
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the stereoselectivity of rac-LA polymerization by chiral recognition induced the formation of homochiral dimeric metal alkoxides†
P.
Horeglad
*a,
M.
Cybularczyk
ab,
A.
Litwińska
b,
A. M.
Dąbrowska
ab,
M.
Dranka
c,
G. Z.
Żukowska
c,
M.
Urbańczyk
ad and
M.
Michalak
e
aCentre of New Technologies, University of Warsaw, Banacha 2c, 02-097 Warsaw, Poland. E-mail: phoreglad@uw.edu.pl
bFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
cFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664, Warsaw, Poland
dFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
eInstitute of Organic Chemistry, Polish Academy of Science, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 13th January 2016
Abstract
Using dimeric dialkylgallium and dialkylindium alkoxide catalysts for the polymerization of rac-lactide (rac-LA), we have shown for the first time that the formation of homochiral dimeric species [Me2MOR]2 (M = Ga, In), induced by chiral recognition of monomeric Me2MOR units in the presence of Lewis base, leads to an increase of the heteroselectivity of the ring opening polymerization (ROP) of rac-LA, and therefore provides a new tool for controlling the stereoselectivity of the polymerization of heterocyclic monomers. To explain the origin of the heteroselectivity of the [Me2Ga(μ-OCH(Me)CO2Me)]2/Lewis base system in the ROP of rac-LA, structure of (S,S)-[Me2Ga(μ-OCH(Me)CO2Me)]2 ((S,S)-1) and rac-[Me2Ga(μ-OCH(Me)CO2Me)]2 (1) in the absence and presence of tertiary amines and pyridines was investigated. Studies were further extended by analysis of the structure/activity data for both (S,S)-[Me2In(μ-OCH(Me)CO2Me)]2 ((S,S)-2) and rac-[Me2In(μ-OCH(Me)CO2Me)]2 (2). Contrary to gallium complex 1, which exists in a solution as equimolar mixture of homo- and heterochiral diastereomers, an excess of homochiral (R*,R*)-2 species was observed in the case of 2. For both the Ga and In complexes, the interaction of amines with the metal center increased the tendency for the formation of homochiral species with retention of the dimeric structure in the solution. This tendency was additionally demonstrated by the structure of model dialkylgallium (3) and indium (4) complexes with monoanionic ligands possessing chiral centers in the α-position to the alkoxide oxygen and pyridine functionalities. The polymerization of rac-LA with gallium and indium catalysts (S,S)-1 and (S,S)-2 resulted in the formation of heterotactically enriched polylactide (PLA) (Pr = 0.50–0.85) and (Pr = 0.54–0.72), respectively. The heteroselectivity of the investigated systems was in line with the excess of the homochiral catalytic species. The higher activity of homochiral species activated by amines resulted in a positive non-linear effect between an excess of homochiral (R*,R*)-1 or (R*,R*)-2 catalysts and the heterotacticity of the obtained PLA. The observed dependence of stereoselectivity of rac-LA polymerization on the excess of homochiral species was similar to the asymmetric amplification in enantioselective organic catalysis; however, it is exceptional in polymerization processes.
1. Introduction
Almost 30 years ago, Kagan described for the first time the positive non-linear dependence between the excess of auxiliary ligand and the enantiomeric excess of a product, observed for the epoxidation of geraniol catalysed by titanium complexes.1 This phenomenon, known in asymmetric organic synthesis as asymmetric amplification, was found to be dependent on the excess of homochiral over heterochiral (meso) catalytic species, which resulted in positive non-linear effects in the cases of several important organic transformations.2 However, only two reports have been concerned with asymmetric amplification in polymerization; in both cases, this amplification led to positive non-linear effects. These are illustrated by the works of Yashima and Nonokawa on the enanatiomeric imbalance in α-amino acids on stereoregular poly(phenylacetylene) with crown ether pendants,3 and the asymmetric copolymerization of epoxides with CO2 shown by Nozaki and coworkers.4 In the latter case, the different catalytic properties of homo- and heterochiral dimeric alkylzinc alkoxides were shown to be decisive for asymmetric amplification. With regard to the latter, it is noteworthy that diastereomeric dimeric species can be formed during polymerization of chiral cyclic esters with metal alkoxides which tend to aggregate in solution (Scheme 1). In this case, the formation of homochiral or heterochiral diastereomers, which could affect the stereoselectivity, may be caused by the supporting ligand (Scheme 1, blue stars) or by the presence of a chiral center located in the growing polymer chain (Scheme 1, red stars). Thus, the polymerization stereoselectivity could be controlled by the relative stereoselectivity, activity or ratio of homo- and heterochiral diastereomers. This new approach for controlling stereoselectivity would be especially important for the ring opening polymerization (ROP) of rac-LA, as the properties of polylactide (PLA) – a biodegradable and bioresorbable polymer5 – are strongly dependent on its tacticity.6 Contrary to this approach, scientists have focused on single site catalysts for stereoselective polymerization of rac-LA, which has resulted in the development of many monomeric metal alkoxides, by the use of appropriate supporting ligands.7–9 Despite the tendency of metal alkoxides to aggregate, reports on dimeric complexes, which can polymerize rac-LA in a controlled manner with a certain degree of stereoselectivity, have been less frequent.7,10,11 However, the effect of catalyst nuclearity was recently addressed for Y,12 Cu13 and In14 complexes, and the formation of dimeric species was shown to be decisive for the stereoselectivity. It is noteworthy that in the case of isoselective indium complexes, the presence of homochiral and heterochiral diastereomers due to terminally bonded chiral supporting ligands was suggested to influence the stereoselectivity.14a Although not indicated by the authors, the effects of the formation of homochiral dimeric Zr and Hf,15 Sc16 and In17 complexes on their stereoselectivity could not be excluded. However, the effect of diastereomeric species, resulting solely from the presence of a chiral centre in the growing PLA chain (Scheme 1, red stars), on the stereoselectivity of rac-LA polymerization has never been reported.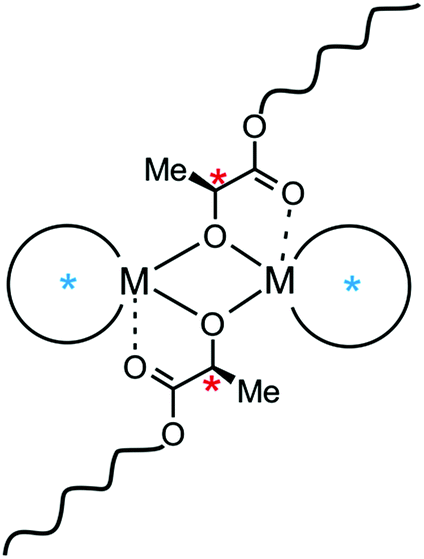 | ||
| Scheme 1 The structure of dimeric metal alkoxides in the polymerization of rac-LA: an example of dimeric active sites in the ROP of chiral cyclic esters. | ||
Recently, we have focused on simple dialkylgallium gallium alkoxides, previously reported by us, which polymerize rac-LA in heteroselective fashion in the presence of Lewis base,18 in order to explain the origin of the stereoselectivity. Herein we show that for simple dialkylgallium and dialkylindium alkoxides, the addition of amines as a Lewis base leads to the chiral recognition of monomeric Me2MOCH(Me)CO2Me (M = Ga, In) units with the formation of excess homochiral (R*,R*)-[Me2M(μ-OCH(Me)CO2Me)]2 over heterochiral (R,S)-[Me2M(μ-OCH(Me)CO2Me)]2 species, and is responsible for the heteroselective rac-LA polymerization. Furthermore, we show for the first time that the heteroselectivity of the investigated catalysts originates from the formation of homochiral dimers with chiral centres located only at the growing PLA chain. This opens new ways for the design of stereoselective catalysts for the polymerization of chiral cyclic esters and other heterocyclic monomers. Due to the higher activity of homochiral dimers activated with amines over heterochiral species, we demonstrate for the first time the non-linear dependence between the excess of homochiral dimers and the tacticity of the obtained PLA.
2. Results and discussion
The trigger to take a closer look at heteroselective dialkylgallium alkoxide/amine catalytic systems previously reported by us18 was our observation concerning the role of Lewis base in the stereoselectivity. The effect of Lewis base on the stereoselectivity of other catalysts, e.g. magnesium alkoxides and amides, for the polymerization of rac-LA has already been noted; the role of the Lewis base was to slow the reaction and/or prevent transesterification reactions.9b Importantly, for dialkylgallium alkoxides, we showed that although (S,S)-1 (Scheme 2) was not stereoselective, the polymerization of rac-LA, even without the addition of Lewis base, was living and fully controlled; essentially no transesterification was observed under mild conditions (up to 40 °C).18 Moreover, the addition of Lewis base resulted in heteroselective polymerization but did not result in deceleration of the polymerization. Therefore, both of the above observations indicated the unknown role of the Lewis base. An additional motivation to revisit and extend our studies concerning this group of complexes, as well as to explain the mechanism of stereocontrol, was the fact that dialkylgallium/amine systems could be used for the synthesis of stereodiblock PLA comprised of isotactically and heterotactically enriched PLA blocks with the use of a facile stereoselectivity switch.19 It must be noted that the switch between heteroselective and isoselective catalytic species has recently attracted the interest of several research groups, including ours,20 and is a prospectively challenging task in the field of ROP.21 Finally, we recently demonstrated that dialkylgallium alkoxides can be used for the synthesis of non toxic PLA for biomedical applications.22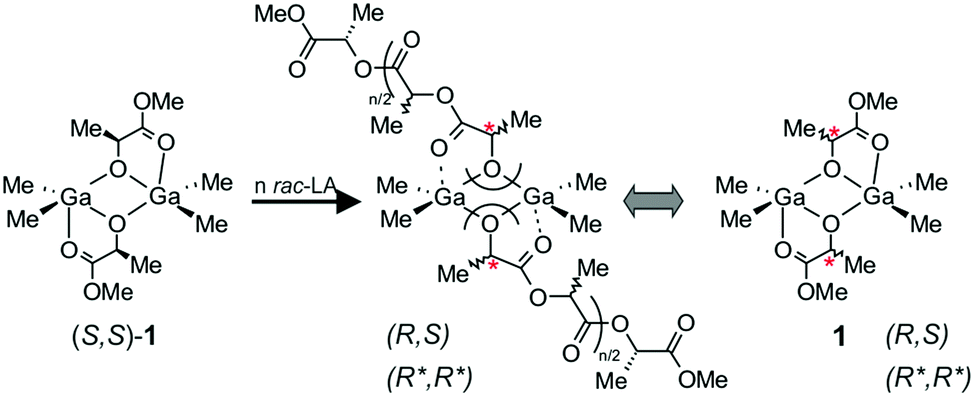 | ||
| Scheme 2 The structure of active species in the heteroselective polymerization of rac-LA with (S,S)-1. | ||
Although in our previous studies on the structure and activity of dialkylgallium alkoxides, (S,S)-[Me2Ga(μ-OCH(Me)CO2Me)]2 ((S,S)-1) was used as a model catalytic species,18 we showed that the structure of propagating species resulting from the insertion of rac-LA into the Ga–OR bond could be better described by the mixture of homochiral (R*,R*)-[Me2Ga(μ-OCH(Me)CO2R)]2 and heterochiral (R,S)-[Me2Ga(μ-OCH(Me)CO2R)]2 species (Scheme 2).23 Therefore, in our further studies described in this article, we used both (S,S)-1 and rac-[Me2Ga(μ-OCH(Me)CO2Me)]2 (1), existing in solution as mixture of homochiral (R*,R*)-[Me2Ga(μ-OCH(Me)CO2Me)]2 ((R*,R*)-1) and heterochiral (R,S)-[Me2Ga(μ-OCH(Me)CO2Me)]2 ((R,S)-1) complexes, as model compounds to investigate the structure of active species in the presence of tertiary amines and pyridines. Indium alkoxides have been shown over the last few years to be promising catalysts for the stereoselective polymerization of rac-LA,11b,14,20e,24 and dialkylindium alkoxides [(Me3SiCH2)2In(μ-OR)]2 were able to polymerize rac-LA in a controlled manner (however, without stereoselectivity);25 thus, we were interested in how dialkylindium alkoxides compared to gallium analogues. Therefore, we used analogous (S,S)-[Me2In(μ-OCH(Me)CO2Me)]2 ((S,S)-2) and rac-[Me2In(μ-OCH(Me)CO2Me)]2 (2) to investigate the relationship between their structure, activity and stereoselectivity in the polymerization of rac-LA.
2.1 The effect of Lewis base on the structure of [Me2Ga(μ-OCH(Me)CO2Me)]2
Prior to further polymerization studies, we examined the structure of (S,S)-[Me2Ga(μ-OCH(Me)CO2Me)]2 ((S,S)-1) and rac-[Me2Ga(μ-OCH(Me)CO2Me)]2 (1) in the absence and presence of a series of amines with different basicities and steric effects. As we demonstrated previously, (S,S)-1 was a homochiral dimer both in solution and in the solid state.18 For 1, synthesized analogously to (S,S)-1 with rac-methyl lactate (rac-(melac)), crystallization from CH2Cl2/hexane solution resulted in the formation of colourless crystals, which were found to be heterochiral dimers – (R,S)-1 due to the presence of chiral centres in the melac ligands (Fig. 1). Although the dimeric structure and coordination sphere of gallium were similar for (S,S)-1 and (R,S)-1, the detailed analysis of their structure in the solid state revealed significant differences between the molecules. While (R,S)-1 is a centrosymmetric dimer, (S,S)-1 reveals C1 point symmetry, contrary to the expected C2 point symmetry, due to the presence of chiral centres with the same absolute configuration. The latter is, among others, caused by the distortion of methine carbon of one of the melac ligands from the GaOCCO plane, which can be represented by significantly different Ga–O–Ga–CH(Me) torsion angles (Fig. 2). Moreover, the tension within the central Ga2O2 ring, evidenced by a Ga(1)O(1)Ga(2)O(2) torsion angle of about 2° and the different lengths of the weak C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Ga chelate bonds (2.475(2) Å and 2.400(2) Å), contributed to the differentiation of the Ga centres and the desymmetrization of (S,S)-1.18 Interestingly, [Me2Ga(μ-OCH(Ph)CH(Me)NH(Me))]2
O⋯Ga chelate bonds (2.475(2) Å and 2.400(2) Å), contributed to the differentiation of the Ga centres and the desymmetrization of (S,S)-1.18 Interestingly, [Me2Ga(μ-OCH(Ph)CH(Me)NH(Me))]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and [Me2Ga(μ-OCH(Ph)CH(Me)NMe2)]2,27 which were synthesized using enantiomerically pure ligands, are the only examples of X-ray characterized homochiral dialkylgallium alkoxides, and essentially possess C2 symmetry.
26 and [Me2Ga(μ-OCH(Ph)CH(Me)NMe2)]2,27 which were synthesized using enantiomerically pure ligands, are the only examples of X-ray characterized homochiral dialkylgallium alkoxides, and essentially possess C2 symmetry.
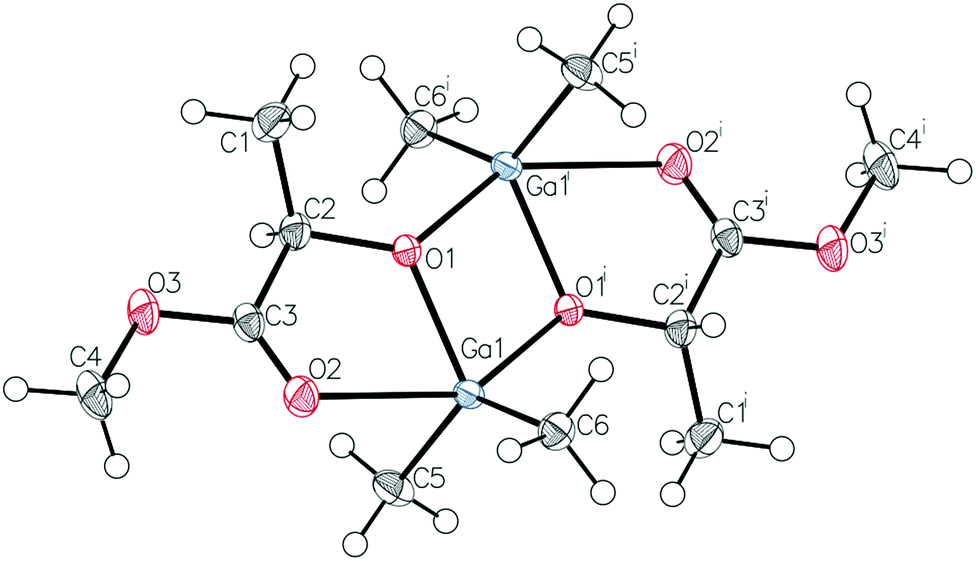 | ||
| Fig. 1 Molecular structure of (R,S)-1 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ga(1)–C(5) 1.9611(17), Ga(1)–C(6) 1.9584(18), Ga(1)–O(1) 1.9469(11), Ga(1)–O(1)i 2.0231(11), Ga(1)–O(2) 2.5220(13), O(2)–Ga(1)–O(1)i 148.09(4). The selected torsion angles (°): Ga(1)–O(1)–Ga(1)i–C(2) 146.08(15), Ga(1)i–O(1)i–Ga(1)–C(2)i −146.69(15). | ||
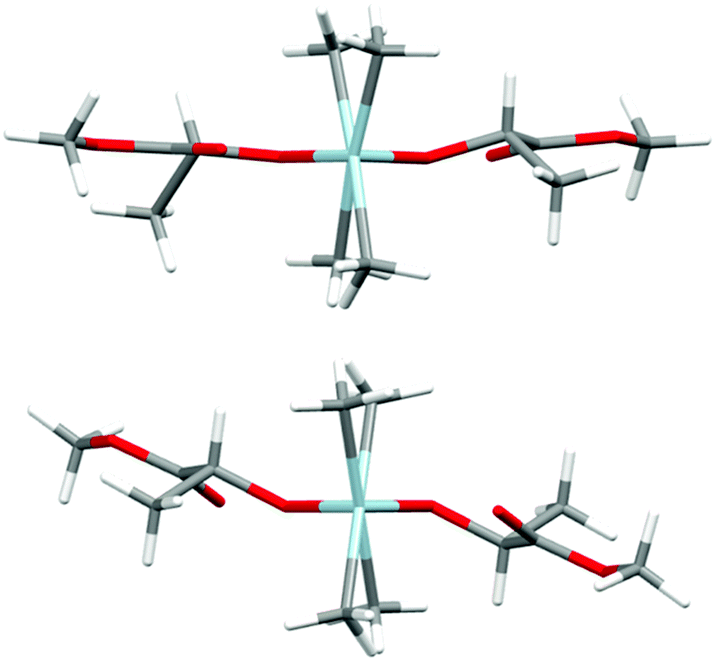 | ||
| Fig. 2 The views of the molecular structures of (S,S)-118 (top) and (R,S)-1 (bottom) along the Ga–Ga axis. Gallium and oxygen atoms are depicted as light blue and red, respectively. | ||
While compound (S,S)-1 remained in solution as a homochiral dimer, the dissolution of (R,S)-1 led to an equimolar mixture of homochiral (R*,R*)-1 and heterochiral (R,S)-1 dimers, which was indicated by three signals corresponding to Ga–Me protons. The appearance of homochiral dimers can be explained only by the fast exchange between homo and heterochiral dimers, despite the presence of strong Ga2O2 bridges and the presence of dimers in solution.28 This exchange was additionally demonstrated by the reaction between (R,R)-1 and (S,S)-1, which resulted in the instant formation of (R*,R*)-1 and (R,S)-1 in a 1:1 molar ratio (see the ESI†). With regard to polymerization studies, elevated temperature during the polymerization should only facilitate the demonstrated exchange. Although the mechanism of this exchange cannot be explained at this stage, it must be noted that it does not necessarily require the dissociation to monomeric species, which was not observed in our case.
In order to investigate in detail the effect of Lewis base on the structure of [Me2Ga(μ-OCH(Me)CO2R)]2 species, we used the tertiary amines with different steric and electronic properties: trimethylamine (TEA), dimethylethylamine (DMEA), and pyridines: 2,4,6-colidine, pyridine, γ-picoline and dimethylaminopyridine (DMAP). Their coordinating properties can be reflected by increasing donor number (in brackets) represented as the BF3 affinity of Lewis bases, and changing in a row: 2,4,6-colidine (101.03 kJ mol−1) < pyridine (128.08 kJ mol−1) < γ-picoline (134.17 kJ mol−1) < TEA (135.87 kJ mol−1) < DMAP (151.55 kJ mol−1).29 The interaction of amines with gallium was reflected by FTIR studies of 1/amine systems and the (R*,R*)-1 to (R,S)-1 ratio revealed by 1H NMR spectroscopy. Previously performed FTIR analysis in CH2Cl2 showed that the addition of 6 equiv. of γ-picoline to (S,S)-1 resulted in an increase in the number of uncoordinated CO groups.18 In the case of investigated (S,S)-1/amine systems, both in CH2Cl2 and toluene, the coordination efficiency of amine to gallium resulted in an increase in the number of free CO groups (Table 1). Although in CH2Cl2, the coordination efficiency was essentially in line with the amine donor number, in toluene a discrepancy between the relatively high donor number of the tertiary amines (TEA and DMEA) and the low coordination efficiency was observed. Moreover, the lower tendency of (S,S)-1 for the formation of dimers with pendant (S)-melac ligands in toluene was reflected by the lack of any uncoordinated CO groups. For aromatic amines, it should be noted that the presence of steric hindrances resulted in lower coordination efficiency, which was demonstrated for sterically hindered 2,4,6-colidine. An increase in the quantity of free CO groups was also observed for the decreased (S,S)-1/amine ratio, which was demonstrated for the 1/pyridine system from ratios of 1:1 to 1:60.
O groups for (S,S)-1 and (S,S)-1/amine calculated on the basis of FTIR spectroscopy
| Amine | (S,S)-1/Amine molar ratio | Free CO (%) (CH2Cl2)a |
Free CO (%) (toluene)a |
|---|---|---|---|
|
a The percentage of coordinated and pendant CO groups was calculated after the deconvolution of the CO band, resulting in two CO bands corresponding to coordinated (1723–1724 cm−1 in CH2Cl2 and 1726 cm−1 in toluene) and free CO (1741 cm−1 in CH2Cl2 and 1744–1749 cm−1 in toluene) (see the ESI).
|
|||
| — | — | 12 | 0 |
| 2,4,6-Collidine | 1:6 |
12 | 6 |
| TEA | 1:6 |
22 | 6 |
| DMEA | 1:6 |
25 | 9 |
| Pyridine | 1:1 |
14 | — |
| Pyridine | 1:2 |
16 | — |
| Pyridine | 1:6 |
21 | 11 |
| Pyridine | 1:60 |
28 | — |
| Pyridine | In pyridine | 41 | |
| γ-Picoline | 1:6 |
34 | 28 |
| DMAP | 1:6 |
42 | 34 |
Most importantly, the influence of the amine on the excess of homochiral dimers (R*,R*)-1 was evidenced for aromatic amines by 1H NMR spectroscopy of the 1/amine systems. The analysis of the Ga–Me signals of 1 showed the presence of one singlet at −0.35 ppm and two singlets at −0.42 and −0.27 ppm, corresponding to the homochiral (R*,R*)-1 and heterochiral (R,S)-1 complexes, respectively, in a 1:1 ratio. Although all investigated amines could interact with gallium, as evidenced by FTIR, the addition of 6 equivalents of TEA, DMEA or 2,4,6-collidine to 1 had no effect on the ratio of (R*,R*)-1 and (R,S)-1. However, for the 1/pyridine (1:6) and 1/γ-picoline (1:6) systems, we observed the broadening of signals corresponding to (R,S)-1 and the excess of homochiral (R*,R*)-1 species, resulting in (R,S)-1/(R*,R*)-1 ratios equal to 45:55 and 41:59, respectively. The excess of homochiral species was also strongly dependent on the excess of pyridine, which was confirmed for 1/pyridine (1:1)–1/pyridine (1:60) (Fig. 3). For 1 in pyridine-d5 and 1/DMAP (1:6), only homochiral (R*,R*)-1 species were essentially present in solution. For 1/DMAP (1:6), the dimeric nature of the gallium species was confirmed by DOSY NMR, with (S,S)-118 used as a reference (Table 2). In light of the results discussed above, the addition of amine shifts the equilibrium between (R*,R*)-1 and (R,S)-1 towards (R*,R*)-1 species, as shown in Fig. 3.
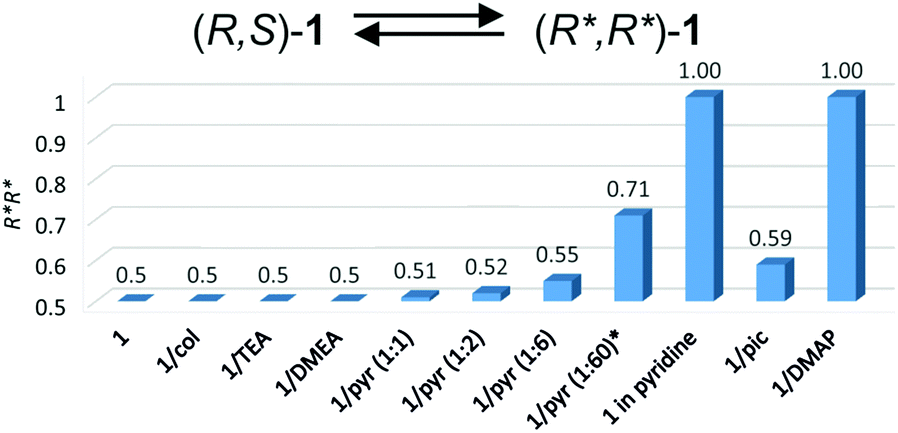 | ||
| Fig. 3 Molar fraction of (R*,R*)-1 for 1/amine systems. *In the presence of 1/pyridine (1:60), the excess of homochiral species was estimated on the basis of the deconvolution of a broad signal corresponding to Ga–Me protons (11.5 Hz) (see the ESI†). The abbreviations ‘col’, ‘pyr’ and ‘pic’ are used for 2,4,6-collidine, pyridine and γ-picoline, respectively. | ||
| System | Compound | M [g mol−1] | D [m2 s−1] × 109 | M found [g mol−1] |
|---|---|---|---|---|
| a M of (S,S)-1 was confirmed by cryoscopic determination of the molecular weight. b The error of M found was calculated on the basis of the error of D, which was estimated at 5%.30 | ||||
| (S,S)-1a | 406a | 1.282 | — | |
| (S,S)-1/DMAP (1:6) |
(S,S)-1 | 406 | 1.157 | 552 (83)b |
| (S,S)-1/DMAP (1:6) |
DMAP | 122 | 1.550 | 227 (34)b |
| (S,S)-2 | 496 | 1.235 | 454 (68)b | |
| (S,S)-2/DMAP (1:6) |
(S,S)-2 | 496 | 1.160 | 540 (81)b |
| (S,S)-2/DMAP (1:6) |
DMAP | 122 | 1.770 | 154 (23)b |
| 3 | 446 | 1.273 | 415 (62)b | |
| 4 | 536 | 1.217 | 475 (71)b | |
2.2 The effect of Lewis base on the structure of [Me2In(μ-OCH(Me)CO2Me)]2
(S,S)-[Me2In(μ-OCH(Me)CO2Me)]2 ((S,S)-2) and rac-[Me2In(μ-OCH(Me)CO2Me)]2 (2) were synthesized in an equimolar reaction between Me3In and (S)-methyl lactate ((S)-melac) and rac-melac, respectively, and were isolated as colourless crystals suitable for X-ray diffraction analysis. While (S,S)-2 (Fig. 4a) crystallized in the form of homochiral dimers, the crystallization of 2 resulted, analogously to 1, in the formation of heterochiral (R,S)-2 (Fig. 4b). For both (S,S)-2 and (R,S)-2, X-ray diffraction analysis revealed dimers with five-coordinate indium centres. The coordination spheres of both indium atoms adopted a distorted trigonal-bipyramidal geometry, with the methyl groups and alkoxide oxygen of melac ligand defining the equatorial plane. The carbonyl oxygen atom of the melac ester functionality and a bridging alkoxide oxygen atom of the second monomeric unit are located in the axial positions. The coordination of CO to indium resulted in the elongation of bridging In–O bonds for both (S,S)-2 and (R,S)-2 (Fig. 4). Similarly to the gallium complexes, the differences between (S,S)-2 and (R,S)-2 were related to their symmetry (Fig. 5). While heterochiral (R,S)-2 was centrosymmetric, the homochiral (S,S)-2 was asymmetric due to the distortion of the methine carbon of one of the melac ligands from the InOCCO plane, and the different lengths of the CO⋯In bonds (2.458(7) and 2.524(7) Å). Tension within central the In2O2 ring was reflected by the In–O–In–O torsion angles of about 5° (Fig. 5) and was slightly higher in comparison with the gallium analogue (R,S)-2. It is noteworthy that among all known homochiral dialkylindium alkoxides [R2In(O,X)]2 (where (O,X) represents a monoanionic alkoxide bidentate ligand with Lewis base functionality) with chiral centres in the α position with respect to the alkoxide oxygen, similar desymmetrization was observed for [Me2In(μ-OCH(Me)CH2NH2)]231 but was essentially absent in the case of [Me2In(μ-OCH(Ph)CH2NMe2)]2,32 which in both cases were synthesized from enantiomerically pure alcohols.
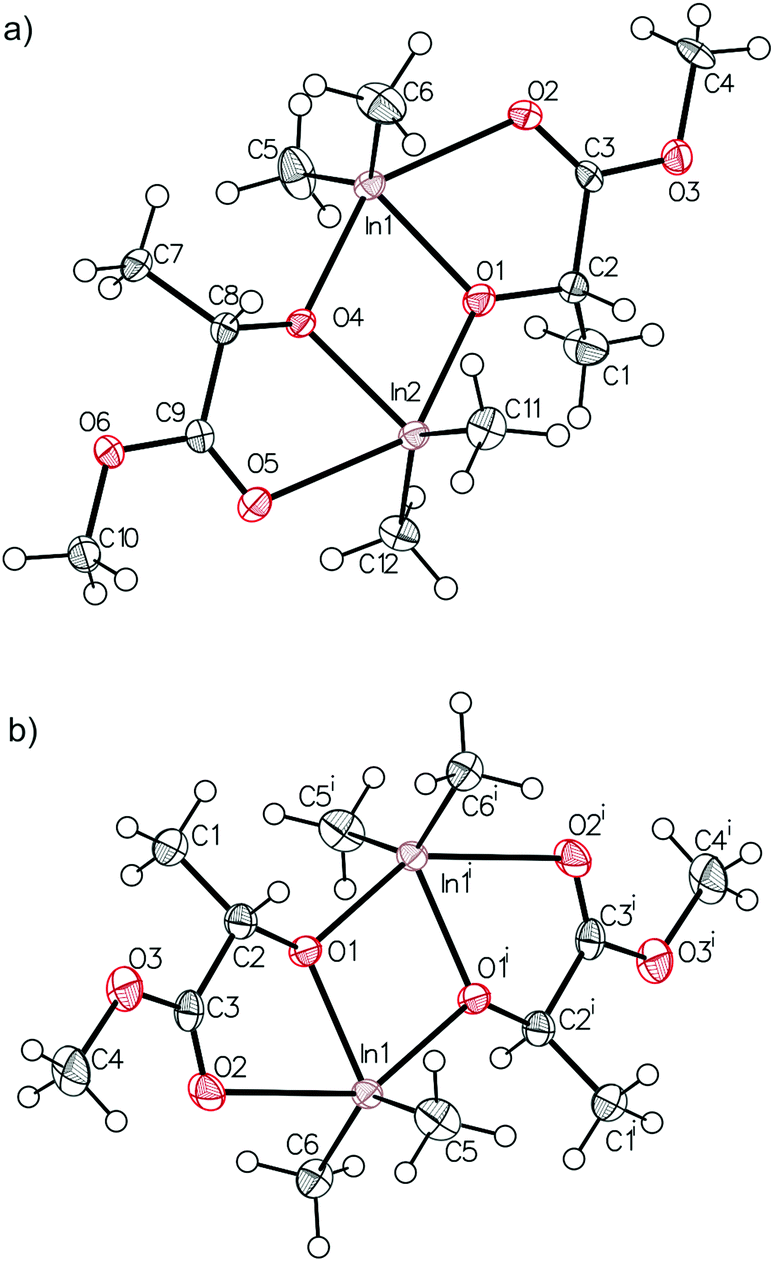 | ||
| Fig. 4 Molecular structure of (S,S)-2 (a) and (R,S)-2 (b) with ellipsoids at the 50% probability level. Selected bond lengths (Å) and angles (°) for (S,S)-2: In(1)–C(5) 2.132(10), In(1)–C(6) 2.163(9), In(1)–O(1) 2.157(7), In(1)–O(4) 2.217(8), In(1)–O(2) 2.458(7), In(2)–C(11) 2.158(11), In(2)–C(12) 2.137(9), In(2)–O(4) 2.156(7), In(2)–O(1) 2.231(8), In(2)–O(5) 2.524(7), O(2)–In(1)–O(4) 143.3(3), O(5)–In(2)–O(1) 142.4(3); and (R,S)-2 In(1)–C(5) 2.142(3), In(1)–C(6) 2.135(3), In(1)–O(1) 2.1566(19), In(1)–O(1)i 2.2161(18), In(1)–O(2) 2.532(2), O(2)–In(1)–O(1)i 142.50(7). | ||
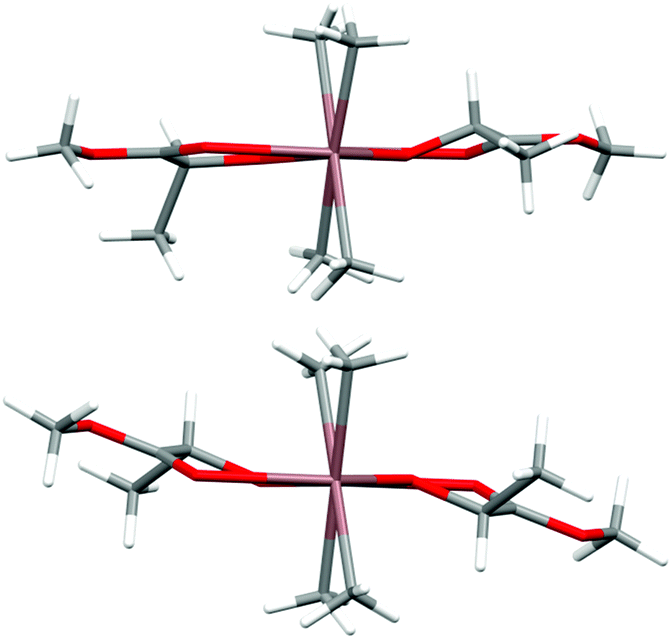 | ||
| Fig. 5 The view of the molecular structures of (S,S)-2 (top) and (R,S)-2 (bottom) along the In–In axis. Indium and oxygen atoms are depicted as brown and red, respectively. The selected torsion angles (°) of (S,S)-2: In(1)–O(1)–In(2)–O(4) 4.9(3), In(1)–O(4)–In(2)–O(1) −4.7(3), In(2)–O(1)–In(1)–O(4) −4.7(3), In(2)–O(4)–In(1)–O(1) 4.9(3), In(1)–O(1)–In(2)–C(2) −177.6(6), In(2)–O(4)–In(1)–C(8) −148.9(6) and (R,S)-2: In(1)–O(1)–In(1)i–C(2) 150.5(2), In(1)i–O(1)i–In(1)–C(2)i −150.5(2). | ||
The dimeric structure of (S,S)-2 was retained in solution, as demonstrated by DOSY NMR (Table 2). For 2, the dimeric structure in solution was reflected by 1H NMR, which revealed, similarly to gallium, three singlets corresponding to the In–Me protons of homochiral (R*,R*)-2 (−0.32 ppm) and heterochiral (R,S)-2 (−0.35 and −0.27 ppm) (Fig. S20†). It is noteworthy that in this case, the excess of homochiral dimers (R*,R*)-2 (57%) was already observed in the absence of amine. The latter could be explained by the much stronger interaction of CO with the fifth coordinate site of indium in comparison with the gallium analogues, and is discussed later in the text. A much stronger CO⋯In interaction in comparison with CO⋯Ga in (S,S)-1 was evidenced by the FTIR spectrum of (S,S)-2. For (S,S)-2, the carbonyl group (νCO = 1706 cm−1, CH2Cl2), was red-shifted by 31 cm−1 (in comparison with νCO of (S)-melacH), more significantly than for (S,S)-1 (12 cm−1). Contrary to the (S,S)-1/amine system, the addition of amine to (S,S)-2 did not cause any shift of the CO band. In the cases of (S,S)-2/pyridine (1:6), (S,S)-2/γ-picoline (1:6) and (S,S)-2/DMAP (1:6), the symmetrical signal at 1706 cm−1 (CH2Cl2) or 1707 cm−1 (toluene) indicated that (S,S)-2 remains in solution in the form of dimeric species with CO⋯In bonds. In the case of (S,S)-2/DMAP (1:6), the presence of an additional trace signal at around 1740 cm−1 may indicate that DMAP competes to a very limited extent with the CO group of the (S)-melac ligand. Although for the investigated indium complexes, the pyridines could barely compete with CO interactions for coordination to the fifth coordinate site, they could still interact with indium, leading to an excess of homochiral species, as evidenced by 1H NMR spectra of 2. The addition of 6 equivalents of 2,4,6-collidine and TEA to 2 had essentially no effect on the excess of homochiral dimers in comparison with 2. However, for 2/DMEA (1:6), 60% homochiral species were observed. For 2/pyridine (1:6) and 2/γ-picoline (1:6) the interaction of the amine led to further slight increases in the fraction of homochiral species (R*,R*)-2 to 62% and 63%, respectively. The addition of 1 equiv. or 3 equiv. of DMAP to 2 resulted in the formation of essentially pure homochiral (R*,R*)-2, which is shown by the presence of one singlet corresponding to the In–Me protons. At the same time, the presence of dimeric species was confirmed by DOSY NMR (Table 2). In light of the above results, the interactions of amines with 2 shift the equilibrium between (R*,R*)-2 and (R,S)-2 towards the (R*,R*)-2 species, as shown in Fig. 6.
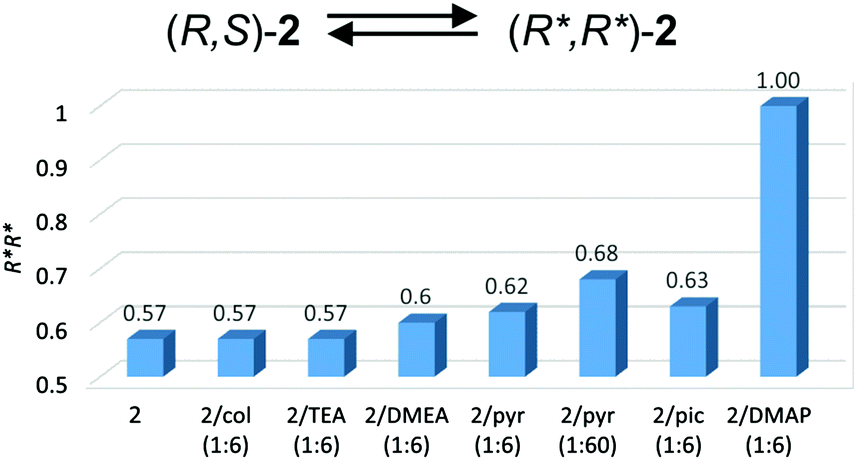 | ||
| Fig. 6 Molar fractions of (R*,R*)-2 for 2/amine systems. The abbreviations ‘col’, ‘pyr’ and ‘pic’ are used for 2,4,6-collidine, pyridine and γ-picoline, respectively. | ||
The presence of an excess of the homochiral indium species (R*,R*)-2 (57%) in comparison with the equimolar mixture of gallium homochiral (R*,R*)-1 and heterochiral (R,S)-1 dimers could be explained by the increase of the strength of the CO⋯M interaction in the case of [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga, In). This reasoning is further supported by the structure of the dimethylaluminum derivative of rac-ethyl lactate, for which exclusive formation of (R*,R*)-[Me2Al(μ-OCH(Me)CO2Et)]2 was evidenced in the solid state and in solution.33 In this case, the interaction of the carbonyl functionality with the Al centre is stronger in comparison with both (S,S)-1 and (S,S)-2, as shown by FTIR measurements, which revealed that the νCO band was shifted by 38 cm−1. Therefore, the increase in the quantity of homochiral gallium species in the presence of amines should also be related to the stronger interaction of the amines with the fifth coordinate site in comparison with the ester functionality. Therefore, the acidic character of the four coordinate metal centre in [Me2M(μ-OR)]2, influencing the strength of the CO⋯M or amine⋯M interactions, plays a key role in the distribution of homochiral and heterochiral [Me2M(μ-OCH(Me)CO2R)]2 complexes. In order to demonstrate the effect of amine on the formation of homochiral dialkylgallium alkoxides, we synthesized and characterized model rac-[Me2M(μ-OCH(Me)C5H4N)]2 (M = Ga (3), In (4)) complexes possessing monoanionic ligands with chiral centres in the α-position to the alkoxide oxygen, as well as pyridine Lewis base functionality.
2.3 The synthesis and structure of model [Me2M(μ-OCH(Me)C5H4N)]2 (M = Ga, In) complexes
The reaction between Me3Ga and a racemic mixture of 1-(2-pyridinyl)ethanol (HOCH(Me)C5H4N) led to the formation of homochiral (R,R)-[Me2Ga(μ-OCH(Me)C5H4N)]2 ((R,R)-3) and (S,S)-[Me2Ga(μ-OCH(Me)C5H4N)]2 ((S,S)-3), which was the only product observed both in solution and in the solid state (Scheme 3).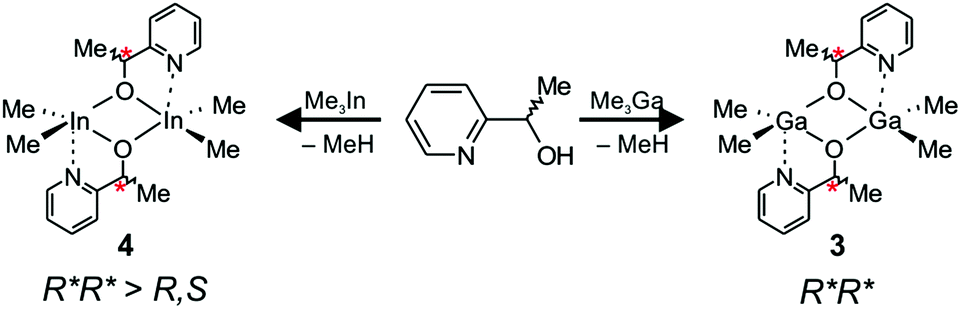 | ||
| Scheme 3 Synthesis and structure of 3 and 4 in the solution. | ||
The X-ray diffraction analysis revealed the presence of (R,R)-3 and (S,S)-3, of C2 symmetry, with equivalent five-coordinate gallium centres (Fig. 7). The coordination spheres of both gallium atoms adopt a distorted trigonal-bipyramidal geometry with the methyl groups and the alkoxide oxygen of OCH(Me)C5H4N defining the equatorial plane. The nitrogen atom of the pyridine functionality and a bridging alkoxide oxygen are located in the axial positions. The bond distances, including bridging Ga–Oalkoxide (1.9263(14) and 2.0931(14) Å) and chelate Ga–N (2.2789(18) Å) are similar to the bond distances in dimeric [R2Ga(μ-OCH2C5H4N)]2 (R = Me, Et) complexes.34 In the case of 3, the coordination of the pyridine functionality to the fifth coordination site of gallium resulted in much stronger interaction in comparison with the CO group of the melac ligand of 1, as shown by significant elongation of the axial bridging Ga–Oalkoxide bonds (2.0931(14) Å) in comparison with (S,S)-1 (2.0269(18) and 2.0349(17) Å)18 and (R,S)-1 (2.0231(11) Å). Interestingly, in the case of 3, the three fused rings formed due to the chelate Ga–N interaction adopted a boat conformation rather than a centrosymmetric planar or slightly distorted planar conformation in the cases of [Me2Ga((μ-OCH2C5H4N))]2 and [Et2Ga((μ-OCH2C5H4N))]2, respectively.35 Importantly, the structure of 3 confirms that the stronger coordination of the pyridine functionality in comparison with other Lewis base functionalities may be responsible for the formation of homochiral dimers of general formula [Me2Ga(μ-O,X)]2. In the case of [R2Ga(μ-OCH(Me)CH2NMe2)]2 (R = Me, Et),35 [R2Ga(μ-OCH(Me)CH2OMe)]236 and [R2Ga(μ-OCH(Me)C6H4OMe)]237 the crystallization of heterochiral dimers was reported, which was associated with much weaker Ga–N bond in comparison with 3. The dimeric structure of 3 in solution and the presence of homochiral (R*,R*)-[Me2Ga(μ-OCH(Me)CH2NMe2)]2 dimers were further evidenced by DOSY NMR (Table 2) and by the presence of one singlet at −0.38 ppm for the Ga–Me protons in the 1H NMR spectrum.
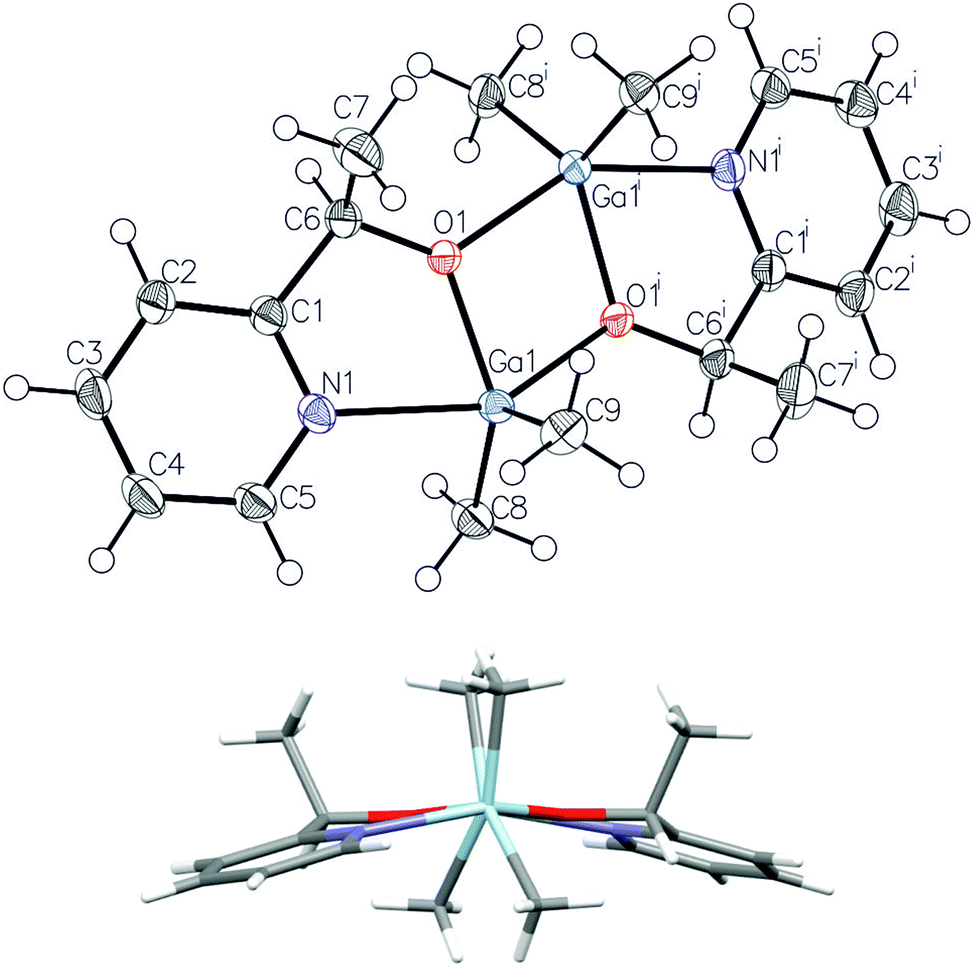 | ||
| Fig. 7 Molecular structure of (R,R)-3 with ellipsoids at 50% probability level and the view along the Ga–Ga axis (below); hydrogen atoms were omitted for clarity. Selected bond lengths (Å) and angles (°): Ga(1)–C(8) 1.976(2), Ga(1)–C(9) 1.969(2), Ga(1)–O(1) 1.9263(14), Ga(1)–O(1)i 2.0931(14), Ga(1)–N(1) 2.2789(18), N(1)–Ga(1)–O(1)i 146.83(6), Ga(1)–O(1)–Ga(1)i–O(1)i 8.61(6), Ga(1)–O(1)i–Ga(1)i–O(1) −7.92(6). | ||
rac-[Me2In(μ-OCH(Me)C5H4N)]2 (4) was synthesized analogously to 3 by an equimolar reaction between Me3In and rac-HOCH(Me)C5H4N (Scheme 3). Although 4 was isolated as a crystalline solid, crystals suitable for X-ray analysis could not be obtained. However, it must be noted that [Me2In(μ-OCH2CH2C5H4N)]2, possessing alkoxide ligands with pyridine functionality, has already been synthesized and characterized by X-ray.38 In this case, the coordination of the pyridine functionality to the fifth coordination site of indium resulted in a much stronger interaction in comparison with the CO group of the melac ligand for 2, as shown by the more significant elongation of bridging In–Oalkoxide bonds (2.239 Å) in comparison with (S,S)-2 (2.217(8) and 2.231(8) Å)18 and (R,S)-2 (2.2161(18) Å). Importantly, in the case of dialkylindium propanolates possessing Lewis base functionality and synthesized using racemic alcohols, only heterochiral complexes were crystallized and characterized by X-ray diffraction.32,39 However, for 4, the presence of three signals corresponding to the protons of the In–Me groups in 1H NMR were indicative of a mixture of homo and heterochiral dimers in solution. The presence of the pyridine functionality instead of the ester group resulted in a higher fraction of homochiral species (67%) in comparison with 2 (57%). Interestingly, although 2 exhibited a higher tendency for the formation of homochiral species than 1, due to the stronger interaction of the CO functionality with indium, the effect of the interaction of the amine with the metal five coordinate site was much more pronounced in the case of 3 in comparison with 4. Finally, the DOSY NMR spectrum was in line with the dimeric structure of 4 (Table 2).
2.4 Polymerization studies
In order to investigate the effect of the excess of homochiral species on the heteroselectivity of dialkylgallium and dialkylindium alkoxides in the polymerization of rac-LA, we selected a series of catalytic systems – 1/amine and 2/amine – with different abilities for the formation of homochiral dimeric species. As the mixture of homochiral (R*,R*)-[Me2M(μ-OCH(Me)CO2R)]2 and heterochiral (R,S)-[Me2M(μ-OCH(Me)CO2R)]2 (M = Ga, In; R = growing PLA chain) species is formed after the insertion of rac-LA into the M–Oalkoxide bond of 1 or 2, irrespectively to the diastereomer (cf. Scheme 2), in our studies we used (S,S)-1 and (S,S)-2 as initiators. In order to generate a different excess of homochiral species during the polymerization, we used the following with gallium: (S,S)-1/TEA (1:6), (S,S)-1/DMEA (1:6), (S,S)-1/2,4,6-colidine (1:6), (S,S)-1/pyridine (1:6), (S,S)-1/γ-picoline (1:6) and (S,S)-1/DMAP (1:6) and indium systems: (S,S)-2/TEA (1:6), (S,S)-2/DMEA (1:6), (S,S)-2/2,4,6-colidine (1:6), (S,S)-2/pyridine (1:6), (S,S)-2/γ-picoline (1:6) and (S,S)-2/DMAP (1:6). Additionally (S,S)-1 and (S,S)-2 with different excesses of pyridine were used (see Tables S4 and S5†). In all cases, polymerization of rac-LA occurred due to the insertion of rac-LA into the Ga–Oalkoxide bond or the In–Oalkoxide bond, which was in each case evidenced by 1H NMR and MALDI TOF analysis (see the ESI†). The latter results, as well as the lack of activity of amines towards rac-LA under the investigated conditions, were in line with the coordination insertion mechanism. The controlled nature of the polymerization was further supported by GPC analysis, which revealed a narrow Mn distribution in most cases, in agreement with living polymerization for both dialkylgallium and dialkylindium alkoxides; however, the increased PDI were in some cases indicative of the presence of transesterification. Dimethylgallium alkoxides, due to the low acidity of their metal centres, were much less susceptible to the transesterification reaction in comparison with indium analogues. For the polymerization of rac-LA with (S,S)-1, transesterification was observed clearly for (S,S)-1/DMAP (1:6) in CH2Cl2 and (S,S)-1 in pyridine (see the ESI†). For the polymerization in toluene, essentially no transesterification was observed. In the case of indium systems, both inter- and intramolecular transesterification was observed for the polymerization of rac-LA in CH2Cl2 and toluene with (S,S)-2/pyridine (1:6), and was massive for (S,S)-2/γ-picoline (1:6) and (S,S)-2/DMAP (1:6). It must be noted that for selected catalytic Ga and In systems, transesterification is more likely to occur in CH2Cl2. Therefore, for our further discussion on the effect of homochiral diastereomers on stereoselectivity, we focused on the polymerization of rac-LA in toluene. Although the solvent effect on the stereoselectivity of rac-LA polymerization is an important issue, which has already been shown in several studies concerning the stereoselectivity of rac-LA polymerization,40 it is too far from the main aspect of this article to investigate it in detail.
2.5 Stereoselectivity of rac-LA polymerization with (S,S)-1/amine catalysts
The polymerization of rac-LA initiated with (S,S)-1, (S,S)-1/2,4,6-collidine (1:6), (S,S)-1/TEA (1:6), (S,S)-1/DMEA (1:6), for which a lack of excess of homochiral dimeric gallium species (R*,R*)-[Me2Ga(μ-OCH(Me)CO2R)]2 during polymerization could be expected on the basis of the distribution of homochiral and heterochiral dimers of 1, resulted in formation of atactic PLA. In light of the results showing that the formation of homochiral dimeric gallium species is essential for heteroselectivity (see below), the above results indicate that heterochiral species could be expected to be isoselective. The excess of homochiral species observed in the case of 1/pyridine (1:6) (55%) and 1/γ-picoline (1:6) (59%) resulted in the formation of heterotactically enriched PLA of Pr equal to 0.69 and 0.75, respectively (Fig. 8). The dependence of heteroselectivity on the presence of the excess of homochiral species was also observed for (S,S)-1/pyridine (1:1–1:60) (Fig. 9). Although for 1 in pyridine, only homochiral species were observed, the lower heteroselectivity (Pr = 0.66) could be explained by the presence of transesterification reactions, as evidenced by MALDI-TOF (see the ESI†). The essential lack of transesterification in the case of 1/DMAP (1:6), resulting in exclusively homochiral dimers in solution, allowed for the formation of highly heterotactic PLA (Pr = 0.85). It is noteworthy that except for the latter case, the heteroselectivity of the investigated systems was much higher in comparison with the fraction of homochiral species. The observed non-linear effect (Fig. 11) can be explained, similarly to zinc catalysts for the copolymerization of epoxides with CO2,4 by the increased activity of the heteroselective homochiral species resulting from the interaction of the amine with gallium. The increased activity of [Me2Ga(μ-OCH(Me)CO2R)]2 in the presence of amines was demonstrated by the increasing rac-LA polymerization rate in a row (S,S)-1 < (S,S)-1/pyridine (1:6) < (S,S)-1/DMAP (1:6) (see the ESI† for Mn:rac-LA conversion graphs). The latter could be rationalized by the elongation of bridging Ga–Oalkoxide bonds and the increased nucleophilicity of alkoxide oxygen. Moreover, the increased polymerization rates of (S,S)-1 with tertiary amines (TEA, DMEA), which could interact with gallium, were indicated by higher rac-LA conversion (Table S4†). It is noteworthy that the heteroselectivity of the essentially purely homochiral 1/DMAP (1:6) system could reflect the heteroselectivity of homochiral dimeric gallium species.
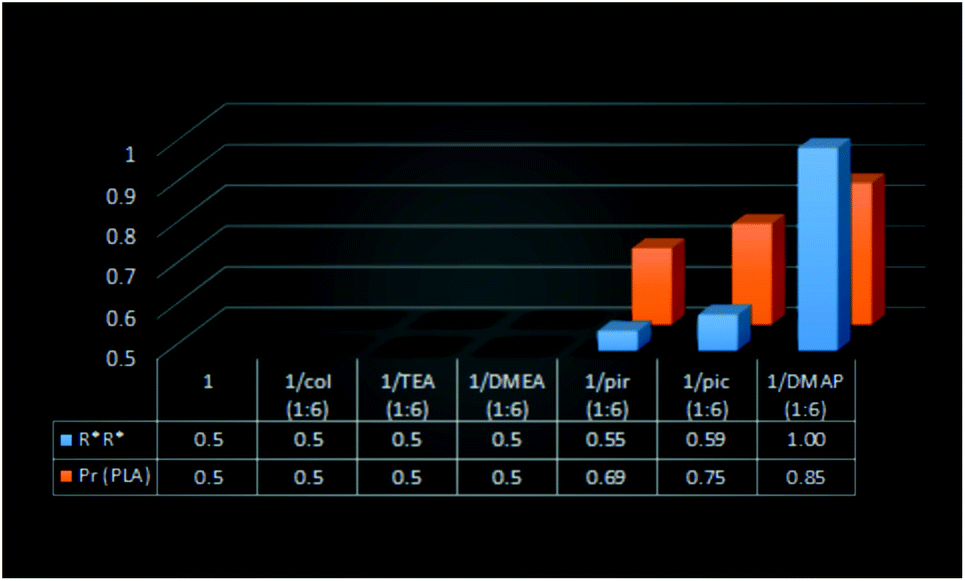 | ||
| Fig. 8 The relation between the fraction of homochiral species for 1/amine and the heteroselectivity of rac-LA polymerization with (S,S)-1 in toluene at 40 °C. | ||
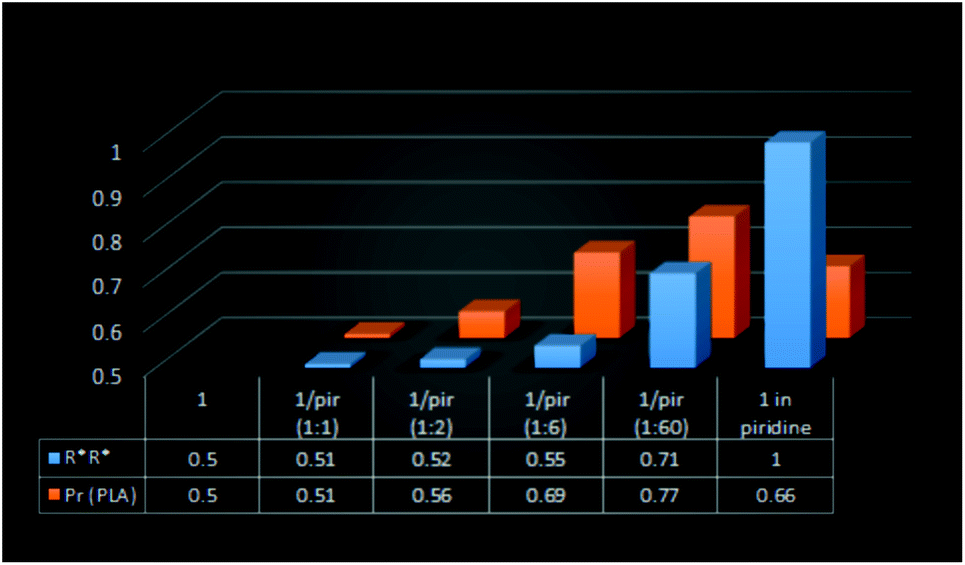 | ||
| Fig. 9 The relation between the fraction of homochiral species for 1/pyridine and the heteroselectivity of rac-LA polymerization with (S,S)-1 in toluene (or pyridine) at 40 °C. | ||
2.6 Stereoselectivity of rac-LA polymerization with (S,S)-2/amine catalysts
Similarly to gallium catalysts, indium complexes were also heteroselective, although the observed heteroselectivities were lower, which is in agreement with the literature.41 However, for the discussion on the influence of homochiral dimers on the hetereoselectivity, it is especially noteworthy that the heterotacticity of PLA obtained with (S,S)-2 in toluene (Pr = 0.55 at 40 °C; Pr = 0.58 at r.t.) was closely correlated with the fraction of homochiral species observed for 2 (R*,R* = 0.57), while the slightly lower heteroselectivity at 40 °C could result from transesterification reactions. This is in agreement with the essentially identical activity of both the homochiral and heterochiral dialkylindium species in the absence of amines, which is in contrast to the zinc catalysts reported by Nakano et al.4 The latter serves as strong evidence that heteroselectivity is dependent on the excess of homochiral species, while the addition of external amine is not necessary to induce heteroselectivity. However, the addition of amine was shown to increase the excess of homochiral [Me2In(μ-OCH(Me)CO2R)]2 species to a smaller extent than in the case of the gallium complexes. For PLA synthesized in toluene with (S,S)-2/DMEA (1:6) (Pr = 0.65), (S,S)-2/pyridine (1:6), (Pr = 0.69), (S,S)-2/pyridine (1:60) (Pr = 0.68), the increase in tacticity of PLA was in line with the excess of homochiral species observed for respective 2/amine systems. However, in contrast to rac-LA polymerization initiated by (S,S)-2, the heteroselectivity was in each case higher than the fraction of homochiral species. The non-linear effect can be explained by the increased activity of heteroselective homochiral species resulting from the interaction of amines with indium, similarly to the rac-LA polymerization with gallium catalysts (Fig. 11). The former was demonstrated by the increasing rac-LA polymerization rates in this order: (S,S)-2 < (S,S)-2/pyridine (1:6) < (S,S)-2/DMAP (1:6) as shown by the Mn:rac-LA conversion graphs (see the ESI†). Notably, a weaker positive non-linear effect should result from the smaller difference between the heteroselectivity of [Me2In(μ-OCH(Me)CO2R)]2 without amine and the maximum heteroselectivity in the presence of amine. The higher tendency of indium complexes to undergo transesterification reactions should result in a decrease of the heteroselectivity, and was demonstrated by the polymerization of PLA with (S,S)-2/γ-picoline (1:6) or (S,S)-2/DMAP (1:6) as evidenced by MALDI-TOF (see the ESI†). In other cases, smaller transesterification effects were supported by a slight increase in the heterotacticity of PLA obtained at room temperature (Fig. 10).
 | ||
| Fig. 10 The relationship between the fraction of homochiral species for 2/amine and the heteroselectivity of rac-LA polymerization with (S,S)-2 in toluene at 40 °C (orange) and room temperature (pale orange). | ||
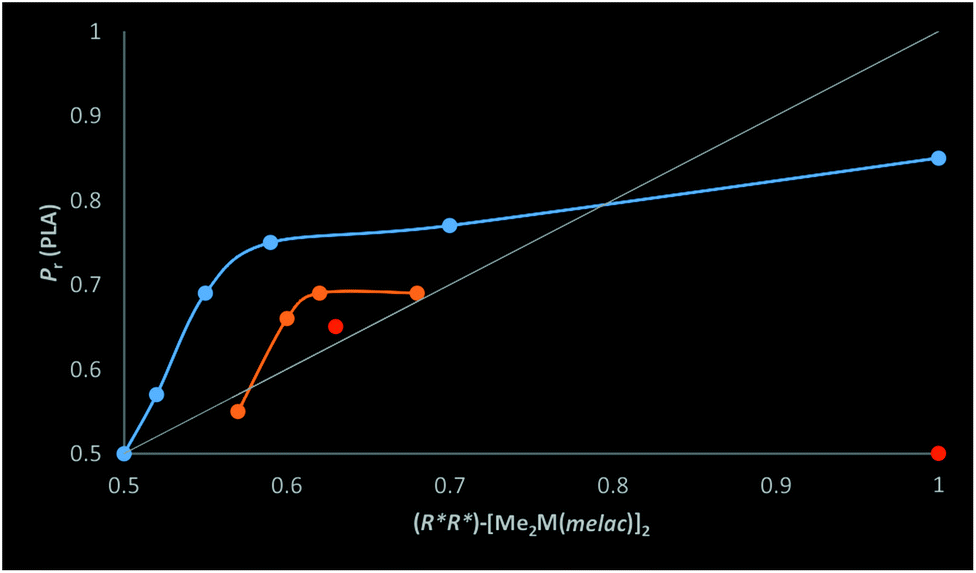 | ||
| Fig. 11 The dependence of the heteroselectivity of [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga and In) on the fraction of homochiral gallium (blue dots) and indium (orange dots) dimers (lines are indicated only for the clarity of the presentation) for the ROP of rac-LA in toluene at 40 °C. Red dots indicate indium catalysts (S,S)-2/γ-picoline (1:6) (R*,R* = 0.63) and (S,S)-2/DMAP (1:6) (R*,R* = 1), for which the transesterification reaction was observed by MALDI-TOF. | ||
2.7 Discussion on the stereoselectivity of rac-LA polymerization with [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga, In)
Although previously, we assigned the increase in the heteroselectivity of dialkylgallium alkoxides upon addition of the amine to the coordination efficiency of the Lewis base,18 our results clearly show that the excess of homochiral gallium [Me2Ga(μ-OCH(Me)CO2R)]2 and indium [Me2In(μ-OCH(Me)CO2R))]2 species is responsible for their heteroselectivity. This observation has no precedence in the literature. However, one should note that the presence of homochiral dimeric metal based catalysts, due to a chiral centre located in the supporting ligand or initiating group, could be responsible for the stereoselectivity of rac-LA polymerization.14a,15,16 Moreover, in the case of aluminum complexes with salan ligands, the presence of different diastereomers due to chiral centres on the nitrogen atoms have been recently shown to have a crucial effect on stereoselectivity, resulting in iso- or heteroselectivity.42 Additionally, the homochiral diastereomers of lanthanides (Y, Eu, Er) – Ln(OC*H(R)CH2P(O)R2)3 (Ln = lanthanide) allowed for the highly isoselective polymerization of rac-LA.43 These examples further support our studies showing that various diastereomers of catalytic species can influence the stereoselectivity of rac-LA polymerization. At this point, it can also be noted that in our case, the excess of stereoselective gallium or indium heterochiral species (potentially isoselective – see section 2.5 above), could allow, analogously to heteroselective homochiral species, control of rac-LA polymerization stereoselectivity. With regard to the examples above, not only is our system a new system to control the stereoselectivity of rac-LA polymerization using a diasteromeric dimeric catalyst, resulting from the presence of chiral centres located only at the growing polymer chain; it also allows for easy control of stereoselectivity during polymerization and, consequently, the microstructure of the PLA. Importantly, this concept has the potential to be implemented for other chiral heterocyclic monomers by choosing an appropriate catalyst which can form homo- and heterochiral aggregates in solution.Although our observations concerning the effects of homochiral [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga, In) species on their heteroselectivity in rac-LA polymerization has no precedence in the literature, it does not contradict well accepted mechanisms for stereoselectivity control.7 However, neither the enantiomorphic site nor the chain end control mechanism can be clearly ascribed to our systems. The heteroselective homochiral dimers (R*,R*)-[Me2M(μ-OCH(Me)CO2R)]2 are asymmetric dimers, for which stereoselectivity could be controlled by an enantiomorphic site mechanism. On the other hand, chiral centres located on the last monomeric unit of the growing PLA chain can control the stereoselectivity. Moreover, for the discussed gallium and indium catalysts, a clear indication of the mechanism is difficult due to the dynamic character of our stereoselective catalytic system. The heteroselectivity of the homochiral dimers, showing strong preference for racemo linkages, imposes the dominant insertion of (R*,R*)-LA into heteroselective (S*,S*)-homochiral dimers with the formation of (R,S)-heterochiral species; the subsequent rearrangement to the preferred homochiral dimers is triggered by the Lewis base (Scheme 4). Therefore, even for (S,S)-1/DMAP (1:6), for which only homochiral species are observed, the stereoerrors during the formation of heterotacticly enriched PLA may occur from the rearrangement rate of heterochiral to homochiral dimers. In this regard, the 1H NMR of mixtures of (R,R)-1 and (S,S)-1 or (R,R)-2 and (S,S)-2 were the same as 1 and 2, respectively, indicating essentially instant exchanges of the gallium and indium centres at ambient temperature (see the ESI†). Therefore, the relationship between polymerization rate and exchange rate would be a crucial factor. However, for the discussed gallium and indium systems, the polymerization rates are relatively slow in comparison with other reported catalysts,7–11 and the exchange should not decrease the polymerization stereoselectivity. Although it is difficult to clearly assign the mechanism of stereocontrol, our system, for which the heteroselectivity depends on the excess of homochiral dimers, is to some extent analogous to the model for enantioselective organic synthesis with the use of dimeric metal complexes.2
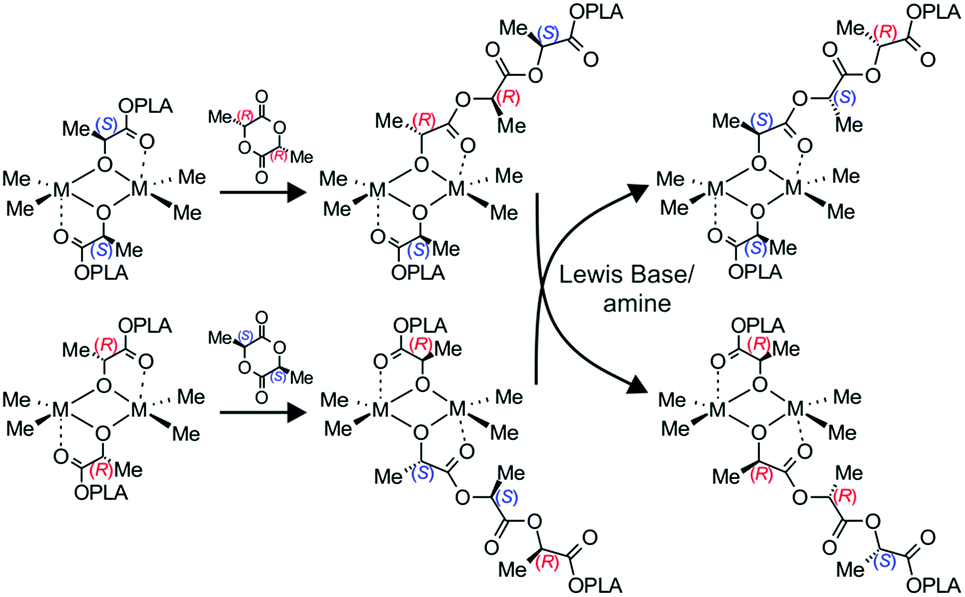 | ||
| Scheme 4 The distribution of active [Me2M(μ-OCH(Me)CO2R)]2 species in the presence of amine, after heteroselective insertion of rac-LA into the M–Oalkoxide bond of (R*,R*)-[Me2M(μ-OCH(Me)CO2PLA)]2 (M = Ga and In, PLA – growing polylactide chain). | ||
2.8 Heteroselectivity in rac-LA polymerization vs. enatioselectivity in organic transformations
The dependence of the heteroselectivity of rac-LA polymerization on the excess of homochiral dimers [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga, In) is analogous to a situation in which the enantiomeric excess of the organic product is dependent mainly on the presence and activity of a homochiral diastereomer.2 In the latter case, an excess of only one homochiral enantiomer is required in order to synthesize only one enantiomer of the product. In the case of polymerization, the presence of a mixture of homochiral (R,R) and (S,S) dimeric species is sufficient, as only the relative arrangement of chiral centres in PLA is important for its properties. According to the model of stereoselectivity control in enantioselective syntheses with metal complexes,2 the stereoselectivity should be mainly dependent on the enantioselectivity of homochiral dimeric species and the ratio of the reaction rates with stereoselective homochiral and non selective heterochiral dimers. In our case, the stereoselectivity of the catalyst can be represented by the probability of racemo linkages (Pr) of PLA. Therefore, Pr reflecting the selectivity of the catalytic system should be treated as analogous to the enantiomeric excess of the organic product (ee). While Pr-max represents maximum selectivity of the whole system, it must be noted that Pr-max is a combination of the stereoselectivity of homochiral dimers and the exchange rate of heterochiral dimers formed after the insertion of rac-LA into (R*,R*)-[Me2M(μ-OCH(Me)CO2R)]2 (M = Ga, In, R = Me or growing PLA chain) to homochiral dimers (Scheme 4). For gallium catalyst, the maximum selectivity (Pr-max) can be assigned to 0.85, which is the Pr value of PLA obtained with an essentially purely homochiral 1/DMAP (1:6) system. In the case of dialkylindium species, Pr-max could not be assigned due to transesterification reactions in the case of rac-LA polymerization with essentially purely homochiral 2/DMAP. Analogously to enantioselective organic transformations, the asymmetric amplification in our system, resulting in a positive non-linear effect, results from the higher activity of heteroselective homochiral dimers interacting with the amines over heterochiral dimers.
3. Conclusions
Using a simple model [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga and In) as catalysts, we focused on the mechanism of heteroselective polymerization of rac-LA with metal alkoxides on the basis of structure reactivity studies. We showed that dialkylgallium and dialkylindium alkoxides form homochiral complexes, which is dependent on the interaction of the Lewis base with the coordination centre. The heteroselectivity of the [Me2M(μ-OCH(Me)CO2Me)]2 (M = Ga and In) species was strongly dependent on the excess of homochiral dimeric active species in the course of polymerization occurring in a controlled manner. Despite a certain similarity between the stereoselectivities of the discussed systems in the ROP of rac-LA and enantioselective organic transformations with metal complexes, the specificity of polymerization imposes certain requirements that must be fulfilled to design catalysts/catalytic systems for the stereoselective polymerization of rac-LA:• The formation of homochiral dimeric or higher multinuclear (e.g. trimeric, tetrameric) species that retain their structure during the polymerization of rac-LA.
• The excess of homochiral species.
• The exchange between homochiral and heterochiral diastereomers, which leads to the transformation of heterochiral species formed after the insertion of rac-LA into homochiral species (Scheme 4), is the prerequisite for heteroselective polymerization. For isoselective polymerization, the latter would not be a requirement.
• The lack of intermolecular transesterification, which strongly affects the tacticity of the resulting PLA.
If the initial requirements are fulfilled, the stereoselectivity is expected to depend on:
• The maximum stereoselectivity of the homochiral species. This parameter may vary depending on the complex used, while the metal coordination centre and ligands should have a strong influence.
• The exchange rate between the heterochiral and homochiral complexes. An exchange rate that is very slow in comparison with the polymerization rate may lead to a decrease in the heteroselectivity.
• The higher activity of homochiral over heterochiral species, which leads to the positive non-linear effect described above.
This is the first report that clearly shows that chiral recognition leading to homochiral metal alkoxides can be a strategy for the formation of stereoselective catalysts for the polymerization of rac-LA. Most importantly, our concept could be extended to stereoselective multinuclear catalysts for other types of polymerization, especially ROP of chiral cyclic esters and other classes of chiral heterocyclic monomers. The use of the effect of homochiral multinuclear species on the stereoselective polymerization of heterocyclic monomers for the design and synthesis of new efficient systems based on the proposed model is currently of scientific interest to us.
4. Experimental
4.1 Materials and methods
All operations were carried out under dry argon using standard Schlenk techniques. Solvents and reagents were purified and dried prior to use. Solvents were either dried over potassium (toluene, hexane) or calcium hydride (CH2Cl2), or purified using MBRAUN Solvent Purification Systems (MB-SPS-800). rac-Lactide was purchased from Aldrich and further purified by crystallization from anhydrous toluene. (S)-Methyl lactate and rac-methyl lactate were purchased from Aldrich, dried over molecular sieves and distilled under argon. Me3Ga and Me3In were purchased from STREM Chemicals, Inc. and used as received. (S,S)-[Me2Ga(μ-OCH(Me)CO2Me)]218 and rac-HOCH(Me)C5H4N44 were synthesized according to the literature. (R,R)-1 was synthesized analogously to (S,S)-1 and its structure was confirmed on the basis of NMR spectroscopy (see the ESI†).
1H and 13C NMR spectra were recorded on Agilent 400-MR DD2 400 MHz and Varian UnityPlus 200 MHz spectrometers with shifts given in ppm according to the deuterated solvent shift. PGSE NMR (DOSY) experiments were performed on an Agilent DD2 600 MHz spectrometer equipped with an HCN probe. FTIR spectra were recorded on a FTIR Perkin Elmer System 2000 instrument. GPC measurements were recorded on a Spectra-Physics chromatograph equipped with two high performance Plgel 5 μm MIXED-C columns and detectors: RI (VE3580 Viscotek) and viscometer (270 Dual Detector Array Viscotek) with universal calibration according to a polystyrene standard. MALDI-TOF spectra were recorded on a Bruker ultrafleXtreme mass spectrometer.
4.2 Syntheses
O) 179.3 (CO).
O).
1H NMR (CD2Cl2, 400 MHz) of 2: −0.35, −0.32, −0.27 (s, 6H, InCH3), 1.31 (d, 3H, 3J(H,H) = 6.8 Hz, CHCH3), 3.75 (s, 3H, OCH3), 4.39 (q, 1H, 3J(H,H) = 6.8 Hz, CHCH3); 13C NMR (CD2Cl2, 100 MHz) of 2: −6.7 (InMe2), −5.7 (InMe2), −4.5 (InMe2), 23.7, 23.8, 53.5, 67.7, 67.9, 183.5 (CO), 183.7 (CO).
Crystal data for (R,S)-1. C12H26Ga2O6 (M = 405.77 g mol−1): monoclinic, space group P21/c, a = 8.15438(13) Å, b = 8.28026(15) Å, c = 12.8297(2) Å, β = 92.0637(14)°, V = 865.70(3) Å3, Z = 2, T = 120.0(1) K, μ(MoKα) = 3.130 mm−1, Dcalc = 1.557 g cm−3, 40
636 reflections measured (7.016° ≤ 2Θ ≤ 53.744°), 1870 unique (Rint = 0.0352, Rsigma = 0.0096) which were used in all calculations. The final R1 was 0.0188 (I > 2σ(I)) and wR2 was 0.0462 (all data).
Crystal data for (S,S)-2. C12H26O6In2 (M = 495.97 g mol−1): triclinic, space group P1, a = 6.9927(3) Å, b = 8.3025(3) Å, c = 8.4128(3) Å, α = 76.086(3)°, β = 83.924(3)°, γ = 87.628(3)°, V = 471.37(3) Å3, Z = 1, T = 100.0(3) K, μ(MoKα) = 2.463 mm−1, Dcalc = 1.747 g cm−3, 20
453 reflections measured (7.806° ≤ 2Θ ≤ 52.744°), 3846 unique (Rint = 0.0283, Rsigma = 0.0180) which were used in all calculations. The final R1 was 0.0158 (I > 2σ(I)) and wR2 was 0.0398 (all data).
Crystal data for (R,S)-2. C12H26In2O6 (M = 495.97 g mol−1): monoclinic, space group P21/c (no. 14), a = 8.2700(2) Å, b = 8.4677(2) Å, c = 13.1476(3) Å, β = 91.014(2)°, V = 920.56(4) Å3, Z = 2, T = 120.0(1) K, μ(MoKα) = 2.522 mm−1, Dcalc = 1.789 g cm−3, 14
092 reflections measured (6.888° ≤ 2Θ ≤ 52.742°), 1885 unique (Rint = 0.0281, Rsigma = 0.0131) which were used in all calculations. The final R1 was 0.0202 (I > 2σ(I)) and wR2 was 0.0461 (all data).
Crystal data for 3. C18H28Ga2N2O2 (M = 443.86 g mol−1): monoclinic, space group P2/n, a = 8.0739(4) Å, b = 8.5229(5) Å, c = 14.4630(8) Å, β = 97.288(5)°, V = 987.20(9) Å3, Z = 2, T = 120.0(1) K, μ(MoKα) = 2.741 mm−1, Dcalc = 1.493 g cm−3, 8468 reflections measured (6.982 ≤ 2Θ ≤ 55.736), 2362 unique (Rint = 0.0237, Rsigma = 0.0171) which were used in all calculations. The final R1 was 0.0283 (I > 2σ(I)) and wR2 was 0.0765 (all data).
Acknowledgements
This study was financially supported by the National Science Centre of Poland (SONATA BIS2 Programme, Grant No. DEC-2012/07/E/ST5/02860). PH and AL also thank the Ministry of Science and Higher Education of Poland (IUVENTUS PLUS Programme, Grant No. IP2010012970). The NMR diffusometry measurements (DOSY) were carried out at the Biological and Chemical Research Centre, University of Warsaw, established within a project co-financed by the European Union through the European Regional Development Fund under the Operational Programme Innovative Economy (2007–2013).Notes and references
- C. Puchot, O. Samuel, E. Dunach, S. Zhao, C. Agami and H. B. Kagan, J. Am. Chem. Soc., 1986, 108, 2353 CrossRef CAS PubMed.
- For selected examples see: (a) D. Guillaneux, S.-H. Zhao, O. Samuel, D. Rainford and H. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430 CrossRef CAS; (b) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef; (c) B. L. Feringa and R. A. van Delen, Angew. Chem., Int. Ed., 1999, 38, 3418 CrossRef; (d) H. B. Kagan, Adv. Synth. Catal., 2001, 343, 227 CrossRef CAS; (e) J. Inanaga, H. Furuno and T. Hayano, Chem. Rev., 2002, 102, 2211 CrossRef CAS PubMed; (f) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456 CrossRef CAS PubMed; (g) D. G. Blackmond, Tetrahedron: Asymmetry, 2010, 21, 1630 CrossRef CAS.
- R. Nonokawa and E. Yashima, J. Am. Chem. Soc., 2003, 125, 1278 CrossRef CAS PubMed.
- K. Nakano, T. Hiyama and K. Nozaki, Chem. Commun., 2005, 1871 RSC.
- M. Vert, Biomacromolecules, 2005, 6, 538 CrossRef CAS PubMed.
- (a) J. M. Becker, R. J. Pounder and A. P. Dove, Macromol. Rapid Commun., 2010, 31, 1923 CrossRef CAS PubMed; (b) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486 RSC.
- For selected reviews see: ref. 6b, (a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC; (b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed; (c) J. Wu, T. L. Yu, C. T. Chen and C. C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS; (d) A. P. Dove, Chem. Commun., 2008, 6446 RSC; (e) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC; (f) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165 RSC; (g) P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520 RSC; (h) S. Słomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436 CrossRef.
- For selected monomeric isoselective catalysts see: Al: (a) C. P. Radano, G. L. Baker and M. R. III Smith, J. Am. Chem. Soc., 2000, 122, 1552 CrossRef CAS; (b) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316 CrossRef CAS PubMed; (c) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938 CrossRef CAS PubMed; (d) Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510 CrossRef CAS; (e) Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291 CrossRef CAS PubMed; (f) K. Majerska and A. Duda, J. Am. Chem. Soc., 2004, 126, 1026 CrossRef CAS PubMed; (g) X. Pang, H. Du, X. Chen, X. Wang and X. Jing, Chem. – Eur. J., 2008, 14, 3126 CrossRef CAS PubMed; (h) M. H. Chisholm, N. J. Patmore and Z. Zhou, Chem. Commun., 2005, 127 RSC; (i) M. Bouyahyi, E. Grunova, N. Marquet, E. Kirillov, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2008, 27, 5815 CrossRef CAS; (j) H. Du, A. H. Velders, P. J. Dijkstra, J. Sun, Z. Zhong, X. Chen and J. Feijen, Chem. – Eur. J., 2009, 15, 9836 CrossRef CAS PubMed; (k) A. Alaaeddine, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2009, 28, 1469 CrossRef CAS. Y: (l) M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem. – Eur. J., 2011, 17, 1872 CrossRef CAS PubMed. Ga: (m) C. Bakewell, A. J. P. White, N. Long and C. K. Williams, Inorg. Chem., 2013, 52, 12561 CrossRef CAS PubMed. Zn: (n) M. Honrado, A. Otero, J. Fernandez-Baeza, L. F. Sanchez-Barba, A. Lara-Sanchez, J. Tejeda, M. P. Carrion, J. Martinez-Ferrer, A. Garces and A. M. Rodriguez, Organometallics, 2013, 32, 3437 CrossRef CAS; (o) H. Wang and H. Ma, Chem. Commun., 2013, 49, 8686 RSC; (p) S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689 CrossRef CAS PubMed. Zr, Hf: (q) B. Rajashekhar, S. K. Roymuhury, D. Chakraborty and V. Ramkumar, Dalton Trans., 2015, 44, 16280 RSC. Na, K: (r) Z. Dai, Y. Sun, J. Xiong, X. Pan and J. Wu, ACS Macro Lett., 2015, 4, 556 CrossRef CAS; (s) Y. Sun, J. Xiong, Z. Dai, X. Pan, N. Tang and J. Wu, Inorg. Chem., 2016, 55, 136 CrossRef CAS PubMed.
- For selected heteroselective monomeric catalysts see: Zn and Mg: (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS PubMed; (b) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785 CrossRef CAS PubMed; (c) D. J. Darensbourg and O. Karroonnirun, Inorg. Chem., 2010, 49, 2360 CrossRef CAS PubMed; (d) L. F. Sanchez-Barba, A. Garces, J. Fernandez-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sanchez and A. M. Rodriguez, Organometallics, 2011, 30, 2775 CrossRef CAS; (e) A. Otero, J. Fernandez-Baeza, L. F. Sanchez-Barba, J. Tejeda, M. Honrado, A. Garces, A. Lara-Sanchez and A. M. Rodriguez, Organometallics, 2012, 31, 4191 CrossRef CAS. Ca: (f) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48 RSC; (g) D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493 CrossRef CAS; (h) M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276 RSC. Y: (i) Y. A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem. – Eur. J., 2006, 12, 169 CrossRef PubMed; (j) L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273 RSC; (k) T. P. A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157 CrossRef CAS PubMed; (l) J. S. Klitzke, T. Roisnel, E. Kirillov, O. D. L. Casagrande and J.-F. Carpentier, Organometallics, 2014, 33, 309 CrossRef CAS. Ge: (m) A. J. Chmura, C. J. Chuck, M. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280 CrossRef CAS PubMed. Zr and Hf: (n) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293 RSC.
- For isoselective dimeric complexes see: In: (a) D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295 RSC. Zn: (b) M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánachez-Barba, A. Garcés, A. Lara-Sánchez and A. M. Rodriguez, Organometallics, 2014, 33, 1859 CrossRef CAS. Zn, Mg: (c) Y. Sun, Y. Cui, J. Xiong, Z. Dai, N. Tang and J. Wu, Dalton Trans., 2015, 44, 16383 RSC.
- For examples of heteroselective dimeric complexes see: Zn: (a) T. R. Jensen, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2004, 2504 RSC. In: (b) A. Pietrangelo, S. C. Knight, A. K. Gupta, L. J. Yao, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2010, 132, 11649 CrossRef CAS PubMed. Ln: (c) M. Sinenkov, E. Kirillov, T. Roisnel, G. Fukin, A. Trifonov and J.-F. Carpentier, Organometallics, 2011, 30, 5509 CrossRef CAS. Zr: (d) S. Pappuru, E. R. Chokkapu, D. Chakraborty and V. Ramkumar, Dalton Trans., 2013, 42, 16412 RSC. Mg: (e) A. Garcés, L. F. Sánachez-Barba, J. Fernández-Baeza, A. Otero, M. Honrado, A. Lara-Sánchez and A. M. Rodriguez, Inorg. Chem., 2013, 52, 12691 CrossRef PubMed. Zn: (f) T. J. J. Whitehorn, B. Vabre and F. Schaper, Dalton Trans., 2014, 43, 6339 RSC.
- R. H. Platel, A. J. P. White and C. K. Williams, Chem. Commun., 2009, 4115 RSC.
- S. Fortun, P. Daneshmand and F. Schaper, Angew. Chem., Int. Ed., 2015, 54, 13669 CrossRef CAS PubMed.
- (a) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290 CrossRef CAS PubMed; (b) I. Yu, A. Acosta-Ramirez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758 CrossRef CAS PubMed; (c) J. Fang, I. Yu, P. Mehrkhodavandi and L. Maron, Organometallics, 2013, 32, 6950 CrossRef CAS; (d) D. C. Aluthge, E. X. Yan, J. M. Ahn and P. Mehrkhodavandi, Inorg. Chem., 2014, 53, 6828 CrossRef CAS PubMed; (e) D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284 RSC.
- M. Hu, M. Wang, H. Zhu, L. Zhang, H. Zhang and L. Sun, Dalton Trans., 2010, 39, 4440 RSC.
- H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818 CrossRef PubMed.
- M. Hu, M. Wang, P. Zhang, L. Wang, F. Zhu and L. Sun, Inorg. Chem. Commun., 2010, 13, 968 CrossRef CAS.
- P. Horeglad, P. Kruk and J. Pécaut, Organometallics, 2010, 29, 3729 CrossRef CAS.
- P. Horeglad, A. Litwińska, G. Z. Żukowska, D. Kubicki, G. Szczepaniak, M. Dranka and J. Zachara, Appl. Organomet. Chem., 2013, 27, 328 CrossRef CAS.
- (a) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. Whiteand and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688 CrossRef CAS PubMed; (b) A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698 CrossRef CAS; (c) P. Horeglad, G. Szczepaniak, M. Dranka and J. Zachara, Chem. Commun., 2012, 48, 1171 RSC; (d) W. Zhao, Y. Wang, X. Liu, X. Chen, D. Cui and E. Y. X. Chen, Chem. Commun., 2012, 48, 6375 RSC; (e) M. Normand, E. Kirillov, T. Roisnel and J.-F. Carpentier, Organometallics, 2012, 31, 1448 CrossRef CAS; (f) W. Zhao, Y. Wang, X. Liu, X. Chen and D. Cui, Chem. – Asian J., 2012, 7, 2403 CrossRef CAS PubMed; (g) C. Bakewell, A. J. P. White, N. Long, X. F. L. Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577 CrossRef CAS PubMed; (h) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226 CrossRef CAS PubMed; (i) H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750 CrossRef CAS; (j) M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892 CrossRef CAS; (k) P. Horeglad, M. Cybularczyk, B. Trzaskowski, G. Z. Żukowska, M. Dranka and J. Zachara, Organometallics, 2015, 34, 3480 CrossRef CAS.
- (a) S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem. – Eur. J., 2015, 21, 7988 CrossRef CAS PubMed; (b) A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2015 DOI:10.1021/acs.chemrev.5b00426.
- E. Oledzka, P. Horeglad, Z. Gruszczyńska, A. Plichta, G. Nałęcz-Jawecki and M. Sobczak, Molecules, 2014, 19, 19460 CrossRef CAS PubMed.
- For examples on the model lactate metal deravitavies for active species in ROP of lactide see: ref. 9l and J. S. Klitzke, T. Roisnel, E. Kirillov, O. D. L. Casagrande and J.-F. Carpentier, Organometallics, 2014, 33, 5693 CrossRef CAS , and references therein.
- (a) A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2009, 2736 RSC. For the review on gallium and indium complexes for the ring-opening polymerizations of cyclic esters, see: (b) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869 CrossRef CAS.
- M. P. Blake, A. D. Schwarz and P. Mountford, Organometallics, 2011, 30, 1202 CrossRef CAS.
- F. Yuan, C. Zhu, J. Sun, Y. Liu and Y. Pan, J. Organomet. Chem., 2003, 682, 102 CrossRef CAS.
- H. Schumann, S. Wernik, F. Girgsdies and R. Weimann, Main Group Met. Chem., 1996, 19, 331 CrossRef CAS.
- C. J. Carmalt and S. J. King, Coord. Chem. Rev., 2006, 250, 682 CrossRef CAS.
- Lewis Basicity and Affinity Scales: Data and Measurement, ed. C. Laurence and J.-F. Gal, John Wiley & Sons Ltd, Chichester, United Kingdom, 2010 Search PubMed.
- M. Urbańczyk, D. Bernin, W. Koźmiński and K. Kazimierczuk, Anal. Chem., 2013, 85, 1828 CrossRef PubMed.
- E. Hecht, T. Gelbrich, K.-H. Thiele and J. Sieler, Main Group Chem., 2000, 3, 109 CrossRef CAS.
- H. Schumann, J. Kaufmann, B. C. Wassermann, F. Girgsdies, N. Jaber and J. Blum, Z. Anorg. Allg. Chem., 2002, 628, 971 CrossRef CAS.
- J. Lewiński, J. Zachara and I. Justyniak, Chem. Commun., 1997, 1519 RSC.
- (a) S. J. Rettig, A. Storr, J. Trotter and K. Uhrich, Can. J. Chem., 1984, 62, 2783 CrossRef CAS; (b) M. Westerhausen, A. N. Kneifel, P. Mayer and H. Nöth, Z. Anorg. Allg. Chem., 2004, 630, 2013 CrossRef CAS.
- (a) K.-H. Thiele, E. Hecht, T. Gelbrich and U. Dümichen, J. Organomet. Chem., 1997, 540, 89 CrossRef CAS; (b) S. Basharat, C. J. Carmalt, R. Palgrave, S. A. Barnett, D. A. Tocher and H. O. Davies, J. Organomet. Chem., 2008, 693, 1787 CrossRef CAS.
- H. Schumann, M. Frick, B. Heymer and F. Grigsdies, J. Organomet. Chem., 1996, 512, 117 CrossRef CAS.
- E. Hecht, Z. Anorg. Allg. Chem., 2000, 626, 1642 CrossRef CAS.
- Y. Shen, Y. Pan, X. Jin, X. Xu, X. Sun and X. Huang, Polyhedron, 1999, 18, 2423 CrossRef CAS.
- (a) J.-Z. Hu, M. Yang, X.-S. Wu, Y. Pan, Y.-J. Liu and X.-Z. Sun, Chin. J. Inorg. Chem., 1999, 15, 347 CAS; (b) C. E. Knapp, L. Pemberton, C. J. Carmalt, D. Pugh, P. F. McMillan, S. A. Barnett and D. A. Tocher, Main Group Chem., 2010, 9, 31 CAS.
- For selected examples of solvent effect on the stereoselectivity of rac-LA polymerization see: Zn: (a) H. Y. Chen, B. H. Huang and C. C. Lin, Macromolecules, 2005, 38, 5400 CrossRef CAS; (b) F. Drouin, P. O. Oguadinma, T. J. J. Whitehorne, R. E. Prud'homme and F. Schaper, Organometallics, 2010, 29, 2139 CrossRef CAS. Mg: 9b, (c) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2005, 44, 8004 CrossRef CAS PubMed; (d) J. C. Wu, B. H. Huang, M. L. Hsueh, S. L. Lai and C. C. Lin, Polymer, 2005, 46, 9784 CrossRef CAS. Al: (e) M. H. Chisholm, N. J. Patmore and Z. Zhou, Chem. Commun., 2005, 127 RSC; (f) M. H. Chisholm, J. C. Gallucci, K. T. Quisenberry and Z. Zhou, Inorg. Chem., 2008, 47, 2613 CrossRef CAS PubMed.
- For the comparison of gallium and indium complexes concerning their stereoselectivity in rac-LA polymerization see for instance ref. 20e.
- K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015, 54, 14858 CrossRef CAS PubMed.
- P. L. Arnold, J.-C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2008, 47, 6033 CrossRef CAS PubMed.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742 CrossRef CAS PubMed.
- CRYSALISPRO Software system, Agilent Technologies, Oxford, UK, 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of (R,S)-1, (S,S)-2, (R,S)-2 and 3 in CIF format; 1H, 13C and PGSE NMR for 1–4; FTIR data for (S,S)-1 and (S,S)-2; details of rac-LA polymerization with (S,S)-1 and (S,S)-2; MALDI-TOF spectra of PLA. CCDC 1438998–1439001. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py02005k |
| This journal is © The Royal Society of Chemistry 2016 |