
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blending block copolymer micelles in solution; obstacles of blending†
Daniel B.
Wright
a,
Joseph P.
Patterson
b,
Nathan C.
Gianneschi
b,
Christophe
Chassenieux
c,
Olivier
Colombani
c and
Rachel K.
O'Reilly
*a
aUniversity of Warwick, Department of Chemistry, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: R.K.O-Reilly@warwick.ac.uk
bDepartment of Chemistry & Biochemistry, University of California, 9500 Gilman Drive, La Jolla, San Diego, CA, USA
cLUNAM Université, Université du Maine, IMMM UMR CNRS 6283 Département PCI, Avenue Olivier Messiaen, 72085 Le Mans Cedex 09, France
First published on 26th January 2016
Abstract
Amphiphilic block copolymers can assemble into a variety of structures on the nanoscale in selective solvent. The micelle blending protocol offers a simple unique route to reproducibly produce polymer nanostructures. Here we expand this blending protocol to a range of polymer micelle systems and self-assembly routes. We found by exploring a range of variables that the systems must be able to reach global equilibrium at some point for the blending protocol to be successful. Our results demonstrate the kinetic requirements, specifically core block glass transition temperature, Tg, and length of the block limiting the exchange rates, for the blending protocol which can then be applied to a wide range of polymer systems to access this simple protocol for polymer self-assembly.
Introduction
Much like small molecule surfactants, amphiphilic block copolymers will spontaneously self-assemble when dispersed in a selective solvent for one of the copolymer blocks.1 Although small molecule surfactants and amphiphilic block copolymers both exhibit similar behavior in solution, once the molecules have assembled into micelles there are some behaviors which are vastly different between the two. The most prevalent difference in solution is that surfactant micelles will typically reach thermodynamic equilibrium mainly through a constant exchange of surfactant molecules between structures,2–4 whereas amphiphilic block copolymers only rarely reach the true Gibbs free energy minima and hence thermodynamic equilibrium.5,6 Predominantly the exchange of polymer chains between structures is often kinetically hindered because the energy barrier for unimer exchange is too great and the concentration of polymer chains free in solution is typically negligible.7,8 Unimer exchange can occur at a reasonable rate only if the solvophobic block is sufficiently mobile (low glass transition temperature, Tg) and has a low solvophobicity (low interfacial tension with respect to the solvent).5,9–11Such out-of-equilibrium behavior often restricts block copolymer developments for an array of applications and prevents the full understanding of the self-assembly of diblock copolymers in solution as the ability to control and form reproducible stable structures at thermodynamic equilibrium can be challenging.12 One method to overcome these challenges is to use block-random block copolymers; polymers consisting of (a) pure homopolymer block(s) and (b) copolymer block(s).13 This development of new amphiphilic block-random block copolymers has opened up a range of new structures in solutions, with some structures reaching equilibrium.14–21 Nevertheless, the sometimes laborious synthesis of diblock copolymers often negatively impacts the large scale implementation of functional diblock copolymers. An alternative is the copolymer blending method, which is based on the blending together of two polymers, that vary in functionality or response to stimuli, to obtain a blended mixture; where the blended structure has the composition and/or response which is an intermediate between the two parent polymers.22–25 As shown previously this blending strategy is a promising new method for block-random diblock copolymer assembly.26 These blends of block-random diblock copolymers formed structures identical to those formed by a single block-random copolymer with the same composition as that of the blend. This offers a simple and accessible method towards a variety of different nanostructures in solution by using only two polymers blended in different stoichiometric ratios.
The ability to successfully mimic the structure of pure spherical micelles through this blending strategy is believed to crucially depend on the system's ability to equilibrate at some point during or after assembly. Thus the dynamic nature of polymer micelles is a precondition for this strategy as hypothesised by Tian et al. who attempted to blend poly(styrene)-block-poly(methacrylic acid) diblock copolymers in water/dioxane solvent mixtures.22 This equilibration is often a misunderstood topic of polymer self-assemblies as it is difficult to deduce how far from equilibrium a system truly is or if equilibrium is indeed reached.5,6,11,27–32 Furthermore, the out-of-equilibrium micelles will typically produce structures that have large pathway dependence and consequently the same polymer could produce an array of different nanostructures in solution.
Consequently, to further understand the ability of the copolymer blending method to produce blend micelles, which structurally mimic pure micelles,26 a series of polymer systems and a range of assembly conditions were studied. Hence the parameters needed for successful blending were investigated. The fundamental understanding of this copolymer blending method is developed and its limitations are elucidated further to expand the copolymer blending protocol towards an array of copolymer systems.
Experimental
Materials
Monomers were filtered through a plug of silica prior to use and stored at 4 °C. AIBN (2,2′-azo-bis(isobutyronitrile)) was recrystallized from methanol and stored in the dark at 4 °C. All other materials were used as received from Aldrich, Fluka, or Acros.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume compared to monomer) was added to a dry ampoule containing a stirrer bar. The solution was degassed using at least 3 freeze–pump–thaw cycles, back filled with nitrogen, sealed and placed in a pre-heated oil bath at 70 °C. After 2 hours the conversion reached more than 99%. The polymerization was quenched by liquid nitrogen and opened to air, the crude reaction was diluted with the minimum amount of dichloromethane (CH2Cl2), the solution was precipitated into cold diethyl ether and the polymer isolated by filtration. Precipitation was repeated two more times to afford a yellow macroCTA polymer powder (see Table 1 for molecular weight data).
:1 volume compared to monomer) was added to a dry ampoule containing a stirrer bar. The solution was degassed using at least 3 freeze–pump–thaw cycles, back filled with nitrogen, sealed and placed in a pre-heated oil bath at 70 °C. After 2 hours the conversion reached more than 99%. The polymerization was quenched by liquid nitrogen and opened to air, the crude reaction was diluted with the minimum amount of dichloromethane (CH2Cl2), the solution was precipitated into cold diethyl ether and the polymer isolated by filtration. Precipitation was repeated two more times to afford a yellow macroCTA polymer powder (see Table 1 for molecular weight data).
| Monomer | Polymer | x | n | m | M n NMRb (kDa) | M n SECc (kDa) | Đ SECc |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by end-group analysis from 1H NMR spectroscopy. c From SEC based on poly(styrene) standards in CHCl3. | |||||||
| EHA | 1 | 0.60 | 120 | 70 | 22.9 | 25.0 | 1.11 |
| 2 | 0.50 | 120 | 69 | 22.2 | 24.9 | 1.20 | |
| 3 | 0.40 | 120 | 71 | 21.3 | 23.2 | 1.20 | |
| 4 | 0.90 | 350 | 250 | 77.3 | 77.9 | 1.11 | |
| 5 | 0.80 | 350 | 228 | 70.5 | 79.1 | 1.18 | |
| 6 | 0.70 | 350 | 260 | 71.3 | 74.6 | 1.14 | |
| IBA | 7 | 0.60 | 120 | 68 | 23.1 | 23.6 | 1.30 |
| 8 | 0.50 | 120 | 71 | 23.3 | 22.7 | 1.27 | |
| 9 | 0.40 | 120 | 66 | 22.0 | 25.8 | 1.24 | |
:1 volume compared to the monomer) and were added to a dry ampoule containing a stirrer bar. The solution was degassed using at least 3 freeze–pump–thaw cycles, back filled with nitrogen, sealed and placed in a pre-heated oil bath at 70 °C. After 5 hours the conversion reached more than 99% and the polymerization was quenched by liquid nitrogen, the crude reaction was diluted with the minimum amount of dichloromethane (CH2Cl2) and the solution was precipitated into diethyl ether and the polymer was isolated by filtration. Precipitation was repeated two more times to afford a yellow powder (see Table 1 for molecular weight data).
:1 volume compared to the monomer) and were added to a dry ampoule containing a stirrer bar. The solution was degassed using at least 3 freeze–pump–thaw cycles, back filled with nitrogen, sealed and placed in a pre-heated oil bath at 70 °C. After 5 hours the conversion reached more than 99% and the polymerization was quenched by liquid nitrogen, the crude reaction medium was diluted with the minimum amount of dichloromethane (CH2Cl2) and the polymer was precipitated into diethyl ether and isolated by filtration. Precipitation was repeated two more times to afford a yellow powder (see Table 1 for molecular weight data).
Reactivity ratios of DMA and hydrophobic monomer
DMA and hydrophobic monomer (IBA or EHA) at different ratios, CMDT and AIBN were dissolved in 1,4-dioxane. The ratio of [monomers]:[CTA]:[AIBN] was 70:1:0.1, the solution was degassed using at least 3 freeze–pump–thaw cycles, back filled with nitrogen, sealed and placed in a pre-heated oil bath at 70 °C. The conversion was kept below 10% and the reaction was quenched by liquid nitrogen and opened to air. Aliquots were taken and characterised by 1H NMR spectroscopy in CDCl3.
Preparation of the aqueous solutions
Results and discussion
Synthesis and molecular characterization of the diblock copolymers
For this blending study a series of diblock copolymers were synthesized (Fig. 1). To explore the copolymer blending method further block-random diblock copolymers, consisting of a random copolymeric solvophobic block whose chemical structure and properties can be varied by copolymerization and a homopolymer solvophilic block,13 were prepared. First two hydrophilic poly(dimethyl acrylamide), P(DMA), macro chain transfer agents (macro-CTAs) were synthesized by RAFT polymerization with a degree of polymerization 120 or 350. These macro-CTAs were then chain extended by a random block consisting of dimethyl acrylamide (DMA) as hydrophilic monomer and a hydrophobic monomer (either isobornyl acrylate (IBA) or 2-ethylhexyl acrylate (EHA) respectively), to afford block random diblock copolymers with a homopolymeric block and a statistical block (see ESI† for reactivity ratio data). All diblock copolymers formed had low dispersity, known hydrophobic monomer incorporation and controlled molecular weight, Table 1.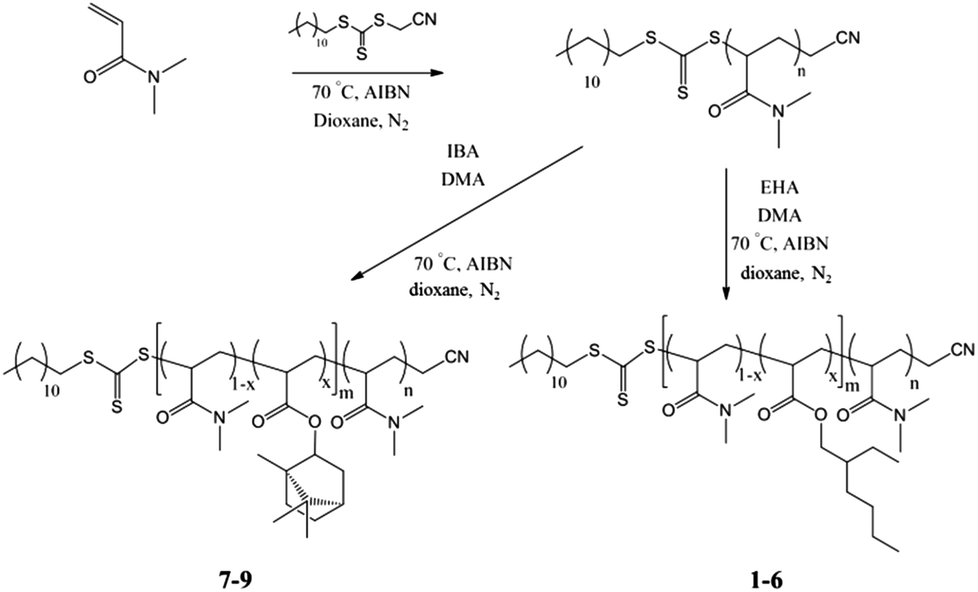 | ||
| Fig. 1 General schematic of the synthesis of the DMA-based amphiphilic block random diblock copolymers via RAFT polymerization. | ||
Blending of diblock copolymers
We investigated the self-assembly of diblock copolymer blends in aqueous solutions, where these blend micelles can structurally match pure micelles with the same average composition. Based on previous reports it is hypothesised that equilibrium must be reached in order to ensure the spontaneous formation of blend micelles.26 To understand this hypothesis and to explore the blending protocol further, a range of different polymers and assembly conditions were utilized.The two block-random diblock copolymers differ in the block random composition and once blended gave an intermediate composition between the two parent polymers. Once assembled these blend micelles can be compared structurally to a pure system where the composition is achieved from synthesis as opposed to blending. For the P(EHA-co-DMA)-b-PDMA diblock copolymer blends the polymers 1 and 3 were blended together to match the composition of 2. In the second system 4 and 6 were blended together to match the composition of 5. For the P(IBA-co-DMA)-b-PDMA diblock copolymer blends the polymers 7 and 9 were blended together to match the composition of 8. Blended systems were all targeted to obtain one of three different compositions but the assembly pathway was varied. To distinguish between polymer systems the notation is as follows; N-M-2, N signifies if the polymer samples are blend (B) or pure (P), M represents the pathway: DD is direct dissolution, TF is thin film rehydration, and SS is solvent switch (see Materials and methods for details), 2 corresponds either to the pure polymer constituting the sample for pure samples or to the polymer composition which is to be targeted in blends; see Table 1. For the molar blending ratios and assembly routes investigated see Tables S3–S5.†
Influence of pathway dependence on blending
It is well known that for amphiphilic diblock copolymers different pathways for assembly can produce a range of structures if these structures are not formed under thermodynamic equilibrium.5,7,12,33 Hence, initially a range of different assembly pathways were employed and the ability to form blend micelles at equilibrium for each was examined. No matter the pathway, all solutions were initially prepared at room temperature here.The first sets of studies were the P(EHA-co-DMA)-b-PDMA polymers 1–3 with blends B-DD-2, B-TF-2, and B-SS-2. Static and dynamic light scattering (SLS and DLS) experiments were conducted at 20 °C and were used to analyze the structure of the blended micelles in water (Fig. 2a and b). First, the results represented in Fig. 2a and b indicate that the three preparation pathways (DD, TF and SS) led to different final states, stronger differences being observed for the 1 and 3 mixtures than for 2.42 This is evidence that unimer exchange was not fast enough at room temperature in aqueous medium for this system, otherwise, all preparation methods would have led to the same final state at thermodynamic equilibrium. Note that the hydrophobic monomer in these copolymers is EHA, leading to P(DMA-co-EHA) copolymers which have low Tg (see ESI† for thermal analysis). Consequently, the hydrophobic block is sufficiently mobile in this set of studies and cannot be the factor limiting unimer exchange.
 | ||
| Fig. 2 Relationship of Rh (a) and Nagg (b) with preparation pathway for the blended and pure solutions of P(EHA-co-DMA)-b-PDMA diblock copolymers “2” obtained at room temperature (RT) and after heating (Heated). X = Direct dissolution (DD), Thin film rehydration (TF), Solvent switch (SS). Error bars indicate 10% error on Nagg and Rh. The dashed line in b) corresponds to the Nagg theoretically expected for a mixture of non-blended aggregates of 1 and 3 at equilibrium, see ESI† for details. | ||
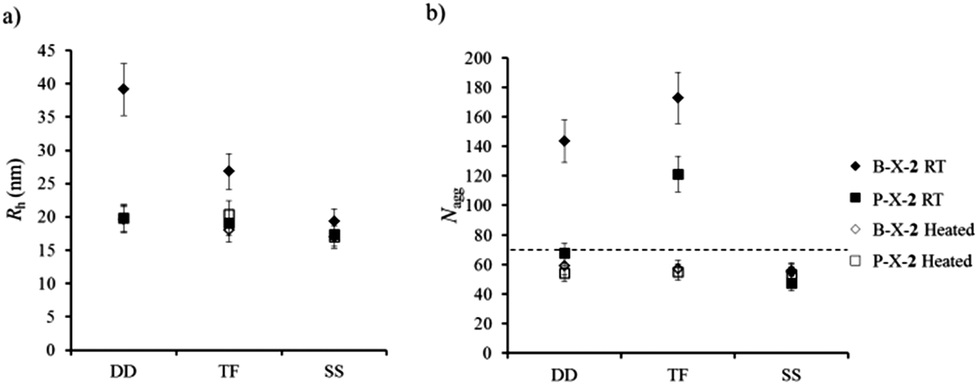
It is understood that for polymeric assemblies there is an energy barrier for the expulsion of the hydrophobic block which is relative to the block length, N, and its interfacial tension with the solvent, γ.10,11,31,34 In an effort to overcome this barrier and to reach thermodynamic equilibrium all polymer solutions were heated to 75 °C for 5 h and were then reanalyzed by SLS and DLS at 20 °C. After the heating cycle the DLS and SLS results showed that blend micelles prepared according to the DD and TF pathways reorganize whereas those prepared by the SS pathway showed no change (Fig. 2a and b). Additionally the blend micelles become notably less disperse after the heating cycle, which can be a strong indication of an equilibrium state being reached, as observed by DLS and cryo-TEM (Fig. 3). More importantly, Nagg and Rh values are identical for all routes of assembly after the heating cycle and whether the system is pure or blend (Fig. 2a and b). It is thus very probable that these structures are close to, or at, equilibrium. It should also be mentioned that the SS method most probably led directly to structures at equilibrium without the need for a heating cycle since no reorganization was observed upon heating in this case and, still, the final state was the same as that observed with the two other pathways. In the remainder of this section, only these equilibrated structures are discussed.
 | ||
| Fig. 3 (a) Distribution of relaxation times from DLS of P-TF-2, B-TF-2 after heating and B-TF-2 without heating. (b) cryo-TEM micrograph for blend micelles from the TF protocol after heating. (c) Distribution of relaxation times from DLS of P-DD-2, B-DD-2 after heating and B-DD-2 without heating. (d) cryo-TEM micrograph for blend micelles from the DD protocol after heating. | ||
The structures observed after heating do not seem to consist of pure aggregates of polymer 1 mixed with pure aggregates of polymer 3, but of blend micelles with both 1 and 3. Indeed the aggregation number of the blends is significantly different from the value expected for a mixture of non-blended aggregates of 1 and 3 (see dashed line in Fig. 2 and ESI† for the details of the calculation of the expected value of Nagg for a solution of non-blended aggregates). Moreover, these blend micelles mimic those obtained with pure polymer 2 with similar aggregation number and hydrodynamic radius, see Fig. 2.
At this point, it may be argued that the difference between the experimental Nagg and the theoretical Nagg expected for non-blended aggregates is not large enough to be sure that blending occurred. Furthermore, assuming that blended micelles were obtained, it is not possible to determine whether they were formed via unimer exchange during the reorganization occurring upon heating or through a more complex scenario not necessarily requiring unimer exchange. In particular, it is very probable that with the TF preparation method, polymer chains of 1 and 3 were intimately mixed within the film so that blended aggregates are necessarily formed even at room temperature upon addition of water, albeit out-of-equilibrium. In this case, these out-of-equilibrium structures may rearrange upon heating without the need for unimer exchange as observed by Bendejacq et al. on PS-b-PAA diblock copolymers.35
In order to prove more clearly that blended aggregates were indeed formed by mixing 1 and 3 and that blending requires unimer exchange, a solution blending route was additionally used. Here, 1 and 3 are assembled and heated individually to form micelles at steady state, Table S7.† It should be highlighted that no matter whether these micelles were formed using the DD, TF or SS method, their characteristics were the same after heating, again indicating that the micelles were probably at equilibrium. The solutions were then mixed at room temperature and analyzed by DLS and SLS. After 10 days, no reorganisation could be observed for all blend systems, Table 2. Moreover, the Nagg and Rh values determined for the mixture at room temperature are equal to the values expected for a solution of independent micelles of 1 and 3. This confirmed that the micelles were frozen at room temperature and could not reorganize to form blended micelles. Moreover, it showed that Nagg is determined with an accuracy of ∼±3.
| Mixed solution | N agg | N agg | N agg | N agg | R h | R h | R h | R h |
|---|---|---|---|---|---|---|---|---|
| Day 0 | Day 10 | Eq. S8 | Post heat | Day 0 (nm) | Day 10 (nm) | Eq. S9 | Post heat | |
| B-TF-2 | 72 | 72 | 70 | 54 | 17 | 17 | 16 | 16 |
| B-DD-2 | 71 | 71 | 67 | 55 | 18 | 18 | 16 | 15 |
| B-SS-2 | 67 | 67 | 72 | 53 | 14 | 13 | 17 | 15 |
| P-SS-2 | 52 | 52 | N/A | 55 | 15 | 17 | N/A | 16 |
These blended solutions were then subjected to the heating cycle and reanalyzed by laser light scattering, Table 2. It can be observed for all solutions regardless of pathway that the structures do reorganize upon heating and reach the same final state as when the two polymers were directly mixed together before assembly. Moreover, the Nagg of the heated system is clearly different from the theoretical value expected for a solution of non-blended micelles of 1 and 3. This experiment definitely proved that polymers 1 and 3 were not able to exchange unimers at room temperature; but that upon heating unimer exchange is made possible leading to blend micelles which mimic micelles of pure polymer 2 with the same monomer composition as the blend. The fact that unimer exchange is too slow at room temperature but becomes possible upon heating was corroborated by variable temperature 1H NMR spectroscopy in order to analyze core mobility (see ESI†).
These experiments demonstrate that the formation of blend micelles of 1 and 3 was thermodynamically favourable and was indeed observed by heating an aqueous solution of 1 and 3 at 75 °C for 5 hours. The same blend structures could directly be obtained at room temperature without heating using the SS method, implying that the SS method probably allows the system to reach thermodynamic equilibrium directly. For this pathway, a non-selective solvent is used, implying that only unimers are initially present in the absence of water. These unimers self-assemble upon addition of water but probably remain in equilibrium with their aggregates at the lowest water contents, allowing the system to reach equilibrium at room temperature, as previously observed for poly(styrene)-block-poly(acrylic acid) diblock copolymers in solvent/water mixtures by Eisenberg and coworkers.36–39 In contrast, the DD and TF methods lead to out of equilibrium structures at room temperature. These structures may still be partially blend micelles but are nevertheless far from equilibrium.
The influence of the pathway on the characteristics of the structures obtained at room temperature or after mixing equilibrated solutions of the parent polymers was a clear indication of the inhibition of the formation of equilibrated blend micelles at room temperature. It is important to emphasise that all routes lead to the same micelle structures once heated (Fig. 1 and Table 2) although for blended micelles to be formed unimer exchange must occur at some point.
Effect of an energy barrier
To explore the effect of a kinetic energy barrier which must be overcome to form blend micelles at equilibrium, a second P(EHA-co-DMA)-b-PDMA diblock copolymer series was synthesized, 4–6. The kinetic energy barrier related to the extraction of the hydrophobic block from the core of the micelles is proportional to the associating block length and interfacial tension between the associating block and the solvent.9,10 Hence, a P(EHA-co-DMA) associating block with a much larger degree of polymerization ca. 250 was synthesized and the EHA incorporation was greater than 70 mol%, Table 1. This generates a theoretically higher energy barrier for molecular exchange and possibly will produce kinetically trapped structures during the blending.Here copolymers 4 and 6 were blended together to give an average composition to match 5, Table 1. In contrast to the previous P(EHA-co-DMA)-b-PDMA diblock copolymers these copolymers showed for the direct dissolution and thin film rehydration protocols polydisperse aggregates displaying a multi populated and broad distribution in size (see ESI†) which meant that quantitative analysis of static light scattering measurements was not possible. These results are confirmed by cryo-TEM analysis which was undertaken to explore the copolymer blending method further. Indeed, the nanostructures formed from these two assembly routes exhibit multiple morphologies (see Fig. 4a and b). The presence of multiple morphologies and the fact that different pathways lead to different structures provide a strong indication that these structures were frozen at room temperature. This was expected since polymers 1 and 3 were shown to be unable to exchange unimers at room temperature despite the shorter P(EHA-co-DMA) block and lower EHA content. The formation of equilibrium blend structures at room temperature was restricted for this system.
 | ||
| Fig. 4 Cryo-TEM micrographs of P(EHA-co-DMA)-b-PDMA blend. (a) Prepared by direct dissolution, B-DD-5. (b) Prepared by thin film rehydration, B-TF-5. (c) Prepared by solvent switch, B-SS-5. | ||
In a similar manner to polymers 1–3, B-DD-5, B-TF-5 and B-SS-5 underwent the same heating cycle and the solutions were reanalyzed. In contrast to blend systems of 2 no change in cryo-TEM or light scattering analysis were observed, highlighting a lack of reorganization even at 75 °C. This frozen behavior even at high temperature was also corroborated by variable temperature 1H NMR spectroscopy analysis (see ESI† for further data).
Conversely, for the solvent switch protocol cryo-TEM images show only one population of spherical micelles (Fig. 4c). Laser light scattering analysis was undertaken and both blend and pure micelles were identical before and after heating for this pathway. This result is consistent with the B-SS-2 and P-SS-2 systems, suggesting that the structures formed by both blend and pure samples are indistinguishable and the structures formed by the solvent switch route are close to equilibrium and likely form before the solvent quality is reduced and chain exchange is significantly reduced.
Effect of the core Tg
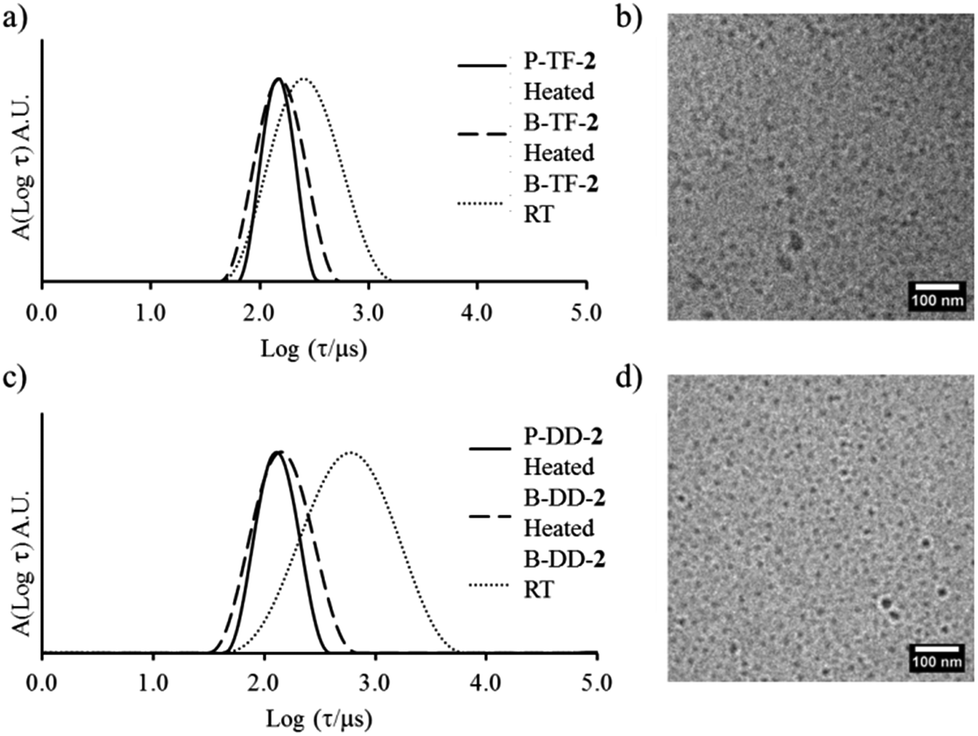
As part of the present study we explored the influence of the glass transition temperature (Tg) on the copolymer blending method. For that the P(IBA-co-DMA)-b-PDMA diblock copolymers 7 and 9 were blended together to match the composition of 8, see Table 1. For the block-random diblock copolymers investigated, the hydrophobic monomer, IBA, forms core blocks with a high Tg (see ESI† for thermal analysis) keeping constant the block lengths compared to the first PDMA-b-P(EHA-co-DMA) series (polymers 1–3). In principle to understand the influence of Tg, the influence of hydrophobicity must also be accounted for. However, surface tension (γ) values of PEHA and PIBA are quite similar, 35 mN m−1 and 32 mN m−1 respectively,40,41 although DMA will reduce these values in the same manner for both IBA-based and EHA-based copolymers. Hence any differences in polymeric behavior can be attributed to the influence of Tg. In the following section it should be highlighted that all the experimental temperatures (room temperature to 75 °C) used were lower than the Tg values of the associating block for the PDMA-b-P(IBA-co-DMA) diblock copolymers, and as a consequence the core block remains glassy throughout. It should be noted that the thin film rehydration pathway did not solubilize the polymer, indicating a lack of hydration in solution even after extended periods of time or heat.Here for this high Tg system it was observed by static and dynamic light scattering at 20 °C that for the solutions left at room temperature the blend and pure systems only structurally match when the solvent switch pathway was used, Fig. 5a. As for the previous systems, there was a strong influence of the preparation pathway on the self-assembly. Moreover, after the heating cycle no changes in the micelle structures were observed for either assembly pathway (see ESI† for cryo-TEM analysis). This lack of rearrangement advocates the system was frozen even at high temperature as expected. This behavior of the P(IBA-co-DMA)-b-PDMA diblock copolymer micelles in water was also highlighted by the distributions of relaxation times observed by DLS, Fig. 5b; broad distributions indicative of structures far from equilibrium, and not perfectly blended, were observed for B-DD-8 and P-DD-8. Moreover, for B-DD-8 we clearly observe a second mode of relaxation, which is proposed to originate from larger insoluble frozen bulk structures. These results indicate that for P(IBA-co-DMA)-b-PDMA diblock copolymers the structures formed in solution were kinetically trapped because of the high Tg of the core-forming block and reorganization was consequently inhibited. (see ESI† for additional analysis of core mobility using variable temperature 1H NMR spectroscopy).
 | ||
| Fig. 5 (a) Relationship of Nagg with preparation pathway for the mixed and pure samples for the P(IBA-co-DMA)-b-PDMA systems. (b) Distribution of relaxation times from DLS of P-DD-8 and B-DD-8 at room temperature. Error bars indicate 10% error on Nagg. | ||
Despite their frozen behavior the blend micelles formed by the solvent switch method still structurally match the pure system at the same composition. These structural similarities indicate that structures formed from the solvent switch protocol are frozen, but were potentially co-assembled under equilibrium conditions, which may have led to blend and pure micelles that can structurally match using this route of assembly.
Conclusions
The considerations and mechanisms for the copolymer blending method have been investigated further in this report. By exploring a range of diblock copolymers and assembly conditions the copolymer blending method can be easily extended using a range of assembly routes to produce blend structures that structurally match those of a pure sample at the same composition.Experimentally, this new blending route is successful depending on the capability of the polymeric micelles reaching equilibrium whilst having compatible core blocks. From both light scattering and cryo-TEM analysis it was understood that the governing factors influencing this are the core mobility with respect to the system globally, which is broken down specifically into two factors (a) a glass transition temperature below that of the experimental temperatures used and (b) an energy barrier low enough for molecular exchange, the latter being influenced by the hydrophobic character of the core-forming block as well as by its length. These two factors allow for a global equilibrium and the lowest free energy blend structure to form, which is identical to the pure system. This report introduced and explored the limitations of the blending method and highlighted the kinetic concerns which must be taken into consideration to produce stable equilibrium micelles. Being able to understand these limitations allows for application advances over conventional polymeric nanostructure assemblies.
Acknowledgements
The ESF P2M, EPSRC, ERC are thanked for financial support. Dr Mathew Robin is thanked for useful discussions. We acknowledge the cryo-TEM was paid for by the NIH grant DP2OD008724-01 and use of the UCSD Cryo-Electron Microscopy Facility, which is supported by NIH grants to Dr Timothy S. Baker and a gift from the Agouron Institute to UCSD.Notes and references
- I. A. Nyrkova and A. N. Semenov, Macromol. Theory Simul., 2005, 14, 569 CrossRef CAS.
- D. D. Bendejacq and V. Ponsinet, J. Phys. Chem. B, 2008, 112, 7996 CrossRef CAS PubMed.
- R. Hadgiivanova, H. Diamant and D. Andelman, J. Phys. Chem. B, 2011, 115, 7268 CrossRef CAS PubMed.
- F. K. von Gottberg, K. A. Smith and T. A. Hatton, J. Chem. Phys., 1998, 108, 2232 CrossRef CAS.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111 RSC.
- J. Stejskal, D. Hlavatá, A. Sikora, Č. Konňák, J. Pleštil and P. Kratochvíl, Polymer, 1992, 33, 3675 CrossRef CAS.
- S. Jain and F. S. Bates, Macromolecules, 2004, 37, 1511 CrossRef CAS.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412 RSC.
- A. Halperin, Macromolecules, 2011, 44, 5072 CrossRef CAS.
- A. Halperin and S. Alexander, Macromolecules, 1989, 22, 2403 CrossRef CAS.
- R. Lund, L. Willner, D. Richter and E. E. Dormidontova, Macromolecules, 2006, 39, 4566 CrossRef CAS.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577 CrossRef CAS.
- C. Tsitsilianis, G. Gotzamanis and Z. Iatridi, Eur. Polym. J., 2011, 47, 497 CrossRef CAS.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, P. Cotanda, C. Chassenieux, O. Colombani and R. K. O'Reilly, Polym. Chem., 2015, 6, 2761 RSC.
- A. Shedge, O. Colombani, T. Nicolai and C. Chassenieux, Macromolecules, 2014, 47, 2439 CrossRef CAS.
- F. Dutertre, O. Boyron, B. Charleux, C. Chassenieux and O. Colombani, Macromol. Rapid Commun., 2012, 33, 753 CrossRef CAS PubMed.
- E. Lejeune, M. Drechsler, J. Jestin, A. H. E. Muller, C. Chassenieux and O. Colombani, Macromolecules, 2010, 43, 2667 CrossRef CAS.
- O. Borisova, L. Billon, M. Zaremski, B. Grassl, Z. Bakaeva, A. Lapp, P. Stepanek and O. Borisov, Soft Matter, 2011, 7, 10824 RSC.
- O. Borisova, L. Billon, M. Zaremski, B. Grassl, Z. Bakaeva, A. Lapp, P. Stepanek and O. Borisov, Soft Matter, 2012, 8, 7649 RSC.
- D. D. Bendejacq, V. Ponsinet and M. Joanicot, Langmuir, 2005, 21, 1712 CrossRef CAS PubMed.
- G. Laruelle, J. François and L. Billon, Macromol. Rapid Commun., 2004, 25, 1839 CrossRef CAS.
- M. M. Tian, A. W. Qin, C. Ramireddy, S. E. Webber, P. Munk, Z. Tuzar and K. Prochazka, Langmuir, 1993, 9, 1741 CrossRef CAS.
- P. Sens, C. M. Marques and J. F. Joanny, Macromolecules, 1996, 29, 4880 CrossRef CAS.
- F. Renou, T. Nicolai, E. Nicol and L. Benyahia, Langmuir, 2008, 25, 515 CrossRef PubMed.
- V. V. Palyulin and I. I. Potemkin, Macromolecules, 2008, 41, 4459 CrossRef CAS.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, A. Lu, N. Kirby, N. C. Gianneschi, C. Chassenieux, O. Colombani and R. K. O'Reilly, Macromolecules, 2015, 48, 6516 CrossRef CAS.
- J. Lu, F. S. Bates and T. P. Lodge, ACS Macro Lett., 2013, 2, 451 CrossRef CAS.
- Z. Li and E. E. Dormidontova, Soft Matter, 2011, 7, 4179 RSC.
- L. Meli and T. P. Lodge, Macromolecules, 2009, 42, 580 CrossRef CAS.
- J. Lu, S. Choi, F. S. Bates and T. P. Lodge, ACS Macro Lett., 2012, 1, 982 CrossRef CAS.
- S.-H. Choi, T. P. Lodge and F. S. Bates, Phys. Rev. Lett., 2010, 104, 047802 CrossRef PubMed.
- T. Rager, W. H. Meyer, G. Wegner and M. A. Winnik, Macromolecules, 1997, 30, 4911 CrossRef CAS.
- S. A. Barnhill, N. C. Bell, J. P. Patterson, D. P. Olds and N. C. Gianneschi, Macromolecules, 2015, 48, 1152 CrossRef CAS.
- T. Nose and K. Iyama, Comput. Theor. Polym. Sci., 2000, 10, 249 CrossRef CAS.
- D. Bendejacq, M. Joanicot and V. Ponsinet, Eur. Phys. J. E: Soft Matter Biol. Phys., 2005, 17, 83 CrossRef CAS PubMed.
- H. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473 CrossRef CAS.
- H. Shen, L. Zhang and A. Eisenberg, J. Phys. Chem. B, 1997, 101, 4697 CrossRef CAS.
- A. Walther, A. S. Goldmann, R. S. Yelamanchili, M. Drechsler, H. Schmalz, A. Eisenberg and A. H. E. Müller, Macromolecules, 2008, 41, 3254 CrossRef CAS.
- Y. S. Yu, L. F. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144 CrossRef CAS.
- Y. Kano, Y. Katayama, S. Akiyama, Y. Kitazaki and T. Kasemura, J. Adhes., 1997, 62, 75 CrossRef CAS.
- Y. Nakata, A. Ichikawa, K. Taguchi and Y. Iwakata, US Patent, 6592973, 2003 Search PubMed.
- This observation is most probably due to the fact that pure 1 suffers from stronger pathway dependency than 2 and 3, which in turn strongly affects the pathway dependency of the 1 + 3 mixture (see ESI† for details about the characteristics of 1 and 3 as pure systems).
Footnote |
| † Electronic supplementary information (ESI) available: Reactivity ratio data, thermal analysis, additional light scattering analysis, cryo-TEM images and 1H NMR spectroscopy with variable temperature. See DOI: 10.1039/c5py02006a |
| This journal is © The Royal Society of Chemistry 2016 |