
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of high-molecular-weight aliphatic polycarbonates by organo-catalysis†
Jingjiang
Sun
and
Dirk
Kuckling
*
University of Paderborn, Chemistry Department, Warburger Str. 100, D-33098 Paderborn, Germany. E-mail: dirk.kuckling@uni-paderborn.de
First published on 20th January 2016
Abstract
Aliphatic polycarbonates have attracted significant attention for biomedical application over the last few years due to their biodegradability, low toxicity and good biocompatibility. However, in most cases, the use of metal-based catalysts is required for the preparation of aliphatic polycarbonates by the polycondensation method, which is difficult to remove completely from the final polymer. For this reason, our work is focused on the synthesis of high-molecular-weight aliphatic polycarbonates using organo-catalysts via a two-step polycondensation of dimethyl carbonate and a linear alkane diol as monomers. A variety of organo-catalysts have been surveyed for the synthesis of aliphatic polycarbonates. The influence of thiourea with mono- or bi-electron acceptor groups as cocatalysts, which were found to activate the carbonyl groups of lactide and trimethylene carbonate in the ring opening polymerization successfully, was investigated in the polycondensation. In summary, high-molecular-weight aliphatic polycarbonates, such as poly(1,4-butylene carbonate) (PBC), poly(1,5-pentamethylene carbonate) (PPC) and poly(1,6-hexamethylene carbonate) (PHC), were successfully prepared with number averaged molar masses (Mn) up to 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, dispersities below 1.8 and high yield of >80% under relatively mild operating conditions (T < 130 °C) using 4-dimethylaminopyridine (DMAP) as the catalyst. At 170 °C the poly(1,4-butylene carbonate) with Mn of 52000 g mol−1 was synthesized. Additionally, hydroxyl group terminated poly(1,4-butylene carbonate) with Mn up to 17000 g mol−1 was obtained and characterized by 1H NMR spectroscopy and ESI-TOF-mass spectrometry. The ratio of end groups (–OH/–OC(O)O–CH3) could be adjusted by using different feed ratios or catalysts.
000 g mol−1, dispersities below 1.8 and high yield of >80% under relatively mild operating conditions (T < 130 °C) using 4-dimethylaminopyridine (DMAP) as the catalyst. At 170 °C the poly(1,4-butylene carbonate) with Mn of 52000 g mol−1 was synthesized. Additionally, hydroxyl group terminated poly(1,4-butylene carbonate) with Mn up to 17000 g mol−1 was obtained and characterized by 1H NMR spectroscopy and ESI-TOF-mass spectrometry. The ratio of end groups (–OH/–OC(O)O–CH3) could be adjusted by using different feed ratios or catalysts.
Introduction
Polycarbonates (PCs) are polymers containing repeating carbonate groups (–O–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–). Aromatic polycarbonates are widely used as engineering plastics1,2 because of their attractive mechanical properties, e.g. low moisture absorption, high impact strength, high elastic modulus, creep resistance and good thermal stability. However, compared with traditional aromatic polycarbonates, aliphatic polycarbonates have received little attention because of their poor thermal stability and high susceptibility to hydrolysis.1,3–7 Over the last few years, aliphatic polycarbonates have attracted significantly increasing attention for biomedical application, e.g. for the composition of biomedical implants and acting as drug delivery devices, due to their biodegradability, low toxicity and good biocompatibility.1,4,8–11 Aliphatic polycarbonates can be prepared through different methods, such as copolymerization of CO2 and an epoxide (Scheme 1a),12,13 which is only suitable for the synthesis of aliphatic polycarbonates in which the carbonate linkages are connected by two carbon atoms, ring opening polymerization of cyclic carbonate monomers (Scheme 1b)1,14–18 and condensation polymerization of dialkyl- or diphenyl carbonate and aliphatic diols (Scheme 1c).4,19–26
O)–O–). Aromatic polycarbonates are widely used as engineering plastics1,2 because of their attractive mechanical properties, e.g. low moisture absorption, high impact strength, high elastic modulus, creep resistance and good thermal stability. However, compared with traditional aromatic polycarbonates, aliphatic polycarbonates have received little attention because of their poor thermal stability and high susceptibility to hydrolysis.1,3–7 Over the last few years, aliphatic polycarbonates have attracted significantly increasing attention for biomedical application, e.g. for the composition of biomedical implants and acting as drug delivery devices, due to their biodegradability, low toxicity and good biocompatibility.1,4,8–11 Aliphatic polycarbonates can be prepared through different methods, such as copolymerization of CO2 and an epoxide (Scheme 1a),12,13 which is only suitable for the synthesis of aliphatic polycarbonates in which the carbonate linkages are connected by two carbon atoms, ring opening polymerization of cyclic carbonate monomers (Scheme 1b)1,14–18 and condensation polymerization of dialkyl- or diphenyl carbonate and aliphatic diols (Scheme 1c).4,19–26
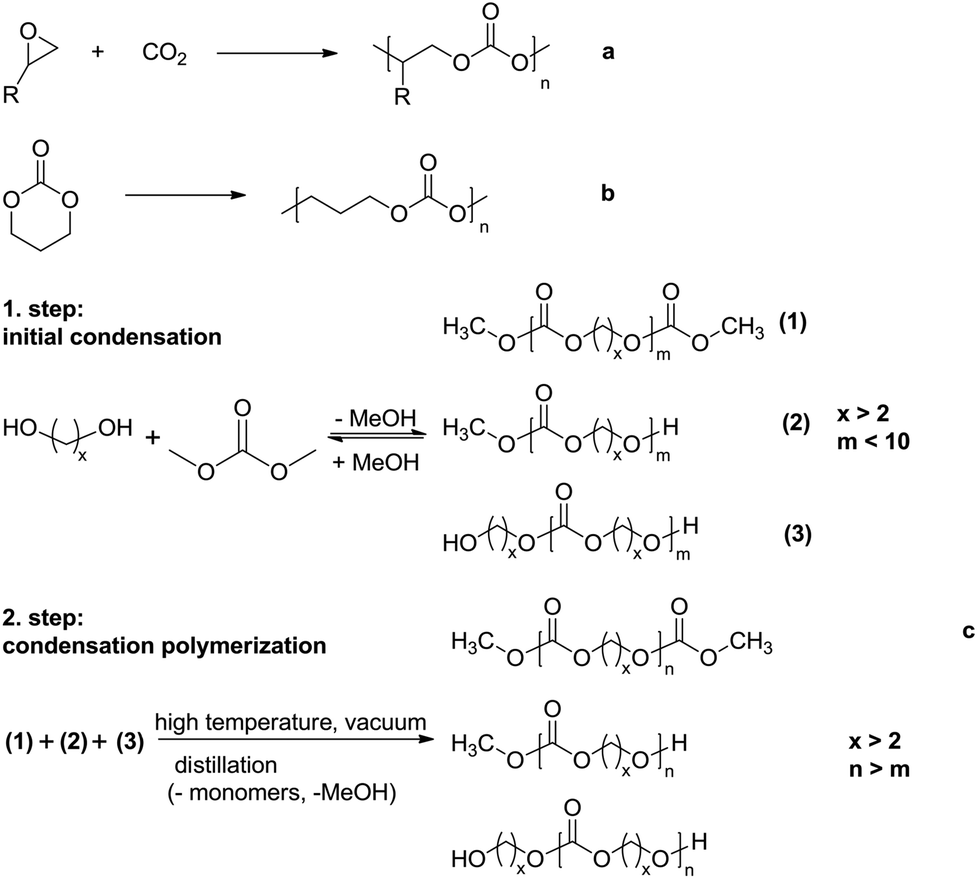 | ||
| Scheme 1 Various methods for the preparation of aliphatic polycarbonates. | ||
The ring opening polymerization of cyclic carbonates is one of the most effective methods to obtain polycarbonates with a high molar mass and low dispersity.20 However, cyclic carbonate monomers are very expensive because of their low synthetic yields. Hence, polycarbonates from ring opening polymerization have been mainly investigated for biomedical application.22,27,28 The best strategy for large-scale preparation of aliphatic polycarbonates is the two-step condensation polymerization of dimethyl carbonate (DMC) and aliphatic diols with more than three carbon atoms. Oligomers with a molar mass lower than 1000 g mol−1 are obtained in the first, initial condensation step, due to the low equilibrium constant. In the second step, polymer chains propagated by transesterification between the hydroxyl and methyl carbonate or two methyl carbonate end groups in the presence of transesterification catalysts, while high temperature and high vacuum are required to remove unreacted monomers and freshly generated byproducts. The resulting oligomers or polymers have three possible end group compositions, hydroxyl end groups, methyl carbonate end groups or a combination of both (Scheme 1c).19,20,22 Recently, Li et al. have reported the preparation of polycarbonates with a high molar mass (Mn up to 94000 g mol−1) using a novel TiO2/SiO2-poly(vinyl pyrrolidone)-based catalyst (TSP-44).21 Lee et al. have used NaH as the catalyst to prepare aliphatic polycarbonates with a high molar mass (Mn up to 150000 g mol−1) successfully with the prerequisite that the [–OH]/[–OCH3] ratio of the oligomers generated in the transesterification step is about 1.0.22 However, in most cases, the use of metal-based catalysts is required for the preparation of aliphatic polycarbonates by the polycondensation method, which is difficult to remove completely from the final polymer.
For this reason, our work is focused on the synthesis of high-molecular-weight polycarbonates using organo-catalysts via a two-step polycondensation of dimethyl carbonate and a linear alkane diol as monomers. Some organo-catalysts such as guanidines, amidines and tertiary amines have been used in the ring opening polymerization of trimethylene carbonate (TMC) and shown to yield poly(trimethylene carbonate) with a high molar mass (Mn up to 72000 g mol−1), with low dispersities (ĐM = 1.04–1.80) and with well-defined terminal groups.15,16,29 Furthermore, thiourea derivatives have been reported for the direct activation of electrophilic substrates via employment of double hydrogen bonding. Hedrick30,31 and Dixon32 demonstrated that thiourea based bifunctional organo-catalysts effectively activated the ring opening polymerization of cyclic esters. Moreover, Hedrick reported that electrophilic thioureas and nucleophilic bases are not required to be linked in the same molecule.31 Kosugi has exploited a 3,5-bis(trifluoromethyl)phenyl and 4-pyrrolidinopyridine (PPY) based zwitter ionic salt organo-catalyst for transesterification reactions.33 However, there have been few reports about the successful synthesis of aliphatic polycarbonates with high-molecular-weight using organo-catalysts through condensation polymerization of DMC and diols. Picquet and Plasseraud described a route to the synthesis of aliphatic polycarbonates (Mn up to 7400 g mol−1) using 1-n-butyl-3-methylimidazol-2-carboxylate (BMIM-2-CO2) as a catalyst.4
In this work, a variety of organo-catalysts (Scheme 2) have been surveyed for the synthesis of aliphatic polycarbonates. The influence of thiourea with mono- or bi-electron acceptor groups as cocatalysts, which were found to activate the carbonyl groups of lactide and trimethylene carbonate in the ring opening polymerization successfully, was investigated in the polycondensation as well.
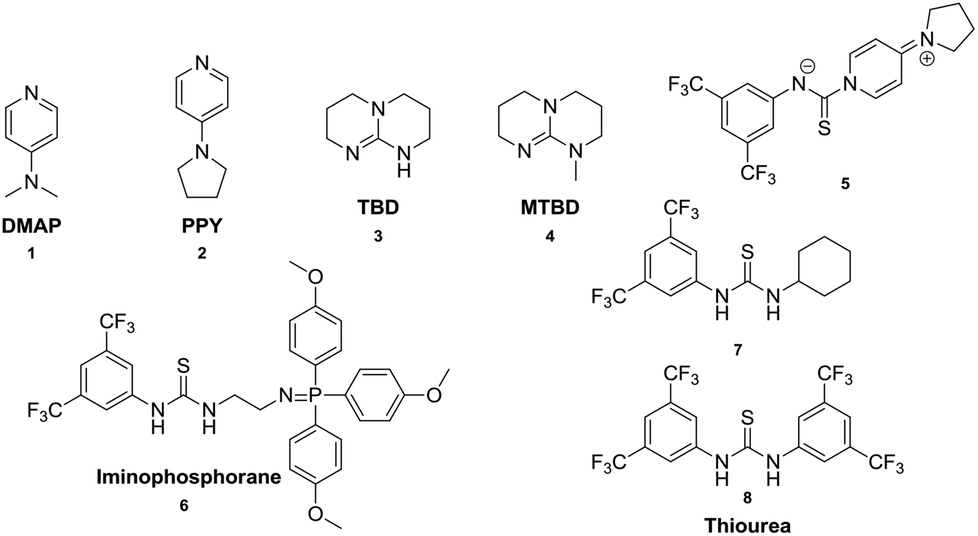 | ||
| Scheme 2 Organo-catalysts for the synthesis of aliphatic polycarbonates via the polycondensation method. | ||
Experimental
Materials
1,4-Butane diol (99+%) was purchased from Acros Organics and vacuum distilled using a short path distillation apparatus and dried with 4 Å molecular sieves before use. Other diols (C > 4) were dried under vacuum at ambient temperature overnight before use. The catalysts arylaminothiocarbonylpyridinium salt (5),33 bifunctional iminophosphorane (6)32 and thioureas (7 and 8)34 were synthesized as previously reported. Other reagents were commercially available and used as received.Measurements
1H and 13C NMR spectra were recorded by using a Bruker AV 500 spectrometer at 500 MHz and 125 MHz, respectively. Chloroform-d (CDCl3, 99.8 D%), dimethylsulfoxide-d6 (DMSO-d6, 99.5 D%) or acetonitrile-d3 (MeCN-d3, 99 D%) were used as solvents for NMR measurements. The molar masses and molar mass distribution (Mw/Mn) were analyzed by employing a size exclusion chromatography (SEC) system equipped with four consecutive columns (PSS-SDV columns filled with 5 μm gel particles with a defined porosity of 106 Å, 104 Å, 103 Å and 102 Å, respectively) and a Shodex RI-detector (RI-101) at 30 °C. The system was operated at a flow of 0.75 mL min−1 with chloroform as the solvent. Polystyrene standards were used for calibration.ESI-TOF-mass spectra in the m/z range 400–4000 were measured on a SYNAPT™ G2 HDMS™ from Waters. The mass spectrometric parameters were the following: capillary voltage: 2.5 kV; sampling cone voltage: 50 V; extraction cone voltage: 1 V; cone gas flow: 30 L h−1; source temperature: 120 °C; desolvation gas flow: 650 L h−1; desolvation temperature: 350 °C; helium cell gas flow: 180 mL min−1; IMS gas flow: 90 mL min−1; IMS wave velocity: 460 m s−1; IMS wave height: 40 V. The PBC sample was dissolved in acetonitrile (2 g L−1) and then mixed with NaI 0.1 g L−1 in methanol in the ratio of 5:5:990. Data were obtained and processed using Drift Scope 2.4 and Polymerix Software.
General procedure for condensation polymerization of diols and DMC
In a two-necked flask connected to a Schlenk line with vacuum and argon gas line diol, DMC and organic catalysts were added under an argon atmosphere. The mixture was stirred in an oil bath at 130 °C until achieving the equilibrium determined by 1H NMR spectroscopy within 2–18 h. Before starting the second step, the flask was equipped with a vacuum distillation apparatus. In the second step the condensation polymerization was carried out under reduced pressure with the oil bath temperature maintained at 130 °C or increased to 170 °C. The condensation polymerization was conducted overnight. The mixture was then cooled to room temperature and dissolved in chloroform. The polymer was isolated by precipitation in ethanol and dried under vacuum to give a white solid.
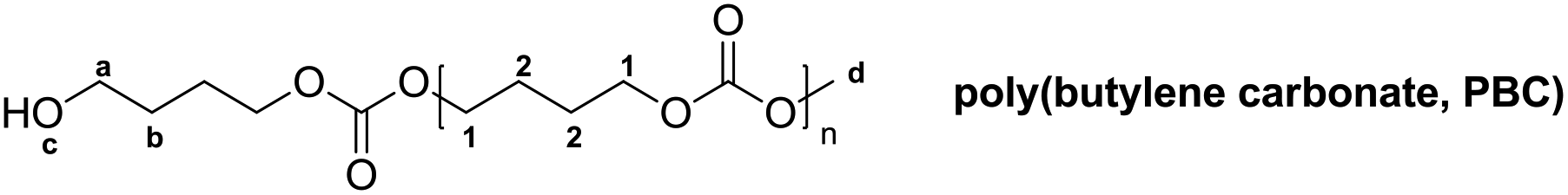
δ (ppm) = 1.65 (m, 3 H, bCH2, cOH), 1.76 (b, 4 H, 2CH2), 3.68 (t, 2 H, JHH = 6.3 Hz, aCH2), 3.77 (s, 3 H, OdCH3), 4.15 (b, 4 H, 1CH2)

δ (ppm) = 1.46 (m, 2 H, 3CH2), 1.70 (m, 4 H, 2CH2), 3.65 (t, 2 H, JHH = 6.5 Hz, aCH2), 3.77 (s, 3 H, ObCH3), 4.13 (t, 4 H, JHH = 6.6 Hz, 1CH2)
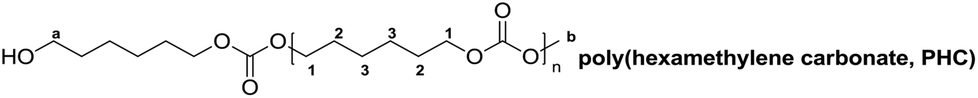
δ (ppm) = 1.40 (m, 4 H, 3CH2), 1.67 (m, 4 H, 2CH2), 3.63 (t, 2 H, JHH = 6.5 Hz, aCH2), 3.76 (s, 3 H, ObCH3), 4.11 (t, 4 H, JHH = 6.8 Hz, 1CH2)
Results and discussion
Catalyst screening
A variety of organo-catalysts such as commercially available pyridines (4-dimethylaminopyridine, DMAP (1) and 4-pyrrolidinopyridine, PPY (2)), guanidines (1,5,7-triazabicyclo[4.4.0]dec-5-ene, TBD (3) and 7-methyl-1,5,7-triazabicyclo[4.4.0] dec-5-ene, MTBD (4)), bifunctional arylaminothiocarbonylpyridinium salt (5) and iminophosphorane (6) have been used in the condensation polymerization of diols and dimethyl carbonate (DMC). Furthermore, the 4-dimethylaminopyridine (1) was also investigated together with thioureas with mono-(7) and bi-(8) electron withdrawing 3,5-bis(trifluoromethyl)phenyl groups as cocatalysts. Such compounds were used in the ring opening polymerization of trimethylene carbonate successfully,15,16,30 and it was of interest to test the influence of thiourea in the transesterification step and in the condensation polymerization. In the first step the transesterification reaction of DMC and diols is an equilibrium reaction. According to the Le Chatelier's principle, the chemical equilibrium could only be affected by a change in the temperature or feed ratio. At the equilibrium point the conversions of –CH2OH to –CH2OC(O)O– groups should be constant, which is shown in the 1H NMR spectrum of the reaction solution at this point as the constant peak area ratio of unreacted dimethyl carbonate (3.70 ppm in DMSO-d6) and generated byproduct methanol (3.19 ppm in DMSO-d6) (Fig. 1).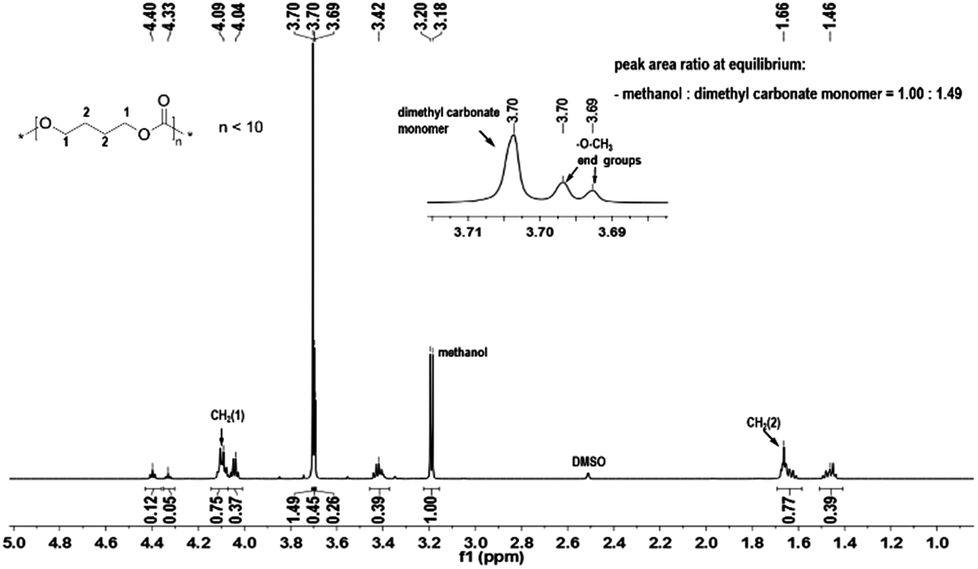 | ||
| Fig. 1 1H NMR spectrum of the reaction solution of BD and DMC using cat. 1 at equilibrium. | ||
The catalytic activities of various organo-catalyst systems with respect to the transesterification step of 1,4-butanediol (BD) and DMC were evaluated by comparing the necessary time to achieve the equilibrium. The fewer times the system needed, the higher the activity of the system. Table 1 summarizes the results of the different catalyst systems in the transesterification step under an argon atmosphere with a constant feed ratio of [BD]:[DMC]:[cat.] = 1:1.2:0.005 at 130 °C.
| Entry | Catalyst systems | Time to achieve equilibriuma | M n (g mol−1) | Đ M |
|---|---|---|---|---|
| BD: DMC:cat. = 1: 1.2:0.005. Reaction time: 1st step: until equilibrium; 2nd step: overnight.a Determined using 1H NMR spectroscopy.b Determined using SEC in chloroform with PS standards. |
||||
| 1 | Cat. 1 | 1.0 h | 16000 |
1.66 |
| 2 | Cat. 2 | 1.0 h | 7900 | 2.03 |
| 3 | Cat. 3 | 0.5 h | 6200 | 2.18 |
| 4 | Cat. 4 | <0.5 h | 17000 |
1.77 |
| 5 | Cat. 5 | 3.0 h | 4100 | 2.40 |
| 6 | Cat. 6 | Overnight | 13000 |
1.68 |
| 7 | Cat. 4 + cat. 7 | 2.5 h | 6900 | 2.16 |
| 8 | Cat. 4 + cat. 8 | 3.0 h | 7500 | 1.80 |
All catalyst systems investigated were active for the transesterification of BD and DMC. It was found that the transesterification reaction was carried out readily (<1 h) in the presence of pyridine (cat. 3 and 4) and guanidine (cat. 5 and 6) catalysts. However, in the same reaction catalyzed either by bifunctional catalysts including thiourea groups (cat. 1 and 2) or by DMAP with mono- or bi-electron withdrawing 3,5-bis(trifluoromethyl)phenyl groups, thiourea (cat. 7 and 8) cocatalysts proceeded much slower.
A proposed mechanism is shown in Scheme 3. The thiourea is able for the direct activation of the carbonyl group by means of double hydrogen bonding. The activation may lead to a more stable intermediate, which may subsequently release the methanol difficultly. This also indicates why the thiourea based catalysts could be used in the ring opening polymerization of cyclic esters or carbonates and inhibiting simultaneously the transesterification side reaction.
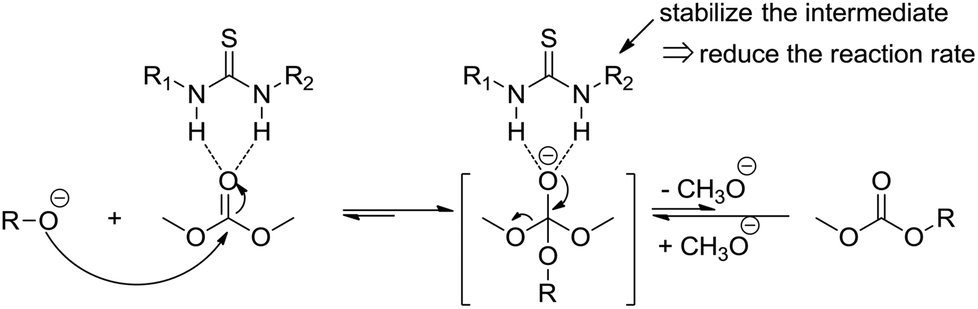 | ||
| Scheme 3 Possible mechanism of a thiourea catalyzed transesterification reaction. | ||
Besides the transesterification step, all catalyst systems were also investigated in the polymerization step after achieving the equilibrium in the 1st step. All catalyst systems were effective for the synthesis of poly(butylene carbonate) from BD and DMC and polycarbonates were obtained with molar masses higher than 4100 g mol−1 and dispersities lower than 2.40. DMAP (cat. 1) and MTBD (cat. 4) showed the best results with the synthesized polycarbonate having a molar mass up to 17000 g mol−1 and dispersity of 1.66. Also in the polycondensation step, thioureas as cocatalysts retarded the polymerization. Moreover, an experiment without any catalyst was also evaluated at 130 °C and 170 °C for 1st and 2nd steps, respectively. However, the 1H NMR spectrum after the first step showed that the transesterification reaction between DMC and BD did not occur. All of the compounds in the reaction flask were distilled off after 30 min in the second step. This also proved the efficiency of all investigated catalyst systems.
Results of polycarbonate synthesis
The polymerization temperature and initial feed ratio of diols and DMC are further two important parameters. They can influence the final polymer properties significantly. In order to obtain polycarbonates with higher molar masses, we optimized the polymerization conditions. The initial [BD]:[DMC] ratio was varied from 1:1.2 up to 1:2.0 and the temperatures in condensation polymerization steps were set from 130 °C up to 170 °C. The aim of using excess dimethyl carbonate is enhancing the conversion of diols in the 1st step and obtaining oligomers with more methyl carbonate end groups, which are more reactive than hydroxyl end groups in the 2nd step for the transesterification reaction between two polymer chains and thus leading to higher molar masses. The excess of DMC was removed in the 2nd step.24
Table 2 summarizes the most significant results of the polycarbonate synthesis under different polymerization conditions. As shown, Mn increased significantly from 5900 g mol−1 to 11000 g mol−1, respectively, while the feed ratio changed from 1:1.5:0.5 mol% to 1:2.0: 1 mol% (entries 1–3), indicating that the methyl carbonate end group is more reactive than the hydroxyl end group in the condensation polymerization step. With the feed ratio of 1: 2.0: 1 mol%, PBC, PPC and PHC samples with relatively high Mn values up to 23000 g mol−1 were obtained in the presence of more reactive catalyst DMAP (entries 9, 11 and 12). Yields were achieved up to 88%, which was calculated by the following equation (eqn (1)).
 | (1) |
| [Diol]:[DMC]:[cat.] |
Cat. | T (2 step) (°C) | Yields (%) | M n (g mol−1) | Đ M | End groupsb [–OCH3]:[–OH] |
|
|---|---|---|---|---|---|---|---|
| a Determined using SEC in chloroform with PS standards. b Determined using 1H NMR spectroscopy. Reaction time: 1st step: until equilibrium; 2nd step: overnight. | |||||||
| PBC 1 | 1:1.5:0.005 |
Cat. 5 | 130 | 60 | 5900 | 1.85 | 2:98 |
| PBC 2 | 1:1.5:0.01 |
Cat. 5 | 130 | 70 | 9000 | 1.69 | 14:86 |
| PBC 3 | 1: 2.0:0.01 |
Cat. 5 | 130 | 65 | 11000 |
1.71 | 80:20 |
| PBC 4 | 1:1.2:0.005 |
Cat. 1 | 130 | 57 | 16000 |
1.66 | 0:100 |
| PBC 5 | 1:1.2:0.005 |
Cat. 2 | 130 | 59 | 7900 | 2.03 | 0:100 |
| PBC 6 | 1:1.2:0.005 |
Cat. 4 | 130 | 61 | 17000 |
1.77 | 0:100 |
| PBC 7 | 1:1.2:0.005 |
Cat. 5 | 130 | 57 | 4100 | 2.40 | 0:100 |
| PBC 8 | 1:1.2:0.005 |
Cat. 6 | 130 | 53 | 13000 |
1.68 | 0:100 |
| PBC 9 | 1:2.0:0.01 |
Cat. 1 | 130 | 85 | 23000 |
1.77 | 32:68 |
| PBC 10 | 1:2.0:0.01 |
Cat. 1 | 170 | 79 | 52000 |
1.77 | 70:30 |
| PPC 1 | 1:2.0:0.01 |
Cat. 1 | 130 | 77 | 22000 |
1.60 | 43:57 |
| PHC 1 | 1:2.0:0.01 |
Cat. 1 | 130 | 88 | 23000 |
1.53 | 61:39 |
In addition, the end group ratio in the resulting polymers could be adjusted by changing the initial feed ratios, catalysts, or polymerization temperatures. The hydroxyl end group content decreases from 86% to 20% with the increasing initial concentration of DMC (entries 2 and 3). When the polymerization was conducted using a lower amount of the catalyst, PBC with a higher hydroxyl content (98% –OH end group) was obtained. The end group composition can also be controlled by using various catalysts due to their different catalytic activities (entries 3 and 9). Using cat. 5 leads to a PBC with a 20% –OH end group, while a PBC with a 68% –OH end group could be prepared at the same feed ratio of [diol]:[DMC]:[cat.] = 1:2:0.01, when DMAP (cat. 1) was used. Besides, polymerization temperature is also an important factor in controlling the end group composition. The hydroxyl content decreased from 68% to 30% with the temperature increasing from 130 °C to 170 °C (entries 9 and 10). By studying the preparation of polycarbonates with defined end group compositions, we found that hydroxyl terminated PBCs, which are of great interest, especially for further terminal group modification, could be obtained by using different catalysts (0.5 mol%) with the initial feed ratio of [BD]:[DMC] < 1:1.2 (entries 4–8). Among them Mn values determined for the samples using DMAP and MTBD as catalysts (entries 4 and 6) were obtained up to 17000 g mol−1 and the dispersities were lower than 1.8. Polymers synthesized using PPY based catalysts (entries 5 and 7) showed lower Mn and higher ĐM (ĐM > 2) in contrast to DMAP and MTBD. The lower catalytic activity of PPY based catalysts in the 2nd step is probably reflective of the decreased nucleophilic properties for the transesterification reaction between two methyl carbonate end groups. According to our research results, polycarbonates with a defined Mn, end group composition and low dispersity could be achieved by using alterable initial feed ratios, polymerization temperatures and catalysts with different activities.
The 1H and 13C NMR spectra of PBC 9 are shown in Fig. 2. Two multiplet signals at 1.76 ppm and 4.15 ppm are attributed to both the CH2–groups in the polymer backbone. The small signals at 1.64 ppm and 3.67 ppm indicated the existence of a terminal butanol group, while the singlet at 3.76 ppm is assigned to the terminal methyl carbonate group. The 1H NMR spectroscopy indicated that no decarboxylation occurred because no ether linkage (CH2–O–CH2) at 3.4–3.5 ppm was detected. In addition, by comparing the peak areas of the terminal butanol and methyl carbonate groups, the hydroxyl content could be calculated. For the samples with pure hydroxyl end groups, only two signals at 1.64 ppm and 3.67 ppm were detected, while the singlet peak at 3.76 ppm for –C(O)OCH3 was not visible. In the 13C NMR spectrum, the peaks around 25.14 ppm and 67.25 ppm correspond to C1 and C2 carbon atoms of the polymer backbone, respectively. The carbonate group is observed at 155.16 ppm. Signals of terminal groups are absent in the 13C NMR spectrum.
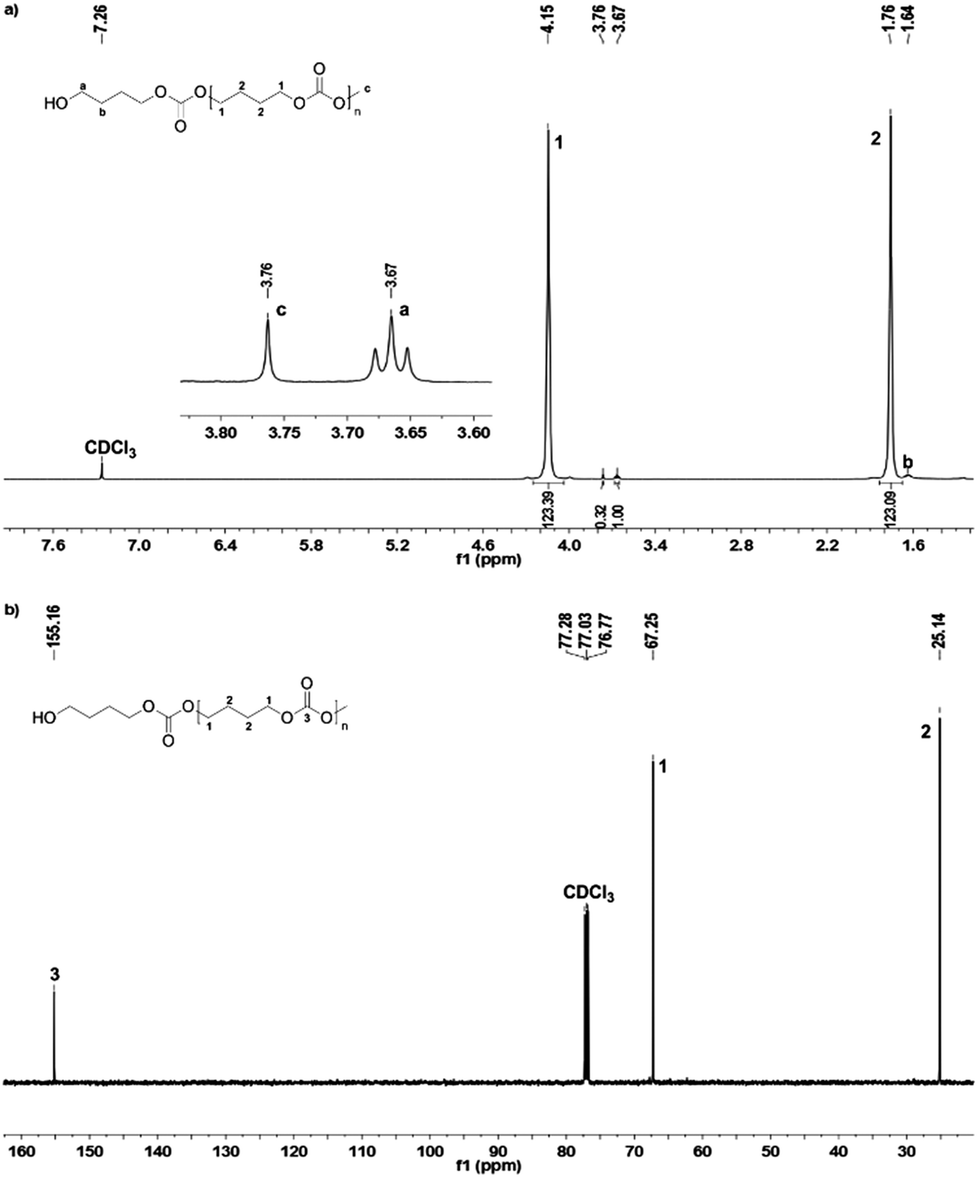 | ||
| Fig. 2 1H (a) and 13C NMR (b) spectra of PBC 9. | ||
To determine the influence of polymerization times on the molar mass of PBC, a kinetic study of PBC 9 ([BD]:[DMC]:[DMAP] = 1:2:0.01, 130 °C) was carried out. Fig. 3 shows the molar mass and molar mass distribution data determined by SEC. The molar mass of the polymer increased rapidly throughout the initial 30 min. After a reaction time of 3 h, a molar mass of 14000 g mol−1 was obtained. When the condensation reaction was further conducted, the molar mass increased slowly and finally up to Mn = 23000 g mol−1 for 24 h reaction time. The dispersity values remained below 1.8 during the condensation reaction.
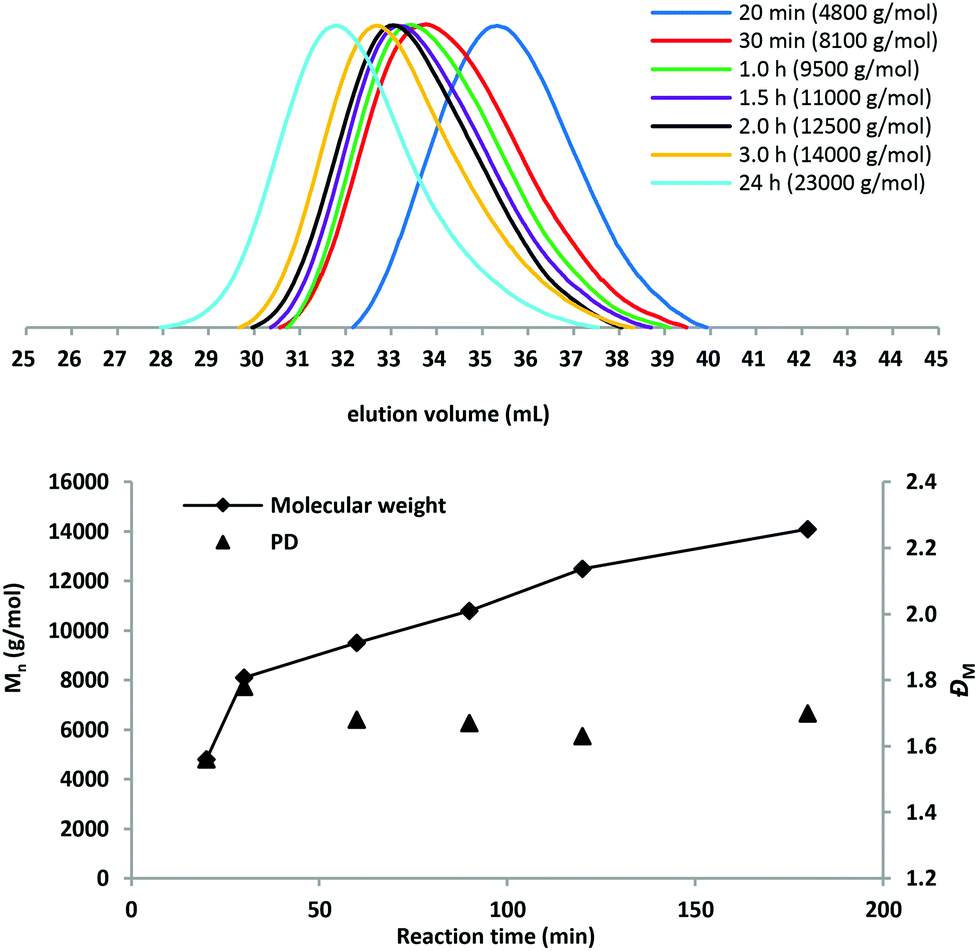 | ||
| Fig. 3 SEC traces and plot Mn (determined by SEC) and the dispersity values of PBC 9 versus the polymerization time in the 2nd step. | ||
Hydroxyl terminated PBCs (PBC 4–9 in Table 2) have also been investigated by ESI-TOF-MS to determine the end groups. Fig. 4a shows a typical ESI-TOF-MS spectrum for hydroxyl terminated PBC. Polymers were multiply charged during the ionization. The different series can be separated by Ion Mobility Separation (IMS).35 The separated spectra of up to tetraly charged polymers are shown in Fig. 4b. Moreover, the pentaly and hexaly charged polymers were also detected but they are distributed with low intensity. The ESI-TOF-MS spectra show the presence of the main series of polymer chains corresponding to (HO-PBC-C4H8OH a × Na)a+ (a = 1, 2, 3 or 4) with repeating units of 116.05 Da, which is the molar mass of the repeating PBC unit. For the doubly charged polymer with n = 20 (Table 3, entry 2), the measured value of 1229.36 Da corresponded to the calculated value of 1229.19 Da using eqn (2). No further series could be seen, indicating that the polymer was only terminated with hydroxyl groups at both the chain ends. Hence, the organo-catalyzed synthesis of polycarbonates proceeded successfully without any side reaction, such as decarboxylation.
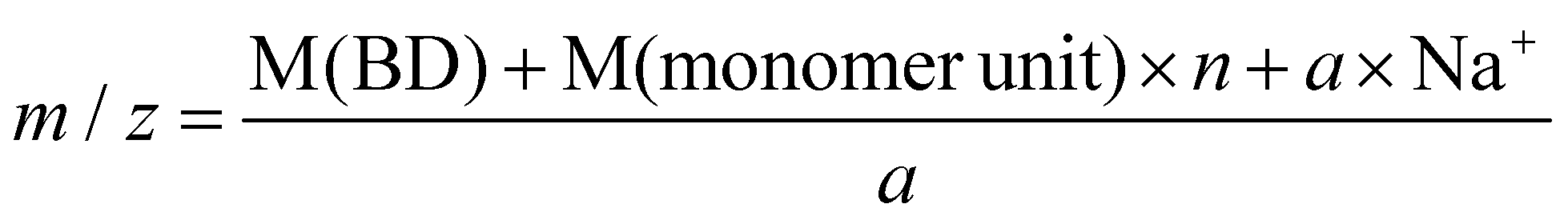 | (2) |
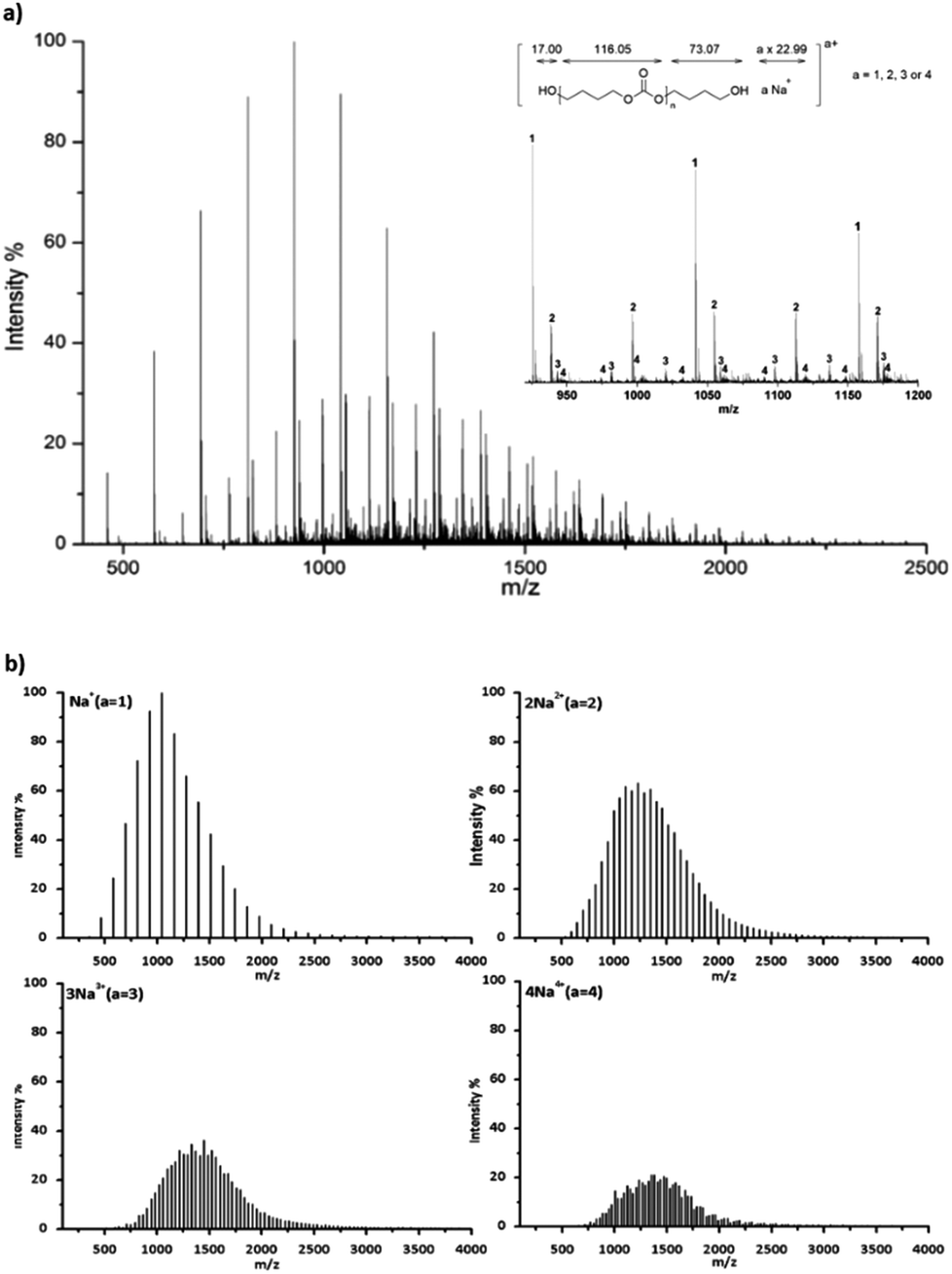 | ||
| Fig. 4 ESI-TOF mass spectrum of PBC 7 (Table 2, entry 7) terminated with the hydroxyl group, (a) complete spectrum and a part of the spectrum distinguished by carrying charges a = 1, 2, 3 and 4 in the region m/z 400 to 2500, and (b) separated spectra with a = 1, 2, 3 and 4. | ||
| a | Calculated [Da] | Found [Da] | Calculated [Da] | Found [Da] | Difference [Da] |
|---|---|---|---|---|---|
| 1 | 1042.01 | 1042.19 (n = 8) | 1158.12 | 1158.33 (n = 9) | 116.14 |
| 2 | 1229.19 | 1229.36 (n = 20) | 1287.24 | 1287.37 (n = 21) | 58.01 |
| 3 | 1214.18 | 1214.38 (n = 30) | 1252.88 | 1253.06 (n = 31) | 38.68 |
| 4 | 1206.67 | 1206.86 (n = 40) | 1235.69 | 1235.91 (n = 41) | 29.05 |
The thermal properties of the PBC, PPC and PHC samples were evaluated by DSC as shown in Table 4. The PBC samples displayed glass transition temperatures (Tg) of −36–31 °C and Tg increases with the increasing molar mass. The Tg of the PHC sample tended to lower Tg due to the higher chain flexibility. The melting temperatures (Tm) were observed at 56–62 °C, while the PPC and PHC showed lower Tm. In our case, the Tg was not visibly affected by the nature of the chain end group compositions.
Conclusions
In summary, we demonstrated that the commercially available organo-catalysts DMAP, PPY, TBD and MTBD were suitable for the synthesis of aliphatic polycarbonates with high molar masses and low dispersities via a two-step condensation polymerization under relatively mild operating conditions. Poly(1,4-butylene carbonate) (PBC), poly(1,5-pentamethylene carbonate) (PPC) and poly(1,6-hexamethylene carbonate) (PHC), were successfully prepared with Mn up to 23000 g mol−1, dispersities below 1.80 and yields of >80% at 130 °C using 4-dimethylaminopyridine (DMAP) as the catalyst. At 170 °C, the molar mass of poly(1,4-butylene carbonate) increased up to 52000 g mol−1.
In addition, according to our results, polycarbonates with a defined Mn, end group composition and low dispersity could be achieved by changing the initial feed ratios, polymerization temperatures and catalysts with different activities. Remarkably, depending on the initial feed ratio ([BD]:[DMC] < 1:1.2), hydroxyl terminated polycarbonates with different molar masses can also be obtained with a high molar mass (up to 17000 g mol−1, ĐM = 1.77). These materials are of great interest, because the combination with other polymerization methods, such as controlled radical polymerization (ATRP, RAFT or NMRP) for further application and thermal property improvement is allowed by end group modification. Additionally, the thiourea based organo-catalysts retarded the transesterification and condensation polymerization steps. On the other hand, this also proves why the thiourea based catalysts could be used in the ring opening polymerization of cyclic esters or carbonates and inhibiting simultaneously the transesterification side reaction.
Acknowledgements
The authors gratefully acknowledge the financial support from the German Ministry of Education and Research (BMBF Grand no. IB-070).References
- J. Feng, R. Zhuo and X. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS.
- H. Schobert and C. Song, Fuel, 2002, 81, 15–32 CrossRef CAS.
- A. Davis and J. H. Golden, J. Macromol. Sci. Polym. Part C: Polym. Rev., 1969, 3, 49–68 CAS.
- P. U. Naik, K. Refes, F. Sadaka, C.-H. Brachais, G. Boni, J.-P. Couvercelle, M. Picquet and L. Plasseraud, Polym. Chem., 2012, 3, 1475–1480 RSC.
- A. Welle, M. Kröger, M. Döring, K. Niederer, E. Pindel and I. S. Chronakis, Biomaterials, 2007, 28, 2211–2219 CrossRef CAS PubMed.
- A. P. Pêgo, D. W. Grijpma and J. Feijen, Polymer, 2003, 44, 6495–6504 CrossRef.
- W. Kuran, M. Sobczak, T. Listos, C. Debek and Z. Florjanczyk, Polymer, 2000, 41, 8531–8541 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- I. Engelberg and J. Kohn, Biomaterials, 1991, 12, 292–304 CrossRef CAS PubMed.
- W. H. Carothers and F. J. Van Natta, J. Am. Chem. Soc., 1930, 52, 314–326 CrossRef CAS.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 1736–1740 CrossRef CAS.
- X. Lu, W. Ren and G. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665 RSC.
- K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772–1782 CrossRef CAS.
- M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chem. – Eur. J., 2010, 16, 13805–13813 CrossRef CAS PubMed.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153–160 CrossRef CAS PubMed.
- F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Polym. Chem., 2011, 2, 528–533 RSC.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- E. Foy, J. B. Farrell and C. L. Higginbotham, J. Appl. Polym. Sci., 2008, 111, 13–17 Search PubMed.
- J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822 Search PubMed.
- W. Zhu, X. Huang, C. Li, Y. Xiao, D. Zhang and G. Guan, Polym. Int., 2011, 60, 1060–1067 CrossRef CAS.
- J. H. Park, J. Y. Jeon, J. J. Lee, Y. Jang, J. K. Varghese and B. Y. Lee, Macromolecules, 2013, 46, 3301–3308 CrossRef CAS.
- S. Bigot, N. Kébir, L. Plasseraud and F. Burel, Polymer, 2015, 66, 127–134 CrossRef CAS.
- Q. Li, W. Zhu, C. Li, G. Guan, D. Zhang, Y. Xiao and L. Zheng, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1387–1397 CrossRef CAS.
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC.
- W. Zhu, W. Zhou, C. Li, Y. Xiao, D. Zhang, G. Guan and D. Wang, J. Macromol. Sci., Pure Appl. Chem., 2011, 48, 583–594 CrossRef CAS.
- H. R. Kricheldorf and A. Mahler, J. Polym.Sci., Part A: Polym. Chem., 1996, 34, 2399–2406 CrossRef CAS.
- H. R. Kricheldorf and A. Mahler, Polymer, 1996, 37, 4383–4388 CrossRef CAS.
- D. Delcroix, B. Martín-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2010, 43, 8828–8835 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 103, 13798–13799 CrossRef PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- A. M. Goldys and D. J. Dixon, Macromolecules, 2014, 47, 1277–1284 CrossRef CAS.
- K. Ishihara, M. Niwa and Y. Kosugi, Org. Lett., 2008, 10, 2187–2190 CrossRef CAS PubMed.
- C. B. Tripathi and S. Mukherjee, J. Org. Chem., 2012, 77, 1592–1598 CrossRef CAS PubMed.
- J. Song, C. H. Grün, R. M. A. Heeren, H.-G. Janssen and O. F. van den Brink, Angew. Chem., Int. Ed., 2010, 49, 10168–10171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for preparation of catalysts and NMR spectra of catalysts and polycarbonates. See DOI: 10.1039/c5py01843a |
| This journal is © The Royal Society of Chemistry 2016 |