
Raman technologies in cancer diagnostics
Lauren A.
Austin
a,
Sam
Osseiran
ab and
Conor L.
Evans
*a
aWellman Center for Photomedicine, Harvard Medical School, Massachusetts General Hospital, 149 13th Street, Charlestown, Massachusetts 02129, USA. E-mail: Evans.Conor@mgh.harvard.edu
bHarvard-MIT Division of Health Sciences and Technology, 77 Massachusetts Avenue E25-519, Cambridge, Massachusetts 02139, USA
First published on 22nd October 2015
Abstract
Despite significant effort, cancer still remains a leading cause of death worldwide. In order to reduce its burden, the development and improvement of noninvasive strategies for early detection and diagnosis of cancer are urgently needed. Raman spectroscopy, an optical technique that relies on inelastic light scattering arising from molecular vibrations, is one such strategy, as it can noninvasively probe cancerous markers using only endogenous contrast. In this review, spontaneous, coherent and surface enhanced Raman spectroscopies and imaging, as well as the fundamental principles governing the successful use of these techniques, are discussed. Methods for spectral data analysis are also highlighted. Utilization of the discussed Raman techniques for the detection and diagnosis of cancer in vitro, ex vivo and in vivo is described. The review concludes with a discussion of the future directions of Raman technologies, with particular emphasis on their clinical translation.
 Lauren A. Austin | Lauren A. Austin received her BS in chemistry from the University of Central Florida where she worked with Dr Qun “Treen” Huo. She then completed her PhD from the Georgia Institute of Technology with Prof. Mostafa A. El-Sayed and focused on using bioconjugated plasmonic nanoparticles and SERS to study and monitor real-time molecular processes in live cells. She is currently a postdoctoral fellow at Harvard Medical School/Massachusetts General Hospital. Her current research includes developing advanced microscopy assays to detect low levels of biomolecule–cell binding as well as developing a methodology to perform genetic sequencing on specific tumor subpopulations. |
 Sam Osseiran | Sam Osseiran earned his B. Eng. in Biomedical Engineering from École Polytechnique de Montréal in Canada. He is currently a PhD candidate at the Harvard-MIT Division for Health Sciences and Technology (HST), where his graduate research focuses on the non-invasive investigation of skin cancer pathogenesis using nonlinear optical techniques. This includes using multiphoton fluorescence, fluorescence lifetime imaging microscopy, second harmonic generation, and coherent Raman scattering to assess oxidative stress, identify skin microstructure, and target specific molecular biomarkers relevant to skin cancer. |
 Conor L. Evans | Conor L. Evans received his degrees from Brown University (Bachelor of Science in Physical Chemistry) and Harvard University (PhD in Chemistry with Dr X. Sunney Xie). He carried out postdoctoral training with Tayyaba Hasan and Johannes de Boer (both at Massachusetts General Hospital) in the application of advanced microscopy to cancer research. He now serves as an Assistant Professor at the Wellman Center for Photomedicine of Harvard Medical School at the Massachusetts General Hospital. His research is focused on the development and clinical translation of optical microscopy and spectroscopy tools, with a specific interest in coherent Raman imaging. |
Introduction
With 14.1 million diagnosed cases and 8.2 million deaths worldwide in 2012, cancer is one of the most prevalent and fatal diseases across the globe.1 While over 60% of these cancer deaths occur in underdeveloped countries and are largely attributed to poor healthcare systems and inadequate medical resources, developed countries (i.e. the United States and United Kingdom) with access to the top medical technologies and treatments are still projected to see more than 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2015 alone.1,2 Significantly reducing these staggering death tolls will, in large part, require improvements to current diagnostic methodologies.
000 deaths in 2015 alone.1,2 Significantly reducing these staggering death tolls will, in large part, require improvements to current diagnostic methodologies.
Cancer diagnoses usually occur through histopathology and biomedical imaging techniques. Histopathology requires the removal of tissue from the suspicious region of interest followed by tissue sample preparation that is often labor and time intensive. Once preparation is complete, slides are evaluated under a microscope by a trained pathologist. Biomedical imaging, such as magnetic resonance imaging (MRI), positron emission tomography (PET), ultrasound, and computed tomography (CT), can provide physicians information about a tumor's three-dimensional volume, structure, and composition.3 These imaging modalities have led to significant advancements in cancer management and have aided in a surgeon's ability to resect a tumor. However, while histopathology and these biomedical imaging techniques are well established, the continual rise in cancer incident rates, the difficulty in incorporating these techniques into intraoperative procedures, and optical resolution and multi-labeling limitations provide motivation to explore alternative diagnostic platforms.3,4
Raman-based technologies present several advantages for the cancer diagnostics community. In particular, Raman tools have the ability to provide sensitive, quantitative and chemically-specific information about important biological components in cellular and tissue milieu.5,6 Raman spectroscopy relies on the inelastic light scattering of a photon after its interaction with a vibrating molecule. The difference in energy between the incident photon and the inelastically scattered photon relates to the energy required to excite a molecule's specific vibration. Therefore, biological molecules with distinct chemical and molecular features (i.e. lipids, proteins, DNA) can be readily identified, quantified without exogenous labels, and changes in these “molecular fingerprints” can provide pathogenic information.5 In addition to molecular specificity, Raman scattering is compatible with NIR excitation sources and can be used for real-time measurements, making this technique extremely advantageous for biomedical, and specifically, in vivo imaging.
While the use of Raman spectroscopy in the diagnosis of cancer has the potential to lead to improvements, it has several well-understood shortcomings that must be overcome in order for it to become a reliable and common diagnostic technique. Standard Raman spectroscopy relies on inelastic light scattering occurring through a spontaneous process. Spontaneous Raman spectroscopy suffers from relatively weak signals (Raman cross section ≈10−30 cm2 per molecule) and is often overwhelmed from elastic light (Rayleigh) scattering signals as well as tissue autofluorescence.7,8 This presents significant hurdles especially when trying to carry out Raman techniques in strongly optically scattering tissue environments. Due to the low signal efficiency, spontaneous Raman spectroscopy often requires high-powered lasers that can, under the wrong conditions, cause damage to biological samples. To address this issue, several Raman enhancement techniques have been developed, including coherent and surface enhanced Raman scattering. Coherent Raman techniques make use of at least one additional laser source for signal enhancement of up to five orders of magnitude, enabling real-time vibrational imaging in vivo. Surface enhanced Raman scattering (SERS), on the other hand, achieves signal enhancement over ten orders of magnitude via the use of plasmonic nanoparticles. All three approaches have been coupled to fiber optic probes for their incorporation into endoscopes and other diagnostic devices to allow for enhanced accuracy and sensitivity in vivo.6
In this review, Raman techniques and their governing fundamental principles will be described. Methods used for Raman spectral analysis on large data sets will also be surveyed. An emphasis will be placed on in vitro, ex vivo and in vivo cancer diagnostic applications that utilize the presented Raman techniques. Lastly, potential improvements and future directions of Raman technologies and their successful translation into the clinic will be discussed.
Raman-based techniques and fundamental principles
Spontaneous Raman scattering
Raman scattering is a non-parametric process related to the interaction of light with a molecule and its associated vibrational states. This process was discovered by Sir Chandrasekhara Venkata Raman in 1928, when he observed the scattering of an entire spectrum of light upon illumination of a sample with monochromatic light of frequency ωP. As he expected, the most prominent feature of the spectrum occurred at the incident light frequency ωP, which is attributed to an elastic interaction known as Rayleigh scattering. However, weak spectral lines were also observed, which were later understood to occur as a result of inelastic interactions between incident photons and the molecular vibrations present in a sample. As energy is transferred during this process, the spectral lines had shifted frequencies ωP ± ωR, corresponding to what are now known as Stokes (ωP − ωR) and anti-Stokes shifts (ωP + ωR). Raman observed that these shifts and their resulting spectra were highly material specific – a discovery that was awarded the 1930 Nobel Prize in physics. It is worth noting that in most use cases, the incident field at ωP is within the visible or infrared range of the electromagnetic spectrum, while the molecular vibrations themselves can be directly probed at ωR that lie in the mid- or long-wavelength infrared ranges.Fundamentally, Raman scattering is derived from interactions with the time-varying electromagnetic field of light and the time-varying electric polarizability caused by molecular vibrations. When a molecule interacts with a photon, it can experience an instantaneous coupling with the photon's electric field, leading to a polarization of the molecule. This coupling can result in the creation of a scattered photon at frequency ωP − ωR if some of the energy from the incident light is converted to drive the molecular vibration; it is scattered at ωP + ωR if the molecule is already in an excited vibrational state, whereupon the scattered photon exits with energy equal to the sum of those of the incident photon and the molecular vibration. These interactions at ωP − ωR and ωP + ωR correspond to Stokes and anti-Stokes shifts, respectively. However, this process is far from efficient, as the creation of a frequency-shifted photon hinges on an infrequent interaction with the vacuum field. Given that a spontaneous emission process is defined by the creation of a photon in a previously unoccupied mode, this Raman process is deemed spontaneous.9
It should be noted that Raman transitions depend on the rate of change of polarizability with respect to the molecule's geometry. This implies that Raman scattering is only possible for certain symmetries of molecular vibrations, which forms the basis for the so-called selection rules for Raman scattering. While the formalism here predicts the existence of Stokes and anti-Stokes frequency components, it fails to consider the strength of individual Raman transitions, which would otherwise require a more elaborate quantum mechanical formalism that is beyond the scope of this review. The reader is encouraged to refer to the textbook by Long for a more rigorous quantum mechanical approach to the principles of Raman scattering.10
Coherent Raman scattering
Coherent Raman scattering is a term that refers to a class of stimulated interactions where Raman processes are driven coherently, leading to the generation of strong scattering signals. This differs from traditional Raman scattering, which is dependent on spontaneous interactions. As was previously mentioned, spontaneous Raman scattering depends on an infrequent interaction with the vacuum field. In coherent Raman scattering, at least two light fields are introduced into a sample that have an energy difference equal to a molecular vibration of interest. These fields act to simulate, or drive, the Raman process, increasing the efficiency of the Raman process by several orders of magnitude.9Though many types of coherent Raman scattering processes have been discovered and developed,11,12 two types of coherent Raman scattering have been specifically developed towards biomedical applications: stimulated Raman scattering (SRS) and coherent anti-Stokes Raman scattering (CARS). Both mechanisms of signal generation are so-called “four-wave mixing” processes, which implies that incident photons interact with the material's vibrational properties to produce scattering contrast that can be used for imaging. Important distinctions exist between CARS and SRS however, resulting in differences in detection schemes and image contrast. Schematically, the energy diagrams for spontaneous Raman scattering, SRS, and CARS are presented in Fig. 1 below.
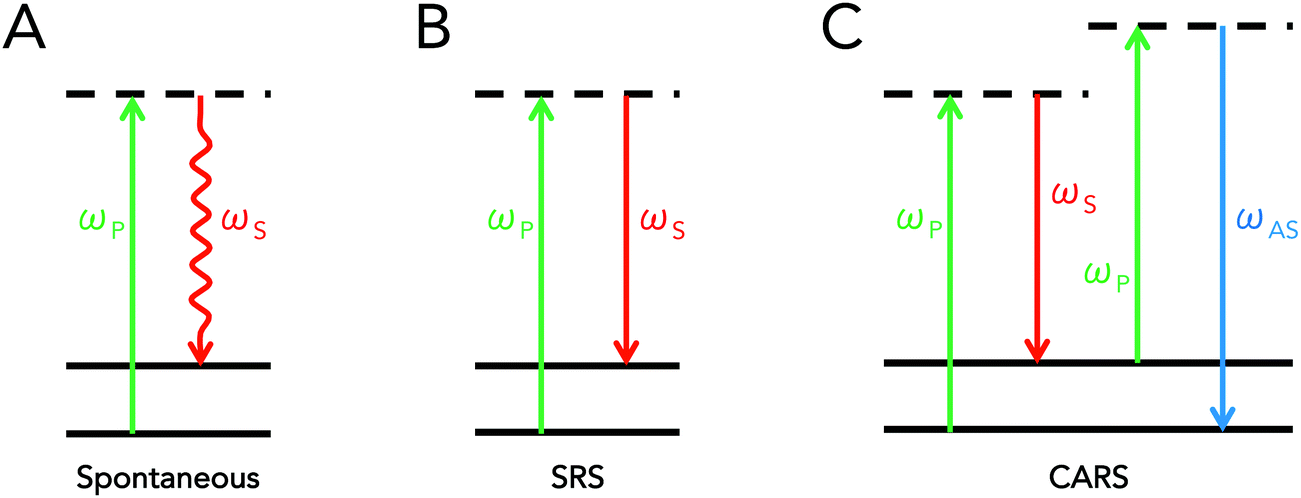 | ||
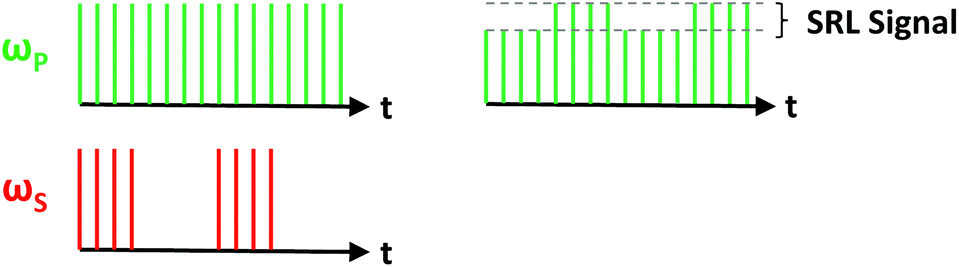
| Fig. 1 Schematic representation of the fields involved in Raman scattering processes. (A) Spontaneous Raman scattering, (B) stimulated Raman scattering (SRS), and (C) coherent anti-Stokes Raman scattering (CARS). The pump, Stokes, and anti-Stokes frequencies are respectively designated by ωP, ωS, and ωAS, while the dashed and solid lines respectively refer to virtual and real molecular states. In spontaneous Raman scattering, a pump photon scatters inelastically, thus generating a Stokes photon. In SRS, both pump and Stokes fields are incident on the sample simultaneously; if there is a vibrational band gap that corresponds to the energy difference between the two beams, the pump field decreases (stimulated Raman loss, SRL), while the Stokes field is amplified (stimulated Raman gain, SRG). Finally, in CARS, a new field is created at the anti-Stokes frequency and can be readily detected via optical filters, as it is blue-shifted relative to the incident pump and Stokes fields. | ||
Conceptually, SRS and CARS typically both involve the use of two incident fields at frequencies ωP and ωS, respectively referred to as the pump and Stokes beams, where the difference between these two frequencies is tuned to match molecular vibrations of interest. In the case of SRS, the introduction of electric fields at both frequencies allows for a transfer of photons at the pump frequency to photons at the Stokes frequency due to coupling with the sample's endogenous molecular vibrations. This implies that the pump beam intensity decreases in the presence of a Raman-active molecule, while that of the Stokes beam increases. In order to pick up this small energy transfer, SRS microscopy is achieved either by detecting the decrease in pump intensity (stimulated Raman loss, SRL) or the increase in Stokes intensity (stimulated Raman gain, SRG).
SRL is achieved through high-frequency modulation (typically on the order of several MHz) of the Stokes beam via an acousto-optic or electro-optic modulator. This implies that the SRS signal will manifest itself as a high-frequency modulation superimposed onto the intensity of the pump beam. Detection thus requires the use of a lock-in amplifier to isolate the high-frequency component of the pump intensity. Conversely, SRG is performed by modulating the pump beam and detecting of the high-frequency fluctuations of the Stokes intensity. Additional information on SRS microscopy is provided later in the instrumentation/microscope design section.
The CARS process, on the other hand, can be conceptually perceived as an initial induction of a coherent vibrational oscillation of the material, achieved by tuning the difference (“beat”) frequency between the pump and Stokes beams to a given vibrational transition. This results in the creation of a macroscopic, coherently driven polarization change in the sample at the excited vibrational state of interest. A subsequent interaction of pump photons with this macroscopic polarizability then results in the strong scattering of photons at the anti-Stokes frequency. In terms of implementation for microscopy, CARS is simpler than SRS in that it does not require signal modulation nor lock-in amplification for detection, since a new wavelength is generated altogether. As such, detection of the anti-Stokes signal simply requires the use of proper short-pass or band-pass filters that can adequately block out any residual pump or Stokes light. Supplementary information on CARS microscopy is described in the instrumentation/microscope design section of this review. For further reading, various reviews on the topic are also available.13,14
One difference between CARS and SRS is the existence of the so-called “non-resonant” background in the CARS signal. This background contribution arises due to the fact that CARS is a parametric process – that is, the energy state of the molecule is unchanged after CARS. This differs from SRS and spontaneous Raman, where molecules gain or lose a quantum of vibrational energy. The parametric nature of CARS enables a second, non-resonant, set of interactions that are derived from purely electronic processes. The end result is that the CARS signal contains both resonant and non-resonant contributions. This set of additional coherent interactions has the practical effects of adding a nonspecific, background contribution to CARS that can also result in shifted and complex vibrational spectra.12
Surface enhanced Raman scattering
Surface enhanced Raman scattering was first reported in 1974 by McQuillan and co-workers when they saw signal enhancement after pyridine adsorbed onto a roughened silver electrode.15 Three years later, two different SERS mechanisms based on charge-transfer and electromagnetic (EM) enhancement were proposed.16,17 Regardless of the enhancement mechanism, utilizing a roughened metal surface or a metallic (i.e. gold or silver) nanoparticle has led to reported signal enhancements of 106 to 1014 when compared to signals acquired by conventional Raman spectroscopy.8,18,19The charge-transfer enhancement mechanism is attributed to changes in a molecule's polarizability due to charge-transfer between the adsorbed molecule and the roughened metal surface. After a molecule adsorbs onto a metal surface, its highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are generally positioned symmetrically about the metal's Fermi level.20 This arrangement allows for electron transfer between the molecule and the metal to occur at much lower energies than that required for the excitation of the molecule itself (Fig. 2a).
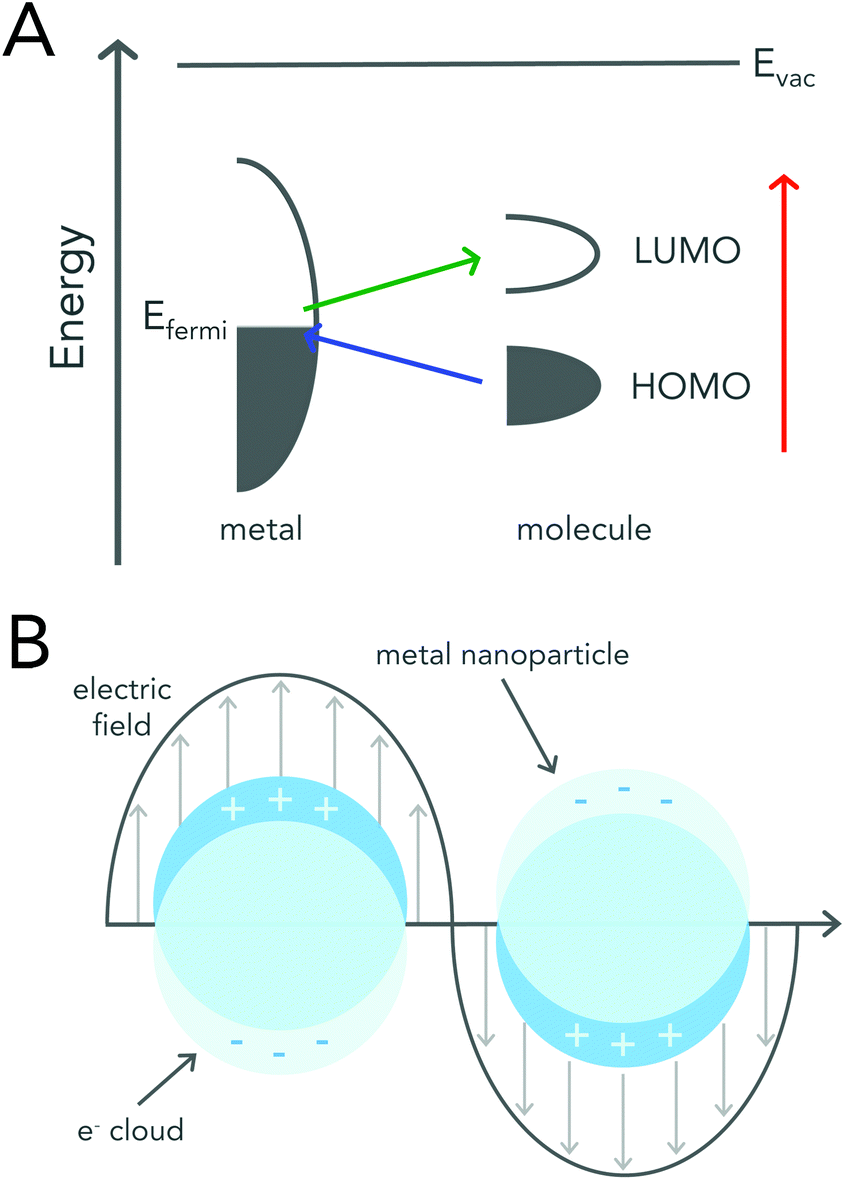 | ||
| Fig. 2 Schematic diagrams describing enhancement mechanisms of SERS. (A) An energy level diagram of a molecule adsorbed onto a metal surface. The HOMO and LUMO levels of the molecule broaden upon adsorption. The colored arrows represent the possible electron transfers. Reproduced from ref. 20 with permission from The Royal Society of Chemistry. (B) Graphical illustration demonstrating the interaction of metal nanoparticles with an external electromagnetic field. Incoming light causes the collective oscillation of the free conduction band electrons, resulting in the localized surface plasmon resonance (LSPR). Excitation of the LSPR leads to an enhanced electromagnetic field at the surface of the nanoparticle. | ||
The electromagnetic mechanism, which is largely regarded as the dominant enhancement mechanism, relies on the enhanced electromagnetic field that surrounds metal nanoparticles.21 Metallic or plasmonic nanoparticles are known for their unique interaction with light that results in the excitation of their localized surface plasmon resonance (LSPR) (Fig. 2b). LSPR excitation, results in not only enhanced light absorption and scattering from the nanoparticle, but also produces an enhanced electromagnetic field around the surface of the nanoparticle that can be readily exploited for Raman enhancement.21–24 When a molecule is adsorbed onto or is within close proximity of the nanoparticle surface the EM field felt by the molecule is that of the incident field and the enhanced EM field at the nanoparticle surface. A more detailed mathematical description of plasmonic nanoparticle Raman signal enhancement can be found in several excellent texts by Van Duyne and colleagues.21,23
Signal enhancement due to the presence of metal nanoparticles can be experimentally determined by comparing the Raman intensity acquired in the presence and absence of the nanoparticle system and is mathematically represented by eqn (1).
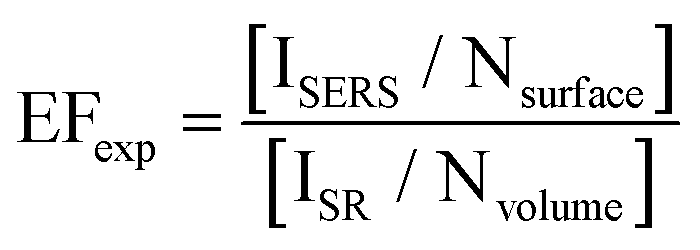 | (1) |
In this equation, ISERS and ISR represent the Raman intensities obtained using metallic nanoparticle Raman enhancement and conventional spontaneous Raman, respectively. The acquired intensities are “normalized” by dividing by the number of molecules present in the sample system.
The degree of Raman enhancement is strongly distance dependent.21,25 A particle's electric field decays from the surface rapidly following 1/r3, and due to the nature of nanoparticle EM amplification, SERS enhancement theoretically scales by 1/r12. However, in practice a distance dependence of 1/r10 is observed as the molecules themselves contribute to the surface area of the SERS system. Taking into account these factors, Raman spectra acquired when using isolated metallic nanoparticles typically results in a 103–104 enhancement, while 1012 enhancements are seen when using aggregated nanoparticle systems.26 In aggregated systems, “hot spots” are formed when neighboring nanoparticles are in close enough proximity to each other causing their LSPRs to couple. It should be noted that hot spot formation is heavily dependent on the size and shape of the nanoparticle as well as the interparticle distance between nanoparticles.27
While metallic nanoparticles can be employed alone to acquire enhanced Raman signals, each class of biomolecules (i.e. proteins, DNA, lipids) exhibit the identical spectral features making specific intraclass delineations impossible. In order to identify a specific biomolecule, the surface of metallic nanoparticles can be functionalized with Raman reporters without interfering with other biological targeting moieties such as peptides or antibodies. Raman reporters are usually dyes or fluorophores that exhibit very sharp and distinct Raman bands. Use of this Raman reporter-nanoparticle system also allows for multiplexed assay or imaging formats to be utilized. With the increased signal enhancements observed when employing metallic nanoparticles or roughened metal surfaces coupled with the ability to conduct multiple analyses in one experimental system, SERS has been heavily utilized in diagnostic applications, especially when working with liquid biopsies. Some of these applications will be highlighted in later sections.
Instrument/microscope design
Raman spectroscopy may be conducted in a variety of forms depending on the type of sample, but the premise remains the same in all cases. Whether probing endogenous chemical species or using nanoparticles for SERS, an external narrow-linewidth laser light source is responsible for excitation, and a sensitive detection system must be in place downstream of a dispersive optical setup for spectroscopic detection. In terms of light sources, near-infrared light is often preferred, owing to its minimal autofluorescence excitation in biological samples, low risk of photodamage, and enhanced penetration depth within tissues. In this regard, 785 and 830 nm diode lasers are most commonly used for Raman spectroscopy, although some multimodal applications requiring pulsed lasers use a 1064 nm Nd:YAG laser. Raman spectra can be rapidly acquired via dispersion of the scattered light from the sample using diffraction gratings, and capturing the resulting spectrum on a high-sensitivity CCD detector. Alternatively, single-element detectors such as photodiodes can also be used, but they require tunable filter systems that progressively scan through the spectral range of interest in order to generate Raman spectra.In particular, in vivo applications of Raman spectroscopy in cancer research have often required the use of fiber optics in order to relay excitation light from the laser source to the sample, and scattered light from the sample back to the detection system. It is not surprising that illumination and collection fibers have been integrated into a variety of endoscopic systems for investigative work on bladder,28 cervical,29–33 colorectal,34,35 esophageal,36–40 gastric,38,41–48 and lung cancers.49 Similarly, handheld fiber-based probes have also been developed for spectroscopic assessment of brain,50,51 breast,52,53 oral,54–56 and skin cancers.57–64 The use of these fibers, however, often requires filtering elements built into the fiber tips to remove the Raman scattered light generated by the fiber material itself. An example of a handheld fiber-based probe for Raman spectroscopy is shown in Fig. 3.
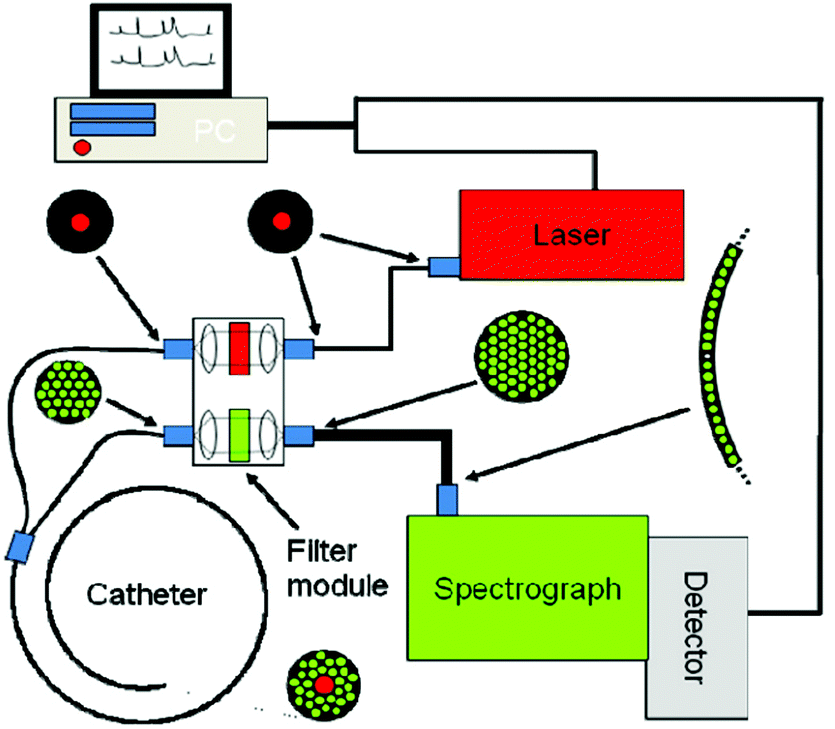 | ||
| Fig. 3 Schematic diagram of a Raman system used for imaging skin. The Raman excitation source is a 785 nm, 500 mW diode laser. The mixed tissue fluorescence and Raman signals are analyzed using a spectrograph equipped with a movable reflection type grating allowing spectra to be obtained from 0 to 3400 cm−1. The intensity of the dispersed light is measured with a thermo-electrically cooled CCD array. A catheter consisting of ultra-low hydroxide fibers is used for delivering excitation light to the tissue surface and collecting the scattered photons. The collection fibers are spread out in a parabolic arc and coupled directly to the spectrometer to reduce CCD image aberrations. Reproduced from ref. 65 with permission from The Optical Society of America. | ||
Coherent Raman scattering technologies, on the other hand, have not seen a development as extensive as that of Raman spectroscopy specifically for in vivo cancer work, given the still young age of the technology. In fact, it was not until 1999 that CARS microscopy was developed by Zumbusch et al., where three-dimensional vibrational imaging of chemical and biological samples was reported for the first time.66 In order to build a CARS imaging system, pulsed laser sources are first required. As with other nonlinear imaging techniques, such as multiphoton fluorescence, the high peak powers of pulsed laser sources are necessary to probe the higher-order optical properties of samples, while their low average powers avoid damaging the sample. Early CARS microscope designs used Ti:sapphire lasers and optical parametric oscillators (OPO) or amplifiers (OPA) that were tuned to emit near-infrared (NIR) wavelengths. More recently, APE, Spectra-Physics, and Coherent have designed automated dual output laser sources, providing a fixed NIR output most often used as a Stokes source, and another that can be selectively tuned over a useful range (generally 680 nm to 1300 nm). The advent of such technology greatly reduces the complexity of the imaging system, since different vibrational bands can be selected by changing the tunable output via the provided laser software in a matter of seconds.
In addition to laser wavelength, pulse duration must also be considered when designing a coherent Raman platform. The shorter a pulse in the temporal domain, the wider its profile becomes in the spectral domain. Thus, the spectral pulse width of picosecond laser sources is orders of magnitude narrower than that of femtosecond pulses. The dependence of pulse duration on spectral resolution thus influences the performance of a coherent Raman imaging system, which is optimized when the spectral pulse widths of the laser sources match those of the Raman peak of interest.
Regarding microscope design, the pump and Stokes beams are combined using a dichroic mirror, and raster-scanned across the sample through a high numerical aperture (NA) objective. Such objectives are preferred for CARS imaging in order to tightly focus the incident pump and Stokes beams into a small focal volume for optimal signal generation. Moreover, they ensure that the so-called “phase-matching condition” is met such that anti-Stokes radiation is constructively generated.66 Finally, as with all nonlinear optical techniques, the CARS signal strictly originates from the focal point of the objective. Therefore, the distribution of a Raman-active compound of interest can be mapped in three dimensions by raster scanning the focus across the sample, and detecting the anti-Stokes light using a photomultiplier tube (PMT) downstream of appropriate optical filters.
Following the success of CARS as a label-free microscopy platform, a branching point in coherent Raman imaging was met in 2008 with the advent of SRS microscopy.67 To this end, Freudiger et al. used essentially the same configuration as a CARS microscope, but modulated the Stokes beam via an electro-optic modulator (EOM) in order to detect a minuscule variation in the pump beam using a high-end lock-in amplifier (Fig. 4). Such a tool is essential in the context of SRS microscopy, since the signal originates from small fluctuations of the pump intensity at the modulation frequency over a significantly larger input power level. As was mentioned earlier, this configuration is referred to as stimulated Raman loss (SRL), while modulation of the pump beam and subsequent detection of modulation amplitude in the Stokes beam is known as stimulated Raman gain (SRG).
 | ||
| Fig. 4 Detection scheme for stimulated Raman loss (SRL). Conversely, stimulated Raman gain (SRG) is detected by modulating the pump beam and detecting the fluctuations of the Stokes beam. | ||
Translation of coherent Raman microscopes from the laboratory to the clinic has recently begun as well. JenLab now offers the MPTflex CARS, a standalone portable system with a pivoting handheld scan head capable of both multiphoton fluorescence and CARS imaging.68 For a more directed use in intra-operative settings, Invenio Imaging offers a state of the art portable SRS/CARS imaging system based on fiber lasers in the form of a compact microscope.69 The advent of such novel technologies is promising for the medical community, in that rapid label-free assessments can be made directly on patients or biopsies in order to improve cancer diagnostic speed and quality leading to increased patient well-being.
Following the above discussion on the fundamentals and implementation of Raman technologies, it is apparent that not all methods are applicable in all contexts of cancer diagnostics. To illustrate their various advantages and limitations, a brief comparison is presented in Table 1.
| Spontaneous Raman | SERS | SRS | CARS | |
|---|---|---|---|---|
| Signal intensity | Weak | Strong | Strong | Strong |
| Signal generation | Endogenous | Exogenous | Endogenous | Endogenous |
| Dependence on concentration of Raman-active molecules | Linear | Linear | Linear | Quadratic |
| Non-resonant background | No | No | No | Yes |
| Ideal for spectroscopy | Yes | Yes | Requires wavelength scanning | No |
| Ideal for imaging | Possible, but inconvenient | Yes | Yes | Yes |
| Cost | Low | Low | High | High |
| Complexity of implementation | Simple | Simple | Complex | Moderate |
| Laser source | 1 (CW or pulsed) | 1 (CW) | 2 (pulsed) | 2 (pulsed) |
| Detection | Spectrometer | Spectrometer or charge-coupled device (CCD) | Diode and lock-in amplifier | Photomultiplier tube (PMT) or avalanche photodiode (APD) |
| Clinical use | Widespread | Emergent | No clinical studies or devices yet available | Emergent |
| Applications/sample platform | Intact tissue, endoscopy, biofluids | Intact tissue, endoscopy, biofluids | Intact tissue, biopsy | Intact tissue, biopsy |
Spectral processing for Raman scattering
Raman spectroscopy methods are powerful tools for the quantitative comparisons between normal, benign and diseased biological specimens. Raman spectral technologies, including spontaneous Raman scattering, SERS, and hyperspectral CARS and SRS provide spectra rich in information that require extensive pre-processing and chemometric analysis to extract relevant analytical parameters. Pre-processing typically involves noise/spike removal, background subtraction and spectral normalization while methods for spectral analysis using chemometric means includes multivariate algorithms and support vector machines (SVMs). Given the importance of these analysis methods to the cancer diagnostic capabilities of Raman spectroscopy, this section will present and review common data analysis methodologies.Pre-processing
Fourier filtering methods are employed when Raman spectra are particularly noisy, which acts to separate the acquired spectra into high, medium and low discrete frequencies.70,71 This spectral truncation is achieved by performing Fourier transform operations using the fast Fourier transform algorithm on the acquired Raman spectra to convert the spectral signals into the weighted sum of sine and cosine functions of increasing frequencies.72 Under this breakdown, spectral noise is extracted within the highest frequency space while the Raman spectra and the spectral background are contained in the medium and low frequency space, respectively (Fig. 5). A drawback when using this method is that often times noise and spectral signals can have similar frequencies, and thus overlap to make for unclear cutoffs for “high”, “medium” and “low” frequency spaces. This can lead to spectral artifacts due to the joint removal of high frequency spectral noise and high frequency components of authentic Raman bands.
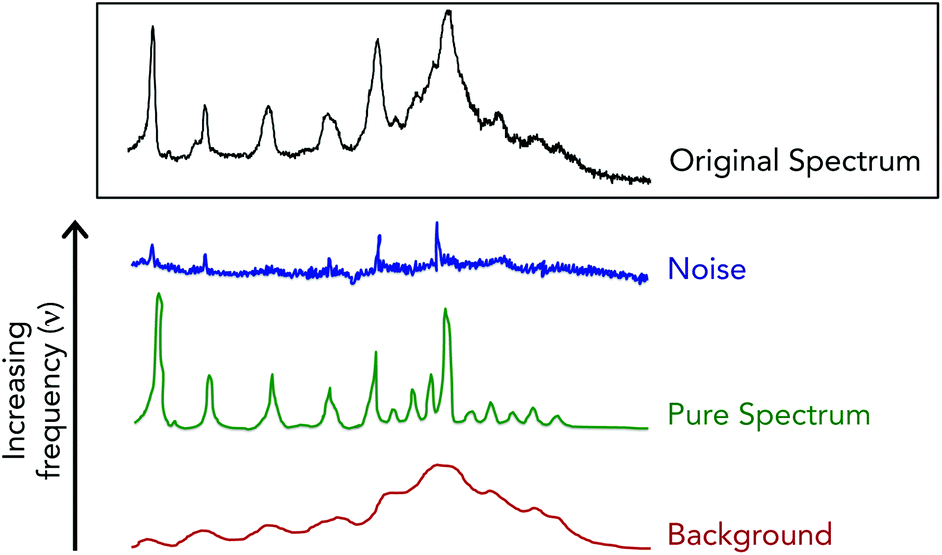 | ||
| Fig. 5 Schematic example of discrete frequency analysis of Raman spectrum. Using the fast Fourier transform algorithm, spectra can be separated into different frequency components allowing for noise removal. | ||
A similar transformation methodology, known as wavelet transformations, can also be performed on acquired Raman spectra to remove spectral noise.71,73,74 Wavelet transformations create basis functions that are concentrated in both time and frequency. Spectra are then transformed into increasing frequency components, just as in Fourier filtering, with high frequency signals being removed. The difference between wavelet and Fourier transforms is that basis functions, due to their inclusion of time and frequency information, act to better preserve Raman band shape. In order for this type of transformation to be effective, noise must be well separated from spectral signals when in the transformed domain.
Smoothing techniques, such as the moving average and Savitzky–Golay polynomial moving average, are another way to reduce spectral noise.70,71,75 These noise reduction practices also rely on the fact that spectral noise occurs at high frequency and therefore its transition from point to point on a spectrum is abrupt and random. When utilizing the moving average technique each individual point on a spectrum is averaged with a chosen number of adjacent points.76 Due to the nature of this technique, spectral points located at the ends of the spectrum are unable to be averaged and are effectively lost. Collectively, the loss of data points and the opportunity for spectral distortion due to the selection of large filter sizes can significantly skew the output (smoothed) spectra. Savitzky–Golay polynomial smoothing can alleviate spectral distortion as this methodology fits a polynomial function to a section of points around the point of interest. This allows for spectral features, such as a band's height and width, to remain conserved.70,75,77 For use of this technique, the number of points required are 2n + 1 where n is the order of the polynomial function. Smoothing with low-order polynomial fits significantly eliminates noise resulting in high signal-to-noise; however, there is a higher propensity for signal distortion. Alternatively, utilizing high-order fits results in reduced signal distortion and allows for abrupt signal variations to be observed. Noise is not removed as effectively when using high-order fits. Choosing the order of a polynomial fit depends on the application, as increased signal-to-noise at the cost of band distortion maybe be acceptable if comparing changes in Raman intensities between different samples. If comparisons require information about a signal's shape, higher ordered fits would be required.
“Cosmic spikes” also contribute to noise in acquired Raman spectra and are a result of cosmic rays and their decay products striking the detector.71 Due to the random nature of cosmic ray arrival and the extreme unlikelihood that a spike will be present at the same wavenumber over multiple measurements, spikes can generally be identified by acquiring repeat measurements. A spectrum where a cosmic spike is thought to be present is then subtracted from a repeat measurement. If a cosmic spike is indeed within the spectra it will appear as a sharp increase in the difference spectra. Recently, methods to remove cosmic rays in a more streamlined, automated fashion have been described.78,79 Using these methods, cosmic spikes were readily distinguishable from both strong and weak Raman bands.
:1 fluorescence to Raman ratio, and the resulting spectra showed impressive band retention when compared to other conventional background subtraction methods. Several variations of this methodology have been published that also incorporate peak removal strategies,83 end-point constraints82 and background contaminants.84 Fluorescence subtraction algorithms based on Savitzky–Golay polynomial fits have also been reported.85–87
PCA is a dimension reducing technique that helps identify data patterns by emphasizing the data's similarities and differences, while reducing data redundancy.89,90 This form of analysis is extremely advantageous for Raman spectroscopy and hyperspectral coherent Raman imaging as the dense spectral information acquired can be deconstructed to allow for spectral and background variations to be more easily visualized. As the theory behind PCA is not the focus of this review and there have been several works that discuss the mathematical foundation of PCA,89,90 we will only provide a brief description of PCA and frame it within the context of Raman spectral analysis.
In PCA, data is represented as a m by n matrix where columns are comprised of the spectral wavenumbers (variables) and rows represent the spectra (observables).91,92 This data matrix can then be decomposed into its pure components (i.e. spectral wavenumbers, pure Raman spectra and noise). However, when conducting Raman experiments, the pure spectra is not known. Therefore to reduce dimensionality, orthogonal linear combinations are used to transform the data into uncorrelated variables termed principle components (PCs). The first PC is determined through a linear combination that maximizes variance leading to the greatest data separation. Each successive PC is a linear combination that maximizes variance in the orthogonal direction respective to the previous PC. PCs that maintain 80% or higher variances are then used to create new axes on which the data can be then be plotted and evaluated. Due to the orthogonal nature between changes in Raman spectral signals and spectral noise, PCA also aids in the elimination of noise. As such, this form of data analysis has been heavily utilized when employing Raman spectroscopy in cancer diagnostic applications, especially when Raman spectroscopic imaging is used.5,71 It should be noted that while PCA is a great tool for identifying trends (i.e. increase or decrease in Raman band intensities) over a complete spectral data set, it is an unsupervised technique and therefore does not provide any information about the different classes of data (i.e. diseased vs. healthy).
Linear discriminate analysis (LDA) is another data reduction analysis technique that relies on the use of linear combinations to describe data. Unlike PCA, which does not account for the class of the input data sets, LDA is a supervised method and thus works to determine the greatest data separation between the different classes of data.5,92 For example, in diagnostic applications data sets may be classified as healthy, benign and diseased, with LDA used to maximize the variation between each set while minimizing the variation within each set. The data separations are expressed as between class and in-between class matrices, and are used to determine the Fisher criterion or the projection that achieves the greatest separation between classes while minimizing the separation within a class. In many diagnostic Raman applications, LDA is often coupled with PCA to minimize the complexity and dimensionality of spectral data sets as PCA reduces spectral features associated with noise. When combining PCA and LDA the number of PCs selected for LDA is critical, as the inclusion of too many PCs will lead to LDA overfitting while too few PCs will remove important spectral information. As a general rule of thumb, a maximum of 10 PCs is used.93
Hierarchical cluster analysis (HCA) is an unsupervised technique that unmixes or separates Raman spectra into different classes based on a distance matrix, and is commonly utilized for Raman hyperspectral image analysis. Clustering is performed using either divisive (“top-down”) or agglomerative (“bottom-up”) approaches to iteratively identify similar Raman spectra until all spectra have been paired. The most common HCA utilizes Euclidian distances to determine spectra similarities and Ward's algorithm for clustering.94 Results of HCA are represented in a two-dimensional dendrogram (“tree diagram”) with branching points depicting data dissimilarities. Once the data has been sufficiently clustered, color assignments are given to each cluster and pseudo-color maps are constructed based on spectral similarities. While HCA is able to accurately segment Raman data into different cellular regions (i.e. nucleus, cytoplasm and mitochondria), it should be noted that due to the nature in which spectral clustering is performed, HCA is computationally taxing and can take up to two hours for a 100 × 100 pixel image.95
Vertex component analysis (VCA) is another unsupervised spectral unmixing technique where each acquired spectrum is assumed to be a linear combination of “pure” spectra.96 Spectra are represented as a n-dimensional vector that spans a n-dimensional Euclidian space, and give rise to a simplex.94 A simplex is a generalized representation of a triangle (2D) or tetrahedron (3D) to arbitrary dimensions. Much like a triangle is a 2-dimensional simplex and has 3 corners, a n-dimensional simplex has n + 1 corners, where each corner corresponds to a pure spectrum. The dimensionality of the data is reduced stepwise with each step resulting in a n − 1 cornered simplex until the predetermined level of “pure” spectra is achieved.95 The linear combinations of the resulting pure spectra are expressed in an abundance map with each color in the map representing a pure component of the hyperspectral data set (i.e. proteins/nucleic acids, water, lipids, etc.). As hyperspectral data sets have large dimensionality, VCA is commonly paired with PCA to first reduce data dimensionality and eliminate noise. This pairing significantly reduces the computational power needed to perform the analysis.
Supervised learning strategies, specifically those that use support vector machine (SVM) algorithms, are becoming increasingly popular for analysis in diagnostic applications due increasing data volumes.97,98 Under supervised learning, test sets or data with known classifications/labels, are used to train a “learning” algorithm that is used to generate a classifier. Then the algorithm and resulting classifiers are tested using new data with unknown labels. The algorithm calculates data projections that maximize the separation between data classes. The performance and accuracy of the supervised learning algorithm is then determined through the repeated construction of data projections by changing the chosen data used to train and test the algorithm. As in other fields, SVMs have seen increased use in Raman spectral analysis for cancer diagnostic applications.5 The growing use of SVM algorithms to streamline spectral analysis will no doubt aid in the clinical translation of Raman spectroscopy as a possible cancer diagnostic technique.
In vitro/ex vivo detection and diagnostics
Biofluids
The ideal biological specimens for diagnostic tests are biofluids (i.e. blood, urine, or saliva) as their collection is non-invasive and they can be repeatedly collected without harm to the patient. Additionally, biofluids are composed of a vast amount of important chemical components, including DNA, hormones, proteins, and metabolites. Thus, a considerable amount of research has been focused on assessing the utility of Raman technologies to analyze biofluids in cancerous and non-cancerous patients.6,92,99 The following discussion involves prostate, oral and breast cancers; however, a list of additional cancers diagnosed using biofluid samples and Raman techniques can be found in Table 2. Table 3 also provides a compilation of common Raman bands identified in these studies.| Type of cancer | Sample platform | ||
|---|---|---|---|
| Biofluid | Ex vivo tissue | In vivo model | |
| Prostate | Blood,100,101,164 urine102 | Human165 | — |
| Oral | Blood,103–105 urine,106 saliva107 | Human166,167 | Human54–56 |
| Breast | Blood,108 urine,109 saliva110 | Human114–118,121,122,124–126,168–170 | Mouse52,53,158–160,171 |
| Skin | — | Human132–139,172–174 | Human59–64,161 mouse58 |
| Brain | — | Human,141,142,144–146,148,149,151,175,176 mouse,147,148,150,151,153,177 pig143,149,178 | Mouse,151,153,154 human50,51 |
| Lung | — | Human127–131 | Human49 |
| Gastrointestinal | Blood179,180 | Human162,163,181 | Human,34,36–39,41–48 pig,35 rat40 |
| Leukemia | Blood182,183 | — | — |
| Head and neck | Blood184 | Human185 | — |
| Cervical | Blood186 | Human187 | Human29–33 |
| Liver | Blood188 | Human189,190 | — |
| Ovarian | — | Human191 | Mouse192 |
| Circulating tumor cells | Blood193,194 | — | — |
| Raman shift (cm−1) | Vibrational mode | Assignment |
|---|---|---|
| 481 | Glycogen | |
| 558 | ν(C–C) | Uric acid |
| 600 |
ν(N–CH3), δ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ring vibrations O), ring vibrations |
Creatinine |
| 621 | t(C–C) | Phenylalanine |
| 650 | t(C–C) | Tryptophan, uric acid |
| 678 |
ν(N–CH2), ν(CO), ring vibrations |
Creatinine |
| 685 | t(C–S) | |
| 692 |
δ(O–CO) |
Creatinine |
| 798 | ν(N–H) | Uric acid |
| 825 | as(O–P–O) | DNA backbone |
| 840 | δ(N–CH2), ring vibrations | Creatinine |
| 903 | ν(C–C–N) | Creatinine |
| 912 | ν(C–C) | Calcium oxalate dihydrate |
| 960 | ν(PO4−3) | Calcium hydroxyapatite |
| 1049 | ν(C–O), ν(C–N) | Proteins |
| 1004 | ν(C–C) ring breathing | Phenylalanine |
| 1155 | β-Carotene | |
| 1176 | ν(C–H) | Tyrosine |
| 1217 | ν(C–C6H5) | Tyrosine, phenylalanine |
| 1265 | ν(C–N), δ(NH) amide III | Proteins, α-helix, collagen |
| 1310 | t(C–H) | Lipids |
| 1340 | w(CH3CH2) | Collagen, lipids |
| 1445 | δ(CH2) | Collagen, lipids |
| 1477 | ν(C–O) | Calcium oxalate dihydrate |
| 1523 | β-Carotene | |
| 1586 |
δ(CC) |
Phenylalanine |
| 1654 |
ν(CC) |
Fatty acids |
| 1655 |
ν(CO) amide I |
Proteins, α-helix, collagen |
| 1739 |
ν(CO) |
Cholesterol esters |
| 2850 | ν(CH2) | Lipids |
| 2940 | ν(CH3) | Proteins, lipids |
Prostate cancer
In 2003, Porter and co-workers reported one of the first immunoassays that utilized SERS for the detection of prostate specific antigen (PSA) in human serum (Fig. 6).100 For specificity to PSA and signal enhancement, Raman probes were prepared by functionalizing 30 nm gold nanoparticles (AuNPs) with an anti-human PSA antibody and the Raman reporter 5,5′-dithiobix(succinimidyl-2-nitrobenzoate) (DSNB). Based upon their previous studies, the authors found that covalently linking the PSA antibodies to the Raman reporter and in turn the gold nanoparticles increased performance over physisorption techniques. Solutions of anti-human PSA-gold nanoparticles and human serum samples containing free PSA were allowed to react, dried onto anti-free PSA antibody treated gold slides, and interrogated by a fiber-optic Raman system equipped with a 632 nm HeNe laser. Using the symmetric nitro stretch of DSNB at 1338 cm−1, PSA levels were detected down to ∼1 pg mL−1 and were comparable to commercially available assays. Raman spectroscopy was also coupled with support vector machine (SVM) analysis to evaluate blood serum from prostate cancer patients and healthy volunteers.101 While the collected serum samples were investigated using both conventional Raman spectroscopy and SERS, SERS with bare silver nanoparticles showed the greatest diagnostic capabilities. The acquired SERS spectra from cancerous and normal patients revealed biochemical changes that correlated to a malignant state. In prostate cancer serum samples, Raman bands at 481 cm−1 (glycogen), 1217 cm−1 (C–C6H5 – phenylalanine, tryptophan) and 1445 cm−1 (CH2 bending – collagen/lipids) were seen to decrease, while increases were seen at 650 cm−1 (C–C twist, tryptophan), 685 cm−1 (C–S twist) and 1586 cm−1 (CC bending – phenylalanine). Spectra classified using the Guassian radial basis function kernel SVM model resulted in diagnostic metrics of 100% sensitivity, 96% sensitivity, and 98% accuracy.
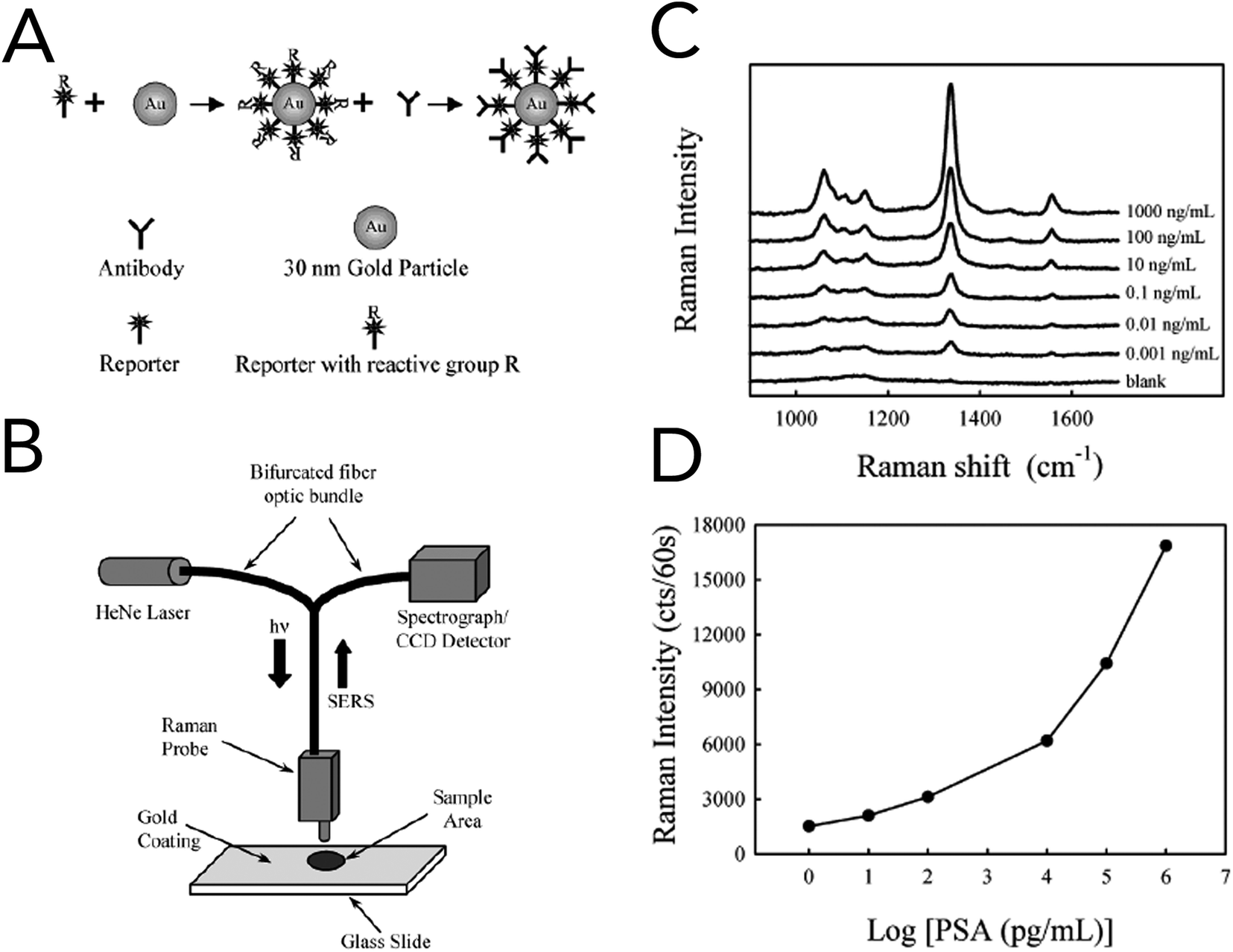 | ||
| Fig. 6 Utilizing SERS for the detection of free PSA in human serum. (A) Raman probes were constructed using 30 nm AuNPs covalently functionalized with DSNB and anti-human PSA antibodies. (B) The experimental set-up for the detection of free PSA in human serum. Using antibody-gold coated glass slides in conjunction with the synthesized AuNP Raman probes creates a sandwich assay format. (C) Acquired SERS spectra at increasing concentrations of free PSA. Spectra show the DSNB characteristic 1338 cm−1 band that correlates to the molecule's symmetric nitro stretching. (D) Dose–response curve of free PSA in human serum using the 1338 cm−1 Raman band. A detection limit of ∼1 pg mL−1 was attained. Reprinted with permission from 100. Copyright 2003 American Chemical Society. | ||
Diagnosis of prostate cancer using SERS has also been extended to urine analysis.102 In this study, filtered urine samples were mixed with gold nanoparticles and then spotted onto a CaF2 microscope slide for measurement. Examination of the difference spectra revealed an increase in Raman bands associated with the metabolite hypoxanthine in prostate cancer samples. Using PCA–LDA for classification, a sensitivity of 100%, a specificity of 89%, and an accuracy of 95% were achieved. While the sample size for this study was limited, this work demonstrates the potential feasibility of urinalysis via SERS and multivariate spectral analysis for cancer diagnoses.
Oral cancers
Blood sample analysis via Raman spectroscopy has also been carried out for oral cancers.103–105 In 2013, Sahu et al. demonstrated that conventional Raman spectroscopy could be used to delineate between serum samples from patients that had been diagnosed with buccal mucoca and tongue cancer, and those from healthy volunteers (Fig. 7).103 An efficacy of ∼85% was found when all spectra were independently analyzed, while ∼78% efficacy was reported when replicate measurements for independent patients were averaged prior to analysis. PCA–LDA was used as the data analysis model for classification generation. Similar to that seen in prostate cancer analysis, changes in Raman bands associated with amino acids and lipids were most significant. This same group demonstrated that Raman spectroscopy on serum samples could potentially predict reoccurrence of oral cancer.105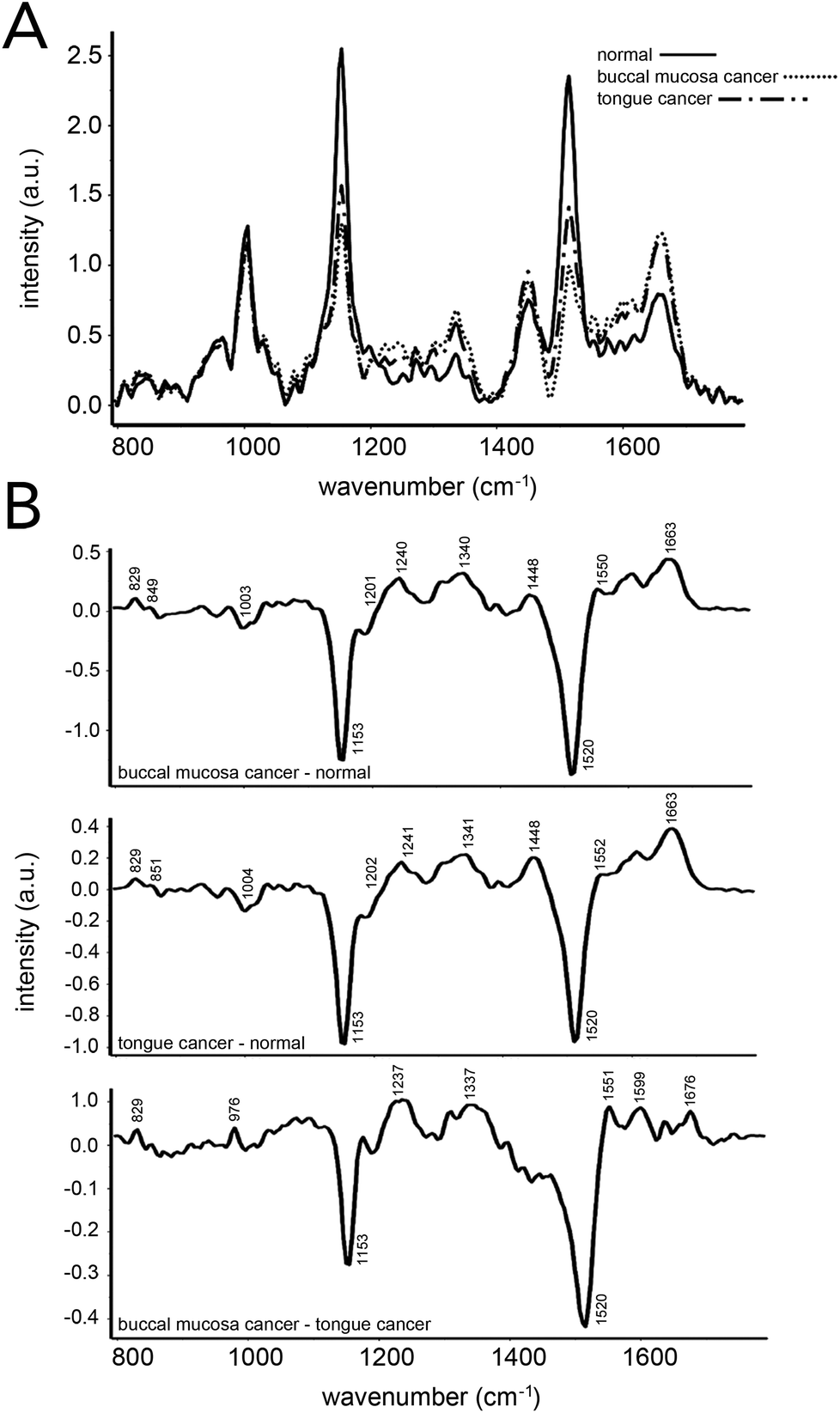 | ||
| Fig. 7 Oral cancer detection using Raman spectroscopy of human serum. (A) Mean Raman spectra acquired from 16 healthy patients, 14 patients with buccal mucosa cancer and 40 patients with tongue cancer. (B) Difference spectra across the three groups evaluated. Spectral differences in proteins, DNA, amino acids, and β-carotene were observed and led to ∼85% and ∼78% accuracies when classifying samples from patients regarded as healthy and those with an oral cancer diagnosis. Accuracies were obtained using a spectra-wise (all individual spectra averaged) and patient-wise (patient spectra averaged) approaches, respectively. Reproduced from ref. 103 with permission from The Royal Society of Chemistry. | ||
Additionally, urine has been investigated for the detection of oral cancer using Raman spectroscopy.106 In this study, voided raw urine was collected from 167 patients (73 healthy and 93 cancerous) and evaluated for its chemical components between 500–1800 cm−1 using 785 nm excitation. Molecular vibrations associated with uric acid, specifically C–C stretching at 558 and 649 cm−1 and N–H stretching at 798 cm−1, showed elevated intensities in cancer patients compared to those that were healthy. Four Raman bands related to creatinine also showed increases, while the band at 692 cm−1 (O–CO deformation) was only present in cancer patients. Other urinary metabolites – urea, DNA, indoxyl sulfate, pteridine, flavin, tryptophan, and phenylalanine – showed differences between cancer and healthy patients. PCA–LDA spectral analysis resulted in a diagnostic sensitivity, specificity, and accuracy of 98.6%, 87.1%, and 93.7%, respectively. Moreover, oral cancer detection has been extended to saliva analysis through coupling Raman spectroscopy and PCA.107
Breast cancer
Diagnostic evaluations using blood, urine and saliva for breast cancer have also been performed. Blood serum analysis using conventional Raman spectroscopy coupled with PCA–LDA was able to distinguish between normal and cancerous patient samples with 97% sensitivity and 78% specificity.108 In order to acquire ample signal, samples were frozen and solid residues were removed for spectral analysis. Multivariate analysis revealed seven Raman band ratios that led to the discrimination between cancerous and non-cancerous samples; these discriminatory band ratios involved the amino acids phenylalanine, tryptophan and tyrosine as well as polysaccharides and β-carotene. Bhattacharjee et al. demonstrated the feasibility of using urine as a sample platform for breast cancer diagnosis.109 Urine was collected from control and breast tumor bearing rats, and was then evaluated with Raman spectroscopy in either an unprocessed or concentrated form. The acquired Raman spectra from unprocessed urine revealed bands associated with urea (1006 cm−1 and 1161 cm−1) and creatinine (680 cm−1 and 850 cm−1). The use of urine concentration led to additional spectral features being revealed. Still, even with additional features present, the Raman bands at 680 cm−1 and 1006 cm−1 showed the greatest change between control and tumor bearing rats. Spectral analysis using PCA–LDA resulted in classification efficiencies of 72% and 91% when using unprocessed and concentrated urine samples, respectively.Saliva has been recently evaluated as biological matrix to diagnosis breast cancer using SERS.110 Saliva was collected from 33 healthy patients and 31 cancerous patients, and then mixed with silver nanoparticles immediately before acquiring the Raman measurement. Difference spectra revealed saliva acquired from cancerous patients had protein band decreases at 621 cm−1 (C–C twist phenylalanine), 1049 cm−1 (C–O, C–N proteins) and 1176 cm−1 (C–H tyrosine) and protein band increases at 1004 cm−1 (C–C phenylalanine), 1208 cm−1 (C–C6H5 phenylalanine) and 1340 cm−1 (CH3CH2 wagging collagen). A partial least square (PLS)-DA algorithm was used to categorize the data and resulted in diagnostic sensitivities above 72%, specificities above 81%, and accuracies above 78%.
Tissue
Ex vivo tissue examination through histopathological techniques is regarded as the gold standard for cancer diagnosis. Histopathology, while reliable, does not allow for in vivo evaluation, which would be extremely beneficial during surgery; the complete removal of tumor margins is heavily correlated to increased survival.51,111,112 Thus, the use of Raman technologies to delineate between cancerous and noncancerous tissues ex vivo has been readily explored as a first step toward the potential intraoperative, real-time evaluation of tissue during surgery.5,6,92 Although the below discussion focuses on breast, lung, skin and brain cancers, a list of other cancers diagnosed using ex vivo tissue samples and Raman techniques can be found in Table 2. Common Raman bands identified in these works can be found in Table 3.Breast cancer
With over 260000 total cases of breast cancer reported each year,1 it is not surprising that breast tissue has been heavily interrogated using Raman techniques. One of the first reports of breast tissue evaluation using Raman spectroscopy was published more than 20 years ago.113 This work focused on the characterization of molecular components in normal breast tissue as well as the optimization of spectroscopic experimental parameters, such as the excitation wavelength, laser power, and the use of a fiber optic probe for spectral acquisition. A year later, the same group used these results to assess the potential spectral differences between normal and cancerous (invasive ductal carcinoma, IDC) breast tissues.114 Using a near-IR excitation source with relatively low power (100–200 mW), the authors reported cancerous tissue having significant differences, especially in bands correlated to lipid content. For statistical analysis, the band area ratios of 1654 cm−1 (CC stretching in fatty acids) and 1439 cm−1 (CH2 scissoring) were chose due to their sensitivity towards histopathological variations. Discrimination between normal and diseased tissue was also capable using a fiber optic probe placed 1 mm into the tumor. It should be noted that both conventional and fiber optic based Raman systems used in this study were unable to distinguish between benign (fibrocystic change) and cancerous samples. Feld and co-workers later published a series of works that reported the discrimination and molecular characterization of breast tissue from normal, fibrocystic change, fibroadenoma and infiltrating carcinoma pathologies through the combination of a Raman microspectroscopic set-up and linear combination models.115–117 Their analysis model revealed that bands associated with fat and collagen were important algorithmic parameters and led to a diagnostic classification sensitivity of 94% and a specificity of 96%. Several works since these foundational studies have been focused on investigating the utility of different combinations of Raman spectral features and multivariate analysis techniques to discriminate between normal, benign and diseased breast tissue.118–122
Raman spectroscopy of calcified species has also been reported as a plausible method to differentiate between normal and cancerous breast tissue. While calcifications are regarded as harmless when found in bones, their presence in soft tissue, especially breast tissue, is often associated with disease. Calcifications are generally composed of either calcium oxalate dihydrate (COD) or calcium hydroxyapatite (HAP), and are extremely difficult to distinguish using traditional pathology techniques.123 Haka et al. reported that bands located at 912 cm−1 (C–C stretching) and 1477 cm−1 (C–O stretching) were indicative of COD calcifications while HAP had a prominent spectral signature at 960 cm−1 that corresponded to phosphate stretching.124 While benign tissues could be distinguished using COD calcification Raman bands alone, differentiation between benign and diseased tissues using HAP band measurements required the use of PCA. PCA resulted in a diagnostic sensitivity of 88% and specificity of 93% when monitoring the increase in bands associated with calcium carbonate and the decrease in protein bands. Stone and Matousek also explored the use of transmission Raman spectroscopy for distinguishing between HAP and COD in breast phantoms at depths translatable to in vivo detection.125 In a push towards rapid clinical translation, Barman et al. demonstrated the feasibility of simultaneously identifying between HAP and COD calcifications and diagnosing breast cancer lesions in real-time during a stereotactic core needle biopsy.126 A combination of Raman spectroscopy and SVM analysis resulted in a 100% positive predicative value, a 95.6% negative predictive value, a 62.5% sensitivity, and a 100% specificity for diagnosing breast cancer tissues with or without calcifications. An 82% overall accuracy for calcification status and breast cancer diagnosis was also reported. However, the most impressive result from this study was the ability of their SVM algorithm to diagnosis ductal carcinoma in situ (DCIS), which was not possible with their previously developed algorithms for biopsy Raman analysis.
While many studies have focused on utilizing conventional Raman spectroscopy, Wong and co-workers employed CARS imaging and quantitative data analysis to distinguish between normal, benign and cancerous breast tissue. In addition, the team demonstrated the ability of CARS to classify cancer subtypes (DCIS, high grade and low grad invasive ductal carcinoma (IDC), and lobular carcinoma). CARS imaging using the CH2 vibration located at 2845 cm−1 revealed normal breast tissue was dominated by adipose and fibrous structures, while malignant tissues showed morphological alterations especially within tumor cells that were confined to the basement membrane and duct space. Diagnostic features from the acquired images were selected according to pathological standards. Using their developed methodology, 80% of intermediate-grade IDC and 85% high-grade IDC samples were accurately separated from each other.
Lung cancer
Ex vivo tissue analysis using Raman spectroscopy has also been performed on lung cancer samples. In 2001, Hamaguchi and co-workers demonstrated the possibility of distinguishing between normal and malignant lung tissues by using near IR Raman spectroscopy.127 By using 1064 nm laser excitation, resolvable biological spectra could be acquired with only minimal tissue autofluorescence. Cancerous tissues were found to have strong bands located at 1448 cm−1 and 1666 cm−1, which corresponded to collagen. Huang et al. also utilized near IR Raman spectroscopy, probing between 700–1800 cm−1 with a 785 nm diode laser, to distinguish between normal and diseased bronchial tissues from 10 patients.128 Their measurements revealed significant spectral differences of bands associated with amino acids, collagen, nucleic acids and phospholipids, with the ratio of Raman band intensities at 1445 cm−1 and 1655 cm−1 serving as a metric of differentiation. The Wong group used CARS microscopy to discriminate between normal, benign, and malignant lung tissues.129,130 Malignancies were categorized using CARS imaging into different subtypes. Distinguishing between normal and cancerous tissues required 11 features and resulted in classification accuracies of 91% and 92%, respectively. In order to separate between adenocarcinoma and squamous cell carcinoma samples, 25 features were required and led to 76% and 72% classification accuracies. Shifted subtracted Raman spectroscopy has also been explored as a diagnostic technique for the evaluation of lung tissue samples.131Skin cancer
Skin cancer can be divided into three subtypes: melanoma, basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), with melanoma accounting for less than 2% of all skin cancer cases, but responsible for ∼75% of skin cancer deaths (not including BCC or SCC cases).1 BCC and SCC are often referred to as nonmelanoma skin cancers (NMSCs) and their detection has been heavily investigated in the Raman community. One of the first reported studies to investigate the utility of Raman spectroscopy to identify NMSCs was conducted by Gniadecka et al. and compared the spectral features of BCC and normal skin biopsies.132 Using a near IR Fourier transform (FT) Raman spectrometer with a 1064 nm Nd:YAG excitation laser, this work revealed alterations in protein, lipid and polysaccharide bands. BCC spectra showed a decrease at 1270 cm−1 (amide III – protein) and 1650 cm−1 (amide I – protein) and were correlated to disturbances in the α-helix secondary structure of the skin. Additional spectral alterations were seen at 850, 870, 950, 1420 and 1450 cm−1 and were associated with changes in protein and lipid structures. The authors employed an artificial neural network analysis technique to achieve complete separation between BCC and normal tissues using spectral changes in the regions of 830–900 cm−1, 900–990 cm−1 and 1220–1360 cm−1. Building upon this work, Puppels and co-workers used a logistic regression model to differentiate BCC from the surrounding normal, healthy epidermis. Their analysis system resulted in 100% sensitivity and 93% selectivity for BCC diagnosis.133 This same group later reported the ability to distinguish between BCC and surrounding healthy tissue using high wavenumber (2800–3125 cm−1) Raman spectroscopy.134 They achieved 100% and 99% prediction accuracies for BCC and healthy tissue, respectively. As high wavenumber analysis eliminates background signals in the fingerprint region that arise from the use of fused-silica-based optical fibers, this work demonstrated the potential feasibility of using a fiber optic probe to diagnose tumor borders in BCC.Multimodal imaging consisting of CARS, second harmonic generation (SHG), and two photon excited fluorescence (TPEF) has also been explored as a potential technique to discriminate between BCC and normal tissues. Multimodal imaging allows for not only structural information (SHG – collagen) but also spatial (TPEF – endogenous fluorophores) and chemical (CARS – lipids) information to be resolved. Vogler et al. used this multimodal imaging strategy in 2010 and revealed that BCC tumor cells could be distinguished due to their undetectable collagen structures and large patches of fat reservoirs.135 Three years later, this group used the same approach to distinguish between BCC and SCC as well as normal tissues.136 BCC and SCC both showed a lack of collagen structure, but BCC also demonstrated a weaker CARS (lipid) signal than that of SCC. Additional morphological features that are traditionally seen in H&E stains could also be visualized using this strategy and added to the strength of diagnosis.
Melanoma and NMSCs have also been able to be distinguished from each other. Wulf and co-workers utilized near IR FT-Raman spectroscopy to delineate melanoma from pigmented nevi, BCC, seborrheic keratoses and normal tissue.137 Visual differentiation was based upon band intensity decreases at 1660 cm−1 (amide I – protein) as well as increases at 1310 and 1330 cm−1 (CH twisting and wagging – lipids) for BCC and SCC, respectively. As proposed in other works, the decrease in the amide I band was attributed to conformational changes in proteins of diseased tissue. When neural network analysis was employed for tissue discrimination, a melanoma diagnostic sensitivity of 85% and specificity of 99% was achieved while a sensitivity of 97% and a specificity of 98% were attained for BCC. These metrics are comparable to that achieved with trained pathologists demonstrating the potential utility and translation of this methodology for melanoma and BCC diagnosis. BCC and melanoma discrimination has also been achieved using PCA for spectral analysis.138,139
Brain cancer
Brain cancer accounts for over 250000 diagnosed cancer cases and 189000 deaths annually worldwide.140 Of these reported cases, approximately 50% are identified as gliomas, which are known for their highly aggressive nature. With this being said, it is not surprising that the majority of work in the Raman community has been geared toward the identification of gliomas. In one of the first reported investigations, Koljenović et al. evaluated 20 unfixed cryosections of glioblastoma by Raman spectroscopy and LDA for separating vital and necrotic tissues.141 The dominating spectral signatures from difference spectra (necrotic minus glioblastoma) resembled that of cholesterol and cholesterol esters, indicating necrotic tissues possess higher concentrations of cholesterol. Subtle band changes, corresponding to carotenoids and calcifications, were also observed. Cluster analysis resulted in 100% diagnostic accuracy when run on 9 independent tissue samples. This group further explored the utility of Raman spectroscopy for brain cancer diagnosis using high wavenumber (2400–3800 cm−1) Raman spectroscopy142 and a single-fiber optical probe Raman system.143
Raman spectroscopy was also compared with IR spectroscopy for the delineation of glioma, meningioma and schwannoma from normal tissues in a study conducted by Krafft et al.144 In this work, both Raman and IR spectroscopic analysis resulted in significant differences between diseased and normal tissue samples; however, Raman spectroscopy was determined to be the better technique due its lack of “contamination” from water and its ability to spatially resolve a greater number of chemical components. For meningioma, Raman spectral decreases were seen at 857, 939, 1246 and 1684 cm−1 when compared to normal tissue. As these bands correspond to collagen, their observed decreases agree well with the notion that tumors have irregular collagen structures. Glioma tissues were evaluated against tissue that had been subjected to a hemorrhage, and spectral analysis showed that hemorrhage tissues had increased band intensities at 661, 751, 1003, 1124, 1258, 1346, 1454 and 1603 cm−1. These bands were correlated to the presence of hemoglobin. When compared to meningioma, close analysis revealed that glioma tissues had greater band intensities at 719 cm−1 indicating differentiations could be based on phosphatidylcholine levels. Schwannomas also showed distinct spectral features at 426 cm−1 and 508 cm−1 when compared to glioma and meningioma tissues. These bands were attributed to the presence of tricalciumphosphate within the lesions. Krafft and colleagues later used Raman microscopic imaging and a spectral unmixing algorithm to investigate histopathological features such as cell density and nuclei.145 Raman signatures in combination with the unmixing algorithm revealed high-grade glioblastomas had increased nucleic acid levels (782, 1099 and 1576 cm−1) over low-grade tissues. Furthermore, this group utilized hyperspectral unmxing of Raman images to correlate morphological (i.e. number and diameter of cell nuclei) and biochemical (i.e. proteins, lipids and nucleic acids) components with the degree of malignancy in brain tissues.146
Coherent Raman imaging has found application in the identification and microscale mapping of gliomas. In 2009, Evans et al. demonstrated the ability of CARS to delineate fresh astrocytoma samples, a type of glioma, from normal brain tissue in mouse models of human brain cancer.147 Images were constructed by using the lipid CH2 stretching mode located at 2845 cm−1 and revealed accurate tumor margins when compared to traditional H&E stains. Kirsch and co-workers have recently used CARS to evaluate primary and secondary brain tumors against normal tissue.148 Primary glioblastoma tumors and brain metastases of breast and melanoma where shown to exhibit decreased CARS signals and were thus distinguishable from normal surrounding tissues (Fig. 8). Tumor infiltrates were also identified by the gradual disappearance in the 2845 cm−1 CARS signal toward the tumor region. Additionally, the use of CARS in a multimodal imaging format similar to that described for skin cancer (BCC) has been used to analyze brain tumors.149
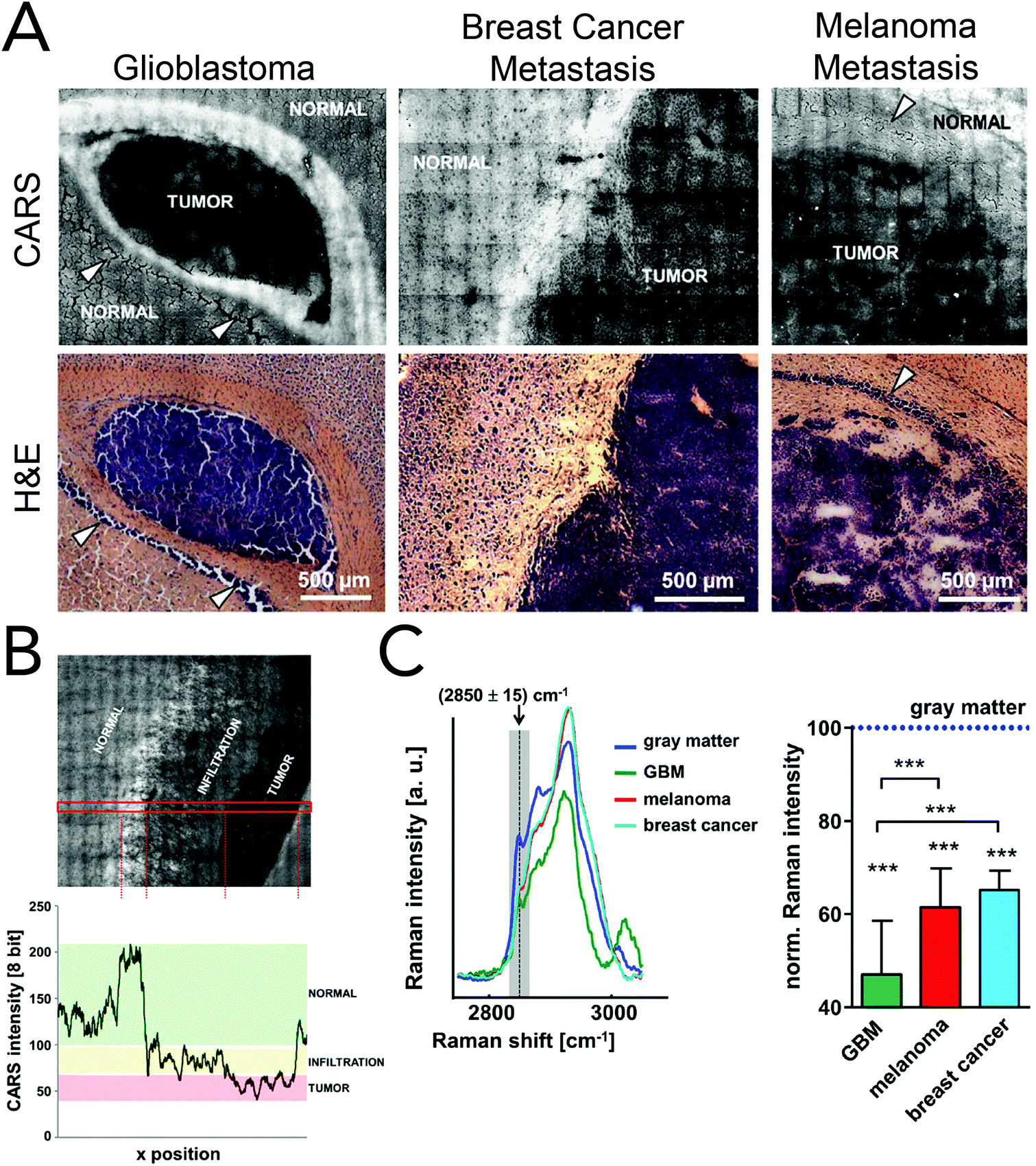 | ||
| Fig. 8 Brain tumor differentiation using CARS microscopy in primary and secondary models. (A) CARS images at 2850 cm−1 (CH2 stretching) and the corresponding H&E stains of whole mouse tumor sections from human (U87MG) glioblastoma, breast cancer (MCF-7) metastasis, and melanoma (A375) metastasis. The reduced CARS signal from the tumor region allows for tumor border identification. Arrows indicate nuclei-rich layers of the hippocampus. (B) Unprocessed CARS image of human (U87MG) glioblastoma (top) and the CARS signal intensity along the area indicated in red (bottom). Tumor infiltrates were distinguishable by tracking CARS signal reduction. (C) CARS spectra (left) and normalized Raman intensity at 2850 cm−1 (right) acquired from normal gray matter, glioblastoma, breast cancer metastasis and melanoma metastasis. Classification was possible from quantifying the Raman intensity at 2850 cm−1. Reproduced with permission from 148. | ||
Gliomas have also been identified through another coherent Raman technique, SRS. Building upon their work in 2008 that demonstrated the ability of two-color SRS imaging to produce high quality brain tissue images,67 Xie and colleagues evaluated the use of this Raman imaging modality to delineate between diseased and normal brain tissues as well as differentiate between primary and secondary tumors.150 Mulitcolored SRS images were created using two Raman vibrational modes: 2845 cm−1 (CH2 stretching – lipids) and 2940 cm−1 (CH3 stretching – lipids and proteins). While the CH2 stretching vibration was used alone to produce images of cytoplasm and myelin sheaths, images of nuclear morphology resulted from the difference image of CH2 stretching (2845 cm−1) subtracted from CH3 stretching (2940 cm−1). Using this type of image acquisition, brain tumor tissues showed increased cellularity compared to normal brain tissue and exhibited similar diagnostic features as conventional H&E tissue analysis. Primary glioma tumors were further distinguishable from brain metastases from the breast by monitoring the tumor margin. Primary glioma tissue samples displayed infiltrating glial cells along the white matter tracts of the corpus callosum, while a defined tumor margin could be seen with tissue samples of brain metastases. This group was later able to quantify and correlate acquired SRS signals at 2845 cm−1 and 2930 cm−1 for the differentiation of cellular regions – solid tumor, cortex and white matter – in human glioblastoma xenografts.151 The ratio of Raman signals (2940 cm−1/2845 cm−1) revealed solid tumor regions had high levels of protein compared to both cortex and white matter regions. By monitoring this Raman signal ratio across the gray matter-tumor interface, tumor infiltration was detectable. Fresh human tumor samples were also analyzed using this multicolor SRS imaging approach. SRS analysis not only revealed hypercellularity (i.e. increased protein content), but also cellular and nuclear pleomorphism, pseudopalisading necrosis and microvascular proliferation. The identified features matched those seen with traditional H&E analysis further. These impressive works demonstrate the potential clinical utility of SRS for interoperative use in brain tumor cytoreduction surgery cases as SRS provides ample information to aid in tumor margin identification.
In vivo detection and diagnostics
Animal models
Animal models are one of the most invaluable components in translating a diagnostic or treatment strategy to the clinic. While they do not fully recapitulate the genetic and epigenetic heterogeneity found in human cancers, animal models allow researchers to investigate harmful and fatal diseases without risking human lives.152 Preclinical studies investigating the utility of Raman spectroscopies for the identification of cancerous lesions have heavily relied on animal models, namely murine models. While several cancer models have been explored (Table 2), the following discussion will focus on brain and breast cancer detection, as these are the most heavily investigated. A list of common Raman bands used for in vivo detection and diagnostics can be found in Table 3.Brain cancer
The gold standard for brain tumor detection is histological analysis on biopsied tissues; however this method does not allow surgeons to differentiate infiltrating lesions from normal tissue in real-time in the operating room. While several groups have focused on characterizing spectral differences of these tissue populations in ex vivo tissues, the use of in vivo Raman detection strategies is still in the beginning stages with the majority of work being conducted on experimental animals. In 2010, Beljebbar et al. demonstrated the potential for in vivo glioblastoma detection using a microprobe coupled to a portable Raman spectrometer (Fig. 9).153 In this study, rat C6 glioma cells were injected into neonatal Wistar rats and intracerebral tumor development was monitored up to 20 days using the Raman microprobe. The microprobe utilized an 830 nm laser excitation source, a fiber optic probe to deliver incident laser light and collect the Raman scattered light, and a spectrograph to record Raman spectra. Alterations in bands associated with proteins, lipids and DNA were observed, and hierarchical cluster analysis of these spectral features showed strong separation between brain tissue pre-injection (i.e. normal) and tissue from the developed tumor. That same year, Krafft and co-workers reported the in vivo detection of brain metastases using a Raman spectroscopic imaging system with a 785 nm laser source.154 Metastatic brain tumors were induced using murine melanoma cells injected into the carotid artery of nude mice. Due to the presence of melanin and its associated Raman signatures (i.e. 587, 976, 1404 and 1595 cm−1), the location of melanoma cells and their developed metastases could be deciphered in cortical and subcortical regions. While the Raman images had lateral resolutions of 250 μm and took several minutes to acquire, this work demonstrated the intraoperative potential of Raman spectroscopic imaging for brain tumor resection.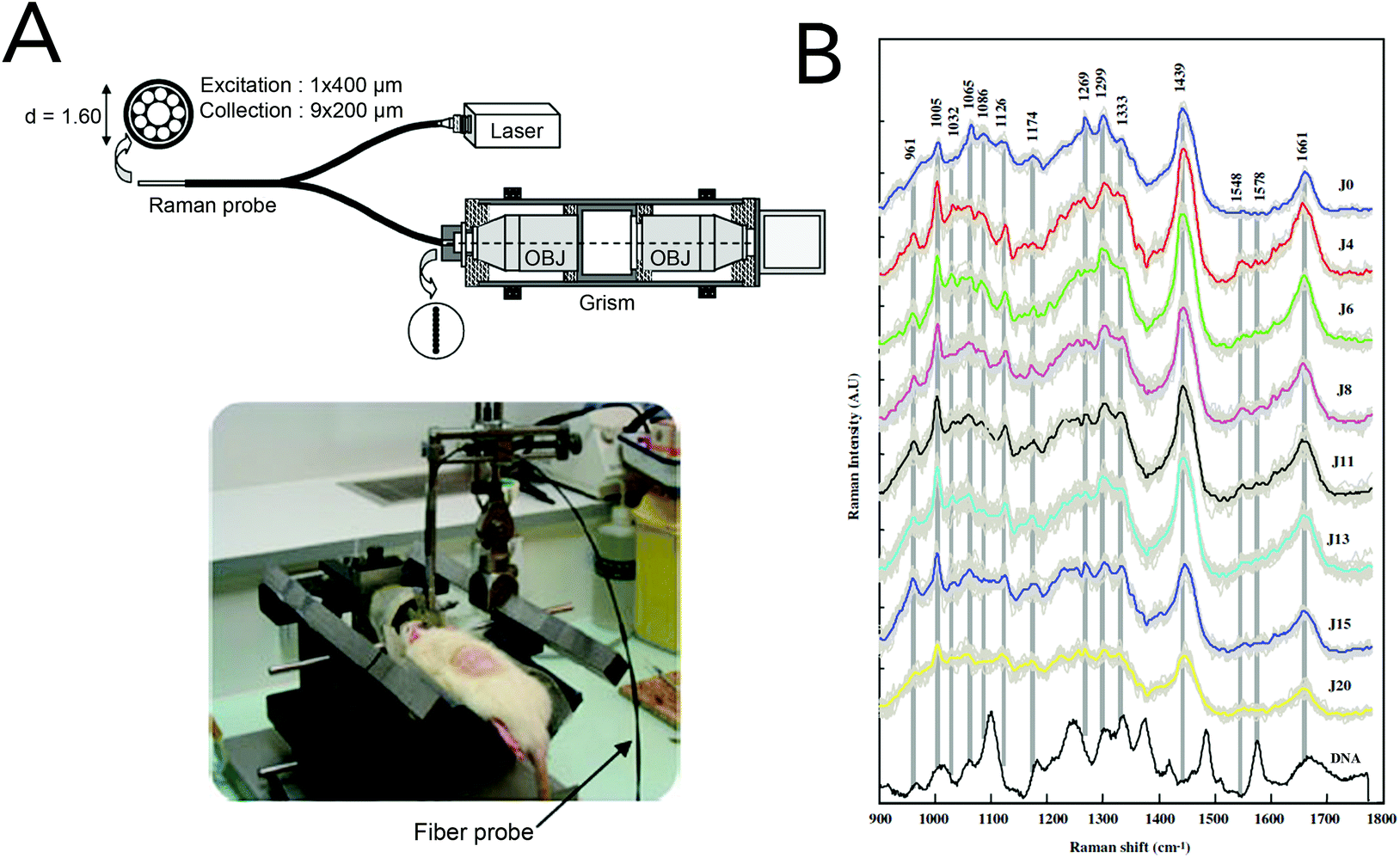 | ||
| Fig. 9 A Raman spectroscopic microprobe for the in vivo detection of brain lesions. (A) A fiber optic probe was used in conjunction with an 830 nm laser and a spectrograph for the acquisition of Raman scattering from glioblastoma tumors in rat models. (B) The acquired Raman spectra after C6 glioma cell injection. Differences in spectra between normal brain tissue (time = 0) and developed gliomas were seen with Raman bands associated with proteins, lipids and DNA. Reprinted with permission from ref. 153. Copyright 2010 Springer-Verlag. | ||
Recently, SERS has been employed for the in vivo identification of brain lesions, specifically for fine margin resection. Kircher et al. described the use of a triple-modality nanoparticle that allowed the combination of MRI, photoacoustic imaging and Raman imaging for the intraoperative identification of brain tumor margins in live mice (Fig. 10).155 This imaging combination resulted in the whole brain tumor's location to be identified (MRI) as well as the tumor's three-dimensional structure at a high spatial resolution to be visualized (photoacoustic imaging). Raman imaging further allowed for brain tumor margins to be delineated as confirmed by histological analysis. To enable the triple imaging strategy, 60 nm spherical gold nanoparticles were functionalized with Raman reporter and MRI contrasting molecules. Under this design the nanoparticles served as a photoacoustic contrasting agent, provided signal enhancement of the Raman tag, and carried contrasting molecules for MRI. The nanoparticles were injected into the tail vein of orthotopic glioblastoma bearing mice, and were able to cross the blood-brain barrier and accumulate in tumor cells via the enhanced permeability and retention (EPR) effect. Tumor site localization allowed for high signal to noise ratios in all imaging modalities and significantly aided in the complete resection of glioblastomas. The use of a handheld Raman scanner in combination with silica coated gold nanoparticles has also been reported.156 The handheld scanner allowed for real-time Raman scans of the tumor region and revealed additional cancer laden regions that were otherwise undetectable by conventional Raman and SERS imaging systems.
 | ||
| Fig. 10 Triple modality imaging agent for the complete resection of brain lesions. (A) Silica coated gold nanoparticles were functionalized with Raman reporter and MRI contrasting molecules to allow for MRI, photoacoustic and Raman imaging. This strategy aided in surgical planning as well as tumor margin removal. (B) The nanoparticle imaging agents showed localization at the tumor site and enabled imaging with high signal to noise using for the three techniques. (C) SERS imaging allowed for complete removal of glioblastoma tumors as well as the tumor margins. Reprinted with permission from ref. 155. Copyright 2012 Nature Publishing Group. | ||
The Xie group has demonstrated the use of SRS for rapid and label-free visualization of glioblastoma multiforme (GBM) brain tumors.151 Accurate resections are of particular importance in the context of brain cancer, given that 85% of recurrent GBM tumors arise at the resection margin where cancer cells were left behind.157 Ji et al. used an infiltrative human GBM xenograft mouse model fitted with a cranial window in order to image the brain tissue non-invasively. Two Raman spectral peaks: 2845 cm−1 and 2930 cm−1, which correspond to lipids and proteins, respectively, were chosen to contrast healthy tissue from the brain tumor. These bands were selected given the marked decrease in lipid content in cancerous tissue, thus offering a source of endogenous contrast. They showed that while the bright-field image of the tumor border appears grossly normal, it is readily distinguishable in SRS. While the authors caution that SRS microscopy still requires rigorous evaluation and validation prior to clinical translation, their work is among the first to introduce in vivo coherent Raman microscopy to the field of cancer research.
Breast cancer
In an effort to better reconcile rodent models of breast cancer with its human counterpart, Bhattacharjee et al. evaluated the Raman spectra from C57, Swiss albino, Swiss bare, and agouti mice as well as Sprague-Dawley rats.158 They first found that white-haired rodents provided better signal-to-noise ratio and less background relative to their colored kin in terms of their Raman spectra. A further investigation of white-haired and hairless mice across various anatomical sites was then carried out in order to evaluate the contributions from skin versus those of the mammary tissue. It was found that the highest quality spectra were obtained from Swiss bare mice, suggesting they are best suited for future transcutaneous spectroscopic investigations of breast cancer in animal models.As for the application of SERS in the context of breast cancer, Qian et al. explored the applicability of using gold nanorod probes to optically detect breast cancer tumors in vivo via tagging with polyethylene glycol (PEG) and 3,3-diethylthiatricarbocyanine iodide (DTTC), a NIR fluorophore and Raman probe.159 Male nude mice were first xenografted with human breast cancer cells and maintained until the tumors reached an approximate size of 5 mm in diameter. The mice were then injected with the functionalized nanoprobes and imaged at several time points post-injection. Their work showcased the potential for using SERS and fluorescence simultaneously on a single platform to map tumors and sentinel lymph nodes in the context of breast cancer.
A later study by Dinish et al. made use of gold nanoparticles tagged with three Raman reporters bound to three antibodies for intrinsic breast cancer biomarkers: Cy5 to TGFβRII, MGITC to CD44, and Rh6G to EGFR.160 Nude female mice were inoculated with a human metastatic breast cancer cell line, and nanoprobes were injected at the center of the tumor once it reached a palpable size. By targeting a single spectral peak from each Raman reporter, the authors observed the multiplex SERS spectra in the tumor region up to 48 hours post-administration, followed by their clearance after 72 hours. This research suggests the possibility of selectively targeting and imaging subtypes of breast cancer in vivo for improved diagnosis and treatment monitoring.
Finally, Jeong et al. recently developed a fluorescence-Raman endomicroscopic system (FRES) using nanoprobes for simultaneously acquiring fluorescence and SERS measurements.52 The nanoprobes consisted of a large silica nanoparticle (200 nm diameter) covered in smaller Raman-labeled silver nanoparticles (10 nm diameter), all coated in a silica shell that was conjugated to AF610, a fluorescent dye (10 nm thickness). To evaluate system performance on a murine breast cancer model, four mice were xenografted with a human breast cancer cell line expressing high levels of HER2 and EGFR, both common biomarkers of breast cancer. The authors report successful molecular detection of the xenografted tumors endoscopically with high sensitivity via multiplexed active targeting. This technology could thus be translated to routine endoscopic procedures where diagnosis of particular cancer subtypes in their early stages is most critical.
Studies in humans
Due to the ethical standards surrounding human research, the exploration of Raman spectroscopy techniques for cancer diagnostics has so far been somewhat limited. However, the works presented here have been conducted on easily accessible regions that do not require invasive procedures or anatomical regions that can be accessed through endoscopic probes. Thus the following discussion focuses on skin, gastrointestinal, cervical and brain cancer detection. Other cancers that have been investigated using in vivo Raman technologies can be found in Table 2. Raman bands associated with these studies can be found in Table 3.Skin cancer
Skin cancer is arguably one of the most studied forms of neoplasia in vivo via Raman spectroscopy, primarily due to the ease of access of suspicious lesions. This avoids the need for endoscopy and favors the use of handheld fiber-based Raman probes for rapid, accurate, and point-of-care diagnosis and classification of suspect skin lesions.The current clinical standard for skin cancer diagnosis relies on visual inspection of suspect lesions, often assisted by dermoscopy. This diagnostic methodology leaves a fair degree of variability in the accuracy of skin cancer diagnosis based on the experience and training of the dermatologist. This subjectivity thus warrants the development of tools that are non-invasive, rapid, and portable for a more objective and rigid framework for evaluating suspicious skin lesions.
To this aim, the Zeng group has been investigating the applicability of Raman spectroscopy for skin cancer diagnostics. In 2012, the group conducted a clinical study investigating various types of skin cancer as well as a variety of benign skin diseases, where the integration time for their studies was set at 1 second or less. They confirmed the ability to distinguish malignant and pre-malignant lesions from benign ones, melanomas from nevi (benign pigmented lesions, commonly referred to as moles), and melanomas from seborrheic keratoses. For highly sensitive detection (ranging from 95% to 99% sensitivity), a specificity ranging from 15% to 54% was reported, suggesting the applicability of Raman spectroscopy as a tool for screening suspicious lesions non-invasively.60 In a related report, it was also shown that intentional photobleaching of skin can be a useful technique to reduce the autofluorescence of the sample, effectively increasing the overall signal-to-noise ratio of the spectroscopic measurements.161 More recently, the group conducted an independent validation of their automatic skin cancer detection methodology, where they used 518 cases from a prior study as a training set and 127 new cases for testing. This study validated prior work, yielding diagnostic accuracy that matching previous findings.64
In parallel, the Meinke group conducted an in vivo clinical study using Raman spectroscopy to discriminate skin cancer from normal tissue via a fiber-coupled probe. In measuring spectra from 104 cases of various skin cancers, accuracies of 73%, 85%, and 91% in distinguishing basal cell carcinoma, squamous cell carcinoma, and malignant melanoma from normal skin, respectively, were found.62
Related work conducted by the Wulf group further revealed that Raman spectroscopy could be used in the context of skin cancer diagnostics in a manner that is independent of skin pigmentation. Specifically, it was reported that while the degree of pigmentation does influence the Raman spectra, proper background correction can negate the contributions from pigments and thus allowing for spectral bands of interest to be interpreted unambiguously.61
Recent efforts have been made into combining imaging and spectroscopic modalities to further improve the accuracy of skin cancer diagnostics. One such effort led by the Tunnell group found that Raman spectroscopy alone was sufficient to distinguish malignant melanoma from benign pigmented lesions with a diagnostic accuracy of 100%. However, in the case of nonmelanoma skin cancers, the diagnostic accuracy was improved when reflectance and fluorescence measurements were combined with the Raman data. In this case, they were able to distinguish actinic keratoses as well as squamous and basal cell carcinomas from normal skin with a sensitivity and specificity of 90% and 85%, respectively.59 Another study conducted by Moryatov and co-workers found that combining measurements from optical coherence tomography (OCT), backscattering, and Raman spectroscopy into a multimodal platform for diagnosing skin cancer increased sensitivity and specificity by 9% and 8%, respectively, when compared to Raman spectroscopy alone. In particular, they were able to distinguish melanoma from nonmelanoma tumors with 89% sensitivity and 93% specificity, and basal cell carcinoma (BCC) from non-BCC tumors with 100% sensitivity and 96% specificity.63 The experimental setup is shown in Fig. 11. These two promising trials illustrate that the advent of multimodal imaging and spectroscopic platforms can be used to improve skin cancer diagnostics over the use of Raman spectroscopy alone.
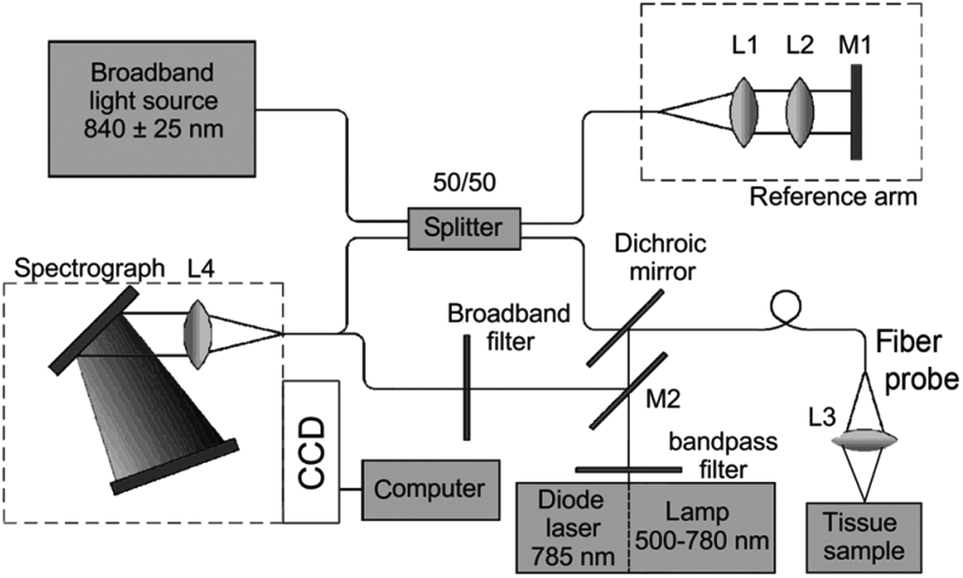 | ||
| Fig. 11 Experimental setup combining principles of OCT, backscattering, and Raman spectroscopy for human tissue studies. The setup includes a diode laser module (785 nm, 150 mW) for excitation of Raman spectra, a broadband laser diode (840 ± 25 nm, 20 mW), and a Michelson interferometer (50/50) based on a fiber optic splitter. The reference arm consists of the collimating lens L1, focusing lens L2, and adjusted mirror M1. The backscattered spectrum is recorded after sample illumination with a broadband light source (500 to 780 nm). Backscattered radiation and the Raman signal were collected by a fiber probe and a collecting lens L3. A focusing lens L4 and mirror M2 were used for radiation delivery to the receiving channel. Reprinted with permission from ref. 63 Copyright 2015 Society of Photo Optical Instrumentation Engineers (SPIE). | ||
Gastrointestinal cancers
Cancers of the gastrointestinal tract can occur anywhere between the oral cavity and the anus, and represent a particularly difficult diagnostic challenge. Raman spectroscopy has been used in several studies investigating mouth or oral cancer, properly known as oral squamous cell carcinoma (OSCC). Considerable progress in this field has been made by the Krishna group, where a study in 2012 on 10 normal and 10 cancerous ex vivo tissues, as well as 10 in vivo measurements, revealed differences in the spectra of normal versus cancerous samples. Moreover, they showed that the spectra from the normal in vivo and ex vivo tissues were similar enough to warrant further in vivo study in the use of Raman spectroscopy as a diagnostic tool for OSCC.55 Indeed, the group later published an investigation of premalignant lesions of the mouth in vivo, finding that premalignant lesions can effectively be distinguished from normal and cancerous sites in patients with and without smoking habits.54 The group further studied the applicability of Raman spectroscopy for OSCC diagnostics by investigating whether early changes associated with cancer – including malignancy-associated changes (MAC) and cancer field effects (CFE) – can be identified spectroscopically. In acquiring 722 spectra from a total of 84 subjects, it was found that early neoplastic transformation can indeed be detected via Raman spectroscopy.56Esophageal and gastric cancers have been another focus of diagnostic research in the field of Raman spectroscopy, with particular emphasis on the precancerous cellular transformation from squamous to gastric cells in the lower esophagus – a condition known as Barrett's esophagus (BE). To this aim, the Huang and Yeoh groups have put forth a number of reports investigating the applicability of Raman spectroscopy for esophageal and stomach cancer diagnoses, beginning with an in vivo assessment of gastric dysplasia. Using narrow-band image-guided Raman spectroscopy, 30 patients were included in a study where 54 spectra were acquired from normal tissues and 18 from dysplastic gastric tissues, yielding 94.4% sensitivity and 96.3% specificity.46 In a related study, the same team examined 67 gastric patients and identified gastric cancer with a diagnostic accuracy of 93.7% (94.0% sensitivity and 93.4% specificity).47 The ability of Raman spectroscopy to distinguish between benign and malignant stomach ulcers was then tested, and sensitivities of 90.8%, 84.7%, and 82.1% as well as specificities of 93.8%, 94.5%, and 95.3% for classification of normal tissue, benign ulcers, and malignant ulcers, respectively, were found.42 Using more elaborate data processing algorithms to improve the overall accuracy, an in vivo gastric cancer diagnostic accuracy of 94.6% was subsequently achieved.43
The Stone group has also significantly contributed to the translation of Raman technologies from the laboratory to the clinic. In 2011, Kendall et al. evaluated a custom Raman probe designed for esophageal diagnostics on excised biopsy samples. They studied normal, low risk (Barrett's esophagus), and high risk (dysplasia and cancer) samples, where 1304 Raman spectra were acquired from a total of 123 samples. In classifying test spectra against a training set, their methodology resulted in sensitivity and specificity of 66–84% and 81–96%, respectively.162 Subsequent work by Almond et al. in 2014 reported the development of a novel custom Raman probe, where a confocal design was implemented to specifically interrogate superficial tissues (∼150 μm) in an attempt to better detect surface lesions that may be cured via endoscopic therapy. While their study was limited to validation on ex vivo resected samples, their endoscopic Raman spectroscopy setup detected Barrett's esophagus-associated high-grade dysplasia (HGD) and esophageal adenocarcinoma with sensitivity and specificity of 86% and 88%, respectively.163 While this technology has yet to be fully tested in vivo, this group is currently focusing on building various probes and adjusting tolerances to maximize performance in a clinical trial evaluating their implementation of endoscopic Raman spectroscopy in patients with Barrett's esophagus.
Extending the use of Raman spectroscopy further into the gastrointestinal tract, Bergholt et al. investigated Raman spectra from tissues of the esophagus and stomach, both ex vivo and in vivo. This study found that there is significant variability between the spectra from the esophagus as compared to those of the stomach, but the spectra from different anatomical sites within a given tissue were fairly similar. Moreover, cancerous tissues from the esophagus and stomach were found to be distinguishable with accuracies of 94.7% and 89.3%, respectively.38 A subsequent study showcased the relevance of combining Raman spectroscopy with autofluorescence measurements in vivo, yielding a diagnostic accuracy for gastric cancer of 92.2%, greater than either of the two approaches evaluated independently (89.7% for Raman spectroscopy alone, and 86.3% for autofluorescence alone).44 A later study by Bergholt et al. showed that esophageal cancer could be diagnosed to an accuracy of 96.0% when the Raman spectroscopic assessment is guided by a wide-field imaging modality such as white light reflectance, narrow-band imaging, or autofluorescence imaging clinical endoscopic examination.37 A fully automated online Raman spectroscopy platform with multimodal image-guided sampling for gastric cancer diagnosis was then developed, where 305 patients were examined yielding a diagnostic accuracy of 85.6%.45 Later work demonstrated that the technology can be further exploited to target biopsy sites more specifically, as well as providing a platform for distinguishing between dysplastic and neoplastic lesions more consistently.41
Subsequently, using a fiber optic confocal Raman endoscope, Bergholt et al. evaluated the potential for in vivo detection of high-grade dysplasia in Barrett's esophagus and identified diagnostic sensitivity and specificity of 87.0% and 84.7%, respectively.36 Different endoscope configurations have also been investigated, where it was found that a beveled probe tip coupled to a ball lens outperformed the standard volume Raman probe via selective interrogation of the superficial tissue and suppression of autofluorescence contributions.48 Even more recently, Wang et al. investigated spectral bands in the fingerprint and high-wavenumber regions of Raman spectra from the esophagus, and distinguished esophageal squamous cell carcinoma (ESCC) from normal tissue with a diagnostic accuracy of 97.3%.39
Finally, Raman spectroscopy has also been investigated for the diagnosis of colorectal tissue for cancer by the Huang group. They developed a novel fiber optic probe capable of acquiring fingerprint and high-wavenumber bands simultaneously from the subsurface of the colorectal tissue and identified a diagnostic accuracy of 88.8% in detecting colorectal cancer, with 93.3% sensitivity and 88.3% specificity.34
Cervical cancer
The diagnostic potential of Raman spectroscopy has also been studied in the context of cervical cancer, where it was found that spectral variations within normal tissue were an obstacle in properly identifying cervical cancer lesions. To this aim, the Mahadevan-Jansen group identified various classes of cervical tissue that can be considered normal: truly normal, previously diseased normal, and adjacent-to-disease normal. The inclusion of these subcategories increased classification accuracy to 97%.33In parallel, the Huang group studied 105 spectra from 29 patients (65 normal and 40 precancerous spectra) using genetic algorithm-partial least squares-discriminant analysis (GA-PLS-DA), yielding 82.9% diagnostic accuracy (72.5% sensitivity and 89.2% specificity).29 As was done in the context of esophageal and gastric cancer described above, they furthered their research by examining the fingerprint and high-wavenumber spectral bands of the cervical Raman spectra. Combining the data from both spectral domains into their analysis, a diagnostic accuracy of 82.6% was identified for the 476 spectra (356 normal and 120 precancerous) collected from 44 patients.30 Duraipandian et al. then evaluated the use of confocal Raman spectroscopy against near-infrared autofluorescence (NIR AF) spectroscopy alone as well as composite NIR AF/Raman spectroscopy. They found confocal Raman spectroscopy to have a diagnostic accuracy of 84.1%, an increase over the composite approach (82.3%), as well as NIR AF spectroscopy alone (59.6%).31
Finally, the Krishna group identified a particular challenge in the diagnosis of cervical cancer in the Indian subcontinent: most cervical cancers in such parts of the world are already advanced upon detection, leaving few intact cervical regions for assessment of normal tissue. In order to find normal baseline tissue, they explored the possibility of using vaginal tissue as an internal control for spectroscopic assessment of the cervical tissue. Indeed, the spectral features of normal vaginal and cervical tissues were found to be closely similar, such that cervical cancer diagnoses based on Raman spectroscopy could be facilitated in conditions where normal cervical tissue is otherwise unavailable.32
Brain cancer
In the context of brain cancer, proper surgical resection of the tumorous mass is critical to minimize the odds of recurrence. To address this need the Leblond group developed a handheld Raman spectroscopy probe for the classification of brain tissue intraoperatively (Fig. 12).50 They conducted a study on 17 patients with gliomas ranging from grade 2 to 4, where a total of 161 spectral measurements were collected. Their study revealed a diagnostic accuracy of 92%, with 93% sensitivity and 91% specificity.51 These results were contrasted with the surgeon's visual inspection of the affected brain tissue using bright-field microscopy and magnetic resonance (MR) guidance, which yielded an accuracy of 73%, a sensitivity of 67%, and a specificity of 86%. This study thus illustrates the utility of Raman spectroscopy in detecting brain tumors intraoperatively, and sets a robust foundation for pursuing clinical trials to further contrast the effectiveness of Raman spectroscopy against the current standard of care for guiding brain cancer resections.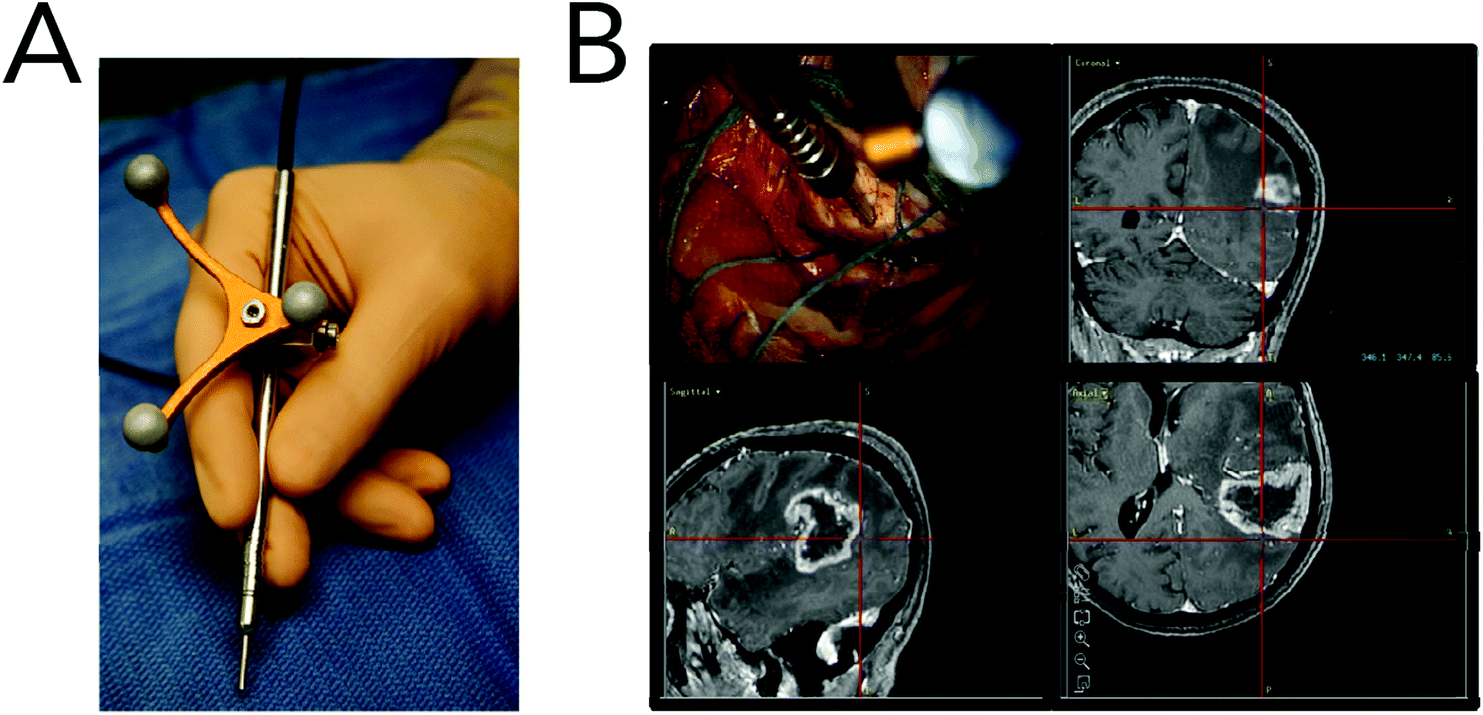 | ||
| Fig. 12 Raman spectroscopy system for intraoperative detection. (A) Photograph of the handheld contact probe, with the attached neuronavigation tracking unit. (B) Illustration of the probe being used intraoperatively, with the neuronavigation system showing the location of the tip of the probe (cross hairs) on the peroperative MR images. Reprinted from ref. 50 with permission from The Optical Society of America. | ||
Conclusions and future perspectives
Raman technologies, from spectroscopy to advanced imaging tools, offer considerable promise for cancer diagnostics in the future. These tools have undergone generations of advances since the Raman process was first discovered, with developments in signal processing, algorithm development, detector technology, and fiber optics driving development. The last decade, in particular, has seen huge strides in Raman technologies, including gains in Raman spectral processing, improvements in SERS-based techniques, and the development of stimulated Raman scattering microscopy.A number of critical advances still under development will continue to push the state-of-the-art of Raman spectroscopy, Raman imaging, coherent Raman imaging, and SERS. In the realm of SERS, considerable effort is underway on optimizing SERS nanoparticles, from their shape to surface chemical modifications. As SERS occurs through a combination of field enhancements, the shape and surface topography of the nanoparticle are being optimized to bias chemical-particle interactions. Parallel efforts in optimizing nanoparticles for the attachment of chemicals, molecules, and biomolecules continue to make strides forward. Customization of chemical attachment conditions to produce “monodentate” nanoparticles that can be used for accurate enumeration are under development. Along the same lines, composite nanoparticles, nanoparticle clusters, and other advancements promise to improve SERS performance.
Engineering efforts at MIT and other institutions are working to push the speed of Raman spectra acquisition. The incorporation of optimized laser setups, the use of multiple simultaneous foci, and improvements in detectors has dramatically improved spectral acquisition. Coherent Raman approaches, in particular SRS, has the potential to dramatically improve spectra acquisition rates. Hyperspectral SRS, where the Stokes beam is scanning over a wavelength range throughout image acquisition, has enabled the rapid, low-noise collection of image stacks, where each image pixel contains an entire SRS spectrum. As SRS and Raman spectra are closely related, hyperspectral SRS tools now under development promise a potential sea change in the field of Raman spectral acquisition. Though SRS systems are currently complex, requiring lock-in detection for example, new advances are rapidly simplifying the technique, making it more widely available. Moreover, SRS can be used in endoscopic tools, enabling in vivo rapid chemical assessment and imaging.
The application of these powerful tools to cancer has taken the Raman and cancer communities many years of effort, but the results are impressive. With specificities and sensitivities exceeding 95% for the detection of a wide array of tumor types, Raman-based systems are now developed to be sufficiently used in demanding clinical applications. This next step – clinical application – may be the most exciting and most difficult yet.
To take Raman technologies and place them in the hands of a physician, nurse, or caregiver, much of the analysis that is commonplace in the Raman community needs to be entirely automated to provide easily interpretable clinical guidance. Though potentially lifesaving, it is unlikely that a doctor or nurse, for example, will learn Raman spectroscopy or the interpretation of Raman spectra. Similarly, a physician will not likely run or operate an SRS endoscope at its current level of complexity. Clinical care can indeed incorporate complex technology when needed – for example, see the impact of CT, MRI, and radiation therapy systems, which are some of the most complex instruments found in hospitals and clinics. Still, these technologies produce either an interpretable image or use computer systems to facilitate their use.
Continued development of Raman technologies, with a focus on clinical needs, interpretability, and simplicity, is essential to the translation of these powerful toolkits. While engineering can and has simplified Raman tools for clinical use, the data analysis must also be well packaged. This does not necessarily mean that the outcome of every Raman measurement must be “red light” or “green light”; probabilities or other indicators could be used as a readout that clinicians can act upon. Outside of numeric indicators or probabilities, it is possible to use Raman technologies to go beyond classification to provide clinicians with new ways of seeing familiar information. For example, recent work in SRS microscopy has produced H&E-like images from purely endogenous, vibrational contrast. As all clinicians are trained in the interpretation of histology, this unique application of Raman technology provides doctors with familiar and easily interpretable information that can one day be used to guide clinical decision-making.
Conflict of Interest
Dr Evans holds patents in CARS technology that have been licensed to major microscope manufacturing companies.Acknowledgements
The authors would like to thank the financial support of the 2015 Ludwig Center at Harvard Award. The authors would also like to thank Dr Hequn (Tracy) Wang for her helpful suggestions and edits.References
- R. L. Siegel, K. D. Miller and A. Jemal, CA-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- C. R. UK, Cancer mortality projections, http://www.cancerresearchuk.org/health-professional/cancer-statistics/mortality/projections.
- L. Fass, Mol. Oncol., 2008, 2, 115–152 CrossRef PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- M. B. Fenn, P. Xanthopoulos, G. Pyrgiotakis, S. R. Grobmyer, P. M. Pardalos and L. L. Hench, Adv. Opt. Technol., 2011, 20 Search PubMed.
- K. Kong, C. Kendall, N. Stone and I. Notingher, Adv. Drug Delivery Rev., 2015, 89, 121–134 CrossRef CAS PubMed.
- R. Petry, M. Schmitt and J. Popp, ChemPhysChem, 2003, 4, 14–30 CrossRef CAS PubMed.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- B. R. Masters and P. T. C. So, Handbook of Biomedical Nonlinear Optical Microscopy, Oxford University Press, New York, 1st edn, 2008 Search PubMed.
- D. A. Long, The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules, John Wiley & Sons Ltd, 1st edn, 2002 Search PubMed.
- R. W. Boyd, Nonlinear Optics, Academic Press, 3rd edn, 2008 Search PubMed.
- Y. R. Shen, The Principles of Nonlinear Optics, John Wiley & Sons Inc, 1st edn, 2003 Search PubMed.
- C. L. Evans and X. S. Xie, Ann. Rev. Anal. Chem., 2008, 1, 883–909 CrossRef CAS PubMed.
- C. H. Camp Jr. and M. T. Cicerone, Nat. Photonics, 2015, 9, 295–305 CrossRef.
- M. Fleischmann, P. J. Hendra and A. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- C. L. Haynes, A. D. McFarland and R. P. V. Duyne, Anal. Chem., 2005, 77, 338A–346A CrossRef CAS.
- M. D. Sonntag, J. M. Klingsporn, A. B. Zrimsek, B. Sharma, L. K. Ruvuna and R. P. Van Duyne, Chem. Soc. Rev., 2014, 43, 1230–1247 RSC.
- A. Campion and P. Kambhampati, Chem. Soc. Rev., 1998, 27, 241–250 RSC.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- C. Louis and O. Pluchery, Gold nanoparticles for physics, chemistry and biology, World Scientific, 2012 Search PubMed.
- G. Wiederrecht, Handbook of nanoscale optics and electronics, Academic Press, 2010 Search PubMed.
- B. Sharma, R. R. Frontiera, A.-I. Henry, E. Ringe and R. P. Van Duyne, Mater. Today, 2012, 15, 16–25 CrossRef CAS.
- B. J. Kennedy, S. Spaeth, M. Dickey and K. T. Carron, J. Phys. Chem. B, 1999, 103, 3640–3646 CrossRef CAS.
- G. Schatz, M. Young and R. Van Duyne, in Surface-Enhanced Raman Scattering, ed. K. Kneipp, M. Moskovits and H. Kneipp, Springer, Berlin Heidelberg, 2006, ch. 2, vol. 103, pp. 19–45 Search PubMed.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- R. O. P. Draga, M. C. M. Grimbergen, P. L. M. Vijverberg, C. F. P. van Swol, T. G. N. Jonges, J. A. Kummer and J. L. H. Ruud Bosch, Anal. Chem., 2010, 82, 5993–5999 CrossRef CAS PubMed.
- S. Duraipandian, W. Zheng, J. Ng, J. J. H. Low, A. Ilancheran and Z. Huang, Analyst, 2011, 136, 4328–4336 RSC.
- S. Duraipandian, W. Zheng, J. Ng, J. J. H. Low, A. Ilancheran and Z. Huang, Anal. Chem., 2012, 84, 5913–5919 CrossRef CAS PubMed.
- S. Duraipandian, W. Zheng, J. Ng, J. J. H. Low, A. Ilancheran and Z. Huang, J. Biomed. Opt., 2013, 18 CrossRef CAS PubMed , 067007.
- R. Shaikh, T. K. Dora, S. Chopra, A. Maheshwari, D. Kedar, K. R. Bharat and C. M. Krishna, J. Biomed. Opt., 2014, 19 CrossRef CAS PubMed , 087001.
- E. Vargis, E. M. Kanter, S. K. Majumder, M. D. Keller, R. B. Beaven, G. G. Rao and A. Mahadevan-Jansen, Analyst, 2011, 136, 2981–2987 RSC.
- M. S. Bergholt, W. Zheng, K. Lin, J. Wang, H. Xu, J.-l. Ren, K. Y. Ho, M. Teh, K. G. Yeoh and Z. Huang, Anal. Chem., 2015, 87, 960–966 CrossRef CAS PubMed.
- E. Garai, S. Sensarn, C. L. Zavaleta, N. O. Loewke, S. Rogalla, M. J. Mandella, S. A. Felt, S. Friedland, J. T. C. Liu, S. S. Gambhir and C. H. Contag, PLoS One, 2015, 10 CAS , e0123185.
- M. S. Bergholt, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So, A. Shabbir and Z. Huang, Gastroenterology, 2014, 146, 27–32 CrossRef PubMed.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So and Z. Huang, Technol. Cancer Res. Treat., 2011, 10, 103–112 CAS.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So and Z. Huang, J. Biomed. Opt., 2011, 16 CrossRef CAS PubMed , 037003.
- J. Wang, K. Lin, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh and Z. Huang, Sci. Rep., 2015, 5, 12957–12957 CrossRef CAS PubMed.
- Y. W. Wang, A. Khan, S. Y. Leigh, D. Wang, Y. Chen, D. Meza and J. T. C. Liu, Biomed. Opt. Express, 2014, 5, 2883–2895 CrossRef CAS PubMed.
- M. S. Bergholt, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So, A. Shabbir and Z. Huang, J. Biophotonics, 2013, 6, 49–59 CrossRef CAS PubMed.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So and Z. Huang, Analyst, 2010, 135, 3162–3168 RSC.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So and Z. Huang, Int. J. Cancer, 2011, 128, 2673–2680 CrossRef CAS PubMed.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So and Z. Huang, Biosens. Bioelectron., 2011, 26, 4104–4110 CrossRef CAS PubMed.
- S. Duraipandian, M. S. Bergholt, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Y. So, A. Shabbir and Z. Huang, J. Biomed. Opt., 2012, 17 CrossRef PubMed , 081418.
- Z. Huang, M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh and K. G. Yeoh, J. Biomed. Opt., 2010, 15 Search PubMed , 037017.
- Z. Huang, S. K. Teh, W. Zheng, K. Lin, K. Y. Ho, M. Teh and K. G. Yeoh, Biosens. Bioelectron., 2010, 26, 383–389 CrossRef CAS PubMed.
- J. Wang, K. Lin, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh and Z. Huang, Anal. Bioanal. Chem., 2015, 407, 8303–8310 CrossRef CAS PubMed.
- M. A. Short, S. Lam, A. M. McWilliams, D. N. Ionescu and H. Zeng, J. Thorac. Oncol., 2011, 6, 1206–1214 CrossRef PubMed.
- J. Desroches, M. Jermyn, K. Mok, C. Lemieux-Leduc, J. Mercier, K. St-Arnaud, K. Urmey, M.-C. Guiot, E. Marple, K. Petrecca and F. Leblond, Biomed. Opt. Express, 2015, 6, 2380–2397 CrossRef PubMed.
- M. Jermyn, K. Mok, J. Mercier, J. Desroches, J. Pichette, K. Saint-Arnaud, L. Bernstein, M.-C. Guiot, K. Petrecca and F. Leblond, Sci. Transl. Med., 2015, 7 CAS , 274ra19.
- S. Jeong, Y.-i. Kim, H. Kang, G. Kim, M. G. Cha, H. Chang, K. O. Jung, Y.-H. Kim, B.-H. Jun, D. W. Hwang, Y.-S. Lee, H. Youn, Y.-S. Lee, K. W. Kang, D. S. Lee and D. H. Jeong, Sci. Rep., 2015, 5, 9455–9455 CrossRef CAS PubMed.
- A. M. Mohs, M. C. Mancini, S. Singhal, J. M. Provenzale, B. Leyland-Jones, M. D. Wang and S. Nie, Anal. Chem., 2010, 82, 9058–9065 CrossRef CAS PubMed.
- S. P. Singh, A. Deshmukh, P. Chaturvedi and C. M. Krishna, J. Biomed. Opt., 2012, 17 CAS , 105002.
- S. P. Singh, A. Deshmukh, P. Chaturvedi and C. M. Krishna, J. Cancer Res. Ther., 2012, 8, S126–S132 CrossRef CAS PubMed.
- S. P. Singh, A. Sahu, A. Deshmukh, P. Chaturvedi and C. M. Krishna, Analyst, 2013, 138, 4175–4182 RSC.
- E. Drakaki, T. Vergou, C. Dessinioti, A. J. Stratigos, C. Salavastru and C. Antoniou, J. Biomed. Opt., 2013, 18 CAS , 061221.
- N. Huang, H. Wang, J. Zhao, H. Lui, M. Korbelik and H. Zeng, Lasers Surg. Med., 2010, 42, 638–648 CrossRef PubMed.
- L. Lim, B. Nichols, M. R. Migden, N. Rajaram, J. S. Reichenberg, M. K. Markey, M. I. Ross and J. W. Tunnell, J. Biomed. Opt., 2014, 19 CrossRef CAS PubMed , 117003.
- H. Lui, J. Zhao, D. McLean and H. Zeng, Cancer Res., 2012, 72, 2491–2500 CrossRef CAS PubMed.
- P. A. Philipsen, L. Knudsen, M. Gniadecka, M. H. Ravnbak and H. C. Wulf, Photochem. Photobiol. Sci., 2013, 12, 770–776 CAS.
- J. Schleusener, P. Gluszczynska, C. Reble, I. Gersonde, J. Helfmann, J. W. Fluhr, J. Lademann, J. Röwert-Huber, A. Patzelt and M. C. Meinke, Exp. Dermatol., 2015, 24, 767–772 CrossRef PubMed.
- V. P. Zakharov, I. A. Bratchenko, D. N. Artemyev, O. O. Myakinin, D. V. Kornilin, S. V. Kozlov and A. A. Moryatov, J. Biomed. Opt., 2015, 20 CrossRef PubMed , 025003.
- J. Zhao, H. Lui, S. Kalia and H. Zeng, Anal. Bioanal. Chem., 2015, 407, 8373–8379 CrossRef CAS PubMed.
- N. Huang, M. Short, J. Zhao, H. Wang, H. Lui, M. Korbelik and H. Zeng, Opt. Express, 2011, 19, 22892–22909 CrossRef CAS PubMed.
- A. Zumbusch, G. R. Holtom and X. S. Xie, Phys. Rev. Lett., 1999, 82, 4142 CrossRef CAS.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322, 1857–1861 CrossRef CAS PubMed.
- M. Weinigel, H. G. Breunig, M. Kellner-Höfer, R. Bückle, M. E. Darvin, M. Klemp, J. Lademann and K. König, Laser Phys. Lett., 2014, 11, 055601–055601 CrossRef.
- C. W. Freudiger, W. Yang, G. R. Holtom, N. Peyghambarian, X. S. Xie and K. Q. Kieu, Nat. Photonics, 2014, 8, 153–159 CrossRef CAS PubMed.
- P. Gemperline, Practical guide to chemometrics, CRC press, 2006 Search PubMed.
- I. R. Lewis and H. Edwards, Handbook of Raman spectroscopy: from the research laboratory to the process line, CRC Press, 2001 Search PubMed.
- J. W. Cooley and J. W. Tukey, Math. Comput., 1965, 19, 297–301 CrossRef.
- P. M. Ramos and I. Ruisánchez, J. Raman Spectrosc., 2005, 36, 848–856 CrossRef CAS.
- H.-W. Tan and S. D. Brown, J. Chemom., 2002, 16, 228–240 CrossRef CAS.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- M. R. Chedekel, S. K. Smith, P. W. Post, A. Pokora and D. L. Vessell, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 5395–5399 CrossRef CAS.
- K. Tsia, Understanding Biophotonics: Fundamentals, Advances, and Applications, CRC Press, 2015 Search PubMed.
- S. Li and L. Dai, Appl. Spectrosc., 2011, 65, 1300–1306 CrossRef CAS PubMed.
- H. G. Schulze and R. F. Turner, Appl. Spectrosc., 2013, 67, 457–462 CrossRef CAS PubMed.
- M. N. Leger and A. G. Ryder, Appl. Spectrosc., 2006, 60, 182–193 CrossRef CAS PubMed.
- C. A. Lieber and A. Mahadevan-Jansen, Appl. Spectrosc., 2003, 57, 1363–1367 CrossRef CAS PubMed.
- A. Cao, A. K. Pandya, G. K. Serhatkulu, R. E. Weber, H. Dai, J. S. Thakur, V. M. Naik, R. Naik, G. W. Auner, R. Rabah and D. C. Freeman, J. Raman Spectrosc., 2007, 38, 1199–1205 CrossRef CAS.
- J. Zhao, H. Lui, D. I. McLean and H. Zeng, Appl. Spectrosc., 2007, 61, 1225–1232 CrossRef CAS PubMed.
- B. D. Beier and A. J. Berger, Analyst, 2009, 134, 1198–1202 RSC.
- K. Chen, H. Wei, H. Zhang, T. Wu and Y. Li, Anal. Methods, 2015, 7, 2770–2778 RSC.
- H. Krishna, S. K. Majumder and P. K. Gupta, J. Raman Spectrosc., 2012, 43, 1884–1894 CrossRef CAS.
- K. Chen, H. Zhang, H. Wei and Y. Li, Appl. Opt., 2014, 53, 5559–5569 CrossRef PubMed.
- J. Popp, V. V. Tuchin, A. Chiou and S. H. Heinemann, Handbook of biophotonics, John Wiley & Sons, 2011 Search PubMed.
- I. Jolliffe, Principal component analysis, Wiley Online Library, 2002 Search PubMed.
- A. C. Rencher, Methods of multivariate analysis, John Wiley & Sons, 2003 Search PubMed.
- R. Gautam, S. Vanga, F. Ariese and S. Umapathy, EPJ Tech. Instrum., 2015, 2, 1–38 CrossRef PubMed.
- Q. Tu and C. Chang, Nanomed. Nanotechnol. Biol. Med., 2012, 8, 545–558 CrossRef CAS PubMed.
- J. G. Kelly, J. Trevisan, A. D. Scott, P. L. Carmichael, H. M. Pollock, P. L. Martin-Hirsch and F. L. Martin, J. Proteome Res., 2011, 10, 1437–1448 CrossRef CAS PubMed.
- M. Hedegaard, C. Matthäus, S. Hassing, C. Krafft, M. Diem and J. Popp, Theor. Chem. Acc., 2011, 130, 1249–1260 CrossRef CAS.
- M. Diem, Raman spectral imaging for the determination of structure and dynamics of human cells, Laboratory for Spectral Diagnosis, Department of Chemistry and Chemical Biology, North-eastern University, Boston, 2009 Search PubMed.
- J. T. Tabarangao and A. D. Slepkov, J. Spectrosc., 2015, 2015, 8 Search PubMed.
- R. G. Brereton and G. R. Lloyd, Analyst, 2010, 135, 230–267 RSC.
- X. Zang, C. M. Jones, T. Q. Long, M. E. Monge, M. Zhou, L. D. Walker, R. Mezencev, A. Gray, J. F. McDonald and F. M. Fernández, J. Proteome Res., 2014, 13, 3444–3454 CrossRef CAS PubMed.
- A. Berger, in Emerging Raman Applications and Techniques in Biomedical and Pharmaceutical Fields, ed. P. Matousek and M. D. Morris, Springer, Berlin, Heidelberg, 2010, ch. 16, pp. 385–404, DOI:10.1007/978-3-642-02649-2_16.
- D. S. Grubisha, R. J. Lipert, H.-Y. Park, J. Driskell and M. D. Porter, Anal. Chem., 2003, 75, 5936–5943 CrossRef CAS PubMed.
- S. Li, Y. Zhang, J. Xu, L. Li, Q. Zeng, L. Lin, Z. Guo, Z. Liu, H. Xiong and S. Liu, Appl. Phys. Lett., 2014, 105, 091104 CrossRef.
- G. Del Mistro, S. Cervo, E. Mansutti, R. Spizzo, A. Colombatti, P. Belmonte, R. Zucconelli, A. Steffan, V. Sergo and A. Bonifacio, Anal. Bioanal. Chem., 2015, 407, 3271–3275 CrossRef CAS PubMed.
- A. Sahu, S. Sawant, H. Mamgain and C. M. Krishna, Analyst, 2013, 138, 4161–4174 RSC.
- D. Lin, J. Pan, H. Huang, G. Chen, S. Qiu, H. Shi, W. Chen, Y. Yu, S. Feng and R. Chen, Sci. Rep., 2014, 4, 4751 Search PubMed.
- A. Sahu, N. Nandakumar, S. Sawant and C. M. Krishna, Analyst, 2015, 140, 2294–2301 RSC.
- B. Elumalai, A. Prakasarao, B. Ganesan, K. Dornadula and S. Ganesan, J. Raman Spectrosc., 2015, 46, 84–93 CrossRef CAS.
- S. Feng, D. Lin, J. Lin, Z. Huang, G. Chen, Y. Li, S. Huang, J. Zhao, R. Chen and H. Zeng, Appl. Phys. Lett., 2014, 104, 073702 CrossRef.
- J. Pichardo-Molina, C. Frausto-Reyes, O. Barbosa-García, R. Huerta-Franco, J. González-Trujillo, C. Ramírez-Alvarado, G. Gutiérrez-Juárez and C. Medina-Gutiérrez, Lasers Med. Sci., 2007, 22, 229–236 CrossRef CAS PubMed.
- T. Bhattacharjee, A. Khan, G. Maru, A. Ingle and C. M. Krishna, Analyst, 2015, 140, 456–466 RSC.
- S. Feng, S. Huang, D. Lin, G. Chen, Y. Xu, Y. Li, Z. Huang, J. Pan, R. Chen and H. Zeng, Int. J. Nanomed., 2015, 10, 537–547 CrossRef CAS PubMed.
- A. S. Haka, Z. Volynskaya, J. A. Gardecki, J. Nazemi, J. Lyons, D. Hicks, M. Fitzmaurice, R. R. Dasari, J. P. Crowe and M. S. Feld, Cancer Res., 2006, 66, 3317–3322 CrossRef CAS PubMed.
- A. Stelling, R. Salzer, M. Kirsch, S. Sobottka, K. Geiger, E. Koch, G. Schackert and G. Steiner, Anal. Bioanal. Chem., 2011, 400, 2745–2753 CrossRef CAS PubMed.
- C. J. Frank, D. C. Redd, T. S. Gansler and R. L. McCreery, Anal. Chem., 1994, 66, 319–326 CrossRef CAS PubMed.
- C. J. Frank, R. L. McCreery and D. C. B. Redd, Anal. Chem., 1995, 67, 777–783 CrossRef CAS PubMed.
- R. Manoharan, K. Shafer, L. Perelman, J. Wu, K. Chen, G. Deinum, M. Fitzmaurice, J. Myles, J. Crowe, R. R. Dasarl and M. S. Feld, Photochem. Photobiol., 1998, 67, 15–22 CrossRef CAS PubMed.
- K. E. Shafer-Peltier, A. S. Haka, M. Fitzmaurice, J. Crowe, J. Myles, R. R. Dasari and M. S. Feld, J. Raman Spectrosc., 2002, 33, 552–563 CrossRef CAS.
- A. S. Haka, K. E. Shafer-Peltier, M. Fitzmaurice, J. Crowe, R. R. Dasari and M. S. Feld, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12371–12376 CrossRef CAS PubMed.
- J. Surmacki, B. Brozek-Pluska, R. Kordek and H. Abramczyk, Analyst, 2015, 140, 2121–2133 RSC.
- M. M. Mariani, L. J. Maccoux, C. Matthäus, M. Diem, J. G. Hengstler and V. Deckert, Anal. Chem., 2010, 82, 4259–4263 CrossRef CAS PubMed.
- B. Brozek-Pluska, J. Musial, R. Kordek, E. Bailo, T. Dieing and H. Abramczyk, Analyst, 2012, 137, 3773–3780 RSC.
- S. Rehman, Z. Movasaghi, A. T. Tucker, S. P. Joel, J. A. Darr, A. V. Ruban and I. U. Rehman, J. Raman Spectrosc., 2007, 38, 1345–1351 CrossRef CAS.
- K. Kenny, Z. Fazliyana, R. Emad, E. Ian, K. Alexey and N. Ioan, Phys. Med. Biol., 2014, 59, 6141 CrossRef PubMed.
- J. E. G. Gonzalez, G. Richard and J. Valaitis, Am. J. Surg. Pathol., 1991, 15, 586–591 CrossRef CAS PubMed.
- A. S. Haka, K. E. Shafer-Peltier, M. Fitzmaurice, J. Crowe, R. R. Dasari and M. S. Feld, Cancer Res., 2002, 62, 5375–5380 CAS.
- N. Stone and P. Matousek, Cancer Res., 2008, 68, 4424–4430 CrossRef CAS PubMed.
- I. Barman, N. C. Dingari, A. Saha, S. McGee, L. H. Galindo, W. Liu, D. Plecha, N. Klein, R. R. Dasari and M. Fitzmaurice, Cancer Res., 2013, 73, 3206–3215 CrossRef CAS PubMed.
- S. Kaminaka, H. Yamazaki, T. Ito, E. Kohda and H. o. Hamaguchi, J. Raman Spectrosc., 2001, 32, 139–141 CrossRef CAS.
- Z. Huang, A. McWilliams, H. Lui, D. I. McLean, S. Lam and H. Zeng, Int. J. Cancer, 2003, 107, 1047–1052 CrossRef CAS PubMed.
- L. Gao, Z. Wang, F. Li, A. A. Hammoudi, M. J. Thrall, P. T. Cagle and S. T. C. Wong, Arch. Pathol. Lab. Med., 2012, 136, 1502–1510 CrossRef CAS PubMed.
- L. Gao, F. Li, M. J. Thrall, Y. Yang, J. Xing, A. A. Hammoudi, H. Zhao, Y. Massoud, P. T. Cagle and Y. Fan, J. Biomed. Opt., 2011, 16, 096004 CrossRef PubMed.
- N. D. Magee, J. S. Villaumie, E. T. Marple, M. Ennis, J. S. Elborn and J. J. McGarvey, J. Phys. Chem. B, 2009, 113, 8137–8141 CrossRef CAS PubMed.
- M. Gniadecka, H. Wulf, N. N. Mortensen, O. F. Nielsen and D. H. Christensen, J. Raman Spectrosc., 1997, 28, 125–129 CrossRef CAS.
- A. Nijssen, T. C. B. Schut, F. Heule, P. J. Caspers, D. P. Hayes, M. H. Neumann and G. J. Puppels, J. Invest. Dermatol., 2002, 119, 64–69 CrossRef CAS PubMed.
- A. Nijssen, K. Maquelin, L. F. Santos, P. J. Caspers, T. C. Bakker Schut, J. C. den Hollander, M. H. A. Neumann and G. J. Puppels, J. Biomed. Opt., 2007, 12, 034004 CrossRef PubMed.
- N. Vogler, T. Meyer, D. Akimov, I. Latka, C. Krafft, N. Bendsoe, K. Svanberg, B. Dietzek and J. Popp, J. Biophotonics, 2010, 3, 728–736 CrossRef CAS PubMed.
- S. Heuke, N. Vogler, T. Meyer, D. Akimov, F. Kluschke, H.-J. Röwert-Huber, J. Lademann, B. Dietzek and J. Popp, Healthcare, 2013164–83 Search PubMed.
- M. Gniadecka, P. A. Philipsen, S. Sigurdsson, S. Wessel, O. F. Nielsen, D. H. Christensen, J. Hercogova, K. Rossen, H. K. Thomsen, R. Gniadecki, L. K. Hansen and H. C. Wulf, J. Invest. Dermatol., 2004, 122, 443–449 CrossRef CAS PubMed.
- B. Bodanese, F. L. Silveira, R. A. Zângaro, M. T. T. Pacheco, C. A. Pasqualucci and L. Silveira, Photomed. Laser Surg., 2012, 30, 381–387 CrossRef CAS PubMed.
- B. Bodanese, L. Silveira Jr., R. Albertini, R. A. Zangaro and M. T. T. Pacheco, Photomed. Laser Surg., 2010, 28, S-119–S-127 CrossRef CAS PubMed.
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J. Cancer, 2015, 136, E359–E386 CrossRef CAS PubMed.
- S. Koljenović, T. C. B. Schut, J. M. Kros, H. J. van den Berge and G. J. Puppels, Lab. Invest., 2002, 82, 1265–1277 CrossRef.
- S. Koljenović, T. C. Bakker Schut, R. Wolthuis, B. de Jong, L. Santos, P. J. Caspers, J. M. Kros and G. J. Puppels, J. Biomed. Opt., 2005, 10, 031116–03111611 CrossRef PubMed.
- S. Koljenovic, T. Bakker Schut, R. Wolthuis, A. Vincent, G. Hendriks-Hagevi, L. Santos, J. Kros and G. Puppels, Anal. Chem., 2007, 79, 557–564 CrossRef CAS PubMed.
- C. Krafft, S. B. Sobottka, G. Schackert and R. Salzer, J. Raman Spectrosc., 2006, 37, 367–375 CrossRef CAS.
- C. Krafft, B. Belay, N. Bergner, B. F. M. Romeike, R. Reichart, R. Kalff and J. Popp, Analyst, 2012, 137, 5533–5537 RSC.
- N. Bergner, A. Medyukhina, K. D. Geiger, M. Kirsch, G. Schackert, C. Krafft and J. Popp, Anal. Bioanal. Chem., 2013, 405, 8719–8728 CrossRef CAS PubMed.
- C. L. Evans, X. Xu, S. Kesari, X. S. Xie, S. T. Wong and G. S. Young, Opt. Express, 2007, 15, 12076–12087 CrossRef CAS PubMed.
- O. Uckermann, R. Galli, S. Tamosaityte, E. Leipnitz, K. D. Geiger, G. Schackert, E. Koch, G. Steiner and M. Kirsch, PLoS One, 2014, 9, e107115 Search PubMed.
- T. Meyer, N. Bergner, C. Bielecki, C. Krafft, D. Akimov, B. F. M. Romeike, R. Reichart, R. Kalff, B. Dietzek and J. Popp, J. Biomed. Opt., 2011, 16, 021113 CrossRef PubMed.
- C. W. Freudiger, R. Pfannl, D. A. Orringer, B. G. Saar, M. Ji, Q. Zeng, L. Ottoboni, W. Ying, C. Waeber, J. R. Sims, P. L. De Jager, O. Sagher, M. A. Philbert, X. Xu, S. Kesari, X. S. Xie and G. S. Young, Lab. Invest., 2012, 92, 1492–1502 CrossRef CAS PubMed.
- M. Ji, D. A. Orringer, C. W. Freudiger, S. Ramkissoon, X. Liu, D. Lau, A. J. Golby, I. Norton, M. Hayashi, N. Y. R. Agar, G. S. Young, C. Spino, S. Santagata, S. Camelo-Piragua, K. L. Ligon, O. Sagher and X. S. Xie, Sci. Transl. Med., 2013, 5 CAS , 201ra119.
- B. A. Ruggeri, F. Camp and S. Miknyoczki, Biochem. Pharmacol., 2014, 87, 150–161 CrossRef CAS PubMed.
- A. Beljebbar, S. Dukic, N. Amharref and M. Manfait, Anal. Bioanal. Chem., 2010, 398, 477–487 CrossRef CAS PubMed.
- M. Kirsch, G. Schackert, R. Salzer and C. Krafft, Anal. Bioanal. Chem., 2010, 398, 1707–1713 CrossRef CAS PubMed.
- M. F. Kircher, A. de la Zerda, J. V. Jokerst, C. L. Zavaleta, P. J. Kempen, E. Mittra, K. Pitter, R. Huang, C. Campos, F. Habte, R. Sinclair, C. W. Brennan, I. K. Mellinghoff, E. C. Holland and S. S. Gambhir, Nat. Med., 2012, 18, 829–834 CrossRef CAS PubMed.
- H. Karabeber, R. Huang, P. Iacono, J. M. Samii, K. Pitter, E. C. Holland and M. F. Kircher, ACS Nano, 2014, 8, 9755–9766 CrossRef CAS PubMed.
- K. Petrecca, M.-C. Guiot, V. Panet-Raymond and L. Souhami, J. Neuro-Oncol., 2013, 111, 19–23 CrossRef PubMed.
- T. Bhattacharjee, P. Kumar, G. Maru, A. Ingle and C. M. Krishna, Lasers Med. Sci., 2014, 29, 325–333 CrossRef CAS PubMed.
- J. Qian, L. Jiang, F. Cai, D. Wang and S. He, Biomaterials, 2011, 32, 1601–1610 CrossRef CAS PubMed.
- U. S. Dinish, G. Balasundaram, Y.-T. Chang and M. Olivo, Sci. Rep., 2014, 4, 4075–4075 CAS.
- H. Wang, J. Zhao, A. M. D. Lee, H. Lui and H. Zeng, Photodiagn. Photodyn. Ther., 2012, 9, 299–302 CrossRef CAS PubMed.
- C. Kendall, J. Day, J. Hutchings, B. Smith, N. Shepherd, H. Barr and N. Stone, Analyst, 2010, 135, 3038–3041 RSC.
- L. M. Almond, J. Hutchings, G. Lloyd, H. Barr, N. Shepherd, J. Day, O. Stevens, S. Sanders, M. Wadley, N. Stone and C. Kendall, Gastrointest. Endosc., 2014, 79, 37–45 CrossRef PubMed.
- G. Wang, R. J. Lipert, M. Jain, S. Kaur, S. Chakraboty, M. P. Torres, S. K. Batra, R. E. Brand and M. D. Porter, Anal. Chem., 2011, 83, 2554–2561 CrossRef CAS PubMed.
- P. Crow, N. Stone, C. A. Kendall, J. S. Uff, J. A. M. Farmer, H. Barr and M. P. J. Wright, Br. J. Cancer, 2003, 89, 106–108 CrossRef CAS PubMed.
- S. Feng, J. Lin, Z. Huang, G. Chen, W. Chen, Y. Wang, R. Chen and H. Zeng, Appl. Phys. Lett., 2013, 102, 043702 CrossRef.
- B. Brozek-Pluska, M. Kopec, I. Niedzwiecka and A. Morawiec-Sztandera, Analyst, 2015, 140, 2107–2113 RSC.
- P. Matousek and N. Stone, J. Biomed. Opt., 2007, 12, 024008 CrossRef PubMed.
- Y. Yang, F. Li, L. Gao, Z. Wang, M. J. Thrall, S. S. Shen, K. K. Wong and S. T. Wong, Biomed. Opt. Express, 2011, 2, 2160–2174 CrossRef PubMed.
- B. Brozek-Pluska, M. Kopec, J. Surmacki and H. Abramczyk, Analyst, 2015, 140, 2134–2143 RSC.
- T. Bhattacharjee, G. Maru, A. Ingle and C. M. Krishna, J. Biomed. Opt., 2013, 18 CrossRef CAS PubMed , 047004.
- S. Fendel and B. Schrader, Fresenius’ J. Anal. Chem., 1998, 360, 609–613 CrossRef CAS.
- L. d. O. Nunes, A. A. Martin, L. Silveira Jr. and M. Zampieri, J. Spectrosc., 2003, 17, 597–602 CrossRef CAS.
- M. Larraona-Puy, A. Ghita, A. Zoladek, W. Perkins, S. Varma, I. H. Leach, A. A. Koloydenko, H. Williams and I. Notingher, J. Biomed. Opt., 2009, 14, 054031 CrossRef PubMed.
- Y. Zhou, C.-H. Liu, Y. Sun, Y. Pu, S. Boydston-White, Y. Liu and R. R. Alfano, J. Biomed. Opt., 2012, 17, 116021 CrossRef PubMed.
- H. Wills, R. Kast, C. Stewart, R. Rabah, A. Pandya, J. Poulik, G. Auner and M. D. Klein, J. Pediatr. Surg., 2009, 44, 386–391 CrossRef PubMed.
- K. Tanahashi, A. Natsume, F. Ohka, H. Momota, A. Kato, K. Motomura, N. Watabe, S. Muraishi, H. Nakahara, Y. Saito, I. Takeuchi and T. Wakabayashi, BioMed Res. Int., 2014, 8 Search PubMed.
- M. Köhler, S. Machill, R. Salzer and C. Krafft, Anal. Bioanal. Chem., 2009, 393, 1513–1520 CrossRef PubMed.
- D. Lin, S. Feng, J. Pan, Y. Chen, J. Lin, G. Chen, S. Xie, H. Zeng and R. Chen, Opt. Express, 2011, 19, 13565–13577 CrossRef CAS PubMed.
- D. Mitra, X. Luo, A. Morgan, J. Wang, M. P. Hoang, J. Lo, C. R. Guerrero, J. K. Lennerz, M. C. Mihm, J. A. Wargo, K. C. Robinson, S. P. Devi, J. C. Vanover, J. A. D'Orazio, M. McMahon, M. W. Bosenberg, K. M. Haigis, D. A. Haber, Y. Wang and D. E. Fisher, Nature, 2012, 491, 449–454 CrossRef CAS PubMed.
- A. Molckovsky, L.-M. W. K. Song, M. G. Shim, N. E. Marcon and B. C. Wilson, Gastrointest. Endosc., 2003, 57, 396–402 CrossRef PubMed.
- C. M. MacLaughlin, N. Mullaithilaga, G. Yang, S. Y. Ip, C. Wang and G. C. Walker, Langmuir, 2013, 29, 1908–1919 CrossRef CAS PubMed.
- J. W. Chan, D. S. Taylor, T. Zwerdling, S. M. Lane, K. Ihara and T. Huser, Biophys. J., 2006, 90, 648–656 CrossRef CAS PubMed.
- A. T. Harris, A. Lungari, C. J. Needham, S. L. Smith, M. A. Lones, S. E. Fisher, X. B. Yang, N. Cooper, J. Kirkham and D. A. Smith, Head Neck Oncol., 2009, 1, 1–8 CrossRef PubMed.
- G. R. Lloyd, L. E. Orr, J. Christie-Brown, K. McCarthy, S. Rose, M. Thomas and N. Stone, Analyst, 2013, 138, 3900–3908 RSC.
- S. Feng, D. Lin, J. Lin, B. Li, Z. Huang, G. Chen, W. Zhang, L. Wang, J. Pan, R. Chen and H. Zeng, Analyst, 2013, 138, 3967–3974 RSC.
- A. Mahadevan-Jansen, M. F. Mitchell, N. Ramanujamf, A. Malpica, S. Thomsen, U. Utzinger and R. Richards-Kortumt, Photochem. Photobiol., 1998, 68, 123–132 CrossRef CAS PubMed.
- I. Taleb, G. Thiefin, C. Gobinet, V. Untereiner, B. Bernard-Chabert, A. Heurgue, C. Truntzer, P. Hillon, M. Manfait, P. Ducoroy and G. D. Sockalingum, Analyst, 2013, 138, 4006–4014 RSC.
- S. R. Hawi, W. B. Campbell, A. Kajdacsy-Balla, R. Murphy, F. Adar and K. Nithipatikom, Cancer Lett., 1996, 110, 35–40 CrossRef CAS PubMed.
- I. J. Pence, C. A. Patil, C. A. Lieber and A. Mahadevan-Jansen, Biomed. Opt. Express, 2015, 6, 2724–2737 CrossRef PubMed.
- K. Maheedhar, R. A. Bhat, R. Malini, N. B. Prathima, P. Keerthi, P. Kushtagi and C. M. Krishna, Photomed. Laser Surg., 2008, 26, 83–90 CrossRef CAS PubMed.
- J. V. Jokerst, A. J. Cole, D. Van De Sompel and S. S. Gambhir, ACS Nano, 2012, 6, 10366–10377 CrossRef CAS PubMed.
- X. Wang, X. Qian, J. J. Beitler, Z. G. Chen, F. R. Khuri, M. M. Lewis, H. J. C. Shin, S. Nie and D. M. Shin, Cancer Res., 2011, 71, 1526–1532 CrossRef CAS PubMed.
- R. Mitra, O. Chao, Y. Urasaki, O. B. Goodman and T. T. Le, BMC Cancer, 2012, 12, 540 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |