
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High precision 142Ce/140Ce stable isotope measurements of purified materials with a focus on CeO2 nanoparticles
Adam
Laycock
*,
Barry
Coles
,
Katharina
Kreissig
and
Mark
Rehkämper
Department of Earth Science & Engineering, Imperial College London, South Kensington, London SW7 2AZ, England, UK. E-mail: a.laycock10@imperial.ac.uk
First published on 10th June 2015
Abstract
Engineered CeO2 nanoparticles (NPs) are becoming increasingly prevalent in consumer products and this has raised concerns about the unknown behaviour and fate of such materials in the environment. Analytical limitations have hampered the detection of CeO2 NPs in natural systems at environmentally relevant levels. This study presents data on the inherent stable isotope composition of commercially available purified Ce materials with a particular focus on CeO2 NPs. The aim of this investigation is to determine whether CeO2 NPs posses a distinct isotopic signature that may be exploited for their detection in natural systems. To achieve this, suitable stable isotope measurement protocols were developed for the precise determination of the 142Ce/140Ce isotope ratio by multiple collector ICP-MS using Ba for external normalisation of the instrumental mass bias. The data presented show that precisions of ±0.01‰ (2se) and ±0.04‰ (2sd) can be routinely achieved with these techniques. The results also demonstrate that commercially available CeO2 NPs do not have a distinct Ce isotope composition that may be exploited for the purpose of stable isotope tracing.
1 Introduction
Engineered CeO2 nanoparticles (NPs) are amongst the most extensively manufactured nanomaterials (NMs). They are widely used as a glass-polishing agent and, more recently, have been employed as an effective diesel fuel borne catalyst, significantly reducing emissions and increasing engine efficiency.1–3 Even though engineered NPs are already used in a wide range and large number of consumer products, the toxicological properties and environmental fate of these materials are still relatively poorly understood.3–5 In particular, such investigations require methods that can detect small quantities of NPs in complex natural samples against the natural background levels of the elements present. One promising tracing approach is based on mass dependent differences in the stable isotope compositions of elements.Mass dependent differences, or fractionations, in stable isotope compositions are generated by processes of physical (e.g., diffusion, adsorption), chemical (e.g., precipitation, redox change) or biological nature. Recent advances in mass spectrometry have allowed the resolution of such isotope effects for numerous metals. This includes the elements Ca, Ba, and U,6–8 which resemble Ce due to their refractory, lithophile nature and moderate to high atomic mass number. Such studies have shown that stable isotope fractionations are common in nature and recorded in many natural materials. Furthermore, industrial processes, such as partial evaporation and diffusion during smelting and purification, have been found to create distinct isotopic signatures for some metals, which have been exploited for tracing anthropogenic emissions to the environment.9
In contrast to many other elements, stable isotope variations for Ce have received only little attention to date, with results presented in just two recent studies.10,11 Whilst the findings of these authors are limited to only a few samples from adsorption and precipitation experiments and 5 natural samples, they do reveal small but clearly resolvable variations in stable Ce isotope compositions. It is therefore conceivable that industrial processing and purification of Ce and the preparation of CeO2 NPs is also associated with isotopic fractionation. If these effects are large enough they may result in an isotopic signature that is sufficiently distinct from the natural background (e.g., soil, water, biological tissue) such that isotopic tracing is feasible. For example, CeO2 NPs that differ in their 142Ce/140Ce isotope ratio from the background value by 10‰ may be suitable for tracing. In this case, detection and quantification of as little as 0.5 mg kg−1 of NP-derived Ce is already possible in a sample with a natural Ce background of 50 mg kg−1, if the isotopic measurements achieve a precision of better than ±0.1‰.
This study investigates the feasibility of the stable isotope tracing approach for CeO2 NPs. To this end, high-precision Ce stable isotope data were acquired by multiple collector inductively coupled plasma mass spectrometry (MC-ICP-MS) for CeO2 nano- and bulk-particles and other purified Ce materials. These samples were chosen, to determine whether the purification of Ce and NP synthesis might result in isotopic signatures that are distinct from ‘normal’ Ce. A sufficiently distinct isotope composition would enable the tracing of CeO2 NMs in natural systems by means of stable isotope measurements. Such an approach would allow investigations into the behavior and fate of CeO2 NPs in real world settings and samples, thereby providing the improved understanding of environmental cycling. This knowledge is needed to investigate whether the use and emission of engineered NMs pose a threat to health or the environment and thus develop informed risk assessments.
2 Experimental
2.1 Samples
Seven commercially available CeO2 NP samples and three CeO2 reference materials (of which two are also in NP form), all provided as dry powders, were analysed in this study (Table 1). The reference materials (JRC NM-211, NM-212, NM-213) are available from the European Commission Joint Research Centre (JRC) whilst the commercial CeO2 NPs were sourced directly from the manufacturers.| Sample type | Supplier/comments | Particle sizea (nm) | n | δ 142Cec (‰) | ±2sdd (‰) | ±2see (‰) |
|---|---|---|---|---|---|---|
| a Particle size as reported by supplier. b Number of individual analyses. c All isotopic data are reported relative to the Alfa Aesar Specpure Ce standard solution, which served as in-house δ142Ce = 0% reference material. d The quoted 2sd uncertainties are based on the reproducibility of n individual measurements. e The quoted 2se uncertainties (standard error of the mean) were calculated as 2se = 2sd/√n. f Bristol Ce solution from Willbold.12 g Supplied directly from Energenics Europe Ltd on request, no product code or particle size data provided. | ||||||
| Ce solutions | Alfa Aesar; in-house δ-zero reference material | ±0 | ||||
| Sigma-Aldrich | 11 | +0.05 | 0.07 | 0.02 | ||
| Bristol Ce solutionf | 9 | +0.14 | 0.07 | 0.02 | ||
| Reference materials | Bulk CeO2; JRC NM-213; Sigma-Aldrich | 600 ± 400 | 11 | +0.07 | 0.05 | 0.01 |
| CeO2 NPs; JRC NM-211; Antaria | 13 ± 3 | 15 | −0.08 | 0.06 | 0.01 | |
| CeO2 NPs; JRC NM-212; Umicore | 28 ± 10 | 10 | +0.07 | 0.05 | 0.01 | |
| Commercial CeO2 NPs | MKnano-CeO2-070 | 70 | 11 | +0.07 | 0.04 | 0.01 |
| Sky Spring nano 2110 CG | 10–30 | 14 | +0.06 | 0.05 | 0.01 | |
| Io-li-tec NO-0029-HP | 15–30 | 6 | +0.03 | 0.02 | 0.01 | |
| Io-li-tec NO-0017-HP | 50–80 | 6 | +0.05 | 0.03 | 0.01 | |
| Sigma-Aldrich 544841-5G | <25 | 8 | +0.04 | 0.04 | 0.01 | |
| Alfa Aesar 44909 | 30 | 7 | −0.16 | 0.04 | 0.01 | |
| Energenics Europe Ltdg | — | 11 | +0.05 | 0.04 | 0.01 | |
| Ce metals (99.9% purity) | Piece 1; RGB research, 99.9% purity | 8 | +0.11 | 0.06 | 0.02 | |
| Piece 2; RGB research, 99.9% purity | 5 | +0.12 | 0.05 | 0.02 | ||
| Ce salts (99.99% purity) | Ammonium Ce(IV) nitrate; Sigma-Aldrich 431338 | 9 | +0.09 | 0.07 | 0.02 | |
| Ce(III) nitrate; Alfa Aesar 11330 | 8 | +0.03 | 0.07 | 0.02 | ||
Furthermore, three different Ce standard solutions were analysed, whereby the Alfa Aesar Specpure solution was used as in-house Ce isotope reference material, relative to which all other Ce isotope compositions were determined. The Ce solution that was prepared by Willbold12 at the University of Bristol (termed ‘Bristol Ce’ hereafter) has also been proposed as a Ce isotope reference material. Additional purified Ce based materials analysed encompass two small chunks of Ce metal that were treated as distinct samples, and the Ce salts ammonium Ce(IV) nitrate (NH4)2Ce(IV)(NO3)6 and Ce(III) nitrate, Ce(III)(NO3)3 (Table 1).
2.2 General procedures and reagents
All sample preparation work was carried out in the ISO class VI clean room facility of the MAGIC Research Centre at the Department of Earth Science and Engineering, Imperial College London. For all blank sensitive procedures ISO class IV laminar flow hoods were utilised. Concentrated 14.5 M HNO3 and 6 M HCl were purified prior to use by sub-boiling distillation in quartz stills, whilst water was from a Millipore Milli-Q Academic system that produces >18.2 MΩ cm−1 water. Suprapur quality 30% H2O2 solution (Merck Millipore) and 99.5% purity NaBrO3 (Alfa Aesar) were used as purchased.2.3 Sample preparation for stable isotope analysis
Working stock solutions with Ce concentrations of 100 μg mL−1 in 0.1 M HNO3 were prepared from the CeO2 powders and the purified Ce based materials (Table 1) as follows. For the three Ce standard solutions, suitable aliquots were diluted with 0.1 M HNO3. Solutions of the two Ce metal pieces were prepared at room temperature by dissolving about 1 g of metal in 200 mL 2 M HNO3. The resulting solutions were diluted with 2 M HNO3 to obtain concentrated Ce solutions of 1000 μg mL−1. Aliquots of these were further diluted to obtain the working stock solutions. For the Ce(III)(NO3)3 and (NH4)2Ce(IV)(NO3)6 salts, about 10–15 mg of material were dissolved in 4 mL 4 M HNO3 at room temperature. The resulting solutions were evaporated to dryness and taken up in 0.1 M HNO3.For the CeO2 powders, 10–15 mg of material were weighed into 15 mL Teflon beakers, 4 mL 14.5 M HNO3 and 1 mL 30% H2O2, to reduce Ce(IV) to Ce(III), were added before placing the beakers on a hotplate at 80 °C. Dissolution was allowed to proceed overnight, whereupon the temperature was increased to 160 °C. The beakers were again left on the hotplate overnight following which the samples were dried down. If complete digestion was not achieved before drying, the above procedure was repeated until CeO2 powders were no longer visible in the solution. The samples were subsequently dried down three times with 100 μL 14.5 M HNO3 at 100 °C, before taking up in 0.1 M HNO3.
2.4 Mass spectrometry
The Ce stable isotope measurements were performed with a Nu Instruments Nu Plasma HR MC-ICP-MS at the Imperial College MAGIC Laboratories. The instrument is fitted with a large capacity (80 L min−1) rotary pump for enhanced evacuation of the expansion chamber to improve sensitivity. A Nu Instruments DSN and a CETAC Aridus I desolvation system were employed for sample introduction, both with glass nebulizers that provided solution uptake rates of about 100 μL min−1. Normal instrumental operating parameters are summarised in Table 2.| Nu Plasma HR MC-ICP-MS | |
|---|---|
| RF Power (W) | 1300 |
| Acceleration potential (V) | ∼6000 |
| Ar coolant gas flow (L min−1) | 13 |
| Ar auxiliary gas flow (L min−1) | 1 |
| Expansion chamber vacuum (mbar) | ∼0.75 |
| Analyzer vacuum (mbar) | ∼2 × 10−9 |
| Typical sample uptake (μl mL−1) | ∼100 |
| Instrument performance | ||
|---|---|---|
| Sensitivity (V/μg mL−1) | Ce | 110–160 |
| Ba | 130–180 | |
| Transmission efficiency (%) | Ce | 0.10–0.14 |
| Ba | 0.11–0.15 | |
For isotopic analyses, the 100 μg mL−1 stock solutions of the purified Ce samples and the in-house standard (Table 1) were diluted with 0.1 M HNO3 to prepare measuring solutions with Ce concentrations of between 50 and 125 ng mL−1. These Ce solutions were doped with a 10 μg mL−1 Ba stock solution (in 0.1 M HNO3) to obtain Ba concentrations of 75 to 350 ng mL−1 and Ce/Ba ratios of between 1.5 and 3.5.
Data collection was carried out by static multiple collection with the Faraday cups of the instrument, which are fitted with 1011 Ω resistors. Each analysis consisted of 60 integrations of 5 seconds each, in 3 blocks of 20, where the ion beams of the atomic masses 135 (Ba-collector L3), 137 (Ba-L2), 139 (La-Axial), 140 (Ce, Nd-H1), 141 (Pr-H2), 142 (Ce-H3), and 145 (Nd-H5) were monitored. Prior to each block, baseline signals were measured for 15 seconds, whilst the ion beam was deflected in the electrostatic analyser. Analyses of mixed Ce–Ba solutions typically yielded sensitivities of about 110 to 160 V μg−1 mL−1 for Ce and 130 to 180 V μg−1 mL−1 for Ba during the course of the study, equivalent to a transmission efficiency of about 0.10–0.15% for both elements (Table 2). Each measurement was followed by a thorough (∼2–3 min) washout of the sample introduction system with 0.1 M HNO3, and this was generally sufficient to reduce the measured signals to the baseline intensities.
To determine variations in the stable isotope composition of Ce, the analyses focused on the two major Ce isotopes and determination of the 142Ce/140Ce isotope ratio. The 135Ba/137Ba isotope ratio of the admixed Ba was monitored for mass bias correction with the exponential law.13 Although only purified Ce materials were analysed, small amounts of Nd were present in the samples and so the 145Nd ion beam was measured for correction of the isobaric interference of 142Nd on 142Ce using a ratio of 142Nd/145Nd = 3.27711.14 The presence of the light rare earth elements (LREE) La and Pr was also monitored, using 139La and 141Pr, even though interference corrections are not necessary. To assess the robustness of the correction for isobaric 142Nd, a series of Alfa Aesar Ce standard solutions were doped with increasing amounts of Nd. This enabled determination of the maximum Nd/Ce ratios that can be tolerated during analyses.
All sample measurements were bracketed with at least two analyses of the in-house Alfa Aesar Ce isotope reference material. Differences between the corrected 142Ce/140Ce ratios of the samples and the reference material are reported in the form of δ142Ce values, which denote the relative deviation of the 142Ce/140Ce isotope ratio of a sample (Rsam) from the in-house Alfa Aesar standard (Rstd) in parts per 1000:
| δ142Ce = ((Rsam − Rstd)/Rstd) × 1000 | (1) |
3 Results and discussion
3.1 Precision and accuracy of Ce stable isotope data for CeO2 and purified Ce samples
The isotope data obtained for the CeO2 powders and purified Ce samples (Table 1) were acquired over a 9 month period that encompassed 11 separate analytical sessions. Each single measurement consumed approximately 50 to 100 ng of Ce and the δ142Ce data for the samples feature 2sd reproducibilities, based on the results of 5 to 15 individual measurements, of ±0.02‰ to ±0.07‰ (Table 1). Based on the multiple analyses, the mean results have 2se uncertainties of only ±0.01‰ to ±0.02‰ (Table 1).These performance characteristics are similar to that achieved in MC-ICP-MS stable isotope analyses for elements of similar mass when normalised to a mass difference of 1 amu (comparison of 2sd/amu results). For example, typical 2sd/amu uncertainties of ±0.01‰ to ±0.03‰ were reported for MC-ICP-MS analyses of Cd and Mo standard solutions by Abouchami et al.15 and Goldberg et al.,16 respectively. Our data also compare favourably to the Ce stable isotope results of Nakada et al.11 and Ohno and Hirata,10 who reported δ142Ce uncertainties (2sd) of ±0.06‰ and ±0.08‰ for multiple analyses of Ce solutions. As in this study, these authors also employed MC-ICP-MS but the samples were doped with Sm, and 149Sm/147Sm was used for external normalisation of the instrumental mass bias.
As the analysed samples were purified Ce materials, it is unlikely that the accuracy of the data is compromised by matrix effects and/or spectral interferences. The absence of the former is confirmed by the observation that none of the samples displayed any unusual mass bias behaviour of Ba during the analyses. In addition, it was observed that the use of two different sample introduction systems and variations in the Ce, Ba concentrations and Ce/Ba ratios between sample and the bracketing standard solutions of up to 460% and 230% respectively, had no discernible effect on the δ142Ce data at the quoted level of precision.
The purity of the Ce solutions was, furthermore, assessed throughout the isotopic analyses by monitoring the LREEs La, Pr and Nd. These elements are likely to be the most prominent contaminants, as they are difficult to separate from Ce. However, only trace amounts of the LREEs were present, with typical LREE/Ce ratios of less than 10 × 10−5 and a maximum Nd/Ce of about 60 × 10−5 for a CeO2 powder and the two metal samples (Table 3). Any Nd contamination is, however, of concern due to the isobaric interference of 142Nd on 142Ce, which was corrected during the analyses based on the monitoring of 145Nd. Table 3 summarises the corrections that were applied to the δ142Ce data of samples to account for the 142Nd interference. The corrections were generally observed to be less than 0.2‰ but with maximum levels of up to 1.0 to 1.4‰ (Fig. 1).
| δ 142Ce correctiona (‰) | Nd/Ce (× 10−5) | La/Ce (× 10−5) | Pr/Ce (× 10−5) | |
|---|---|---|---|---|
| a Change in δ142Ce after correction for the isobaric 142Nd. | ||||
| Purified Ce materials | ||||
| Alfa Aesar Ce solution | 0.2 | 8.3 | 3.2 | 5.7 |
| Other Ce solutions | 0.06–0.3 | 2.4–12 | 0.3–3.5 | 0.3–2.8 |
| CeO2 powders | 0.03–1.4 | 1.2–62 | 0.2–32 | 0.2–52 |
| Ce metals | 1 | 44 | 23 | 1.5 |
| Ce salts | 0.03–0.05 | 1.1–2.2 | <0.01–0.8 | <0.01–4.5 |
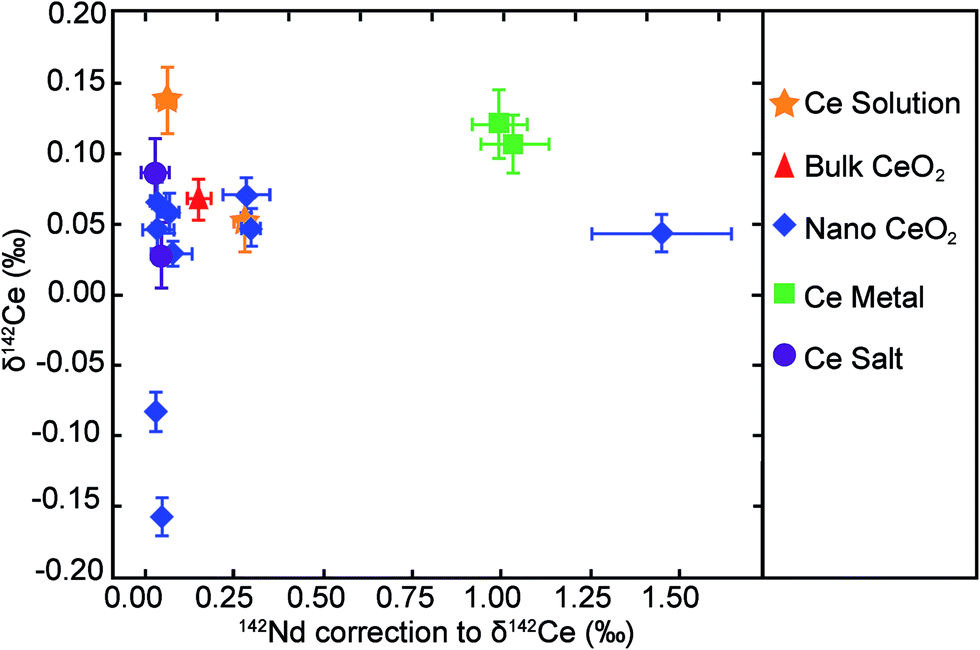 | ||
| Fig. 1 The δ142Ce values of the purified Ce materials (Table 1) are plotted against the correction to the δ142Ce values for the presence of isobaric 142Nd (Table 3). The error bars associated with the x-axis denote the range of corrections applied to the δ142Ce values for the presence of isobaric 142Nd (Table 3), whereas the y-axis error bars denote the 2se uncertainties of the δ142Ce values based on multiple analyses (n = 5 to 15; Table 1). | ||
The controlled doping of the Alfa Aesar Ce standard solution with Nd was used to investigate the maximum Nd/Ce ratios that can be tolerated for reliable Ce isotope measurements. Our data demonstrate that correction of the 142Nd interference on 142Ce resulted in 142Ce/140Ce ratios that were identical, within 2sd uncertainty, to data for non-doped Alfa Aesar Ce solutions at Nd/Ce ratios of up to 0.01 (Fig. 2). As all purified Ce materials analysed here were characterised by Nd/Ce of less than 0.001 (Table 3), the results obtained for these samples can be considered as accurate, as the presence of isobaric 142Nd did not generate analytical artefacts. This conclusion is underlined by additional observations. First, a plot of δ142Ce versus magnitude of the 142Nd correction shows no obvious correlation (Fig. 1). Second, seven of the nine CeO2 powders are characterised by a very narrow range of δ142Ce values, even though they feature variable levels of Nd. It is highly unlikely that the isotopic homogeneity of the CeO2 powders is a fortuitous result of an inaccurate Nd correction. Third, the two isotopically distinct CeO2 samples are subject to only relatively small δ142Ce corrections of 0.03‰ and 0.05‰, which implies that the negative δ142Ce values are unlikely to be an artefact. In summary, these observations indicate, that the applied corrections on δ142Ce for the presence of isobaric 142Nd were accurate even at the highest observed Nd/Ce ratios of about 60 × 10−5 (Table 3).
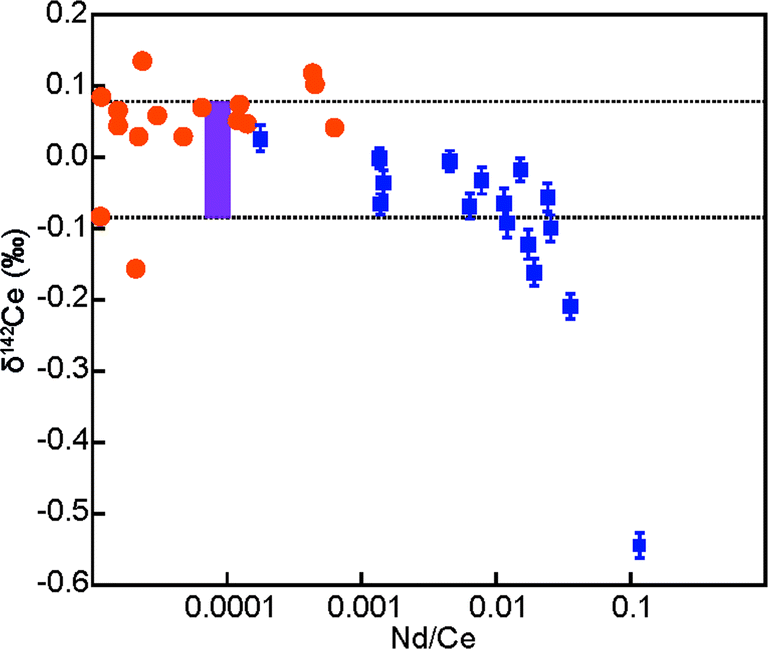 | ||
| Fig. 2 Plot of measured δ142Ce values versus the Nd/Ce ratios of the sample solutions. The purple bar and dotted lines denote the 2sd uncertainty determined from multiple analyses of the Alfa Aesar Ce standard solution (±0.09‰, n = 25). The data obtained for Alfa Aeasar Ce standard solutions doped with varying levels of Nd (blue squares with internal 2sd uncertainties) demonstrate that accurate δ142Ce values can be readily obtained at Nd/Ce ratios of up to 0.01. The orange circles represent the Ce samples that were analysed in this study. | ||
3.2 Isotopic variability
The analysed CeO2 NPs show a total variability in δ142Ce of 0.23‰, with values of between −0.16‰ and 0.07‰. However, 7 of the 9 samples display a very narrow range of δ142Ce values of 0.03‰ to 0.07‰, whilst two outliers are characterised by δ142Ce of −0.08‰ and −0.16‰ (Table 1, Fig. 3). The CeO2 bulk particles are essentially indistinguishable from most of the CeO2 NP powders, with a δ142Ce value of 0.07 ± 0.01‰ (2se). The remaining purified Ce samples also show only very limited variability in δ142Ce. The two Ce salts and the Sigma-Aldrich Ce standard solution are hereby isotopically identical, within the quoted 2se uncertainties of only 0.01‰ to 0.02‰, to the CeO2 particles. The two metal samples are slightly heavier with δ142Ce values that are identical within uncertainty at 0.11‰ and 0.12‰ (Table 1, Fig. 3).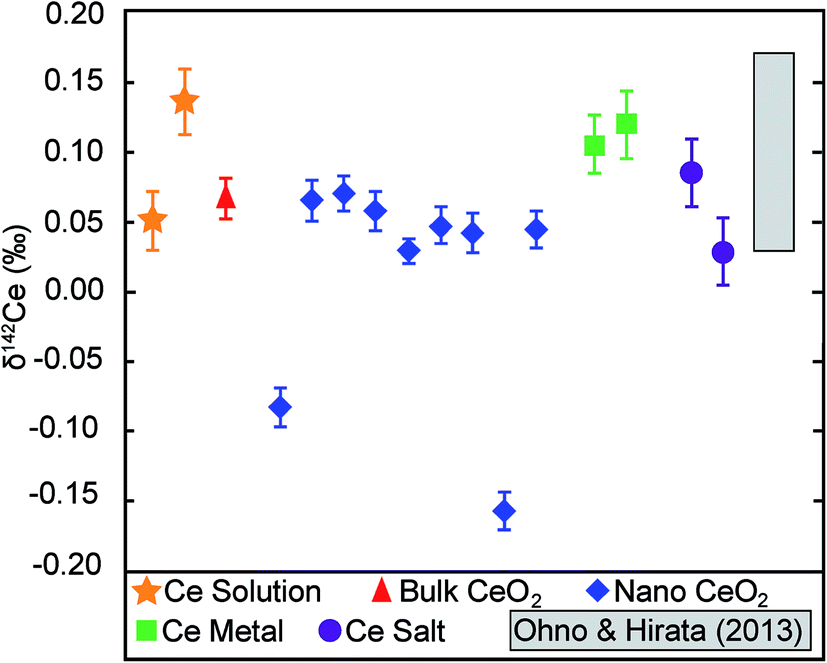 | ||
| Fig. 3 δ 142Ce data for the Ce samples analysed in this study. Error bars denote the 2se uncertainties that were calculated based on multiple analyses (n = 5 to 15; Table 1). The grey field denotes the range of δ142Ce values determined by Ohno and Hirata10 for 5 rock samples. However, the position of this field in relation to our data may not be accurate, as a different Ce reference material were applied to define δ142Ce = 0. | ||
No information was provided by the manufacturers on the provenance of the Ce or the methods that were used for element purification and NP synthesis. It is therefore conceivable that the limited isotopic variability observed here for various forms of purified Ce is simply a consequence of similar origin and similar production methods. It is notable, however, that our results appear to be in good accord with the Ce data published by Ohno and Hirata.10 Even though the two investigations cannot be compared directly, as they are expressed relative to different δ142Ce = 0 reference materials, Ohno and Hirata10 also found only a limited range of δ142Ce values of 0.14‰ for two igneous and three sedimentary rock samples (Fig. 3). This may indicate that, even though some samples display resolvable isotopic differences, the overall variability of Ce stable isotope compositions may be quite limited for most terrestrial materials.
Considering the aims of our study, the observed 0.3‰ variability in δ142Ce is too small to be exploitable for routine tracing purposes. This is demonstrated by a best-case scenario, which assumes that CeO2 NPs have a δ142Ce value that differs from a constant natural background by 0.3‰. In this instance, an increase in the background Ce concentration of 20%, from the addition of CeO2 NPs, would result in a change of the δ142Ce value by 0.05‰. This example demonstrates that isotopic tracing methods for CeO2 NPs which are based on inherent differences in δ142Ce are of no practical value because they do not provide detection capabilities that are clearly superior to those offered by less costly and less time consuming concentration measurements.
4 Conclusions
We present a robust and novel analytical protocol for the determination of 142Ce/140Ce isotope ratios by MC-ICP-MS, which offer a reproducibility (2sd) of about ±0.05‰. The techniques are, in principle, suitable for analyses of natural materials if used in conjunction with an appropriate procedure for the separation and purification of Ce from the sample matrix.Our analyses show that commercially available CeO2 NPs do not have a distinct isotope signature and their detection in natural samples by stable isotope tracing is, therefore, not feasible. However, the stable isotope tracing approach may still be successfully applied if an enriched Ce isotope is used to produce isotopically labelled CeO2 NPs.17
Acknowledgements
Two anonymous referees are thanked for their constructive comments. This research was supported by funding from DEFRA and EPSRC and was carried out as part of PROSPEcT, a public–private partnership between DEFRA, EPSRC, TSB and the Nanotechnology Industries Association (NIA Ltd.) and its members.References
- V. Arul Mozhi Selvan, R. B. Anand and M. Udayakumar, ARPN J. Eng. Appl. Sci., 2009, 4, 1–6 Search PubMed.
- B. Park, P. Martin, C. Harris, R. Guest, A. Whittingham, P. Jenkinson and J. Handley, Part. Fibre Toxicol., 2007, 4, 12 CrossRef PubMed.
- F. R. Cassee, E. C. van Balen, C. Singh, D. Green, H. Muijser, J. Weinstein and K. Dreher, Crit. Rev. Toxicol., 2011, 41, 213–229 CrossRef PubMed.
- R. D. Handy, R. Owen and E. Valsami-Jones, Ecotoxicology, 2008, 17, 315–325 CrossRef CAS PubMed.
- K. Savolainen, L. Pylkkanen, H. Norppa, G. Falck, H. Lindberg, T. Tuomi, M. Vippola, H. Alenius, K. Hameri, J. Koivisto, D. Brouwer, D. Mark, D. Bard, M. Berges, E. Jankowska, M. Posniak, P. Farmer, R. Singh, F. Krombach, P. Bihari, G. Kasper and M. Seipenbusch, Saf. Sci., 2010, 48, 957–963 CrossRef.
- J. L. Druhan, C. I. Steefel, K. H. Williams and D. J. DePaolo, Geochim. Cosmochim. Acta, 2013, 119, 93–116 CrossRef CAS.
- K. von Allmen, M. E. Bottcher, E. Samankassou and T. F. Nagler, Chem. Geol., 2010, 277, 70–77 CrossRef CAS.
- G. A. Brennecka, L. E. Borg, I. D. Hutcheon, M. A. Sharp and A. D. Anbar, Earth Planet. Sci. Lett., 2010, 291, 228–233 CrossRef CAS.
- A. E. Shiel, D. Weis and K. J. Orians, Sci. Total Environ., 2010, 408, 2357–2368 CrossRef CAS PubMed.
- T. Ohno and T. Hirata, Anal. Sci., 2013, 29, 47–53 CrossRef CAS PubMed.
- R. Nakada, Y. Takahashi and M. Tanimizu, Geochim. Cosmochim. Acta, 2013, 103, 49–62 CrossRef CAS.
- M. Willbold, J. Anal. At. Spectrom., 2007, 22, 1364–1372 RSC.
- M. Rehkämper, M. Schönbächler and C. H. Stirling, Geostand. Newsl., 2001, 25, 23–40 CrossRef.
- J. K. Bohlke, J. R. de Laeter, P. De Bievre, H. Hidaka, H. S. Peiser, K. J. R. Rosman and P. D. P. Taylor, J. Phys. Chem. Ref. Data, 2005, 34, 57–67 CrossRef CAS.
- W. Abouchami, S. J. G. Galer, T. J. Horner, M. Rehkämper, F. Wombacher, Z. C. Xue, M. Lambelet, M. Gault-Ringold, C. H. Stirling, M. Schönbächler, A. E. Shiel, D. Weis and P. F. Holdship, Geostand. Geoanal. Res., 2013, 37, 5–17 CrossRef CAS.
- T. Goldberg, G. Gordon, G. Izon, C. Archer, C. R. Pearce, J. McManus, A. D. Anbar and M. Rehkämper, J. Anal. At. Spectrom., 2013, 28, 724–735 RSC.
- F. Larner and M. Rehkämper, Environ. Sci. Technol., 2012, 46, 4149–4158 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |