
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-stoichiometric compositions arising from synergistic electronic and size effects. Synthesis, crystal chemistry and electronic properties of A14Cd1+xPn11 compounds (0 ≤ x ≤ 0.3; A = Sr, Eu; Pn = As, Sb)†
Julien P. A.
Makongo
,
Gregory M.
Darone
,
Sheng-Qing
Xia
and
Svilen
Bobev
*
Department of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716, USA. E-mail: bobev@udel.edu; Fax: +1-302-831-6335; Tel: +1-302-831-8720
First published on 29th July 2015
Abstract
The structural variability in terms of existence of stoichiometric and nonstoichiometric compounds in the well-known “14–1–11” family has been investigated. Four such compounds, Eu14CdAs11, Sr14Cd1.06(1)As11, Eu14Cd1.27(1)Sb11, and Sr14Cd1.30(1)Sb11, synthesized by both flux and traditional solid-state methods, have been structurally characterized by single-crystal X-ray diffraction. They are all isotypic, forming with the Ca14AlSb11 structure type (Pearson code tI208, tetragonal space group I41/acd). Their average structure can be described as space-filling packing of distorted [APn6] octahedra, [APn5] trigonal bipyramids, and [CdPn4] tetrahedra, where Pn denotes As or Sb, and A stands for Eu or Sr. Interstitial sites are partially occupied by Cd atoms, giving rise to the chemical formula A14Cd1+xPn11. The interstitial atom can be seen as forming a cluster, reminiscent of the empty substructure unit in the gamma-brass cluster (also known as “tetraederstern”). Robust cation–cation interactions exist in the structure and might be a natural response of the electronic structure to compensate for the apparent electron deficiency and in order to optimize the bonding interactions. The electronic structure calculations for idealized Sr14CdSb11 revealed that the Fermi level falls near the edge of the valence band, which suggests poor metallic conductivity. The analysis of the volume of coordination polyhedron and that of the Voronoi cell indicates that the content of Cd atoms in the interstitial site is limited by the available “free volume” within the complex tetrahedral unit. This free volume is induced by the instability of the hypervalent [Pn3]7− anion, which in turn is related to the increasing size of pnictogen atoms, and to some extent, by the type/size of the countercations.
1 Introduction
The recent discovery of interesting magnetic, electronic and thermal transport properties in the family of tetragonal Zintl compounds A14MPn11 (where A stands for the divalent electropositive metals Ca, Sr, Ba, Eu, or Yb, M is one of the triel elements Al, Ga, In or transition metal such as Mn, and Pn = pnictogen, i.e., P, As, Sb, Bi) has sparked considerable interest in this class of materials. Not only do these compounds provide a basis for understanding the structural flexibility as a function of the size of the pnictogen atoms, the electropositive A metal, and the M metal atoms, but they also represent a unique class of materials having the ability to integrate, in a single class of compounds, a wide variety of physical properties including ferromagnetic or antiferromagnetic ordering, colossal magnetoresistance, as well as (semi)metallic and semiconducting properties with direct implication for high temperature thermoelectric power generation.1 This profusion of unusual physics can be ascribed to the complex crystal structure of these compounds, which adopt the Ca14AlSb11 structure type.2 In terms of the Zintl–Klemm concept,1a,3 the Ca14AlSb11 structure can be rationalized as consisting of 14 Ca2+ cations and three types of anions—the [AlSb4]9− tetrahedral species, isolated [Sb]3−, and the linear hypervalent [Sb3]7−. The latter seems to be the hallmark of the structure, impacting strongly the bonding and the properties of these compounds. Surveying the literature, we have noticed that not all linear [Pn3]7− units are created equal, and the hypervalent description might be an oversimplification, particularly in the case of [P3]7− and [As3]7−, where crystallographic disorder appears to be prevalent. Take for example the Ca14GaP11 structure,4 where the central atom in the presumed [P3]7− anion is offset from its equilibrium position, resulting in a splitting with a distance between the split sites of ca. 0.8 Å. A similar disorder involving [As3]7− has been reported for Sr14MnAs115 and Sr14GaAs11,6 among others, where the split atomic positions are ca. 0.7 Å apart. This raises the question about the stability of this electron-rich three-center bonding, which Munzarová and Hoffmann7 addressed, in part,‡ in their computational work studying the hypervalent [Pn3]7− anions in Ca14GaAs11,8 Eu14MnSb11,9 Ca14MnBi11 and Ba14MnBi11.10 A very recent experimental/DFT paper on Ca14MgSb11 and Yb14MgSb11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 provided yet another look at the bonding in [Sb3]7−. From the literature, one might conclude that the electronic requirements of the hypervalent bonding in [Pn3]7− are a function of both the type of constituent atoms and the type of counter-cations within the crystal structure. If that is the case, one can then ask the question: if an interplay of atomic sizes and electronegativities does exist, what are the conditions that could make one effect override the other? More than a decade-old study offered a glimpse into this problem, but surprisingly, has not inspired follow-up work—the Kauzlarich group identified A14MSb11+x (A = Ca, Sr; M = Zn, Cd),12 where the structure is shown to accommodate an additional metal atom, which the early study assigned as Sb. With this paper we provide further insight into the interstitial chemistry of the “14–1–11” structure and suggest the formulation A14M1+xSb11 to be a more plausible scenario, from both geometric and electronic arguments (vide infra).
11 provided yet another look at the bonding in [Sb3]7−. From the literature, one might conclude that the electronic requirements of the hypervalent bonding in [Pn3]7− are a function of both the type of constituent atoms and the type of counter-cations within the crystal structure. If that is the case, one can then ask the question: if an interplay of atomic sizes and electronegativities does exist, what are the conditions that could make one effect override the other? More than a decade-old study offered a glimpse into this problem, but surprisingly, has not inspired follow-up work—the Kauzlarich group identified A14MSb11+x (A = Ca, Sr; M = Zn, Cd),12 where the structure is shown to accommodate an additional metal atom, which the early study assigned as Sb. With this paper we provide further insight into the interstitial chemistry of the “14–1–11” structure and suggest the formulation A14M1+xSb11 to be a more plausible scenario, from both geometric and electronic arguments (vide infra).
Herein, we report the synthesis, and an account on the crystal chemistry and electronic properties of the compounds Eu14Cd1+xAs11, Sr14Cd1+xAs11, Eu14Cd1+xSb11, and Sr14Cd1+xSb11, which are referred to hereafter, for simplicity, by their idealized stoichiometric formulae. The structures of these compounds can be viewed as space-filling packing of distorted [APn6] octahedra and [APn5] trigonal bipyramids, where Pn = As, Sb; A = Eu, Sr. The extra Cd atoms are embedded within part of the residual space near the center of a tetrahedral unit consisting of an inner [CdPn4] and an outer [CdA4] tetrahedron. This polyhedral unit is reminiscent of the empty tetrahedral substructure of the gamma-brass cluster. Each [CdPn4] tetrahedron shares faces with distorted [APn6] octahedra and [APn5] trigonal bipyramids. The description of the structure in terms of such complex polyhedral packing is an important element toward the understanding of the origin of the robust Cd–A bonding, furthering the notion that the “free volume” of the interstitial site is related to the electronic instability of the linear hypervalent [Pn3]7− (Pn = As, Sb) anion. By judiciously choosing the two counter-cations (Eu and Sr) and the two pnictogen atoms (As and Sb), we show using the Voronoi cell volume analysis that the propensity of the structure toward additional atom uptake increases with increasing the size of the pnictogen, and to some extent, with the size of the counter-cations. This allows for the accommodation of a small fraction of electron-donating atoms (i.e., Cd, which can fit into the sparse “free volume” within the tetrahedral unit, lending support to the notion that Cd2+ will be preferred over the much bigger Sb3−, at the minimum, for geometric reasons).
Our study on “14–1–11” phases came into being over the course of our careful exploratory studies of ternary pnictides for potential application as thermoelectric materials. This area of research has been one of the core activities in our research group over the past 10 years, and as a result, a large number of new compounds with unique structures have been synthesized and characterized. Several noteworthy examples include, but are not limited to, Ca2CdSb2 and Yb2CdSb2,13 Ba11Cd8Bi14,14 Eu10Cd6Bi12,15 A11Cd6Sb12,16 A21Cd4Pn18,17 A9Cd4+xPn9, and A9Zn4+xPn918 (A denotes Ca, Sr, Ba, Eu, or Yb; Pn stands for As, Sb, Bi). The two latter examples belong to the “A9M4+xPn9” (M = Mn, Zn, Cd) family, which is closely related, at the structural level, to the title A14M1+xSb11 compounds. Coincidentally, the “9–4–9” phases have also been gaining considerable interest due to the recent reports of interesting electronic and thermal transport properties in Yb9Mn4.2Sb9 and its Zn-substituted variant.19
2 Experimental
2.1 Synthetic procedures
All reagents with stated purity greater than 99.9% of the metal basis were used as purchased (Alfa or Aldrich). Single-crystals of the title compounds were grown from the starting elements. The reagents were loaded in 2 cm3 alumina crucibles, topped with lead metal as a flux medium, and subsequently sealed in fused silica tubes under a vacuum of 10−4 Torr. All sample manipulations (prior and post synthesis) were performed inside an argon-filled glove box with oxygen and moisture levels below 0.1 ppm.The best heat treatment routes were found to be as follows: (1) Eu14CdAs11 samples (10-fold excess of Pb) were slowly heated to 1000 °C at a rate of 100 °C h−1, dwelled at this temperature for 16 h for homogenization, and then cooled successively to 900 °C at a rate of 10 °C h−1, to 750 °C at a rate of 2 °C h−1, and finally to 600 °C at a rate of 50 °C h−1, the temperature at which the sample was centrifuged to separate the molten Pb from the crystals. Multifaceted air-stable crystals were obtained and used for single-crystal structure determination. (2) The Sr14CdAs11 samples were heated to 1100 °C in 72 h, the temperature at which a dwelling time of 30 h was necessary for sample homogenization before cooling down to 700 °C over 58 h for centrifugation. Irregular, dark-grey crystals were obtained and found to be air-sensitive; (3) Sr14CdSb11 samples were heated to 1100 °C in 4 h, held there for 8 h, cooled to 900 °C in 30 h, where another dwelling step of 32 h was introduced before cooling the sample to 700 °C over 20 h for removal of the molten lead by centrifugation. Powder X-ray diffraction analysis revealed the “14–1–11” phase as the major product with minor, unknown impurity phases.
For the Eu14CdSb11 samples, which were found to be air-stable, two different strategies were pursued: (1) single-crystals of Eu14Cd1.27(1)Sb11 were grown by a reaction of the reagents in the molar ratio Eu:27:Cd:100:Sb:18, with Cd having a dual role of reagent and flux. The sample was subsequently heated to 1100 °C over 8 h, equilibrated at this temperature for 30 h, and finally cooled to 700 °C over 50 h for centrifugation. Powder X-ray diffraction revealed the product to be a mixture of Eu9Cd4.5Sb9, Eu11Sb10, and Eu14CdSb11. Bulk material of the same composition was prepared by direct fusion of the reagents in the exact molar ratio (i.e., with a stoichiometric amount of Cd) and at the same temperature. The synthesized material was poorly crystalline. Small single-crystals were identified from a similar reaction of Sr, Cd, and Sb and subjected to crystallographic analysis—the synthetic method apparently does not affect the structure, since the refinements are almost identical (ESI†).
Last, we note that attempts to make more isotypic phases were made, but were unsuccessful. For example, we looked into the possibility to synthesize the Ca-, Yb-, and/or Ba-analogs of the herein presented arsenides and antimonides, but succeeded in making Yb14CdSb11, Ba14CdSb11, and the solid-solution (Yb,Ca)14CdSb11 in limited quantities. The structures of these phases remain to be established from single-crystal X-ray diffraction work.§ The synthetic routes described above did not allow us to make any of possible Zn-analogs, although Yb14ZnSb11 is known in the literature.1f The recurring products of these experiments were the stable binary A11Pn10 phases (A = Ca, Sr, Eu, Yb; and Pn = As, Sb), as well as A2ZnSb2 (A = Sr, Eu).20
2.2 Crystallography
The powder X-ray diffraction data were recorded on a Rigaku MiniFlex powder diffractometer operating with a filtered Cu Kα radiation (λ = 1.54056 Å) and enclosed inside a nitrogen-filled glove-box. This enables one to work with samples prone to decomposition in ambient air. The JADE 6.5 software package was employed for the phase analysis and identification.Single-crystals were selected inside the argon-filled glove box and cut in Paratone-N oil to a desired shape and size under an optical microscope, then placed on glass fibers, which were immediately mounted on the goniometer of a Bruker SMART CCD-based diffractometer. An inert atmosphere and constant temperature (200 K) were maintained by flowing a cold nitrogen stream over the crystals. A monochromatized Mo Kα radiation (λ = 0.71073 Å) was used for data collection. Four different batches of ω scans with a scan width of 0.40° and an exposure time of 10 s per frame were collected in the 2θ range up to 60° at four different φ settings. The data collection was processed and monitored employing the Bruker SMART software package.21 The global unit cell refinements, data reduction and integration were completed using the SAINT program.22 The intensities of the data sets were merged and sorted for space group determination on the basis of systematic absences using the subprogram XPREP in SHELXTL.23 Semi-empirical absorption corrections based on symmetrically equivalent reflections were applied to all data using SADABS.24 The structure was solved by direct methods using SIR-92,25 and refined against F2 using SHELXL-97, as implemented in WinGX.26
The centrosymmetric space I41/acd was uniquely identified based on systematic absences, and the structure solution in this space group, in all cases, yielded all nine atomic positions, known in the archetype Ca14AlSb11.2 Isotropic full-matrix least-squares refinement of this model, with only one Cd position, converged to reasonable (albeit somewhat high) residual values for all 4 structures within just a few refinement cycles. At this stage, almost all thermal parameters had isotropic values in the range of 0.02–0.03 Å2 × 10−2. Independent refinements of the occupancy factor of each atomic position showed all to be fully occupied (within 5–6σ). Noteworthily, freeing occupancy factors did not lead to improved R1 values, (which for the Sb-compounds in particular, were about 10%), and the high electron density left in the Fourier map (which was above 20 e− Å−3). The extra density was at reasonable distances to heavy atoms, Sb3, Sr1/Eu1 and Sr4/Eu4 being the closest.
All of the above suggested deficiency in the model. In an attempt to account for this electron density, a provisional partially occupied atom was included; for reasons impacting the valence electron count (vide infra), this position was assigned as Cd2 and it was refined with a low occupancy factor (ca. 1/10 of full). The improvement of the residuals and the electron density map was noticeable—the result was 30% lower R1 and wR2 residuals, while the “extra” electron density dropped by ca. 60%. This provided a hint that the “interstitial” site must be intrinsic to the structure, even though the As-compounds show very low occupation at the Cd2 site.
Refinements of the models with the “interstitial” Cd2 included and with anisotropic displacement parameters (ADP) yielded further lowering of the R-values, but not all issues pertaining to the Fourier maps were resolved by treating the atoms anisotropicaly (the magnitudes of the highest peaks and the deepest holes decreased significantly). For illustration, we provide the contour plots of the observed Fourier maps in ESI.† From the plots, it is apparent that the As4/Sb4 position might be subject to a positional disorder. Without modelling the disorder (split positions), the thermal ellipsoids are grossly elongated (see Fig. S1, ESI†). The disorder is somewhat obscure in the Sb-compounds (cigar-shaped ADP), while for the two As-compounds, the dumbbell-shaped ADP clearly pointed at a split site (the 8b site is slightly shifted to 16f, i.e., two-fold split with an occupancy of 0.5/ea). Indeed, when the As4/Sb4 site was split, the refinement confirmed this notion—the distance between the pair of atoms in the split model measures ∼0.4 Å for Sb and ∼0.7 Å for As.
The volumes of the polyhedra and the Voronoi cells were then calculated to investigate the appropriate disordered model adopted by the structure in the Sb-compounds. From the results, discussed later on, one can conclude that the splitting does not impact the volume of the polyhedra and Voronoi cells. Therefore, A14Cd1+xSb11 (A = Eu, Sr) were refined treating Sb4 at its idealized position, while for the As-analogs, the site was split. Where applicable (non-zero occupation above 3σ), the extra Cd2 was also included in the refinements. All calculations converged smoothly to low R1 values and with no significant electron density left in the difference Fourier map. Exception here is the Eu14CdSb11 structure, where there is a peak of 6 e− Å−3 in close proximity of Eu2 (<0.8 Å), the origins of which are still not understood.
The final positional parameters were standardized using the STRUCTURE TIDY program.27 Selected crystallographic data, atomic coordinates and isotropic equivalent displacement parameters, and important interatomic distances are presented in Tables 1–6. Additional information on the crystal structure refinements can be found from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: +49-7247-808-666; Email: crysdata@fiz-karlsruhe.de), on quoting the depository number CSD-429719 (for Sr14CdAs11); CSD-4329720 (for Eu14CdAs11); CSD-429721 (for Sr14CdSb11); and CSD-429722 (for Eu14CdSb11).
| Empirical formula | Sr14Cd1.06(1)As11 | Eu14CdAs11 |
|---|---|---|
| a R 1 = ∑|Fo| − |Fc|/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2, where w = 1/[σ2Fo2 + (AP)2 + BP] and P = (Fo2 + 2Fc2)/3; A and B are weight coefficients. For additional information, refer to the CIF in the ESI. | ||
| Formula weight | 2169.52 | 3063.96 |
| Space group, Z | I41/acd (No. 142), 8 | |
| a (Å) | 16.615(2) | 16.3057(8) |
| c (Å) | 22.321(6) | 21.860(2) |
| V (Å3) | 6162(2) | 5812.0(7) |
| ρ cal (g cm−3) | 4.68 | 7.00 |
| μ (cm−1) | 364.4 | 428.4 |
| Goodness-of-fit on F2 | 1.06 | 0.95 |
| R 1(I > 2σI)a | 0.028 | 0.027 |
| wR2(I > 2σI)a | 0.066 | 0.045 |
| Empirical formula | Sr14Cd1.30(1)Sb11 | Eu14Cd1.27(1)Sb11 |
|---|---|---|
| a R 1 = ∑|Fo| − |Fc|/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2, where w = 1/[σ2Fo2 + (AP)2 + BP] and P = (Fo2 + 2Fc2)/3; A and B are weight coefficients. For additional information, refer to the CIF in the ESI. | ||
| Formula weight | 2712.05 | 3609.72 |
| Space group, Z | I41/acd (No. 142), 8 | |
| a (Å) | 17.616(2) | 17.403(3) |
| c (Å) | 23.203(6) | 22.862(7) |
| V (Å3) | 7200(2) | 6924(3) |
| ρ cal (g cm−3) | 5.00 | 6.93 |
| μ (cm−1) | 293.7 | 340.9 |
| Goodness-of-fit on F2 | 1.04 | 1.10 |
| R 1(I > 2σI)a | 0.029 | 0.034 |
| wR2 (I > 2σI)a | 0.066 | 0.072 |
| Atom | Site | x | y | z |
U
eq.
(Å2) |
|---|---|---|---|---|---|
| a U eq. is defined as one-third of the trace of the orthogonalized Uij tensor. b Occupancy of 1.6(2)%. c As3 experiences a small positional disorder, modelled as a majority site (above, 91%) and a minority site (As3B, x = 0.1602(9), y = 0.0370(6), z = 0.0739(8), 9%). d Split sites (8b to 16f) with an occupancy of 50%. | |||||
| Sr14CdAs11 | |||||
| Sr1 | 32g | 0.04283(4) | 0.07250(4) | 0.17177(3) | 0.0237(2) |
| Sr2 | 32g | 0.02378(4) | 0.37463(4) | 0.00601(3) | 0.0261(2) |
| Sr3 | 16e | 0.35067(4) | 0 | 1/4 | 0.0194(2) |
| Sr4 | 32g | 0.34883(4) | 0.07405(4) | 0.09541(3) | 0.0284(2) |
| Cd1 | 8a | 0 | 1/4 | 3/8 | 0.0197(2) |
| Cd2b | 32g | 0.4673(3) | 0.0181(2) | 0.0828(2) | 0.023(2) |
| As1 | 16f | 0.13431(4) | 0.38431(4) | 1/8 | 0.023(2) |
| As2 | 32g | 0.36136(4) | 0.25449(4) | 0.05714(3) | 0.0189(2) |
| As3c | 32g | 0.1276(8) | 0.02533(5) | 0.04611(4) | 0.0228(4) |
| As4d | 16f | 0.01666(7) | 0.26666(7) | 1/8 | 0.0174(4) |
| Eu14CdAs11 | |||||
| Eu1 | 32g | 0.04393(2) | 0.07268(2) | 0.17149(2) | 0.0115(9) |
| Eu2 | 32g | 0.02365(2) | 0.37581(2) | 0.00433(2) | 0.0129(2) |
| Eu3 | 16e | 0.35087(3) | 0 | 1/4 | 0.0120(2) |
| Eu4 | 32g | 0.34726(3) | 0.07239(2) | 0.09466(2) | 0.0144(1) |
| Cd1 | 8a | 0 | 1/4 | 3/8 | 0.0105(2) |
| As1 | 16f | 0.1328(5) | 0.38283(5) | 1/8 | 0.0127(2) |
| As2 | 32g | 0.3588(5) | 0.25399(5) | 0.05673(3) | 0.0113(2) |
| As3 | 32g | 0.1289(5) | 0.02624(5) | 0.04755(3) | 0.0135(2) |
| As4d | 16f | 0.0133(9) | 0.26328(9) | 1/8 | 0.0103(7) |
| Atom | Site | x | y | z |
U
eq.
(Å2) |
|---|---|---|---|---|---|
| a U eq. is defined as one-third of the trace of the orthogonalized Uij tensor. b Occupancy of 7.5(2)%. c Occupancy of 6.8(3)%, refined isotropically. | |||||
| Sr14CdSb11 | |||||
| Sr1 | 32g | 0.04254(4) | 0.07391(4) | 0.17206(3) | 0.0188(2) |
| Sr2 | 32g | 0.02350(4) | 0.37470(4) | 0.00245(3) | 0.0249(2) |
| Sr3 | 16e | 0.35384(5) | 0 | 1/4 | 0.0129(2) |
| Sr4 | 32g | 0.3434(4) | 0.07215(4) | 0.09414(3) | 0.0236(2) |
| Cd1 | 8a | 0 | 1/4 | 3/8 | 0.0157(3) |
| Cd2b | 32g | 0.4737(3) | 0.0059(3) | 0.0776(3) | 0.0131(2) |
| Sb1 | 16f | 0.13437(2) | 0.38437(2) | 1/8 | 0.0139(2) |
| Sb2 | 32g | 0.35911(2) | 0.25529(2) | 0.06068(2) | 0.0152(2) |
| Sb3 | 32g | 0.12931(3) | 0.02694(3) | 0.04578(2) | 0.0198(2) |
| Sb4 | 8b | 0 | 1/4 | 1/8 | 0.0335(3) |
| Eu14CdSb11 | |||||
| Eu1 | 32g | 0.04302(3) | 0.07384(3) | 0.17185(2) | 0.0174(2) |
| Eu2 | 32g | 0.02292(3) | 0.37498(4) | 0.00142(3) | 0.0237(2) |
| Eu3 | 16e | 0.35437(4) | 0 | 1/4 | 0.0127(2) |
| Eu4 | 32g | 0.34214(4) | 0.07138(3) | 0.09403(2) | 0.0229(2) |
| Cd1 | 8a | 0 | 1/4 | 3/8 | 0.0152(3) |
| Cd2c | 32g | 0.4740(6) | 0.0063(6) | 0.078(4) | 0.009(3) |
| Sb1 | 16f | 0.13401(4) | 0.38401(4) | 1/8 | 0.0133(2) |
| Sb2 | 32g | 0.35773(4) | 0.25492(4) | 0.06036(3) | 0.0148(2) |
| Sb3 | 32g | 0.12978(5) | 0.02680(4) | 0.04565(3) | 0.0183(2) |
| Sb4 | 8b | 0 | 1/4 | 1/8 | 0.027(4) |
| Sr14CdAs11 | Eu14CdAs11 | ||
|---|---|---|---|
| Atom pair | Distance | Atom pair | Distance |
| a As3 experiences a small positional disorder, modelled as a majority site (As3A, 91%) and a minority site (As3B, 9%). | |||
| Sr1–As3A/Ba | 3.213(2)/2.837(1) | Eu1–As3 | 3.111(1) |
| Sr1–As3A/Ba | 3.226(2)/2.982(1) | Eu1–As3 | 3.136(1) |
| Sr1–As1 | 3.204(1) | Eu1–As1 | 3.141(1) |
| Sr1–As2 | 3.189(1) × 2 | Eu1–As2 | 3.114(1) × 2 |
| Sr1–As4 | 3.418(2) | Eu1–As4 | 3.308(2) |
| Sr1–Sr1 | 3.791(2) | Eu1–Eu1 | 3.689(1) |
| Sr1–Sr3 | 3.833(2) | Eu1–Eu3 | 3.776(1) |
| Sr1–Sr4 | 3.625(1) | Eu1–Eu4 | 3.543(1) |
| Sr2–As1 | 3.233(1) | Eu2–As1 | 3.184(1) |
| Sr2–As2 | 3.101(1) | Eu2–As2 | 3.066(1) |
| Sr2–As3Aa | 3.147(2) | Eu2–As3 | 3.103(1) |
| Sr2–As3Aa | 3.257(1) | Eu2–As3 | 3.201(1) |
| Sr2–As4 | 3.208(2) | Eu2–As4 | 3.217(1) |
| Sr2–Sr1 | 3.953(1) | Eu2–Eu1 | 3.908(1) × 2 |
| Sr2–Sr3 | 3.783(1) | Eu2–Eu3 | 3.711(1) |
| Sr2–Sr4 | 3.782(2) | Eu2–Eu4 | 3.697(1) |
| Sr3–As3A/Ba | 3.093(1)/3.197(1) × 2 | Eu3–As3 | 3.046(1) × 2 |
| Sr3–As1 | 3.397(1) × 2 | Eu3–As1 | 3.345(1) × 2 |
| Sr3–As2 | 3.293(1) × 2 | Eu3–As2 | 3.207(1) × 2 |
| Sr3–Sr4 | 3.664(1) × 2 | Eu3–Eu4 | 3.595(1) × 2 |
| Sr4–As3Aa | 3.283(1) × 2 | Eu4–As3 | 3.222(1) × 2 |
| Sr4–As1 | 3.233(2) | Eu4–As1 | 3.178(1) |
| Sr4–As2 | 3.124(1) | Eu4–As2 | 3.081(1) |
| Sr4–As2 | 3.325(1) | Eu4–As2 | 3.299(1) |
| Cd1–As2 | 2.758(1) × 4 | Cd1–As2 | 2.745(1) × 4 |
| Cd1–Sr2 | 3.605(1) × 4 | Cd1–Eu2 | 3.514(1) × 4 |
| As1–As4 | 2.764(1) | As1–As4 | 2.757(2) |
| As3B–As3Ba | 2.552(3) | ||
| Sr14CdSb11 | Eu14CdSb11 | ||
|---|---|---|---|
| Atom pair | Distance | Atom pair | Distance |
| a Occupancy of 7.5(2)%. b Occupancy of 6.8(3)%. | |||
| Sr1–Sb3 | 3.369(1) | Eu1–Sb3 | 3.322(2) |
| Sr1–Sb3 | 3.407(1) | Eu1–Sb3 | 3.358(2) |
| Sr1–Sb1 | 3.383(1) | Eu1–Sb1 | 3.343(1) |
| Sr1–Sb2 | 3.382(2) × 2 | Eu1–Sb2 | 3.327(2) × 2 |
| Sr1–Sb4 | 3.373(1) | Eu1–Sb4 | 3.333(1) |
| Sr1–Sr1 | 3.980(2) | Eu1–Eu1 | 3.915(2) |
| Sr1–Sr3 | 4.002(1) | Eu1–Eu3 | 3.952(1) |
| Sr1–Sr4 | 3.809(2) | Eu1–Eu4 | 3.747(2) |
| Sr2–Sb1 | 3.454(1) | Eu2–Sb1 | 3.427(1) |
| Sr2–Sb2 | 3.293(1) | Eu2–Sb2 | 3.267(1) |
| Sr2–Sb3 | 3.355(1) | Eu2–Sb3 | 3.317(2) |
| Sr2–Sb3 | 3.452(1) | Eu2–Sb3 | 3.405(2) |
| Sr2–Sb4 | 3.617(1) | Eu2–Sb4 | 3.588(1) |
| Sr2–Sr1 | 4.186(2) | Eu2–Eu1 | 4.137(1) |
| Sr2–Sr3 | 4.008(1) | Eu2–Eu3 | 3.968(1) |
| Sr2–Sr4 | 3.995(2) | Eu2–Eu4 | 3.941(2) |
| Sr3–Sb3 | 3.310(1) × 2 | Eu3–Sb3 | 3.268(1) × 2 |
| Sr3–Sb1 | 3.550(1) × 2 | Eu3–Sb1 | 3.505(1) × 2 |
| Sr3–Sb2 | 3.440(1) × 2 | Eu3–Sb2 | 3.379(1) × 2 |
| Sr3–Sr4 | 3.838(2) × 2 | Eu3–Eu4 | 3.782(2) × 2 |
| Sr4–Sb3 | 3.428(1) × 2 | Eu4–Sb3 | 3.372(2) × 2 |
| Sr4–Sb1 | 3.407(1) | Eu4–Sb1 | 3.363(2) |
| Sr4–Sb2 | 3.329(1) | Eu4–Sb2 | 3.297(1) |
| Sr4–Sb2 | 3.522(1) | Eu4–Sb2 | 3.502(1) |
| Cd1–Sb2 | 2.898(1) × 4 | Cd1–Sb2 | 2.885(1) × 4 |
| Cd1–Sr2 | 3.707(1) × 4 | Cd1–Eu2 | 3.639(1) × 4 |
| Cd2a–Sb1 | 3.068(6) | Cd2b–Sb1 | 3.035(10) |
| Cd2a–Sb2 | 3.187(6) | Cd2b–Sb2 | 3.124(10) |
| Cd2a–Sb3 | 2.898(6) | Cd2b–Sb3 | 2.869(10) |
| Cd2a–Sb3 | 3.410(1) | Cd2b–Sb3 | 3.375(10) |
| Cd2a–Sr1 | 2.872(6) | Cd2b–Eu1 | 2.826(10) |
| Cd2a–Sr2 | 2.896(7) | Cd2b–Eu2 | 2.879(10) |
| Cd2a–Sr2 | 2.940(6) | Cd2b–Eu2 | 2.881(10) |
| Cd2a–Sr4 | 2.603(6) | Cd2b–Eu4 | 2.585(10) |
2.3 Property measurements
Temperature dependent electrical resistivity was measured from 5 to 300 K with an excitation current of 10 mA using a Quantum Design PPMS. Four platinum wires were connected to single crystals of Eu14CdAs11 using EPO TEK H20 silver epoxy. The measurements were carried out on both heating and cooling for two different crystals to ensure reproducibility.Field-cooled direct current (dc) magnetization (M) in the temperature range of 2–300 K and an applied field of 500 Oe (H) was measured also using the PPMS. The measurements were done for two different batches to ensure reproducibility. Several flux-grown Eu14CdAs11 crystals were collected and enclosed in gel-capsules with the top filled with cotton to prevent the crystals from moving under the magnetic field. The raw magnetization data were converted to molar susceptibility (χm = M/H). The effective magnetic moment (μeff) and Weiss temperature (θp) were calculated from a linear fit of the inverse magnetic susceptibility versus temperature.
2.4 Computational details
The electronic structure calculations for idealized Sr14CdSb11, i.e., neglecting the disorder, were performed using the linear muffin-tin orbital (TB-LMTO) method28 within the Stuttgart TB-LMTO 4.7 program.29 The experimental unit cell parameters and atomic coordinates were employed for the calculation. The program package employs the atomic sphere approximation (ASA), in which space is filled with overlapping Wigner–Seitz (WS) atomic spheres.30 Exchange and correlation were treated by the local density approximation (LDA).31 All relativistic effects except spin–orbit coupling were taken into account by using a scalar relativistic approximation.32 In the ASA method, the symmetry of the potential is considered spherical inside each WS sphere, and a combined correction is used to take into account the overlapping part. The radii of WS spheres were obtained by requiring that the overlapping potential be the best possible approximation to the full potential, and were determined by an automatic procedure. This overlap should not be too large because the error in kinetic energy introduced by the combined correction is proportional to the fourth power of the relative sphere overlap. No empty spheres were used. The following WS radii—Sr = 2.06–2.08 Å, Cd = 1.65 Å, Sb = 1.81–1.98 Å—were used for the calculation. The basis sets included 5d, 6s, and (6p) orbitals for Sr; 4d, 5s, and 5p orbitals for Cd; 5s, 5p, and (5d) orbitals for Sb. The downfolded orbitals (in parentheses) were treated by the Löwdin downfolding technique.33 The Cd 4d orbitals were treated as core wave functions. For bonding analysis, the energy contributions of all filled electronic states for selected atom pairs were calculated by the crystal orbital Hamilton population (COHP) method.34 The k-space integrations were conducted by the tetrahedron method,35 and the self-consistent charge density was obtained using consistent irreducible k-points in the Brillouin zone.3 Results and discussion
3.1 Crystal chemistry
During our continuous effort to provide insight into the interplay between the size and electronic effects in the “A9M4+xPn9” family18 (A = Ca, Sr, Eu, or Yb; Pn = Sb, Bi; and M = Mn, Zn, Cd), we serendipitously (re)discovered Sr14Cd1.30(1)Sb11. This structure is the very same one which the Kauzlarich group identified as Sr14CdSb11.37,12 with the difference being the interpretation of the additional interstitial atoms. We assigned the interstitial atoms as Cd, much like M is filled in A9M4+xPn9, whereas the original report on the Sr–Cd–Sb phase assumed the atom at the interstitial site to be antimony.12 Since this structure is familiar, the following discussion will focus primarily on an alternative description and the arguments in favor of the notion that from both geometric and electronic standpoints, Cd2+ will be preferred over Sb3− as an interstitial species.The stoichiometric and non-stoichiometric compounds in the series A14Cd1+xPn11 (A = Sr, Eu; Pn = As, Sb) adopt the Ca14AlSb11 structure type2 which crystallizes in the tetragonal unit cell with space group I41/acd (Pearson code tI208).36 The structure of these compounds is not simple, but can be readily visualized as made of [CdPn4] tetrahedra, linear Pn3-units and isolated Pn atoms as shown in Fig. 1 and 2. Based on the split of the As4 site in particular, as described above, and as shown in Fig. S1 (ESI†), one can argue that the structure of A14CdAs11 (A = Sr, Eu) does not have trimeric As3, and instead features an As2-dimer and an isolated As ion. This split atomic position is less apparent in A14Cd1+xSb11 (A = Sr, Eu)—recall that the distance between the split sites is less than 0.4 Å for the antimonides and over 0.7 Å for the As-analogs.
 | ||
| Fig. 1 (A) A representation of the structure of Sr14CdAs11 (Sr atoms omitted for clarity) showing [CdAs4] tetrahedra, isolated As atoms and the linear hypervalent [As3]7− anion. (B) Cutouts of a ball and stick representation (top) and observed Fourier electron density (bottom) from the refined atomic positions of the hypervalent anion in the structure of Sr14CdAs11 (see Fig. S1 in ESI†). (C) Positional disorder involving the isolated As3 atom. Taking the disorder into consideration, a possible formation of As2-dumbbell can be envisioned. (D) Coordination polyhedra of As1 and As4 atoms of the hypervalent anion (top); coordination polyhedron of Cd1 atoms (bottom). | ||
 | ||
| Fig. 2 (A) A projection of the crystal structure of Sr14CdSb11 viewed approx. along the a-axis showing a framework of [CdSb4] tetrahedra, isolated Sb atoms and the linear hypervalent [Sb3]7− anion. (B) Isolated Cd1 tetrahedra. (C) Framework of edge- and corner-sharing [Cd(2)Sb4] tetrahedra. The Sr atoms are omitted for clarity. | ||
The average As–As distance in the dumbbell measures 2.761(1) Å and compares well with similar distances in Ca14GaAs118 and Eu14MnAs11.37 However, these homoatomic interactions appear to be longer than the sum of the covalent radii between As atoms (2.42 Å),38 or other previously reported compounds, Ba2Cd2As3 (2.49 Å)39 and Ba4Li2Cd3As6 (2.51 Å).40 The As4 and As1, which are atoms of the dumbbell, are located near the center of a tri-capped trigonal prism coordinated with eight Sr or Eu atoms and one As atom (Fig. 1D). The As–A (A = Sr, Eu) distances within these polyhedra are distributed between 3.204(1) and 3.397(1) Å (average 3.30(1) Å) for Sr, and between 3.141(1) and 3.345(1) Å (average 3.243(1) Å) for Eu. These distances are of the same order with similar contacts as in other previously reported compounds.12,17,37
Having discussed the bonding interactions within the As1 and As4 polyhedra, we turn now our attention to the Sb1 and Sb4 local environments. Recall that these two atoms form the [Sb3]7− unit. Sb1 and Sb4 atoms are located at the centers of polyhedra of ten and eleven Sr and Cd neighbors, as shown in Fig. 3. The Sb1–Sb4 bond in the trimer is very long (3.34 Å), which is not typical for the Sb–Sb single bond (expected value ca. 2.8 Å).38 The Sb1–A (A = Sr, Eu) contacts range from 3.383(1) to 3.550(1) Å and from 3.343(1) to 3.505(1) Å for the Sr- and the Eu-analog, respectively. The Sb1–Cd2 bonds measure 3.068(6) Å and 3.035(10) Å in Sr14CdSb11 and Eu14CdSb11, respectively. These bonds match the expected heteroatomic single-bond value (2.9 Å).38 The Sb4–Sr and Sb4–Eu distances fall in the range 3.373(1)–3.617(1) Å and 3.333(1)–3.588(1) Å, respectively.
 | ||
| Fig. 3 (A) Cutouts of a ball and stick representation (top) and observed Fourier electron density (bottom) from the refined atomic positions of the hypervalent anion in the structure of Sr14CdSb11 (see details in Fig. S1 in ESI†). (B) Coordination polyhedra of Sb1 and Sb4 atoms of the hypervalent anion. (C) Coordination polyhedron of Cd1 and Cd2 atoms (bottom). (D) Contour plot scaled in 8 × 8 Å of the observed Fourier electron density (3–6 e− Å−3) from the refined positions of Sr atoms around Cd2. | ||
The coordination polyhedra of the two types of Cd atoms are worthy of a special mention too. One should note here that we included exclusively atoms of the first coordination sphere involving only covalent bonds. Under this approximation, the Cd atoms form a polyhedron consisting of an inner [CdPn4] and an outer [CdA4] tetrahedron (Fig. 1D and 3C). This polyhedron bears resemblance to the tetrahedral unit substructure of the gamma-brass cluster (Fig. 4A), which is a structural building block in many intermetallic compounds.41–45 This tetrahedral unit is also known as “tetraederstern”.41 Let us recall that in the gamma-brass cluster, four interpenetrating icosahedra (each centered on the atom of the inner tetrahedron) share a common tetrahedron. Yet, the icosahedra of the outer tetrahedron offer a different geometrical configuration—they share face with each other and with a central tetrahedron. Such atomic arrangement is known as augmented gamma-brass or Pearce cluster.41,46–50 It is important to note that the bond distance between the Zn atoms (2.7 Å) in this subunit of the gamma-brass cluster compares well with the homoatomic single-bond (2.66 Å).38 Importantly, those clusters are well known to have the ability to host many interstitial atoms, particularly light elements such as C, H, O.
 | ||
| Fig. 4 Alternative description of the crystal structure of Sr14CdSb11 using a small set of coordination polyhedra of the electropositive metal atoms and taking advantage of the resemblance of the Cd polyhedra (A, right) to the “tetraederstern” substructure of the gamma-brass cluster (A, left). (B) First coordination sphere of Cd atoms forming a filled distorted “tetraederstern” consisting of an inner [CdSb4] and an outer [CdSr4] tetrahedron. (C) A projection of the structure of Sr14CdSb11 regarded as a space-filling packing of first coordination spheres of Sr atoms forming distorted octahedra and trigonal bipyramids shown in (D). (E) A complex packing showing how the inner [CdSb4] tetrahedron shares faces with distorted octahedra and trigonal bipyramids of Sr atoms. Two such polyhedra are omitted for the sake of clarity. Not only does this packing help to study the interplay between the size of the “free volume” to accommodate the excess Cd atoms (Cd2) and the instability of the linear hypervalent, but also to explain the relatively short cation–cation contacts in the structure with respect to the Pauling's third rule. | ||
Let us now discuss the bonding interactions within the Cd polyhedron. First, we should note that there is no close contact between the As or Sb atoms within the sphere of the inner tetrahedron. Their homoatomic distances measure 4.44 and 4.61 Å, respectively, which is almost the double of their corresponding single-bond distance.38 The average Cd1–As2 and Cd1–Sr2/Eu2 distances are 2.750(1) Å and 3.605(1) Å for Sr14CdAs11, and 2.745(1) Å and 3.595(1) Å for Eu14CdAs11, respectively. Similarly, the average Cd2–Sb distances are 3.239(5) Å in Sr14CdSb11 and 3.205(10) Å in Eu14CdSb11, respectively. The average Cd2–Sr and Cd2–Eu distances, however, are unreasonably short, ca. 2.7 Å, which must be an artefact of the very low occupancy of the Cd2 position.¶ Apparently, the distances involving Cd2 cannot be established reliably by single-crystal X-ray diffraction, as demonstrated previously on the examples of A9M4+xPn9, where A denotes Ca, Sr, Eu, or Yb and Pn = Sb, Bi,18 which are structurally related in terms of containing additional M-atoms (M = Mn, Zn or Cd) at the interstitial sites. It was therefore imperative to look for an alternative description of this structure in order to shed some light on the origin of those bonding interactions.
If we consider the [CdPn4] and [CdA4] tetrahedra (Fig. 4B) and the first coordination sphere polyhedra of the A2+ cations shown in Fig. 4D—a distorted [APn6] octahedron, [APn5] trigonal bipyramid—the structure can be described in a different way. This structural description model is not only based on a simple geometrical arrangement of atoms, which, of course, is essential to understanding (or visualizing) crystal structures and structural relationships, but also takes into account the covalent bonding interaction. The structure can then be regarded as space-filling packing of distorted [APn6] octahedra and [APn5] trigonal bipyramids. The Cd atoms are located in the interstitial sites forming a complex unit of inner [CdPn4] and outer [CdA4] tetrahedra, as depicted in Fig. 4C.
The [Cd(1)Pn4] tetrahedra (Fig. 1D and 3C) share edges and corners with the [APn5] (A = Eu, Sr) trigonal bipyramid (Fig. 4D and Fig. S3, ESI†), whereas the [Cd(2)Pn4] tetrahedra share edges with distorted [APn6] octahedra and [APn5] trigonal bipyramids (Fig. 4D and Fig. S3, ESI†). This structural topology of face and edge sharing polyhedra highlighted in Fig. 4E helps to understand the origin of the robust cation–cation bonds. After all, these findings seem to be in accordance with the Pauling's third rule.38b As we noted earlier, such interactions are not without precedent and were already observed in other previously reported compounds such as A9Zn4+xPn9 and A9Cd4+xPn9,18,51 where A denotes Ca, Sr, Eu, or Yb and Pn = Sb, Bi, Yb9Mn4+xPn9,52 and Ca9Mn4+xSb9,53 among others. Of course, these structures can also be regarded as space-filling arrangement of distorted [APn6] octahedra and [APn5] square pyramids. An illustration is shown in ESI† (Fig. S4 and S5). The Cd atoms occupy the interstitial sites forming distorted [CdBi4] tetrahedra sharing edges and faces with the neighboring distorted [CaBi6] octahedra and [CaBi5] square pyramids. Finally, it is important to note that the structure of the A9M4+xPn9 family offers more structural flexibility to accommodate new cationic species leading to stuffed variant compounds as evidenced by the recent report of Kazem et al.54 In the next sections we will attempt to gain further insights into two main questions: (1) why the interstitial chemistry in A14M1+xPn11 appears to be limited to the Sb-compounds, while the As-analogs show very little or no occupation at the interstitial site (but show different types of structural distortions/variances)? and (2) given the very low electron density at the interstitial position and the lack of conclusive evidence from the interatomic distances,¶ would the formula A14M1+xPn11 (i.e., interstitial site filled by Cd2+) or A14MPn11+x12 (i.e., assign Sb3− to the interstitial site) be more reasonable?
From simple structural considerations, as illustrated above, it seems reasonable that the geometric factors might have more implication in the formation of these nonstoichiometric compounds. In order to provide rigorous support to this argument, it will be instructing to evaluate how the volume of the unit cell should be divided up between the different constituents, Cd, Pn and A atoms (Pn = As, Sb; A = Eu, Sr), of the structure. A hard sphere approach can be immediately rejected since it accounts for less than 50% of the unit cell volume. A more reasonable method used to evaluate the effective atomic volume is based on the Voronoi cell approach.|| This method55 has been successfully used as a standard method for (1) the calculation of volumes of protein constituents, the description of protein motions, and the analysis of cavities in proteins,56 (2) for climate system and global modeling,57 (3) the analysis of computational molecular simulations,58 and (4) for allocation of space among atoms.59 In this study, the volume of Voronoi domains (or cell) and that of coordination polyhedra were accessed employing the NRCVAX software package.60
The calculated volumes of coordination polyhedra and the Voronoi cell are presented in Table 7 for some selected atoms. The results are quite consistent with the expected trends, and more specifically, for the Cd2 atoms, the volume of the eight-coordinated polyhedron can be seen to decrease slightly from 73.75 to 70.87 Å3 and that of the Voronoi cell from 22.22 to 21.39 Å3 with the decrease in the size of the cations in Sr14CdSb11 (Sr2+ with r = 1.18 Å)61 and Eu14CdSb11 (Eu2+ with r = 1.17 Å),61 respectively. In contrast, when considering Sr14CdSb11 and Sr14CdAs11, the volume of the coordination polyhedron drastically decreases from 73.75 Å3 to 56.43 Å3, and the Voronoi cell volume decreases from 22.22 Å3 to 19.40 Å3 with the decrease in the size of the pnictogen atom. It should be noted that the Cd2 position was found empty (i.e., 0% occupancy) in Eu14CdAs11 meaning that there is insufficient volume to accommodate even a small fraction of Cd2 atoms, as seen for the other three compounds. This supports the argument that the filling of the interstitial site in this structure is very sensitive to the size of the pnictogen atoms and to some extent the counter-cation (Eu, Sr). We should also mention here that these arguments call for additional structural work, and perhaps, for the revision of some older results—notice that Zn and Mg have similar ionic size, Zn2+ (r = 0.60 Å); Mg2+ (r = 0.57 Å),61 yet variable stoichiometry is only reported for the Ca14MSb11 compound with M = Zn, while the Ca14MSb11 compound with M = Mg displays no additional interstitial atom. One will also notice that Yb14ZnSb11 is reported to be stoichiometric (albeit Yb2+/Yb3+ mixed-valent),1f while its Ca-counterpart, despite the virtually identical radii for Ca2+ (r = 1.01 Å)61 and Yb2+ (r = 1.00 Å),61 is sub-stoichiometric. On the other hand, the pair Yb14MgSb11 and Yb14ZnSb11 is reportedly isotypic, but not isoelectronic, as only the latter is mixed-valent.
| Atom | Volume | Sr14CdAs11 | Eu14CdAs11 | Sr14CdSb11 | Eu14CdSb11 |
|---|---|---|---|---|---|
| a Taking into account the possible split position. | |||||
| Cd1 | CP | 99.48 | 95.32 | 116.30 | 112.70 |
| CN8 | VC | 25.07 | 24.12 | 28.50 | 27.67 |
| Cd2 | CP | 56.43 | 73.75 | 70.87 | |
| CN8 | VC | 19.40 | 22.22 | 21.39 | |
| As1 | CP | 67.32 | 64.20 | ||
| CN9 | VC | 26.43 | 25.15 | ||
| As4 | CP | 77.50 | 73.96 | ||
| CN9 | VC | 26.23 | 25.05 | ||
| Sb1 | CP | 86.95 | 84.10 | ||
| CN11 | VC | 28.56 | 27.55 | ||
| Sb4 | CP | 92.22 | 89.33 | ||
| 92.22a | 89.33a | ||||
| CN10 | VC | 31.44 | 30.42 | ||
| 31.36a | 30.37a | ||||
One can also notice that the [CdPn4] tetrahedra appear more distorted for the Sb-compounds than in their As-analogs. For example, the largest Pn–Cd–Pn angle in Sr14Cd1.06(1)As11 is 113.4°, while the corresponding Sb–Cd–Sb angle is 118.0° (in the antimonide with the same cation, i.e., in Sr14Cd1.30(1)Sb11). These differences can be linked to small structural distortions in response to the occupation of the available “free volume” with a small fraction of Cd atoms. This seems to be a natural response of the structure to optimize both the crystal packing and the Cd–Sb covalent bonding. A remarkably similar effect was recently reported for the series RELixSn2 (RE = La–Nd)62 in which it was found that the decrease of Li content in the RESn2 sub-structure (i.e., trigonal prism cavities) was due to the limited available “free volume” within these cavities as a result of the lanthanide contraction. In contrast, the structural analysis of the series Ba4Cd3Li2Pn6 (Pn = P, As, Sb)40 recently reported by us revealed structural vacancies at the Cd and Li sub-lattice promoting thereby an interplay between ionicity and covalency among the electronegative components.
In summary, we have shown that the size of the pnictogen and highly electropositive metal atoms is an important factor for the formation of non-stoichiometric compositions. One should remember that, in principle, the occupation of a given empty position will be determined by the match between the volume of the guest atom and the site volume. This holds true also when contact areas are greater between adjacent atoms of elements that bind strongly with each other. On the basis of volume calculations, one can argue for instance that the Voronoi cell volume of Cd2+ (0.78 Å)61 in Sr14CdAs11 and A14CdSb11 (A = Eu, Sr) is clearly at its maximum. Therefore, filling the interstitial site with Sb3− (much larger than Cd2+), as it was assigned in an early report for A14CdSb11+x (A = Ca, Sr),12 seems not appropriate because these atoms will not fill all the available space. This suggests that Cd2+ should be preferred to Sb3− as a “filler” because of the space requirement and the minimization of the electrostatic effects, which is consistent with cumulative size and electronic effects recently reported for in Sr9Cd4.49Sb9.18 On the other hand, an accumulation of Sb3− may render the [SbSb4] tetrahedron unstable because of the electrostatic effects, which need to be minimized in order for the crystal to achieve minimum energy. All of the above raise concerns about the nature of chemical bonding involving the interstitial atoms.
3.2 On the electronic structure and the origin of interstitial solid solutions
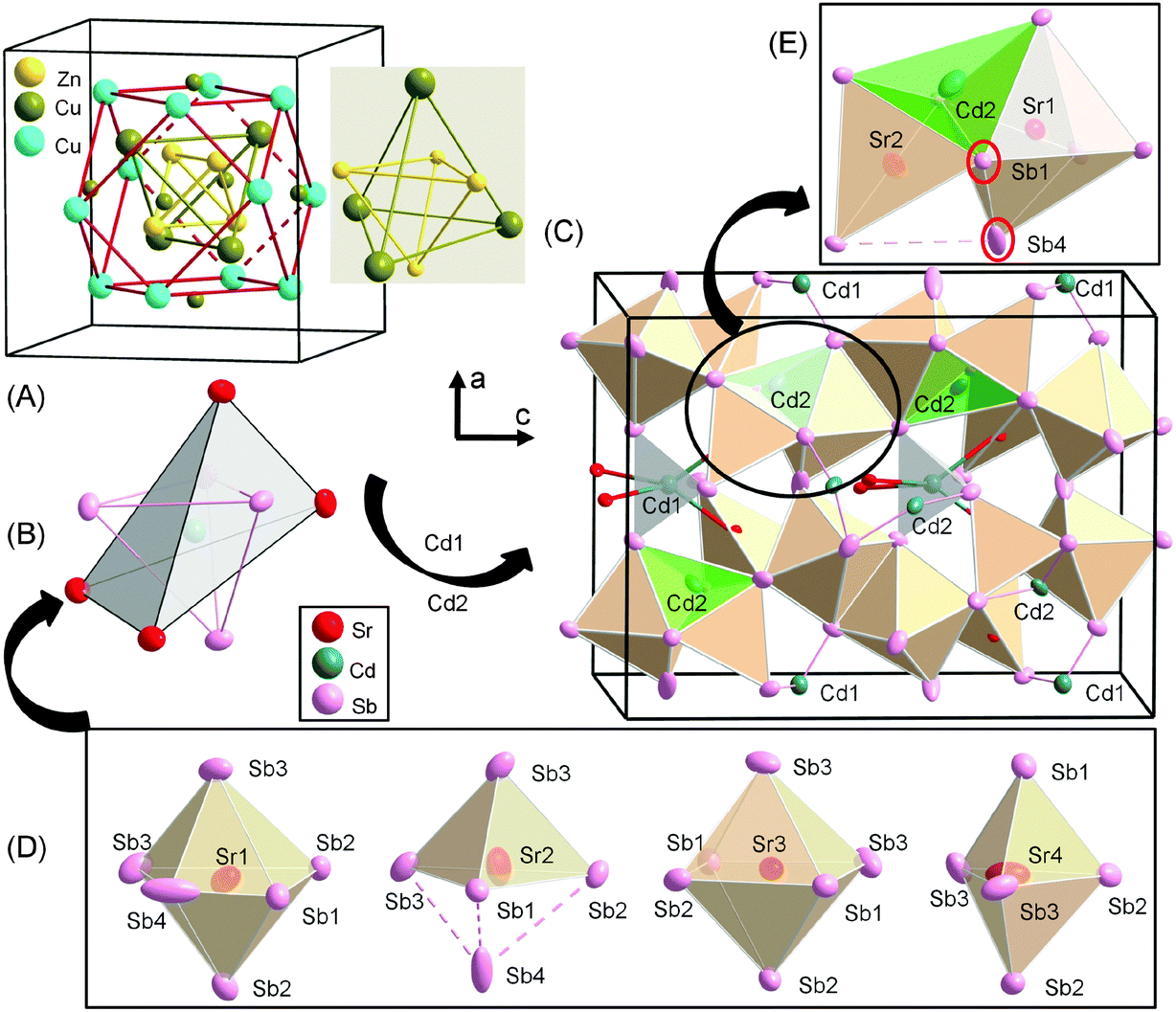
According to the Zintl concept,3 and following the valence electron count in Ca14AlSb11,2 the overall formula unit of idealized Sr14CdSb11 (neglecting the partially occupied interstitial) can be partitioned in four different entities as follows: 14x[Sr2+] + [CdSb4]10− + [Sb3]7− + 4x[Sb]3−. Such electron counting reveals a charge-imbalance, since there is one missing positive charge to counterbalance the 29 electrons needed for covalent bonding within the anionic substructure. The electronic structure calculations (Fig. 5) for stoichiometric Sr14CdSb11 indicate that such structure will not be electronically stable since the Fermi level is at a substantial density of states (DOS) level, while it appears that the top of the valence band could be shifted up (by virtue of filling an extra electron) to the edge of an energy gap. A similar electronic structure was also reported for the isostructural and isoelectronic Ca14MgSb11.11 Consequently, Sr14CdSb11 will not have fully optimized bonding and will be expected to be (semi)metallic (or degenerate semiconductor due to intrinsic disorder). The Fermi level for the disorder-free Sr14CdSb11 falls within the top of the valence band in a region of relatively high DOS (Fig. 5). The corresponding number of valence electrons is 85 per formula unit (14 × 2 provided by the Sr atoms, 1 × 2 provided by the Cd atom, and 11 × 5 provided by the Sb atoms). The full and partial DOS diagrams suggest that increasing the number of electrons to 86 per formula will lead to an optimal number of valence electrons. This valence electron count can come about by the addition of exactly one-half Cd atom, i.e., Sr14Cd1.5Sb11. This also means that Sr14Cd1+xSb11 (x ≈ 0.3) will be slightly electron-deficient, but very close to being a valence-precise compound.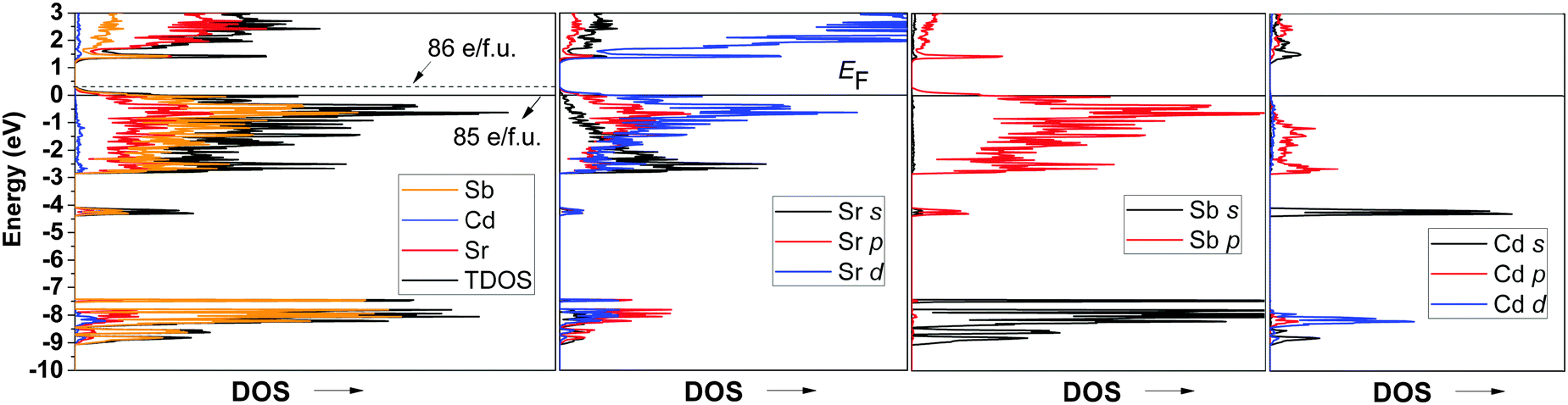 | ||
| Fig. 5 Calculated total density of states (DOS) and partial density of states (PDOS) curves for the idealized compound Sr14CdSb11. The Fermi level is the energy reference at 0 eV. The three PDOS plots show the s, p, and d bands contribution for Sr, Sb (s and p bands), and Cd. | ||
The partial DOS for Cd and Sb (Fig. 5) shows that adding excess Cd will reduce more Sb, which in turn will push the Fermi level up towards the edge of the valence band. Notice as well that according to the partial DOS of the Sr atoms, this shift might also require a slight doping (i.e., dumping electrons) at the Sr sub-lattice. In either case, covalent bonding, both Cd–Sb and Sr–Sb, is optimized, as evident from the COHP diagram depicted in Fig. 6.
 | ||
| Fig. 6 Calculated total COHP curves for the average Sb–Sb, Cd–Sb, and Sr–Sb interactions in Sr14CdSb11. Since –COHP is plotted, the positive and negative signs represent bonding and antibonding states, respectively. | ||
According to the DOS diagrams (Fig. 5), increasing the number of electrons to 86 per formula pushes the Fermi level toward the edge of the valence band. This means that if one were to obtain A14Cd1.5Sb11, an intrinsic semiconductor material should be realized; any other composition will be close, but will not correspond to a true valence compound. Clear support of the notion that A14Cd1+xPn11 (x ≪ 0.5) is not a semiconductor is obtained from the metallic electrical resistivity of Eu14Cd1.06(1)As11 (Fig. 7—note that the measurement was performed on a very small single-crystal). Let us recall that the single-crystals of Eu14Cd1.06(1)As11 were grown from a flux reaction, and although the ideal composition Eu14Cd1.5As11 was intended, it could not be attained. This means that using the Zintl–Klemm concept, one could attempt other dopants to sway the structure–property relationships in A14Cd1+xPn11 in a targeted fashion towards obtaining more or less conductive material.
 | ||
| Fig. 7 Resistivity versus temperature of Eu14CdAs11. | ||
3.3 Magnetic susceptibility
The temperature dependent molar magnetic susceptibility χ and the inverse susceptibility 1/χ of Eu14CdAs11 are shown in Fig. 8. Eu14CdSb11 was not phase-pure and could not be measured. The magnetic susceptibility gradually increases upon cooling to approximately 30 K. As the temperature decreases further, the susceptibility exponentially increases up to around 8 K, at which point a sharp downturn is observed. This steep increase in magnetic susceptibility can be tentatively ascribed to a ferromagnetic (FM) ordering transition with a Curie temperature TC = 30 K (obtained from the mid-point of the first derivative plot of χ vs. T). The second feature in the magnetic susceptibility data could be attributed to a successive ordering transition, which is likely antiferromagnetic (AFM). There is only limited data gathered below the speculated AFM transition, but we could argue that the χ vs. T seems to be mirroring the susceptibility data for the isotypic Eu14MnP11 and Eu14MnAs11,37 which also show similar magnetic transition. The high-temperature regime of the inverse susceptibility can be fitted to the Curie–Weiss law, χ = C/(T − θp). The fit results in an effective magnetic moment, μeff = 28.0 μB/f.u. (μeff = 7.5 μB/Eu), and a paramagnetic Weiss temperature, θp = 13 K. | ||
| Fig. 8 Field-cooled magnetic susceptibility versus temperature of Eu14CdAs11 (data normalized per Eu). The plot shown in the inset is the temperature dependence of the inverse magnetic susceptibility and the linear fit to the Curie–Weiss law. | ||
The calculated effective magnetic moment is very close to the expected theoretical value (7.94 μB) of Eu2+ free-ions, and is consistent with the previously reported results for Eu14MnAs11.37 The magnetic ordering of the Eu spins confirms that the atoms participate in “clustering”, which is consistent with our structural description of a “tetraederstern” cluster consisting of inner CdAs4 and outer CdEu4 tetrahedra as described above. The positive value of θp agrees with the presumed ferromagnetic ordering, and one can notice that θp for Eu14CdAs11 is lower than the one obtained for Eu14MnAs11 (θp = 71 K).37 This may be suggestive of weakened ferromagnetic Eu–Eu interactions in Eu14CdAs11, due to the smaller unit cell parameters of Eu14MnAs11, and correspondingly, shorter Eu–Eu contacts. Of course, it is also possible that Eu14CdAs11 lacks the Mn-sublattice, which might synergistically mediate the Eu–Eu interactions.
Such complex packing can result in a cooperative ferromagnetic order due to geometrical spin frustration in the magnetic subunit. More electronic transport properties and magnetic data are needed to better understand the origin of the ferromagnetism and antiferromagnetism in this material.
4 Conclusions
In this study, we have reported on the synthesis and structural characterization of three new compounds, isostructural, but not isoelectronic with the thermoelectric material Yb14MnSb11. They are Eu14CdAs11, Sr14Cd1.06(1)As11, and Eu14Cd1.27(1)Sb11. The structure of Sr14Cd1.30(1)Sb11 could be considered as a re-assessment of the previously reported Sr14CdSb11.37.12 Both antimonides display a conspicuous feature of this structure—the presence of additional interstitial Cd atoms. The corresponding arsenides show lesser tendency to be stabilized interstitially, and their structures show the signatures of positional disorder on at least one of the As sites. By judiciously choosing the counter-cations (Eu and Sr) and the two pnictogen atoms (As and Sb), we showed using the Voronoi cell volume that the “free volume” is greater in the antimonides than in the corresponding arsenide counterparts. In turn, the greater available space can be correlated with the higher fraction of Cd atoms filling that site. The robust cation–cation bonds observed in the structure might be a natural response of the electronic structure to compensate for the apparent electron deficiency and in order to optimize the bonding interactions. The electronic structure calculations for Sr14CdSb11 showed that the Fermi level falls near the edge of the valence band, which agrees well with the metallic-like conductivity. The thermoelectric properties of the air stable compounds warrant to be investigated since the additional interstitial metal atoms could regulate efficiently both the electronic and thermal transport properties.Acknowledgements
Svilen Bobev acknowledges financial support from the US DOE (Basic Energy Sciences) through a grant DE-SC0008885. The authors are indebted to Dr. N.-T. Suen for his help with the electronic structure calculations.Notes and references
- (a) S. M. Kauzlarich, in Chemistry, Structure, and Bonding of Zintl Phases and Ions, ed. S. M. Kauzlarich, VCH Publishers, New York, 1996, p. 245 Search PubMed; (b) J. Y. Chan, M. M. Olmstead, S. M. Kauzlarich and D. J. Webb, Chem. Mater., 1998, 10, 3583 CrossRef CAS; (c) J. Y. Chan, M. E. Wang, A. Rehr and S. M. Kauzlarich, Chem. Mater., 1997, 9, 2131 CrossRef CAS; (d) J. Y. Chan, S. M. Kauzlarich, P. Klavins, R. N. Shelton and D. J. Webb, Chem. Mater., 1997, 9, 3132 CrossRef CAS; (e) J. Y. Chan, S. M. Kauzlarich, P. Klavins, R. N. Shelton and D. J. Webb, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 8103 CrossRef; (f) I. R. Fisher, S. L. Bud'ko, C. Song, P. C. Canfield, T. C. Ozawa and S. M. Kauzlarich, Phys. Rev. Lett., 2000, 85, 1120 CrossRef CAS; (g) R. F. Gallup, C. Y. Fong and S. M. Kauzlarich, Inorg. Chem., 1992, 31, 115 CrossRef CAS.
- G. Cordier, H. Schäfer and M. Stelter, Z. Anorg. Allg. Chem., 1984, 519, 183 CrossRef CAS PubMed.
- (a) E. Zintl, Angew. Chem., 1939, 52, 1 CrossRef CAS PubMed; (b) H. Schäfer, B. Eisenmann and W. Müller, Angew. Chem., Int. Ed. Engl., 1973, 12, 694 CrossRef PubMed; (c) H. Schäfer and B. Eisenmann, Rev. Inorg. Chem., 1981, 3, 29 Search PubMed , and references cited therein.
- J. T. Vaughey and J. D. Corbett, Chem. Mater., 1996, 8, 671 CrossRef CAS.
- A. Rehr, T. Y. Kuromoto, J. DelCastillo, D. J. Webb and S. M. Kauzlarich, Chem. Mater., 1994, 6, 93 CrossRef CAS.
- S. M. Kauzlarich and T. Y. Kuromoto, Croat. Chem. Acta, 1991, 64, 343 CAS.
- M. L. Munzarová and R. Hoffmann, J. Am. Chem. Soc., 2002, 124, 4787 CrossRef PubMed.
- S. M. Kauzlarich, M. M. Thomas, D. A. Odink and M. M. Olmstead, J. Am. Chem. Soc., 1991, 113, 7205 CrossRef CAS.
- A. Rehr and S. M. Kauzlarich, J. Alloys Compd., 1994, 207, 424 CrossRef.
- T. Y. Kuromoto, S. M. Kauzlarich and D. J. Webb, Chem. Mater., 1992, 4, 435 CrossRef CAS.
- Y. Hu, J. Wang, A. Kawamura, K. Kovnir and S. M. Kauzlarich, Chem. Mater., 2015, 27, 343 CrossRef CAS.
- D. M. Young, C. C. Torardi, M. M. Olmstead and S. M. Kauzlarich, Chem. Mater., 1995, 7, 93 CrossRef CAS.
- S. Q. Xia and S. Bobev, J. Am. Chem. Soc., 2007, 129, 4049 CrossRef CAS PubMed.
- S. Q. Xia and S. Bobev, Inorg. Chem., 2006, 45, 7126 CrossRef CAS PubMed.
- S. Q. Xia and S. Bobev, Chem. – Asian J., 2007, 2, 619 CrossRef CAS PubMed.
- S. Q. Xia and S. Bobev, J. Comput. Chem., 2008, 29, 2125 CrossRef CAS PubMed.
- S. Q. Xia and S. Bobev, Inorg. Chem., 2008, 47, 1919 CrossRef CAS PubMed.
- S. Q. Xia and S. Bobev, J. Am. Chem. Soc., 2007, 129, 10011 CrossRef CAS PubMed.
- (a) S. K. Bux, A. Zevalkink, O. Janka, D. Uhl, S. Kauzlarich, J. G. Snyder and J.-P. Fleurial, J. Mater. Chem. A, 2014, 2, 215 RSC; (b) S. Ohno, A. Zevalkink, Y. Takagiwa, S. K. Bux and G. J. Snyder, J. Mater. Chem. A, 2014, 2, 7478 RSC.
- D. Wilson, B. Saparov and S. Bobev, Z. Anorg. Allg. Chem., 2011, 637, 2018 CrossRef CAS PubMed.
- SMART, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- SHEXLTL, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- (a) A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- L. M. Gelato and E. Parthe, J. Appl. Crystallogr., 1987, 20, 139 CrossRef.
- (a) O. K. Andersen, Phys. Rev. B: Solid State, 1975, 12, 3060–3083 CrossRef CAS; (b) O. K. Andersen and O. Jepsen, Phys. Rev. Lett., 1984, 53, 2571–2574 CrossRef CAS; (c) O. K. Andersen, Z. Pawlowska and O. Jepsen, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5253 CrossRef CAS; (d) H. L. Skriver, The LMTO Method, Springer, Berlin, 1984 Search PubMed.
- O. Jepsen and O. K. Andersen, The Stuttgart TB-LMTO Program, Version 4.7, Max-Planck-Institut für Festkörperforschung, Stuttgart, Germany, 1998 Search PubMed.
- O. K. Andersen, O. Jepsen and D. Glötzel, in Highlights of Condensed Matter Theory, ed. F. Bassani, F. Fumi and M. Tosi, North-Holland, New York, 1985 Search PubMed.
- U. von Barth and L. Hedin, J. Phys. C: Solid State Phys., 1972, 5, 1629 CrossRef CAS PubMed.
- D. D. Koelling and B. N. Harmon, J. Phys. C: Solid State Phys., 1977, 10, 3107 CrossRef CAS PubMed.
- W. R. L. Lambrecht and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 2439 CrossRef CAS.
- R. Dronskowski and P. E. Blöchl, J. Phys. Chem., 1993, 97, 8617 CrossRef CAS.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef.
- Pearson's Handbook of Crystallographic Data for Intermetallic Phases, ed. P. Villars and L. D. Calvert, American Society for Metals, Materials Park, OH, 2nd edn, 1991 Search PubMed.
- A. C. Payne, M. M. Olmstead, S. M. Kauzlarich and D. J. Webb, Chem. Mater., 2001, 13, 1398 CrossRef CAS.
- (a) L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed; (b) L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010 CrossRef CAS.
- B. Saparov, H. He, X. Zhang, R. Greene and S. Bobev, Dalton Trans., 2010, 39, 1063 RSC.
- J. P. A. Makongo, T.-S. You, H. He, N.-T. Suen and S. Bobev, Eur. J. Inorg. Chem., 2014, 5113 CrossRef CAS PubMed.
- J. P. A. Makongo, Yu. Prots, R. Niewa, U. Burkhardt and G. Kreiner, Z. Kristallogr. - New Cryst. Struct., 2005, 220, 291 CAS.
- P. Duwez and J. L. Taylor, AIME Trans., 1950, 188, 1173 CAS.
- M. V. Nevitt, Miscellaneous Structures of Fixed Stoichiometry, in Intermetallic Compounds, ed. J. H. Westbrook, Wiley, New York, 1967, pp. 217–299 Search PubMed.
- R. E. Marsh, Acta Crystallogr., 1954, 7, 379 CrossRef CAS.
- J. K. Brandon, R. Y. Brizard, W. B. Pearson and D. J. N. Tozer, Acta Crystallogr., 1977, B33, 527 CrossRef CAS.
- P. Pearce, Structure in Nature is a Strategy for Design, MIT Press, Cambridge, 1990 Search PubMed.
- E. A. Lord and S. Ranganathan, J. Non-Cryst. Solids, 2004, 334 & 335, 121 CAS.
- G. Kreiner and M. Schäpers, J. Alloys Compd., 1997, 259, 83 CrossRef CAS.
- G. Kreiner and H. F. Franzen, J. Alloys Compd., 1995, 221, 15 CrossRef CAS.
- S. Thimmaiah and G. J. Miller, Chem. – Eur. J., 2010, 16, 5461 CrossRef CAS PubMed.
- S. Bobev, D. J. Thompson, J. L. Sarrao, M. M. Olmstead, H. Hope and S. M. Kauzlarich, Inorg. Chem., 2004, 43, 5044 CrossRef CAS PubMed.
- S. Q. Xia and S. Bobev, Chem. Mater., 2010, 22, 840 CrossRef CAS.
- X.-C. Liu, Z. Wu, S. Q. Xia, X. T. Tao and S. Bobev, Inorg. Chem., 2015, 54, 947 CrossRef CAS PubMed.
- N. Kazem, A. Hurtado, B. Klobes, R. P. Hermann and S. M. Kauzlarich, Inorg. Chem., 2015, 54, 850 CrossRef CAS PubMed.
- G. F. Voronoi, J. Reine Angew. Math., 1908, 134, 198 Search PubMed.
- (a) C. Chothia, Nature, 1974, 248, 338 CrossRef CAS PubMed; (b) F. M. Richards, Annu. Rev. Biophys. Bioeng., 1977, 6, 151 CrossRef CAS PubMed; (c) Y. Harpaz, M. Gerstein and C. Chothia, Structure, 1994, 2, 641 CrossRef CAS; (d) J. Janin and C. Chothia, J. Biol. Chem., 1990, 265, 16027 CAS; (e) F. M. Richards, Carlsberg Res. Commun., 1979, 44, 47 CrossRef CAS; (f) F. M. Richards, Methods Enzymol., 1985, 115, 440 CAS; (g) M. Gerstein, A. M. Lesk, E. N. Baker, B. Anderson, G. Norris and C. Chothia, J. Mol. Biol., 1993, 234, 357 CrossRef CAS PubMed; (h) J. L. Finney, J. Mol. Biol., 1975, 96, 721 CrossRef CAS; (i) J. L. Finney, B. J. Gellatly, I. C. Golton and J. Goodfellow, Biophys. J., 1980, 32, 17 CrossRef CAS; (j) S. J. Hubbard and P. Argos, Curr. Opin. Biotechnol., 1995, 6, 375 CrossRef CAS.
- T. Ringler, L. Ju and M. Gunzburger, Ocean Dynamics, 2008, 58, 475 CrossRef.
- (a) C. H. Rycroft, Chaos, 2009, 19, 041111 CrossRef PubMed; (b) C. H. Rycrof, Z. Martin, M. Z. Bazant, G. S. Grest and J. W. Landry, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73, 051306 CrossRef; (c) K. Kamrin, C. H. Rycroft and M. Z. Bazant, Modell. Simul. Mater. Sci. Eng., 2007, 15, S449 CrossRef CAS; (d) J. P. Shih, S. Y. Sheu and C. Y. Mou, J. Chem. Phys., 1994, 100, 2202 CrossRef CAS PubMed; (e) M. Gerstein, J. Tsai and M. Levitt, J. Mol. Biol., 1995, 249, 955 CrossRef CAS PubMed.
- (a) R. Berthold, M. Mihalkovic, U. Burkhardt, Y. Prots, A. Amarsanaa and G. Kreiner, Intermetallics, 2014, 53, 67 CrossRef CAS PubMed; (b) R. Berthold, G. Kreiner, U. Burkhardt, S. Hoffmann, G. Auffermann, Y. Prots, E. Dashjav, A. Amarsanaa and M. Mihalkovic, Intermetallics, 2013, 32, 259 CrossRef CAS PubMed.
- E. J. Gabe, Y. le Page, P.-P. Charland, F. L. Lee and P. S. White, J. Appl. Crystallogr., 1989, 22, 384 CrossRef CAS.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925 CrossRef CAS.
- J. P. A. Makongo, N. T. Suen, S. P. Guo, S. Saha, R. Greene, J. P. Paglione and S. Bobev, J. Solid State Chem., 2014, 211, 95 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic information files (CIF), crystallographic data for Sr14Cd1.4(1)Sb11, obtained via a stoichiometric reaction of the elements, relevant plots of the electron density from the refined atomic positions of the hypervalent [Pn3]7−, first coordination sphere of the cations in the “14–1–11” structure, alternative description of the structure of Ca9Cd4+xBi9 (data taken from ref. 18). See DOI: 10.1039/c5tc01605c |
| ‡ Munzarová and Hoffmann (ref. 7) discuss a subtle but important trend with regard to the isoelectronic X3 linear systems and the strength of the 3-center 4-electron bonding. They argue that the stability of the X3 species decreases when moving from right to left in the periodic table, but the group trends in s, p mixing are more complex. For the electron-rich tripnictides, the authors argue that the ability to accommodate a hypervalent electron count is the largest in the middle than at the end of group 15. |
| § Tetragonal space group I41/acd assumed in all three cases. Unit cell parameters for Yb14CdSb11: a = 16.605(3) Å, c = 22.144(7) Å; (Yb,Ca)14CdSb11: a = 16.617(3) Å, c = 22.355(9) Å; Ba14CdSb11: a = 18.527(4) Å, c = 24.102(6) Å. Further synthetic and structural work is currently in progress. |
| ¶ In Sr14CdSb11.37 (ref. 12), which we argue is the same material as Sr14Cd1.30(1)Sb11 reported herein, the disorder is the same, but the interpretation is different. The distances of the reported interstitial Sb to its closest neighbors are of the same order as the distances we refine between Cd2 and its closest neighbors. Apparently, assigning Cd2+ or Sb3− to the interstitial site cannot be made based on crystallographic data. In this work, we argue that Cd2+ is the likely interstitial species, i.e., the formula of the compound is Sr14Cd1+xSb11. |
| || The Voronoi cell method consists of partitioning the entire space among a collection of equal-sized atoms. Each atom is surrounded by a polyhedron with its volume assigned to the atom. The faces of Voronoi polyhedra are derived from the separating planes perpendicular to the interatomic vectors, whereas the edges of the polyhedra are formed by the intersection of these planes. The Voronoi procedure requires the location of all atomic neighbors and a definition of the division points on the interatomic vectors. In this article, the volume of Voronoi Cell was calculated using the NRCVAX software package. The unit cell parameters and the atomic coordinates for all the compounds were used for precise volume calculations. As indicated in the manuscript, we seek to fill the space so that the sum of Voronoi cell volumes matches with the unit cell volume. To achieve accurate volume calculations, emphasis has been placed on the influence of the radii to assess avoidable errors since there are atoms showing different radii and regions with varying local atomic density. The total volume was computed with atomic radii in the range of 1.2–1.8 Å. |
| This journal is © The Royal Society of Chemistry 2015 |