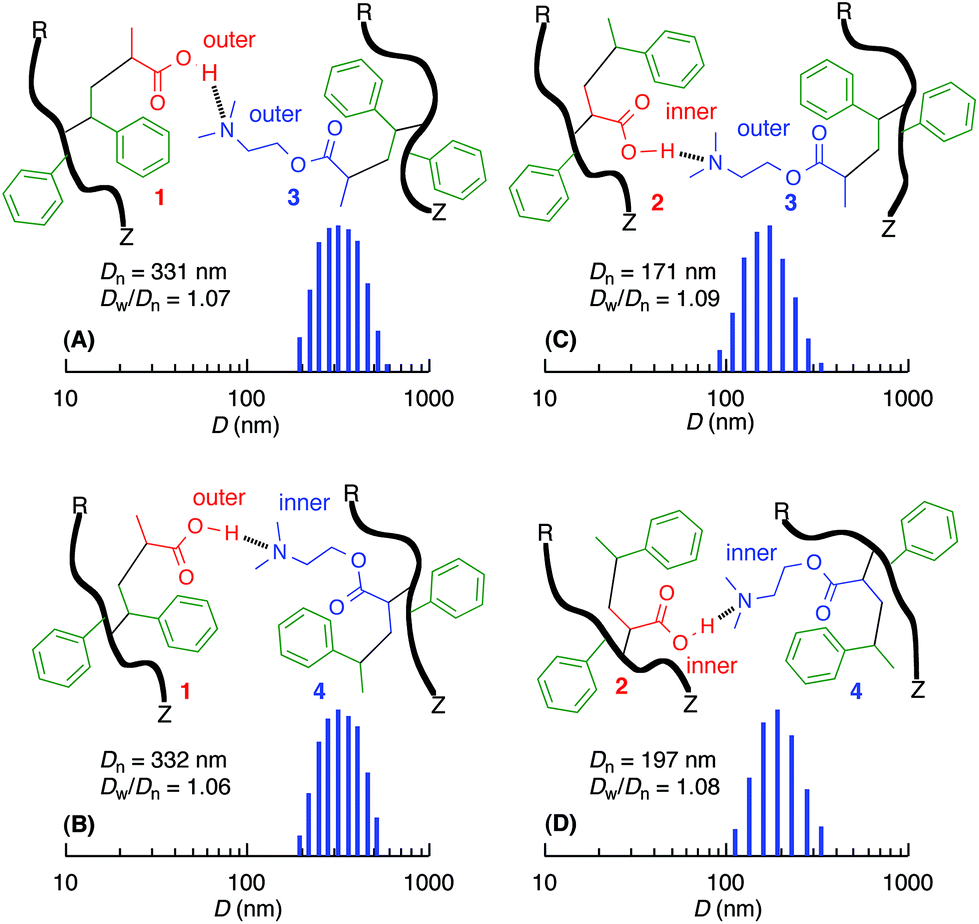 | ||
| Fig. 5 DLS analysis of mixture of acid- and base-functionalized sequence-regulated vinyl copolymers in CHCl3 at 20 °C (0.028 wt%): [–CO2H] = [–NMe2] = 0.50 mM: (A) 1 and 3, (B) 1 and 4, (C) 2 and 3, (D) 2 and 4. | ||
To further analyze the effects of the monomer sequences on the complexation behaviors, the mixing ratios of the acid and base-functionalized copolymers were gradually varied. Fig. S4† shows the dependence of the particle sizes on the ratio of the acid concentration to total concentration of the acid and base; in this figure, values lower than the middle point (0.50) indicate excess amine functions, and values higher than 0.50 indicate excess acid functions. The particle size was the smallest at the same concentration for all the mixtures and increased as the concentration of the amine or acid became larger than that of the counterpart. For the copolymers with outer acrylic acid units, the particle size gradually increased with an excess of both base and acid. However, for the inner acrylic acid units, the particle size abruptly increased and ultimately resulted in insoluble products (>1000 nm) in the presence of excess acid. However, almost no significant dependence on the sequence of the amine parts was observed.
To investigate the interactions between the acid and base groups in the copolymers, the association constants (K) between the acid or base groups and the complimentary model compounds (Me2NCH2CH2OC(O)CH3 (5) or CH3CO2H (6)) were analyzed by 1H NMR titration (Fig. S5†) because a mixture of the complimentary polymers resulted in precipitation in some cases. The K value of 5 for the inner acrylic acid unit (2) was higher (94.1 M−1) than that for the outer one (1) (20.7 M−1) under the same conditions. This result may be due to differences in the monomer sequence or the inductive effect of the adjacent imide group. The latter possibility was indicated by the larger K value (141.8 M−1) of 5 for the model carboxylic acid with the adjacent imide group (7) compared to that (17.4 M−1) for CH3CO2H (6). However, the K values of 6 for the outer (3) (19.8 M−1) or inner (4) (20.0 M−1) DMAEA unit were nearly the same and comparable to that of the model DMAE compound (5) (17.4 M−1). Therefore, the acid–base interactions between the copolymers were affected by the sequences of the functional monomer units in the sequence-regulated vinyl copolymers.
Conclusions
A series of maleimide-ended sequence-regulated oligomonomers possessing a carboxylic acid- or amine-functionalized vinyl monomer unit were successfully prepared by ATRA followed by the introduction of a maleimide group at the chain end. The maleimide-ended oligomonomers underwent alternating radical copolymerization with styrene to afford main- and side-chain sequence-regulated vinyl copolymers with sequenced acid- and base functions in the side chains. The polymers with inner functionalized monomer units in the side chains exhibited slightly higher Tgs and lower Td5s than those of the outer ones. A mixture of acid- and base-functionalized polymers resulted in submicron particles due to acid–base interactions, and the size was affected by the carboxylic acid monomer sequences but not by the amine sequences. This study revealed that the sequence of the functionalized monomer units in vinyl copolymers affects the polymer properties and aggregation behaviors between polymer chains, and these insights will lead to understanding and developing functional materials based on monomer sequence control.Experimental section
Materials
Styrene (S) (Kishida, 99.5%), tert-butyl acrylate (tBA) (Tokyo Kasei, >98%), 2-(dimethylamino)ethyl acrylate (DMAEA) (Tokyo Kasei, >97%), triethylamine (Tokyo Kasei, >99%), 1-phenylethylchloride (Cl–S) (Tokyo Kasei, >97%), 1-phenylethylbromide (Br–S) (Tokyo Kasei, >95%), and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (Aldrich, >99%) were distilled over calcium hydride under reduced pressure before use. tert-Butyl α-chloropropionate (Ap–Cl) (Wako, 97%), 2-chloropropionyl chloride (Tokyo Kasei, >95%), and 2-dimethylaminoethanol (Tokyo Kasei, >99%) were used as received. CuBr (Aldrich, 99.999%), [Cp*Ru(μ3-Cl)]4 (Aldrich, 95%), and triphenylphosphine (Aldrich, 99%) were used as received and treated in a glove box (MBRAUN LABmaster sp) under an oxygen- and moisture-free argon atmosphere (O2, <1 ppm). Toluene (Kanto, >99.5%; H2O <10 ppm) was dried and deoxygenized by passage through columns of Glass Contour Solvent Systems before use. Furan-masked-maleimide was synthesized according to the literature.17 α,α-Azobisisobutyronitrile (AIBN) (Kishida, >99%) was purified by recrystallization from methanol. 2,2′-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) (Wako, 95%) was purified by washing with acetone at −15 °C and was evaporated to dryness under reduced pressure. Cumyl dithiobenzoate (CDB) was synthesized according to the literature.59–61Synthesis of M–SAp
A maleimide-ended styrene–tert-butyl acrylate oligomonomer (M–SAp) was synthesized by copper-catalyzed ATRA followed by SN2 and retro Diels–Alder reactions as follows. CuBr (0.77 g, 5.4 mmol), PMDETA (1.0 ml, 5.4 mmol), toluene (3.0 mL), styrene (25 mL, 22 mmol), and tert-butyl α-bromopropionate (79 mL, 480 mmol) were placed in a 200 mL round-bottomed flask at room temperature. The flask was put into an oil bath at 60 °C under vigorous stirring. The styrene conversion was calculated from the vinyl group of the residual styrene measured by 1H NMR. After 3 h, the styrene conversion reached 71% to from the 1:1 adduct (Br–SAp; 65%). The reaction mixture was passed through a silica gel (Silica Gel 60N, Kanto) column eluted with n-hexane/ethyl acetate (6/4) to remove the residual copper catalyst. After evaporation of the solvents, the products were roughly distilled to yield Br–SAp (44 g, purity >80%).
To a DMF solution (150 mL) of furan-masked maleimide (24 g, 166 mmol) and K2CO3 (24 g, 190 mmol), crude Br–SAp (44 g, 120 mmol) was added at room temperature. The mixture was heated at 50 °C and kept stirred for 12 h. After the SN2 reaction, the mixture was washed with water. The solvents of the organic layer were removed by evaporation. The crude product was purified by a silica-gel column eluted with n-hexane/ethyl acetate (9/1–7/3) and then evaporated under vacuum to yield the furan-masked maleimide-ended oligomonomer of S and Ap units (22 g, 57 mmol). The furan-masked maleimide was diluted with toluene and stirred for 12 h at 115 °C for the retro Diels–Alder reaction. After evaporation of volatiles, the crude product was purified by column chromatography (Silica Gel 60N, n-hexane/ethyl acetate = 7/3) to yield pure M–SAp as a colorless viscous oil (total yield 13%, purity >98%). 1H NMR (CDCl3, r.t.): δ 1.12–1.16 (dd, CH3–CH2), 1.42–1.46 (d, (CH3)3C–O), 2.10–2.71 (m, 3H), 5.11–5.25 (m, CHN), 6.51–6.65 (d, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.16–7.49 (m, 5H).
CH), 7.16–7.49 (m, 5H).
Synthesis of M–ApS
A maleimide-ended tert-butyl acrylate–styrene oligomonomer (M–ApS) was synthesized by ruthenium-catalyzed ATRA followed by SN2 and retro Diels–Alder reactions as follows. [Cp*Ru(μ3-Cl)]4 (0.50 g, 0.50 mmol), PPh3 (1.0 g, 4.0 mmol), and toluene (5.0 mL) were placed in a 300 mL round-bottomed flask at room temperature. The reaction mixture was kept stirred for 12 h at 80 °C, and then tBA (109 mL, 750 mmol), Br–S (136 mL, 760 mmol), and Et3N (0.70 ml, 5.0 mmol) were added at room temperature. The flask was put into an oil bath at 60 °C under vigorous stirring. The tBA conversion was calculated from the vinyl group of the residual tBA measured by 1H NMR. After 65 h, the tBA conversion reached 26% to form the 1:1 adduct (Br–ApS; 19%). The reaction mixture was passed through a silica gel (Silica Gel 60N, Kanto) column with n-hexane/ethyl acetate (7/3) to remove the ruthenium catalyst. After evaporation of the solvents, the products were roughly distilled to yield Br–ApS (58 g, purity >66%).
To a DMF solution (190 mL) of furan-masked maleimide (28 g, 170 mmol) and K2CO3 (28 g, 200 mmol) crude Br–ApS (58 g, 126 mmol) was added at room temperature. The reaction mixture was heated at 50 °C and kept stirred for 12 h. After the SN2 reaction, the mixture was washed with water. The organic layer was evaporated to remove the solvents. The crude product was purified by a silica-gel column eluted with n-hexane/ethyl acetate (9/1–7/3) and then evaporated under vacuum to obtain the furan-masked maleimide-ended oligomonomer of Ap and S units (26 g, 66 mmol). The furan-masked maleimide was diluted with toluene and stirred for 12 h at 115 °C for the retro Diels–Alder reaction. After evaporation of volatiles, the crude product was purified by column chromatography (Silica Gel 60N, n-hexane/ethyl acetate = 7/3) and by recrystallization to yield pure M–ApS as colorless crystals (total yield 5%, purity >99%). 1H NMR (CDCl3, r.t.): δ 1.23–1.29 (dd, CH3–CH2), 1.36–1.42 (d, (CH3)3C–O), 2.27–2.76 (m, 3H), 4.22–4.32, 4.59–4.66 (m, CH–N), 6.41, 6.69 (d, CHCH), 7.05–7.35 (m, 5H).
Synthesis of M–SB
A maleimide-terminated styrene–2-(dimethylamino)ethyl acrylate oligomonomer (M–SB) was synthesized by ATRA followed by SN2 and retro Diels–Alder reactions as follows. 2-(Dimethylamino)ethyl α-chloropropionate was synthesized from 2-chloropropionyl chloride and 2-dimethylaminoethanol. 2-Chloropropionyl chloride (82 mL, 880 mmol) was added dropwise with stirring to a mixture of 2-dimethylaminoethanol (81 mL, 800 mmol) and triethylamine (123 mL, 840 mmol) in dry THF (340 mL) at 0 °C. The reaction mixture was stirred for 5 h at 0 °C and then for 5 h at room temperature. After dilution with n-hexane, the mixture was washed with the aqueous solution of NaOH and then NaCl followed by evaporation to remove the solvents. The product was distilled under reduced pressure to give pure 2-(dimethylamino)ethyl α-chloropropionate (58 g, 320 mmol, 40%).Then, ruthenium-catalyzed ATRA of styrene to the obtained product was conducted. [Cp*Ru(μ3-Cl)]4 (0.11 g, 0.10 mmol), PPh3 (0.21 g, 0.80 mmol), and toluene (5.0 mL) were placed in a 300 mL round-bottomed flask at room temperature. The reaction mixture was kept stirred for 12 h at 80 °C, and then styrene (11 mL, 93 mmol), 2-(dimethylamino)ethyl α-chloropropionate (35 mL, 254 mmol), and Et3N (0.15 ml, 3.2 mmol) were added at room temperature. The flask was put into an oil bath at 60 °C under vigorous stirring. The styrene conversion was calculated from the vinyl group of the residual styrene measured by 1H NMR. After 18 h, the styrene conversion reached 81% to form the 1:1 adduct (Cl–SB; 75%). The reaction mixture was passed through a silica gel (Silica Gel 60N, Kanto) column with diethyl ether to remove the ruthenium catalyst. After evaporation of the solvents, the products were roughly distilled to yield Cl–SB (17 g, 58 mmol, 63%).
To a DMF solution (91 mL) of furan-masked maleimide (9.2 g, 56 mmol) and K2CO3 (9.2 g, 67 mmol), crude Cl–SB (17 g, 58 mmol) was added at room temperature. The reaction mixture was heated at 65 °C and kept stirred for 90 h. After the SN2 reaction, the mixture was washed with water. The solvents of the organic layer were removed by evaporation. The crude product was purified by a silica gel column eluted with acetone and then evaporated under vacuum to yield the furan-masked maleimide-ended oligomonomer of S and B units (14 g, 34 mmol). The furan-masked maleimide was diluted with toluene and stirred for 11 h at 115 °C for the retro Diels–Alder reaction. After evaporation of volatiles, the crude product was purified by column chromatography (Silica Gel 60N, diethyl ether/acetone/Et3N = 9.0/0.5/0.5) to yield pure M–SB as a clear and viscous oil (total yield 7%, purity >99%). 1H NMR (CDCl3, r.t.): δ 1.22–1.30 (dd, CH3–CH2), 2.10–2.75 (m, 5H), 2.19–2.23 (d, (CH3)2–N), 4.10–4.28 (m, CH2–O), 4.36–4.44, 4.69–4.78 (m, CH–N), 6.39, 6.69 (d, CHCH), 6.99–7.34 (m, 5H).
Synthesis of M–BS
A maleimide-ended 2-(dimethylamino)ethyl acrylate–styrene oligomonomer (M–BS) was synthesized by ruthenium-catalyzed ATRA followed by SN2 and retro Diels–Alder reactions as follows. [Cp*Ru(μ3-Cl)]4 (0.6 g, 0.50 mmol), PPh3 (1.1 g, 4.0 mmol), and toluene (10 mL) were placed in a 300 mL round-bottomed flask at room temperature. The reaction mixture was kept stirred for 12 h at 80 °C, and then DMAEA (76 mL, 500 mmol), Cl–S (163 mL, 1230 mmol), and Et3N (0.7 ml, 8.0 mmol) were added at room temperature. The flask was immersed in an oil bath maintained at 60 °C under vigorous stirring. The DMAEA conversion was determined from the vinyl group of the residual DMAEA measured by 1H NMR. After 20 h, the DMAEA conversion reached 51% to form the 1:1 adduct (Cl–BS; 12%). To remove the ruthenium catalyst, the reaction mixture was drained through a silica gel (Silica Gel 60N, Kanto) column eluted with diethyl ether. After evaporation of the solvents, the products were roughly distilled to yield Cl–BS (14 g, 48 mmol, 10%).
To DMF solution (63 mL) of furan-masked maleimide (6.3 g, 38 mmol) and K2CO3 (6.3 g, 47 mmol) was added crude Cl–BS (11 g, 46 mmol) at room temperature. The mixture was heated at 65 °C and kept stirred for 15 h. After the SN2 reaction, the mixture was washed with water. The organic layer was evaporated to remove the solvents. The crude product was purified by a silica gel column eluted with acetone and then evaporated under vacuum to yield the furan-masked maleimide-ended oligomonomer of B and S units (11 g, 28 mmol). The furan-masked maleimide was diluted with toluene and stirred for 10 h at 115 °C for the retro Diels–Alder reaction. After evaporation of volatiles, the crude product was purified by column chromatography (Silica Gel 60N, diethyl ether/acetone/Et3N = 9.0/0.5/0.5) to yield pure M–BS as clear and viscous oil (total yield 7%, purity >99%). 1H NMR (CDCl3, r.t.): δ 1.19–1.24 (dd, CH3–CH2), 2.18–2.75 (m, 5H), 2.26–2.89 (d, (CH3)2–N), 4.08–4.25 (m, CH2–O), 5.15–5.25 (m, CH–N), 6.61–6.64 (d, CHCH), 7.18–7.49 (m, 5H).
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 alternating free radical copolymerization
:1 alternating free radical copolymerization
1:1 alternating free radical copolymerization was conducted under dry nitrogen in sealed glass tubes by the syringe technique. A representative example for the polymerization procedure is provided below. Toluene, M–SAp (0.65 g, 2.0 mmol), styrene (0.23 mL, 2.0 mmol), and toluene solution of AIBN (0.20 mL of 200 mM solution, 0.04 mmol) were placed in a 25 mL round-bottomed flask at room temperature. The total volume of the reaction mixture was 3.0 mL. Instantly after mixing, aliquots (0.2 mL each) of the solution were divided by syringe into baked glass tubes, which were then sealed by flame under a nitrogen atmosphere. The tubes were put into thermostatic oil bath at 60 °C. In certain intervals, the reaction mixture was cooled to −78 °C to terminate the polymerization. Conversions of oligomonomers and styrene were determined from the residual M–SAp and styrene of concentrations, respectively, by 1H NMR. The quenched reaction solutions were evaporated dryness to yield the product polymers (5 h, 92% for M–SAp and 91% for styrene, Mn = 243600, Mw/Mn = 2.28).
1:1 alternating RAFT copolymerization
RAFT copolymerization was conducted under dry nitrogen in sealed glass tubes by syringe technique. A representative example for the copolymerization with CDB in the presence of AIBN in toluene is provided below. Toluene, M–SAp (0.65 g, 2.0 mmol), styrene (0.23 mL, 2.0 mmol), toluene solution of AIBN (0.10 mL of 100 mM solution, 0.01 mmol), and another toluene solution of CDB (0.49 mL of 408 mM solution, 0.04 mmol) were placed in a 50 mL flask at room temperature. The total volume of the reaction mixture was 2.0 mL. Instantly after mixing, aliquots (0.4 mL each) of the solution were divided by syringe into baked glass tubes, which were then sealed by flame under a nitrogen atmosphere. The tubes were put into a thermostatic oil bath at 60 °C. In certain intervals, the reaction mixtures were cooled to −78 °C to terminate the polymerization. Conversion of oligomonomers and styrene were determined from the residual M–SAp and styrene of concentrations, respectively, by 1H NMR. The quenched reaction solutions were evaporated to dryness to give the product polymers (24 h, 95% for M–SAp and 96% for styrene, Mn = 15900, Mw/Mn = 1.16).
Hydrolysis of tert-butyl ester
Hydrolysis of the tert-butyl ester of the obtained polymers was conducted using CF3COOH as follows. The polymer with the tert-butyl ester (400 mg) was dissolved in CHCl3 (12 mL), and then CF3COOH (6 mL) was added dropwise with vigorous stirring at room temperature for 12 h. The reaction mixture was evaporated to dryness and the product dissolved in THF, and precipitated from n-hexane to give the polymer with carboxylic acid as a colorless powder.Measurement
1H NMR spectra were measured in CDCl3 at room temperature on a JEOL ESC-400 spectrometer, operating at 400 MHz. MALDI-TOF-MS spectra were recorded on a Shimadzu AXIMA-CFR Plus mass spectrometer (linear mode) with trans-2-[3-(4-tert-buthylpheyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) as the ionizing matrix and sodium trifluoroacetate as the ion source. The number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) of the product polymers were determined by SEC in THF at 40 °C on two polystyrene gel columns [Tosoh Multipore HXL-M (7.8 mm i.d. × 30 cm) × 2; flow rate 1.0 mL min−1] or in DMF containing 100 mM LiCl at 40 °C on two polystyrene gel columns [Shodex K-805L (pore size: 20–1000 Å; 30 cm × 8.0 mm i.d.), flow rate 1.0 mL min−1] connected to a JASCO PU-2080 precision pump and a JASCO RI-2031 detector. The columns were calibrated against standard polystyrene samples (Varian; Mp = 580–3053000, Mw/Mn = 1.02–1.23). The glass transition temperature (Tg) of the polymers was recorded on a Q200 differential scanning calorimeter (TA Instruments Inc.). Samples were first heated to 220 °C at 10 °C min−1, equilibrated at this temperature for 10 min, and cooled to −50 °C at 10 °C min−1. After being held at this temperature for 5 min, the samples were then reheated to 220 °C at 10 °C min−1. All Tg values were thus obtained from the second scan. Thermogravimetric analyses (TGA) were performed on a Q500 system (TA Instruments Inc.) at 5 °C min−1 under a N2 gas flow. DLS measurements were performed on a DLS-8000DL (Otuska Electronics) with a 632.8 nm He–Ne laser, where the scattering angle was fixed at 90°.
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research (A) (no. 15H02181) for M. K. by the Japan Society for the Promotion of Science and Program for Leading Graduate Schools “Integrative Graduate Education and Research Program in Green Natural Sciences”.Notes and references
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- J.-F. Lutz, Acc. Chem. Res., 2013, 46, 2696–2705 CrossRef CAS PubMed.
- ACS Symposium Series 1170, Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, ed. J.-F. Lutz, T. Meyer, M. Ouchi and M. Sawamoto, American Chemical Society, Washington DC, 2014 Search PubMed.
- Control of Polymer Sequences, M. Kamigaito and K. Satoh, in McGraw-Hill Yearbook of Science and Technology 2015, McGraw-Hill, New York, 2015, http://www.accessscience.com/content/control-of-polymer-sequences/YB150543 Search PubMed.
- J. M. G. Cowie, Alternating Copolymers, Plenum Press, New York, 1985 Search PubMed.
- C. Hagiopol, Copolymerization: Toward a Systematic Approach, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS PubMed.
- M. Zamfir and J.-F. Lutz, Nat. Commun., 2012, 3, 1138 CrossRef.
- J.-F. Lutz, B. V. K. J. Schmidt and S. Pfeifer, Macromol. Rapid Commun., 2011, 32, 127–135 CrossRef CAS PubMed.
- S. Srichan, H. Mutlu and J.-F. Lutz, Eur. Polym. J., 2015, 62, 338–346 CrossRef CAS.
- K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS PubMed.
- M. Matsuda, K. Satoh and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1774–1785 CrossRef CAS.
- M. Matsuda, K. Satoh and M. Kamigaito, Macromolecules, 2013, 46, 5473–5482 CrossRef CAS.
- K. Satoh, Polym. J., 2015, 47, 527–536 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, ACS Symp. Ser., 2014, 1170, 189–200 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, ACS Macro Lett., 2015, 4, 745–749 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2016, 138, 944–954 CrossRef CAS PubMed.
- A. Kanazawa and S. Aoshima, ACS Macro Lett., 2015, 4, 783–787 CrossRef CAS.
- L. R. Hutchings, P. P. Brooks, D. Parker, J. A. Mosely and S. Sevinc, Macromolecules, 2015, 48, 610–628 CrossRef CAS.
- K. Takemoto and Y. Inaki, Adv. Polym. Sci., 1981, 41, 1–51 CrossRef CAS.
- Y. Y. Tan, Prog. Polym. Sci., 1994, 19, 561–588 CrossRef CAS.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef CAS.
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Polym. Chem., 2015, 6, 1944–1951 RSC.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS PubMed.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2010, 132, 14748–14750 CrossRef CAS PubMed.
- S. Ida, M. Ouchi and M. Sawamoto, Macromol. Rapid Commun., 2011, 32, 209–214 CrossRef CAS PubMed.
- Y. Hibi, S. Tokuoka, T. Terashima, M. Ouchi and M. Sawamoto, Polym. Chem., 2011, 2, 341–347 RSC.
- Y. Hibi, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed., 2011, 50, 7434–7437 CrossRef CAS PubMed.
- R. McHale, J. P. Peterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- T. Uemura, S. Mochizuki and S. Kitagawa, ACS Macro Lett., 2015, 4, 788–791 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028–8033 CrossRef CAS.
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. M. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. S. Pappas, G. Zhang, C. Wan, P. Wilson, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Sci., 2014, 5, 3536–3542 RSC.
- A. Anastasaki, V. Nikolaou, N. W. McCaul, A. Simula, J. Godfrey, C. Waldron, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 1404–1411 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 Search PubMed.
- M. Minoda, M. Sawamoto and T. Higashimura, Macromolecules, 1990, 23, 4889–4895 CrossRef CAS.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 6 Search PubMed.
- X. M. Tong, B. H. Guo and Y. B. Huang, Chem. Commun., 2011, 47, 1455–1457 RSC.
- C.-H. Wang, Z.-Y. Song, X.-X. Deng, L.-J. Zhang, F.-S. Du and Z.-C. Li, Macromol. Rapid Commun., 2014, 35, 474–478 CrossRef CAS PubMed.
- B. Quiclet-Sire, G. Revol and S. Z. Zard, Tetrahedron, 2010, 66, 6656–6666 CrossRef CAS.
- S. Houshyar, D. J. Keddie, G. Moad, R. J. Mulder, S. Saubern and J. Tshanaktsidis, Polym. Chem., 2012, 3, 1879–1889 RSC.
- E. G. Williams, B. Fairbanks, G. Moad, R. J. Mulder, E. Rizzardo and S. H. Thang, Polym. Chem., 2015, 6, 228–232 RSC.
- Y. Hibi, M. Ouchi and M. Sawamoto, Nat. Commun., 2016, 7, 11064 CrossRef CAS PubMed.
- J. Li and J. He, ACS Macro Lett., 2015, 4, 372–376 CrossRef CAS.
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 5411–5417 CrossRef CAS.
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 8963–8970 CrossRef CAS.
- T. V. Baughman, J. C. Sworen and K. B. Wagener, Macromolecules, 2006, 39, 5208–5036 Search PubMed.
- T. V. Baughman, E. van der Aa and K. B. Wagener, Macromolecules, 2006, 39, 7015–7021 CrossRef CAS.
- T. V. Baughman, C. D. Chan, K. I. Winey and K. B. Wagener, Macromolecules, 2007, 40, 6564–6571 CrossRef CAS.
- G. Rojas and K. B. Wagener, Macromolecules, 2009, 42, 1934–1947 CrossRef CAS.
- S. Kobayashi, L. M. Pitet and M. A. Hillmyer, J. Am. Chem. Soc., 2011, 133, 5794–5797 CrossRef CAS PubMed.
- J. Zhang, M. E. Matta and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 3507–3532 Search PubMed.
- J. Zhang, M. E. Matta, H. Martinez and M. A. Hillmyer, Macromolecules, 2013, 46, 2535–2543 CrossRef CAS.
- H. Martinez, N. Ren, M. E. Matta and M. A. Hillmyer, Polym. Chem., 2014, 5, 2535–2543 RSC.
- W. J. Neary and J. G. Kennemur, Macromol. Rapid Commun., 2016, 37, 975–979 CrossRef CAS PubMed.
- J. Chiefari, R. T. A. Mayadunnem, G. Moad, E. Rizzardo and S. H. Thang, PCT Int. Appl. WO99/31144, 1999 Search PubMed.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00965d |
| This journal is © The Royal Society of Chemistry 2016 |