
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater decontamination with hydrogen production using microwave-formed minute-made ruthenium catalysts†
Leo E.
Heim
,
Simona
Vallazza
,
Dominic
van der Waals
and
Martin H. G.
Prechtl
*
Department of Chemistry, Institute of Inorganic Chemistry, University of Cologne, Greinstraße 6, 50939 Cologne, Germany. E-mail: martin.prechtl@uni-koeln.de; Web: http://catalysis.uni-koeln.de Fax: +49 221 4701788
First published on 6th November 2015
Abstract
A method for the decontamination of water, with concomitant hydrogen formation, is herein described. Formaldehyde is an impurity that is often present in industrial wastewater in significant quantities. The formaldehyde decomposition is possible with a series of ruthenium catalysts which are accessible within minutes via microwave-assisted synthesis.
Introduction
Formaldehyde is a ubiquitous product in natural environments due to its high solubility in water (∼400 g L−1) and its production from the bio-degradation of organic matter. Whilst formaldehyde occurs naturally, there are many industrial processes that can increase the levels of formaldehyde in the wastewater to dangerous levels including in particular; wood processing, paint and resin industries and paper manufacturing.1,2 Removal of this formaldehyde contamination is essential to mitigate environmental damage and to ensure the availability of safe, non-toxic water supplies. Challenges remain however to identify an inexpensive method for this water purification, this is especially important as formaldehyde has been shown to be particularly toxic to aquatic organisms.3Whilst many different routes to mitigate the impact of formaldehyde release have been investigated including; sulphite trapping4,5 heterogeneous catalytic wet oxidation6 as well as aerobic7 and anaerobic2 bio-reactors; there has been relatively little research into the use of homogeneous or supported molecular catalysts. Investigating the ability to remove the toxic formaldehyde from wastewater using a highly active, and ideally recyclable system, is therefore of great interest. The conversion of the hydrogen-rich waste effluence into the valuable commodity of hydrogen gas would additionally be beneficial.
Ruthenium arene complexes have been widely used in the field of homogeneous catalysis for many years and have been employed for applications as varied as asymmetric transfer hydrogenation8 through to the functionalization of aqueous carbon dioxide.9 One of the key reasons for their ubiquitous nature in catalysis is the robustness of the compounds formed; this is due to the strong arene–ruthenium bond. This stability coupled with a plethora of inexpensive and readily available aryl ligands, many of which are frequently sourced from natural products, gives rise to a wide availability of versatile potential catalysts.
Our current interest, within the field of homogeneous ruthenium catalysis, was initiated by the discovery that the commercially available catalyst, [Ru(p-cymene)Cl2]2, could be used for the rapid release of hydrogen gas from aqueous formaldehyde solutions as depicted in Fig. 1.10 Of note from this report was that methanediol, favourably produced from the addition of formaldehyde to water, could release a high quantity (8.4 wt%) of hydrogen. The requirement for novel and effective methods for hydrogen production and storage is an expanding research area due to the need for alternatives to fossil fuels in order to mitigate the damage of anthropologic climate change. Reviews into the area of hydrogen storage-and-release systems in general11–13 and others that focus specifically on the use of C1 organic compounds as suitable storage materials have recently been published.14–17 Whilst many of the methodologies covered in these reviews require harsh reaction conditions or have considerable technical obstacles hindering the facile release of hydrogen; our previously reported catalytic system allows for hydrogen production from aqueous formaldehyde solutions at low temperatures without the need for any additives.10
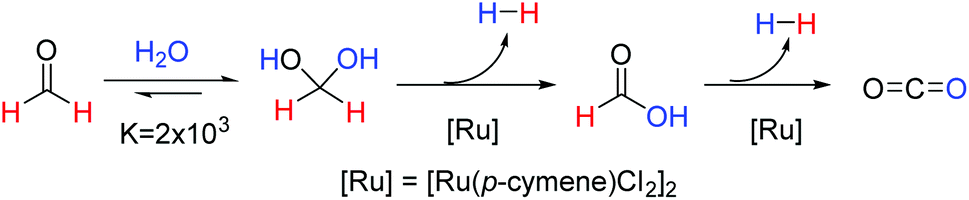 | ||
| Fig. 1 Ruthenium catalysed hydrogen release from aqueous formaldehyde. | ||
Having noted the rapid dehydrogenation of aqueous formaldehyde, it has been speculated whether the methodology could be expanded to become applicable for the catalytic removal of formaldehyde contamination, in wastewater, with the concomitant production of hydrogen. In order to identify the optimum arene–ruthenium system however, a series of analogous tailored ruthenium catalysts was needed. Current established methodologies for the formation of these catalysts, whilst effective, can require long reaction times and high temperatures to achieve only moderate yields in some cases.18 This has encouraged us to investigate the advantages that can be attained through the application of microwave irradiation for the synthesis of these compounds.
Microwave heating has long been used in organic synthesis and is considered to be of synthetic value due to the considerably shorter reaction times required. It has been used for diverse reactions such as; nanoparticle formation,19,20 polymer synthesis21 and application in various bio-reactions.22,23 The use of microwave irradiation has the effect of significantly reducing the energy required to conduct a reaction, an effect that is reported to increase upon scale-up towards industrial-level usage.24 In addition, further fulfilment of the green chemistry principals25 can be achieved as the reactions are generally cleaner, due to the short reaction times hindering the progression of side reactions; this thus produces less waste products in addition to requiring less purification.24
The use of microwave enhanced reactions for the production of ruthenium arene ligands is a much under researched field. An initial report in the area by Mingos et al. described the production of only the [Ru(p-cymene)Cl2]2 aryl-ruthenium compound in moderate yields with the best results being reported with the use of a microwave autoclave.26 A recent paper by Severin and co-workers has built upon the work to describe a methodology for the formation of [Ru(p-cymene)Cl2]2 without the need for the degassing of the solvent and with better reported yields.27 The production of ruthenium arene complexes with alternative aromatic character and modifications to the bridging functionalities has, however, yet to be explored using microwave assisted heating techniques.
We report herein a rapid and efficient route for the microwave assisted synthesis of a range of arene ruthenium catalysts and the result of investigations into their activity towards the decomposition of aqueous formaldehyde down to trace amounts, <30 ppm.
Results and discussion
Synthesis of [RuX2(arene)]2 complexes
The microwave assisted synthetic methodology described below, offers a considerable improvement for the synthesis of a variety of [RuX2(arene)]2 complexes. It has been shown to expedite the formation of the catalysts through three key routes as depicted in Fig. 2, namely; via the well established reductive diene addition methodology,18 through thermally driven arene-exchange reactions and additionally through the complexation of aryl ethers produced in situ.28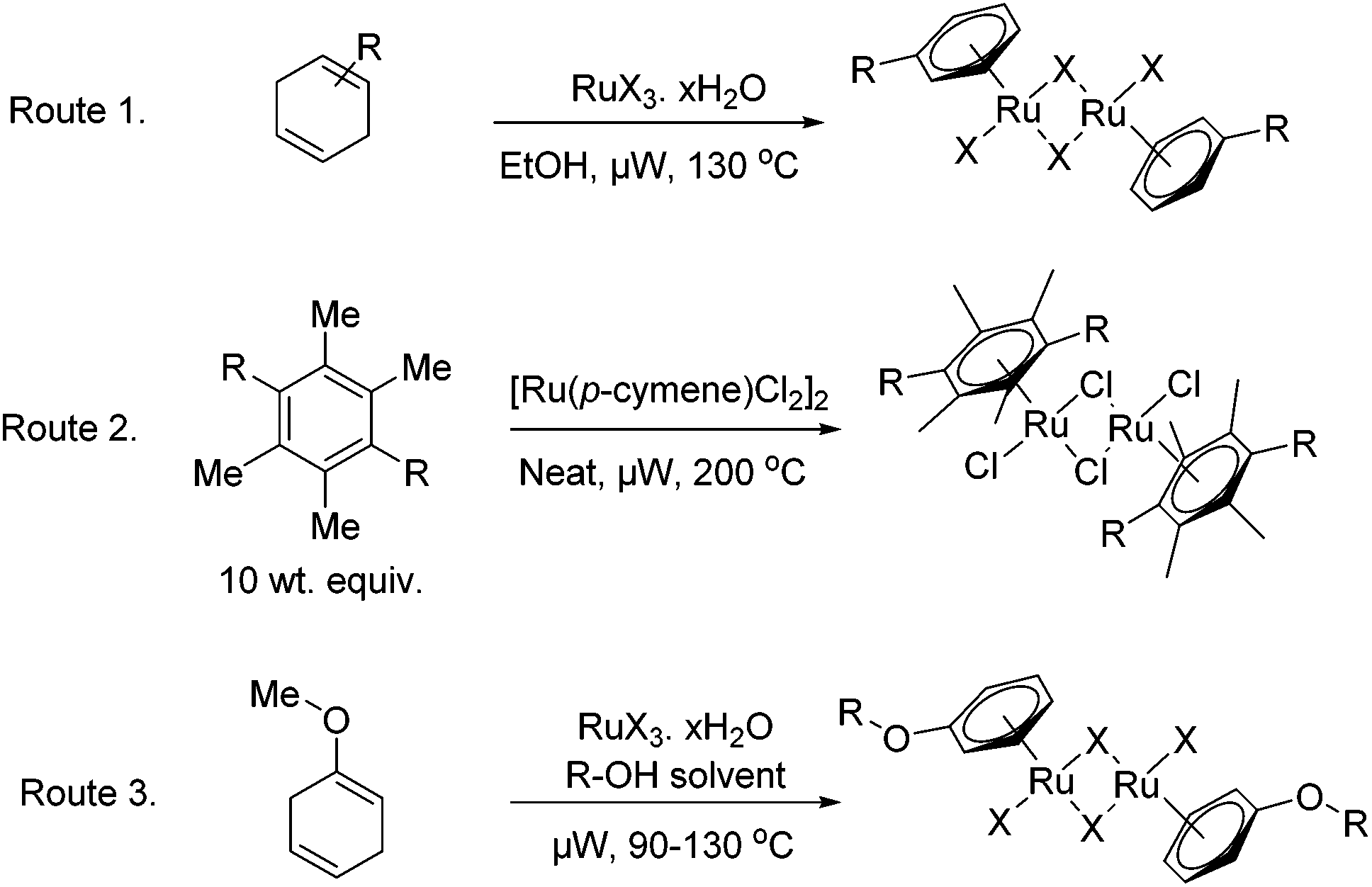 | ||
| Fig. 2 Synthetic routes for the formation of aryl ruthenium halide dimers. | ||
The chloro–ruthenium–arene complexes; entries 1–5 in Table 1, have been synthesised in a microwave reactor by heating the correspondent cyclohexadiene derivatives, obtained commercially or via Birch-reductions, with ethanolic hydrated ruthenium chloride. Good to excellent yields were reported after short reaction times of four minutes. The synthesis of analogous bromo- and iodo-versions of the [Ru(p-cymene)Cl2]2 catalyst was also attempted, entries 6 and 7 in Table 1. For the bromo-compound, hydrated ruthenium bromide was used as precursor and only a moderate yield of the desired catalyst was recovered. Ruthenium iodide arene complexes by comparison are commonly formed in a two step process involving production of the ruthenium aryl chloride from hydrated ruthenium chloride along with a cyclohexadiene; subsequent halogen exchange is then conducted using an excess of sodium iodide to give the product.18 The use of this two step process can be much expedited by microwave irradiation as shown in Table 1, entry 7; reducing heating times from 5 h to 34 minutes. Attempts to produce the Ru(p-cymene)I2]2 in a one-step procedure led only to intractable mixtures.
| Entry | Complex | Temp. [°C] | Time [min] | Yield [%] |
|---|---|---|---|---|
| a From hydrated ruthenium(III)–chloride or bromide and cyclohexadienes. b Ethanol as solvent, 10 equiv. 1,4-diene. c From hydrated ruthenium(III)–chloride and phellandrene heated for 4 min, subsequent addition of 10 equiv. of NaI and heating for a further 30 min. d Methanol as solvent, 10 equiv. 1-methoxycyclohexa-1,4-diene. e Glycol as solvent, 10 equiv. 1-methoxycyclohexa-1,4-diene. f Ligand exchange from [RuCl2(p-cymene)]2 with 10 wt equiv. of arene. | ||||
| 1 | [RuCl2(benzene)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,b a,b |
130 | 4 | 88 |
| 2 | [RuCl2(toluene)]2a,b |
130 | 4 | 93 |
| 3 | [RuCl2(mesitylene)]2a,b |
130 | 4 | 77 |
| 4 | [RuCl2(C6H5-CH2CH2OH)]2a,b |
130 | 4 | 100 |
| 5 | [RuCl2(p-cymene)]2a,b |
130 | 4 | 83 |
| 6 | [RuBr2(p-cymene)]2a,b |
130 | 90 | 41 |
| 7 | [RuI2(p-cymene)]2a,b,c |
130 | 34 | 30 |
| 8 | [RuCl2(anisole)]2a,d |
130 | 20 | 42 |
| 9 | [RuCl2(C6H5-OCH2CH2OH)]2a,e |
90 | 30 | 70 |
| 10 | [RuCl2(1,2,4,5-tetramethylbenzene)]2f |
200 | 30 | 69 |
| 11 | [RuCl2(C6Me6)]2f |
200 | 30 | 64 |
An alternative route for the formation of functionalized arene ruthenium complexes, first reported through conventional heating by White and co-workers, concerns the complexation of aryl ethers. In this interesting paper they note the use of different alcohols, other than methanol, as the reaction medium, when attempting to complex the reduced anisole diene to ruthenium chloride. This led to the incorporation of the solvent alcohol into the aryl unit of the ruthenium catalyst in the place of the methoxy group. Entry 8 in Table 1 shows that the methyl–aryl ether complex can be formed in a mediocre isolated yield. Interestingly when conducting the reaction in glycol the 2-phenoxyethanol ligated complex was formed; this leaves a versatile chemical handle for further potential modifications, such as immobilization, as well as improving the water solubility of the complex.
Whilst the diene-route is the most commonly used methodology for arene–ligand attachment, it is not suitable for complexation of ligands with a high degree of substitution. This is due to the increased electron density of the aromatic ring impeding the Birch-reduction. In some instances direct arene ligand exchange reactions were instead required to form the catalytic complexes. Following an adapted protocol from Bennett et al.,18 the [RuCl2(arene)]2 complexes seen in entries 10 and 11 in Table 1, were produced by heating the [Ru(p-cymene)Cl2]2 substrate with 10 weight equivalents of the respective ligands. All of the reactions were performed without the use of protective gas atmospheres or dry solvents.
Notably the required reaction times were substantially shorter, than for the classic method of refluxing the reagents, whilst the yields were comparable or in many cases better (‡comparative examples are given in the Notes and reference section).18 An example, in which microwave irradiation can be seen to be beneficial is in the case of the formation of Complex 8; this requires 30 hours of conventional heating under reflux to give an isolated yield of 25% and by the formation of the well known ruthenium (para-cymene) dichloride dimer requiring 4 hours of heating, again under reflux, by traditional means, to yield only 64% of the isolated product.18
The decomposition of aqueous formaldehyde solutions
With these compounds in hand, attention was turned towards their application as catalysts for the release of hydrogen from aqueous formaldehyde solutions. For the evaluation of the catalysts 1–11, their performance was initially examined at 95 °C with different catalyst loadings. With the exception of the bromo- and iodo-substituted analogues, all the investigated catalysts were shown to be able to reduce the formaldehyde content of diluted solutions (5 wt%) to levels below 45 ppm (conv. >99.9%) within 24 h, using catalyst loadings of both 1.0 mol% and only 0.1 mol%. For better comparison of the catalyst activities we conducted the decomposition reactions catalysed by 1–11 at 70 °C (Table 2).| Entry | Complex | Conv. [%] with 1 mol% cat.a | Conv. [%] with 0.1 mol% cat.a |
|---|---|---|---|
| a Measured after 24 h, starting concentration: 5 wt% HCHO. | |||
| 1 | [RuCl2(benzene)]2 | 85 | 50 |
| 2 | [RuCl2(toluene)]2 | 90 | 50 |
| 3 | [RuCl2(mesitylene)]2 | 85 | 50 |
| 4 | [RuCl2(C6H5-CH2CH2OH)]2 | 85 | 60 |
| 5 | [RuCl2(p-cymene)]2 | 90 | 70 |
| 6 | [RuBr2(p-cymene)]2 | 85 | 70 |
| 7 | [RuI2(p-cymene)]2 | 20 | 20 |
| 8 | [RuCl2(anisole)]2 | 85 | 50 |
| 9 | [RuCl2(C6H5-OCH2CH2OH)]2 | 85 | 50 |
| 10 | [RuCl2(1,2,4,5-tetramethylbenzene)]2 | 80 | 20 |
| 11 | [RuCl2(C6Me6)]2 | 70 | 50 |
The least efficient catalyst was seen to be the iodo-ruthenium(p-cymene) analogue, Table 2 entry 7. This is thought to be due to solubility problems with the catalyst as even at 0.1 mol% loadings the complex was not completely soluble. With the reaction solution already saturated at 0.1 mol%, the addition of more catalyst does not have a significant effect; precluding direct comparison between the bridging ligands.
Among the arene ligands it was noted that deviations, away from the standard p-cymene ligated complex, including both increases and reductions in the number of substituents were seen to retard the reaction. The electronic nature of the arene substituent does not appear to play a significant role as there are no notable differences in catalytic activity seen after the duration of the reaction between the complexes with the toluene ligand and the ether–ligands. One potential benefit from this wide reaction tolerance, with relation to ligand modifications could be the ability to use the ligands for additional purposes, such as catalyst immobilisation, without interfering with the reaction.
To further investigate the reaction profiles of the catalysts the initial released gas-flow was monitored. From these initial gas production rates the initial rate of formaldehyde decomposition for each catalyst could be determined, allowing for direct comparison as shown in Table 3.
| Entry | Complex | TOFa (h−1) | TON with 0.1% cat.b |
|---|---|---|---|
| a Measured after 3 min. b Measured after reaction completion, starting concentration: 5 wt% HCHO. c Purchased from Sigma Aldrich. | |||
| 1 | [RuCl2(benzene)]2 | 1284 | 500 |
| 2 | [RuCl2(toluene)]2 | 2694 | 500 |
| 3 | [RuCl2(mesitylene)]2 | 2092 | 500 |
| 4 | [RuCl2(C6H5-CH2CH2OH)]2 | 1546 | 600 |
| 5 | [RuCl2(p-cymene)]2 | 3142 | 700 |
| 6 | [RuBr2(p-cymene)]2 | 2132 | 700 |
| 7 | [RuI2(p-cymene)]2c |
613 | 200 |
| 8 | [RuCl2(anisole)]2 | 1057 | 500 |
| 9 | [RuCl2(C6H5-OCH2CH2OH)]2 | 738 | 500 |
| 10 | [RuCl2(1,2,4,5-tetramethylbenzene)]2 | 1612 | 200 |
| 11 | [RuCl2(C6Me6)]2 | 1508 | 500 |
The turnover frequencies (TOFs) of the catalysts were calculated shortly after initiation of the reaction, the results of which along with the turnover numbers (TONs) after the reaction had run to completion are given in Table 3. These clearly shows that the Ru(p-cymene)Cl2]2 complex exhibits both the highest turnover number along with the greatest turnover frequency which, in addition to the benefit of the catalyst being the least expensive, has lead to subsequent investigations focusing primarily on the use of the [Ru(p-cymene)Cl2]2 catalyst.
Further optimisation for the [Ru(p-cymene)Cl2]2 catalyst was undertaken, the results of which are reported in Table 4. As might be expected it was seen that the higher reaction temperatures yielded the greatest levels of formaldehyde decomposition with even the lower catalyst loadings reducing the contaminant to less than 0.5 mg mL−1 within 24 hours. Conversely reducing the temperature increased the contamination levels seen, particularly at low catalyst loadings. One particularly salient result however is reported in entry 5 of Table 4; this shows that almost complete removal of formaldehyde can be achieved at room temperature, both higher catalyst loadings and longer reaction times were however required.
As mentioned it was seen that the iodo-analogue of the ruthenium(p-cymene) dichloride catalyst showed considerably less reactivity for the decomposition of formaldehyde; this is despite having a similar electronic configuration to the chloride catalyst. It was decided therefore to investigate the robustness of the catalytic system and to determine whether the addition of halogen ions had any negative effect upon the ability of the system to decompose aqueous formaldehyde. The results, reported in Table 5, clearly show that whilst the addition of potassium halides do impair the decomposition, particularly in the case of potassium iodide; decomposition can proceed to low concentrations of formaldehyde when given sufficient reaction times. A highly important parameter to be considered in the treatment of wastewater is the ability to use the methodology at a variety of solution acidities. In Table 6. the results for decontamination tests, conducted at different pH-levels so as to determine both the versatility and robustness of the reaction, are shown. The results report the concentrations of formaldehyde remaining when the optimised [Ru(p-cymene)Cl2]2 catalyst was used at 95 °C over both short and longer reaction frames in various pHs. It is apparent that the catalytic system is stable, even at the higher reaction temperature, across a wide range of pH values although efficacy decreases in more acidic conditions. The utilisation of the more accurate method for determining contamination at very low concentrations of formaldehyde showed that the system is both highly efficient as well as robust enough to endure the prolonged reaction times in both acidic and basic medium.
| Entry | Additivea | Reaction time (h) | Conversion [%] |
|---|---|---|---|
| a Starting concentration: 5 weight% HCHO; 1.0 equiv. of additive used. b Analysed with the most sensitive formaldehyde test (see ESI); this conversion correspondence to trace amounts of 35–40 ppm formaldehyde. | |||
| 1 | No additive | 2 | 50 |
| 2 | No additive | 24 | 99.92–99.93b |
| 3 | KCl | 2 | 40 |
| 4 | KCl | 24 | 99 |
| 5 | KBr | 2 | 10 |
| 6 | KBr | 24 | 98 |
| 7 | KI | 2 | 10 |
| 8 | KI | 24 | 82 |
Catalyst separation and recyclability
One of the significant issues to address within the field of water decontamination is the immobilization or separation of the catalyst. This can be important due to the potential toxicity of the catalyst as well as the economical benefit of being able to reuse the metal catalyst.Two primary methods were used to analyse the recyclability of the catalyst; namely sample recharge reactions and biphasic separation.
Recharge-experiments were employed to determine the ability for the catalyst to maintain its efficacy after several uses and at high catalyst turnover numbers. Table 7 details the results obtained using the optimum catalytic system over multiple catalytic cycles with removal of the purified water and recharging with formaldehyde solution. Whilst very high conversions could be seen over the initial three cycles; beyond this extent of recycling the decomposition of the catalyst led to unreliable inconsistencies between duplicated reactions. The cycle TON increases in each cycle to account for the estimated catalyst loss due to the removal of aliquots for analysis.
| Catalyst cycle | Detected FA conc. | Conversiona | Cycle TONb | Cumulative TON |
|---|---|---|---|---|
| a Measured after 24 h at 95 °C, starting concentration: 5 wt% HCHO. 0.0240 g (1 mol%) [Ru(p-cymene)Cl2]2 catalyst used. b 0.1 mL aliquots taken after each run. | ||||
| 1 | <300 ppm | >99% | 99 | 99 |
| 2 | <300 ppm | >99% | 103 | 202 |
| 3 | <300 ppm | >99% | 108 | 310 |
For the investigation into the applicability of a biphasic system, numerous common organic solvents that are immiscible with water were investigated including 2-Me-THF which has been reported by Leitner et al. to enable the separation of a cationic ruthenium triphos complex from the aqueous reaction phase.29 Disappointingly in all these attempts significant catalyst leaching, into the aqueous phase, was noted upon cooling and separation. So as to avoid this ruthenium loss, the use of ionic liquids as solvents for the catalyst was investigated.
Initial investigations revealed that the use of particularly hydrophobic ionic liquids (ILs) was required and those with a triflimide (−NTf2) anion showed the best separation from the aqueous phase whilst remaining active. Imidazolium cations with highly hydrophilic side chains, such as ones containing vicinal alcohols did not, however, form biphasic mixtures upon addition to water, regardless of the counterion. Four suitable ionic liquids were compared for their applicability in the reaction for the decomposition of formaldehyde, the results of which are reported in the ESI† and reveal N-ethyl-N-methyl-imidazolium triflimide (EMIM NTf2) to be the optimal ionic liquid.
Taking the most effective biphasic catalyst system in hand, recyclability experiments were conducted using a greater catalyst loading to determine the potential for the repeated use of the biphasic system. After pre-forming what is postulated to be the active ruthenium-formiato catalyst system in the N-ethyl-N-methyl imidazolium triflimide ionic liquid using formic acid, the initial aliquot of 5 wt% formaldehyde solution was added. This sample was then heated at 95 °C for 24 h, the aqueous phase separated, analysed and replaced with a fresh aliquot of formaldehyde solution.
A background reaction, with the exclusion of the ruthenium catalyst, was conducted to ensure that the formaldehyde was not simply leaching into the IL and it was found that in the absence of catalyst the aqueous phase remains at 5 wt% formaldehyde solution after heating for 24 h. Table 8 shows the high conversions seen throughout the repeat use of the catalyst suspended in the ionic liquid phase. Further research is focusing on the optimisation of the use of ionic liquids as well as investigating alternative methods for catalyst immobilization which allow for water purification down to the ppm levels seen with the homogenous monophasic systems.
Conclusions
We have described herein a robust and efficient method for the decontamination of formaldehyde containing wastewater with the concomitant production of hydrogen gas. Formaldehyde impurities can be decomposed down to trace amounts as low as 10–25 ppm, thus below hazardous levels. The optimised system makes use of arene ruthenium halide complexes which are rapidly formed through the use of microwave irradiation in solvents from renewable sources. The tolerance of the system, with respect to halogen sources as well as a variety of temperatures and pH values, has been demonstrated. Further investigation into the recyclability of these molecular catalysts has shown that repeated use through recharge experiments result in the maintenance of high levels of catalyst efficiency. Ionic liquids have been revealed to be highly effective for creating a biphasic system for catalytic formaldehyde decomposition with negligible levels of catalyst leaching seen even after multiple recycling cycles. Improved immobilization of the catalysts through various routes is currently being investigated to determine the optimum method for catalyst retention with high catalytic efficiency.Experimental
Catalyst synthesis
Experimental procedure for the formation of catalysts entries 1–9 in Table 1. A 30 mL microwave vessel was filled with 0.96 mmol of hydrated ruthenium(III) halide, 10 equiv. of the desired cyclohexadiene and 15.5 mL ethanol. For entries 8 & 9 in Table 1 1-methoxy-1,4-cyclohexadiene was used as the diene along with the corresponding alcohol. The reaction mixture was heated to 130 °C, except for 9 which required 90 °C, for the reaction times indicated in Table 1. Afterwards the reaction mixture was cooled down to −78 °C, the precipitated complex was filtered off and washed with pentane (20 mL) and dried in air. The yields for complexes 1–9 are 30–100%. For the catalysts formed in entries 10 & 11; a 30 mL microwave vessel was filled with 0.2 grams of [Ru(p-cymene)Cl2]2, 10 wt equiv. (2.0 grams) of the desired arene was added without solvent and the reaction mixture was heated to 200 °C for 30 minutes. Excess arenes were removed by soxhlet extraction with pentane overnight and the catalyst recovered from DCM in vacuo in 64–69% yield.Catalyst evaluation experiments
Exemplary experiment: 4.0 μmol (0.1 mol%) [RuCl2(p-cymene)]2 was added along with 2.5 g (4 mmol) of an aqueous formaldehyde solution (5.0 wt% formaldehyde) into an open reaction vessel. The reaction mixture was heated with stirring to 95 °C for 24 h. An aliquot (200 μL) of the reaction mixture was used to determine the residual formaldehyde content after dilution using the Merck Millipore formaldehyde tests. For the biphasic reactions; 0.0481 g (0.08 mmol) of [Ru(p-cymene)Cl2]2 was dissolved in 1 mL of ionic liquid. 0.645 mL of formic acid was added to the reaction vial and heated at 70 °C for 3 h to form the active catalyst within the IL phase. 2.5 mL of 5 wt% aqueous formaldehyde solution was added and the reaction mixture heated to 95 °C for 24 h. Upon completion the aqueous phase of the mixture was separated, once the reaction had cool to room temperature, then submitted to the same formaldehyde quantification procedure. Catalyst Turnover Frequencies (TOFs) were measured using a flow meter to determine the initial volumes of gas release with time. These could then directly be converted into catalyst TOFs as it has previously been seen that the initial gas release is solely due to the dehydrogenation of methanediol to formic acid.10Acknowledgements
We gratefully acknowledge financial support provided by the Ministerium für Innovation, Wissenschaft und Forschung (NRW-returnee award 2009 to M.H.G.P.), the Heisenberg-Program (Deutsche Forschungsgemeinschaft), the COST Action “Catalytic Routines for Small Molecule Activation (CARISMA)” and the Ernst-Haage-Prize 2014 of the Max-Planck Institute for Chemical Energy Conversion. A patent has been filed under 10 2013 011 379.2 at the German Patent Office DMPA and extended to PCT in 2014.Notes and references
- C. Zheng, A. Li, L. Zhao, X. Zhou and Z. Fu, Treatment technologies for organic wastewater, 2013 Search PubMed.
- S. V. W. B. Oliveira, E. M. Moraes, M. T. Adorno, M. B. Varesche, E. Foresti and M. Zaiat, Water Res., 2004, 38, 1685–1694 CrossRef CAS PubMed.
- D. W. Hohreiter and D. K. Rigg, Chemosphere, 2001, 45, 471–486 CrossRef CAS PubMed.
- H. R. Lotfy and I. G. Rashed, Water Res., 2002, 36, 633–637 CrossRef CAS PubMed.
- Z. Jiao, P. Luo, Y. Wu, S. Ding and Z. Zhang, J. Hazard. Mater., 2006, 134, 176–182 CrossRef CAS PubMed.
- A. M. Silva, I. M. Castelo-Branco, R. M. Quinta-Ferreira and J. Levec, Chem. Eng. Sci., 2003, 58, 963–970 CrossRef CAS.
- S. Ebrahimi and M. Borghei, Sci. Iran., 2011, 18, 1372–1376 CrossRef CAS.
- R. Noyori and T. Ohkuma, J. Am. Chem. Soc., 1987, 5856–5858 CrossRef CAS.
- T.-T. Thai, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2009, 694, 3973–3981 CrossRef CAS.
- L. E. Heim, N. E. Schlörer, J.-H. Choi and M. H. G. Prechtl, Nat. Commun., 2014, 5, 3621 Search PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735–8751 RSC.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698 CAS.
- S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207 CAS.
- P. Alsabeh, D. Mellmann, H. Junge and M. Beller, Top. Organomet. Chem., 2014, 48, 45–79 CrossRef.
- B. Loges, A. Boddien, F. Gärtner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS.
- D. van der Waals, Recyclable Catal., 2015, 1–11 Search PubMed.
- M. Bennett and A. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–237 RSC.
- M. T. Kessler, M. K. Hentschel, C. Heinrichs, S. Roitsch and M. H. G. Prechtl, RSC Adv., 2014, 4, 14149 RSC.
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chemistry, 2005, 11, 440–452 CrossRef CAS PubMed.
- R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2007, 28, 368–386 CrossRef CAS.
- K. M. Rahman and D. E. Thurston, Chem. Commun., 2009, 1, 2875–2877 RSC.
- D. D. Young, J. Nichols, R. M. Kelly, A. Deiters and N. Carolina, J. Am. Chem. Soc., 2008, 10048–10049 CrossRef CAS PubMed.
- J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- D. R. Baghurst and D. M. P. Mingos, J. Organomet. Chem., 1990, 384, C57–C60 CrossRef CAS.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Eur. J. Inorg. Chem., 2013, 2013, 4558–4562 CrossRef.
- J. Soleimannejad and C. White, Organometallics, 2005, 2538–2541 CrossRef CAS.
- S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. Vom Stein, U. Englert, M. Hölscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC.
- M. A. Bennett, T.-N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson, Inorg. Synth., 1982, 21, 72–78 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols and data. See DOI: 10.1039/c5gc01798j |
| ‡ Literature precedent for the thermal preparation of exemplary complexes and the given reaction conditions (T [°C], t [h]) and yields (%): Complex 1 (78 °C, 4 h, 95%);18 3 (78 °C, 16 h, 90%);18 5 (78 °C, 4 h, 65%);18 6 (78 °C, 4 h, 53%);18 8 (65 °C, 30 h, 25%);18 9 (80 °C, 3 h, 66%);23 11 (185 °C, 2 h, 80%).30 |
| This journal is © The Royal Society of Chemistry 2016 |