
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulation of the ligand-based fluorescence of 3d metal complexes upon spin state change†‡
Charles
Lochenie
a,
Kristina G.
Wagner
b,
Matthias
Karg
*b and
Birgit
Weber
*a
aInorganic Chemistry II, Universität Bayreuth, Universitätsstrasse 30, NW I, 95440 Bayreuth, Germany. E-mail: weber@uni-bayreuth.de
bPhysical Chemistry I, Universität Bayreuth, Universitätsstrasse 30, NW I, 95440 Bayreuth, Germany. E-mail: matthias.karg@uni-bayreuth.de
First published on 1st June 2015
Abstract
Two new Schiff base-like ligands bearing a heteroaromatic fluorophore were synthesised and converted into the corresponding Ni(II), Cu(II) and Zn(II) square planar complexes. The Ni(II) complexes were studied with regard to a coordination change-induced spin state change upon addition of pyridine in solution. An inverse correlation between the fluorescence properties and the spin state of the metal centre was observed, and investigated with steady state fluorescence and time-resolved spectroscopy.
Introduction
Spin crossover (SCO) complexes are switchable molecules where the change of the spin state can be triggered by a wide range of physical or chemical stimuli such as temperature, pressure, light irradiation or absorption/desorption of guest molecules. This switching process is accompanied by magnetic, optical and structural changes that can be coupled to additional properties (e.g. liquid crystal phase transition).1 Due to their high variability with regard to the factors triggering the spin transition and the accompanying changes that can be “read out”, SCO complexes are one of the most important molecule-based switchable materials and excellent candidates for technological application.2,3 Iron(II) is the most widely used metal centre for the synthesis of SCO complexes. For those complexes wide hysteresis loops around room temperature4 – even at nanoscale3,5 or photoinduced spin state changes6 at high temperatures could already be realised. However, the phenomenon itself is not limited to iron(II) or other 3d4–7 metal centres with an octahedral coordination sphere.7 The coordination induced spin state change of nickel(II) complexes from diamagnetic (S = 0) square planar to paramagnetic (S = 1) square pyramidal or octahedral coordination sphere shifted recently back into focus. With respect to potential applications for example in the field of smart contrast agents for magnetic resonance imaging, it is of interest to realise ligand systems which allow for light induced switching between these states. Ideally the transitions can be induced at room temperature in solution and on a single molecule level.8The combination of the spin transition with luminescence, if possible in a molecular system, would provide another “read-out” feature with a high application potential in the field of drug delivery, biomarkers or thermometry. Several attempts were already reported for the realisation of such bifunctional materials, mostly with iron(II). One possibility to achieve such systems is the synthesis of composite materials such as thin films doped with SCO complexes for electroluminescence,9 functionalised SCO-core–luminescence-shell nanoparticles10,11 or SCO complexes with fluorescent counter anions.10,12 Another possibility is to covalently link the fluorophore to the SCO centre through ligand design.11,13 However, this approach is not always successful with respect to a coupling between spin transition and fluorescence.14 So far only one example for a nickel(II) based fluorescent molecular thermometer is known, where the emission colour and intensity can be switched through a spin state change.15
Here we present a new ligand system that shows a modulation of the fluorescence intensity upon a spin state change. The fluorescence properties of the free ligands, the diamagnetic zinc(II) complexes, the paramagnetic copper(II) complexes and the S = 0 ↔ S = 1 switchable nickel(II) complexes were investigated with steady-state extinction and fluorescence spectroscopy as well as time-resolved fluorescence spectroscopy.
Results
Syntheses
All metal complexes were synthesised in two steps. Starting with the diamines 1 and 4, firstly a condensation with a keto–enol ether forms the chelate cycles, which then react with metal acetate to give the respective complexes [ML1] and [ML2] (M = NiII, CuII, ZnII), the counter anionic acetates acting as bases for deprotonation of the ligand. The molecular structures and the synthetic pathway are given in Scheme 1. All complexes were obtained as pure powder with the general formula [ML1] or [ML2]. All ligands and intermediates were characterised with IR, CHN and 1H-NMR. All complexes were characterised with IR, CHN analysis and mass spectrometry. | ||
| Scheme 1 Pathway of synthesis of the metal complexes described in this work and used abbreviations. | ||
Crystal structure analysis
Single crystals suitable for X-ray diffraction analysis of [NiL1] and [CuL1] were obtained from a vapour–vapour slow diffusion setup between a trichloromethane solution of the complex and ethanol. All crystallographic data are given in the ESI:‡ Table S1. While the complex [NiL1] crystallises with the same composition as the bulk material, the structure of compound [CuL1] was determined as [CuL1(EtOH)]·2CHCl3. Both compounds crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and the asymmetric units contain one complex molecule. ORTEP drawings of the asymmetric units are displayed in Fig. 1. The nickel(II) centre in [NiL1] lies in a N2O2 square planar coordination sphere. Ni–N (1.83 Å) and Ni–Oeq (1.85 Å) bond lengths are in agreement with other complexes with similar coordination sphere.16,17 The sum of the angles is with Σ = 717°, not far from a perfect square planar coordination sphere (Σ = 720°). In the case of [CuL1(EtOH)]·2CHCl3, the copper(II) lies in a N2O3 square pyramidal geometry. Cu–N (1.92 Å), Cu–Oeq (1.92 Å) and Cu–OEtOH (2.378(5) Å) bond lengths are generally longer than for the nickel complex, due to the different geometry of the coordination sphere and the increase of the covalent radius from 124 pm (Ni) to 132 pm (Cu). Selected bond lengths and angles are presented in Table 1.
, and the asymmetric units contain one complex molecule. ORTEP drawings of the asymmetric units are displayed in Fig. 1. The nickel(II) centre in [NiL1] lies in a N2O2 square planar coordination sphere. Ni–N (1.83 Å) and Ni–Oeq (1.85 Å) bond lengths are in agreement with other complexes with similar coordination sphere.16,17 The sum of the angles is with Σ = 717°, not far from a perfect square planar coordination sphere (Σ = 720°). In the case of [CuL1(EtOH)]·2CHCl3, the copper(II) lies in a N2O3 square pyramidal geometry. Cu–N (1.92 Å), Cu–Oeq (1.92 Å) and Cu–OEtOH (2.378(5) Å) bond lengths are generally longer than for the nickel complex, due to the different geometry of the coordination sphere and the increase of the covalent radius from 124 pm (Ni) to 132 pm (Cu). Selected bond lengths and angles are presented in Table 1.
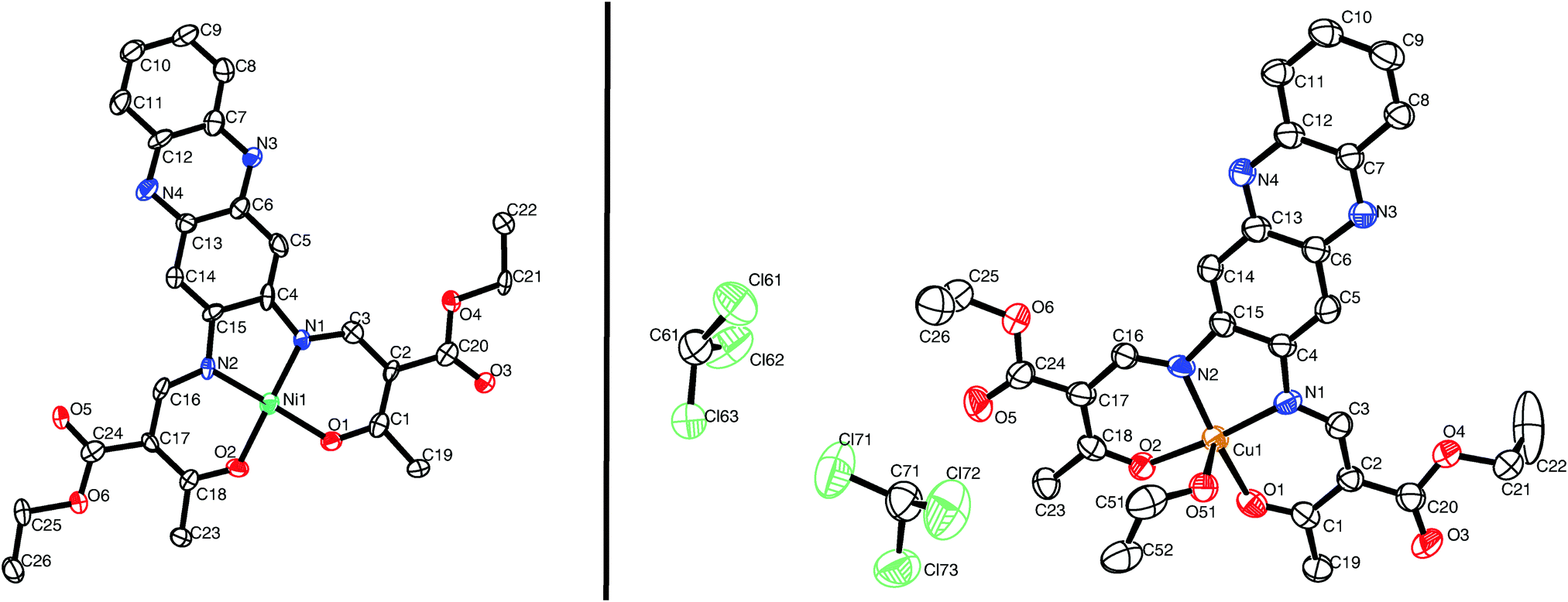 | ||
| Fig. 1 ORTEP drawing of [NiL1] (left) and [CuL1(EtOH)]·2CHCl3 (right). Thermal ellipsoids are shown at 50% level. Hydrogen atoms are omitted for clarity. | ||
| Compound | M–N | M–Oeq | M–OEtOH | Oeq–M–Oeq | N–M–N | N–M–Oeq | N/Oeq–M–OEtOH |
|---|---|---|---|---|---|---|---|
| [NiL1] | 1.837(5) | 1.856(4) | — | 85.32(17) | 87.1(2) | 94.17(19) | — |
| 1.825(4) | 1.848(4) | 93.4(2) | |||||
| 178.3(2) | |||||||
| 178.73(19) | |||||||
| [CuL1(EtOH)]·2CHCl3 | 1.930(7) | 1.905(5) | 2.378(5) | 90.8(2) | 84.8(3) | 92.5(2) | 91.8(2) |
| 1.921(6) | 1.943(5) | 90.9(3) | 92.0(2) | ||||
| 171.8(3) | 95.3(2) | ||||||
| 171.9(2) | 96.1(2) |
The crystal packing of [NiL1] shows the complexes stacked over each other, forming columns along the vector [100]. π–π interactions between the aromatic rings of the ligand, as well as metal–aromatic interactions between the nickel centre and the chelate rings of neighbouring complexes lead to the formation of the columns in the packing. Illustrations of the packing are shown in Fig. 2, and selected distances of the π–π interactions are presented in Table 2. The crystal packing of [CuL1(EtOH)]·2CHCl3 shows a similar stacking than the one observed for [NiL1], with the formation of pairs through π–π interactions. However, since an ethanol molecule is coordinated axially at the copper centre, no metal–aromatic interactions are observed. Only the aromatic rings of the ligands are interacting, with the copper centres looking in opposing directions in a “head-to-toes” fashion. Furthermore, hydrogen bonds are present between the trichloromethane solvent molecules and the complexes, as well as between the coordinating ethanol and neighbouring complex molecules. Illustrations of the packing are shown in Fig. 3, selected distances and angles of the π-interactions and of the hydrogen bonds are presented in Tables 2 and 3, respectively.
 | ||
| Fig. 2 Illustrations of the crystal packing of [NiL1] along [010] (top) and [100] (bottom left); scheme of the π–π and M–π interactions involved in the packing (bottom right). | ||
| Cg(I) | Cg(J) | Cg–Cg | α | β | γ |
|---|---|---|---|---|---|
| a = −1 + x, y, z; b = 1 + x, y, z; c = 3 − x, 1 − y, 1 − z. | |||||
| [NiL1] | |||||
| C4–C5–C6–C13–C14–C15 | C4–C5–C6–C13–C14–C15a | 4.084(3) | 0 | 33.42 | 33.42 |
| C4–C5–C6–C13–C14–C15 | C4–C5–C6–C13–C14–C15b | 4.084(3) | 0 | 33.42 | 33.42 |
| C6–N3–C7–C12–N4–C13 | C6–N3–C7–C12–N4–C13a | 4.084(3) | 0 | 32.92 | 32.92 |
| C6–N3–C7–C12–N4–C13 | C6–N3–C7–C12–N4–C13b | 4.084(3) | 0 | 32.92 | 32.92 |
| C7–C8–C9–C10–C11–C12 | C7–C8–C9–C10–C11–C12a | 4.084(4) | 0 | 32.65 | 32.65 |
| C7–C8–C9–C10–C11–C12 | C7–C8–C9–C10–C11–C12b | 4.084(4) | 0 | 32.65 | 32.65 |
| Ni1–O1–C1–C2–C3–N1 | Ni1b | 3.301 | — | 12.75 | — |
| Ni1–O2–C18–C17–C16–N2 | Ni1a | 3.258 | — | 11.40 | — |
| [CuL1(EtOH)]·2CHCl3 | |||||
| C4–C5–C6–C13–C14–C15 | C7–C8–C9–C10–C11–C12c | 3.837(5) | 1.1(4) | 28.66 | 29.54 |
| C6–N3–C7–C12–N4–C13 | C6–N3–C7–C12–N4–C13c | 3.807(5) | 0 | 28.68 | 28.68 |
| C7–C8–C9–C10–C11–C12 | C4–C5–C6–C13–C14–C15c | 3.838(5) | 1.1(4) | 29.54 | 28.66 |
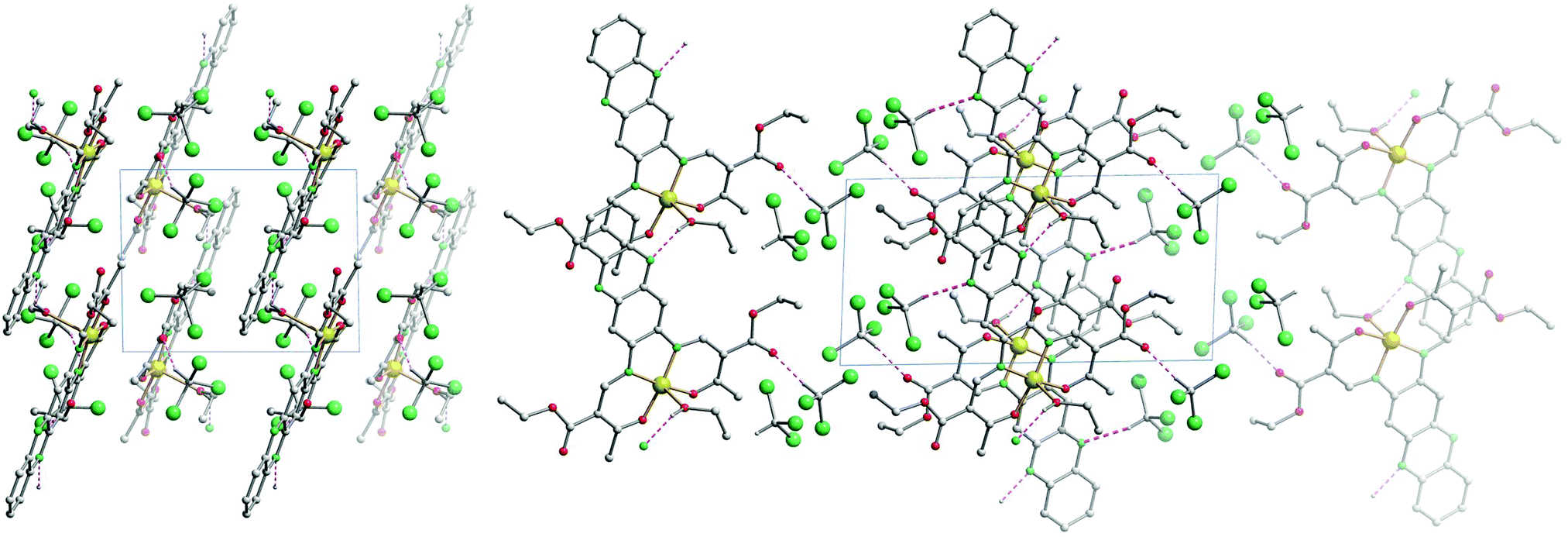 | ||
| Fig. 3 Illustrations of the crystal packing of [CuL1(EtOH)]·2CHCl3 along [001] (left) and along [010] (right). | ||
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| a = −1 + x, y, z; b = x, y, 1 + z. | ||||
| O51–H51⋯N4a | 0.84 | 2.11 | 2.820(8) | 142 |
| C61–H61⋯N3b | 1.00 | 2.35 | 3.297(14) | 158 |
| C71–H71⋯O5 | 1.00 | 2.07 | 3.007(14) | 155 |
Powder diffraction patterns of all investigated complexes were measured; the results are displayed in the ESI,‡ Fig. S1. The diffraction pattern of the powder sample [CuL1] differs significantly from the calculated one for the single crystals of [CuL1(EtOH)]·2CHCl3. This is not unexpected since the additional solvent molecules will strongly influence the packing pattern. The diffraction patterns of the three solvent-free complexes of L1 show only little similarities. The packing of the molecules in the crystals is influenced by the metal centre. In contrast to this the diffraction patterns of [CuL2] and [NiL2] are very similar, for [ZnL2] the differences are more pronounced. For the ligand L2 with the extended aromatic system the packing of the molecules in the crystal is mostly influenced by the ligand and less by the metal centre.
Steady state spectroscopy
The optical properties of the ligands and the complexes were studied by extinction and fluorescence spectroscopy. First of all the pure ligand systems were investigated in trichloromethane. Fig. 4A compares the absorbance and emission spectra of H2L1 and H2L2. The spectral features of both ligands are very similar. Both ligands show a pronounced absorption peak at approx. 440 nm and emission with a maximum at approx. 470 nm. Furthermore the absorption peaks show a weak shoulder at lower wavelength, as well as a weak shoulder in the emission spectra at higher wavelength as compared to the peak maximum. These shoulders can be attributed to the equilibrium between imino–enol/keto–enamine tautomers of the ligands in solution.17,18 | ||
| Fig. 4 Absorption and emission spectra. (A) Pure ligands. (B) Cu(II) complexes. (C and D) Zn(II) complexes. | ||
The spectral analyses of the metal complexes (Cu and Zn) are shown in Fig. 4B–D. In order to investigate the influence of the spin state on the absorption and fluorescence, spectra of the complexes were measured in trichloromethane and pyridine. Fig. 4B compares the absorbance spectra for the Cu(II) complexes [CuL1] and [CuL2]. Comparing the spectra measured in trichloromethane similar absorption spectra are observed with the same number of bands. In contrast to this the absorption peaks are red-shifted when pyridine is used as solvent (solvatochromism). In case of [CuL2], a weak band appears at 585 nm in the pyridine solution, that can be attributed to d–d transitions by comparison with similar complexes.17 In agreement with many examples for fluorophores in literature, fluorescence was not observed for the copper complexes indicating strong quenching due to the presence of the metal centre.19,20
The absorption spectra of the zinc(II) complexes [ZnL1] in trichloromethane and pyridine shown in Fig. 4C resemble the spectra of the pure ligand systems. In addition almost no difference in band position and the number of bands is observed comparing the spectra in the two solvents. In contrast to this the complex emission is significantly influenced by changing the solvent from trichloromethane to pyridine. In trichloromethane the emission maximum is at 473 nm, whereas the emission shifts to 560 nm in pyridine. The absorption spectrum of [ZnL2] in trichloromethane resembles also the spectrum of the corresponding ligand, however changes in the number of bands and their position appear in pyridine solution. The emission spectrum also displays a red-shift as observed for sample [ZnL1], but with a smaller difference between trichloromethane solution (λem = 475 nm) and pyridine solution (λem = 529 nm). The optical properties of the nickel(II) complexes [NiL1] and [NiL2] were investigated by adding a pyridine solution of the complex into an equimolar trichloromethane solution of the same complex, as the nickel centre undergoes a spin state change upon coordination change (see Scheme 2).7,15 Respective absorbance and emission spectra are shown in Fig. 5. The trichloromethane solutions of the nickel complexes show three absorption bands at 365, 414, and 448 nm for [NiL1], and at 412, 440, and 465 nm for [NiL2]. Upon progressive coordination of pyridine molecules onto the nickel centre, the absorption spectra dramatically change. In the case of [NiL1], the bands at 414 and 448 nm tend to disappear whereas the band at 365 nm increases and a new band at 493 nm appears. For the sample [NiL2] the intensity of the bands at 412 and 440 nm decreases and two new bands at 375 and 500 nm appear. The drastic changes in the absorbance spectra of the nickel complexes confirm the change of geometry and by this the spin state upon coordination with pyridine. The spin state change also affects the fluorescence properties of the complexes (Fig. 5B and D). As pyridine is added to the complex [NiL1], its emission band at 478 nm progressively red-shifts and its intensity dramatically decreases. The redshift seems in good agreement with the emergence of a new band at higher wavelength (493 nm) in the absorption spectrum upon addition of pyridine. For the complex [NiL2], a completely different effect is observed. Upon addition of the first equivalents of pyridine, the emission band at 491 nm is shifted to 538 nm, and its intensity increases until ≈400 equivalents of pyridine are added. Further addition of pyridine leads to a decrease of the emission intensity.
 | ||
| Scheme 2 Electronic configuration, spin number and geometry of the different complexes in equilibrium in solution upon addition of pyridine. | ||
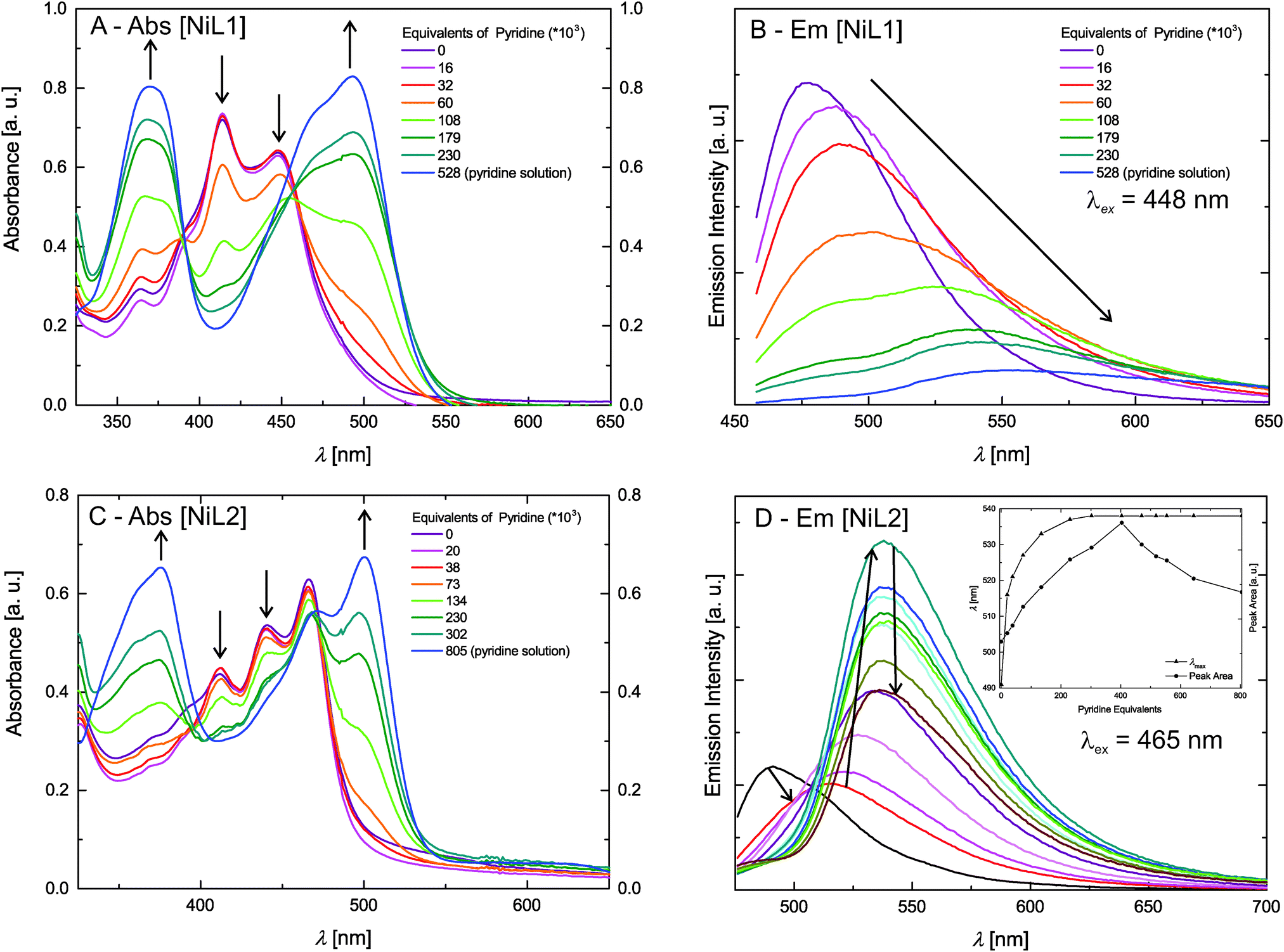 | ||
| Fig. 5 Absorption and steady state fluorescence measurements of the Ni(II) complexes. The inset in D gives the integrated peak area and λmax as guide for the reading of the graph. | ||
The reversibility of the coordination of the pyridine molecules was investigated with absorption spectroscopy. First, a pyridine solution of the [NiL1] complex was progressively added to an equimolar trichloromethane solution of the complex. In the next step, the pyridine concentration of this solution was reduced by addition of the original trichloromethane solution. The intensity of the characteristic absorption bands of the complex in pyridine (λ = 365 nm) and in trichloromethane (λ = 414 nm) varies in agreement with a reversible coordination of the pyridine molecules to the metal centre. Fig. S2 in the ESI‡ illustrates those results. The absorption spectra of the fluorescent Ni(II) and Zn(II) complexes were also measured in trichloromethane solutions containing triethylamine (Et3N) as non-coordinating base, or formic acid (HCOOH), in order to rule out possible effects due to (de)protonation of the complexes. The corresponding UV-vis spectra are presented in the ESI:‡ Fig. S3. The spectra show that in all cases, no significant changes are observed for the CHCl3–Et3N solutions, proving that the changes observed for the pyridine addition are due to coordination of pyridine at the axial positions of the nickel centre. Spectra of the complexes in CHCl3/HCOOH solutions show a pronounced red-shift upon protonation of the heteroaromatic N-atoms of the complex. This is not surprising as phenazine-derivatives are used as pH-indicators (e.g. neutral red). As the effects of the spin state change on the intensity of the emission properties of [NiL1] and [NiL2] are extremely different and intriguing, lifetime measurements of the fluorescence were performed and are discussed further.
Fluorescence lifetime analysis
In addition to the steady-state fluorescence spectra shown in Fig. 5B and D for the complexes [NiL1] and [NiL2], lifetime measurements were performed on the same samples at 298 K. The fluorescence decays were analysed using deconvolution fitting with double exponential decay functions. Fig. 6 compares selected fluorescence decays for complex solutions in trichloromethane and pyridine. It is clearly visible that the lifetime depends strongly on the type of solvent. In pyridine a significantly faster decay is observed as compared to trichloromethane. Table 4 summarises the results of the lifetime analysis for all measured solutions. The fluoresence decays for the complexes show monoexponential behavior for trichloromethane as solvent with relatively short lifetimes of τ = 0.7 ns for [NiL1] and τ = 1.3 ns for [NiL2]. For small equivalents of added pyridine the lifetime decreases in case of [NiL1] and the decays remain monoexponential. For higher amounts of pyridine, monoexponential decays could not be used to satisfyingly describe the decay profiles as a second longer lifetime component emerges. However this second contribution has very small amplitudes except for the highest amount of pyridine used. In the latter case the data analysis will be less reliable since the fast component provides lifetimes which are significantly smaller than the half width of the instrument response function. Therefore no conclusions will be drawn from the latter analysis result. As a general trend, it can be observed that the first lifetime component for [NiL1] systematically shortens with increasing amount of added pyridine.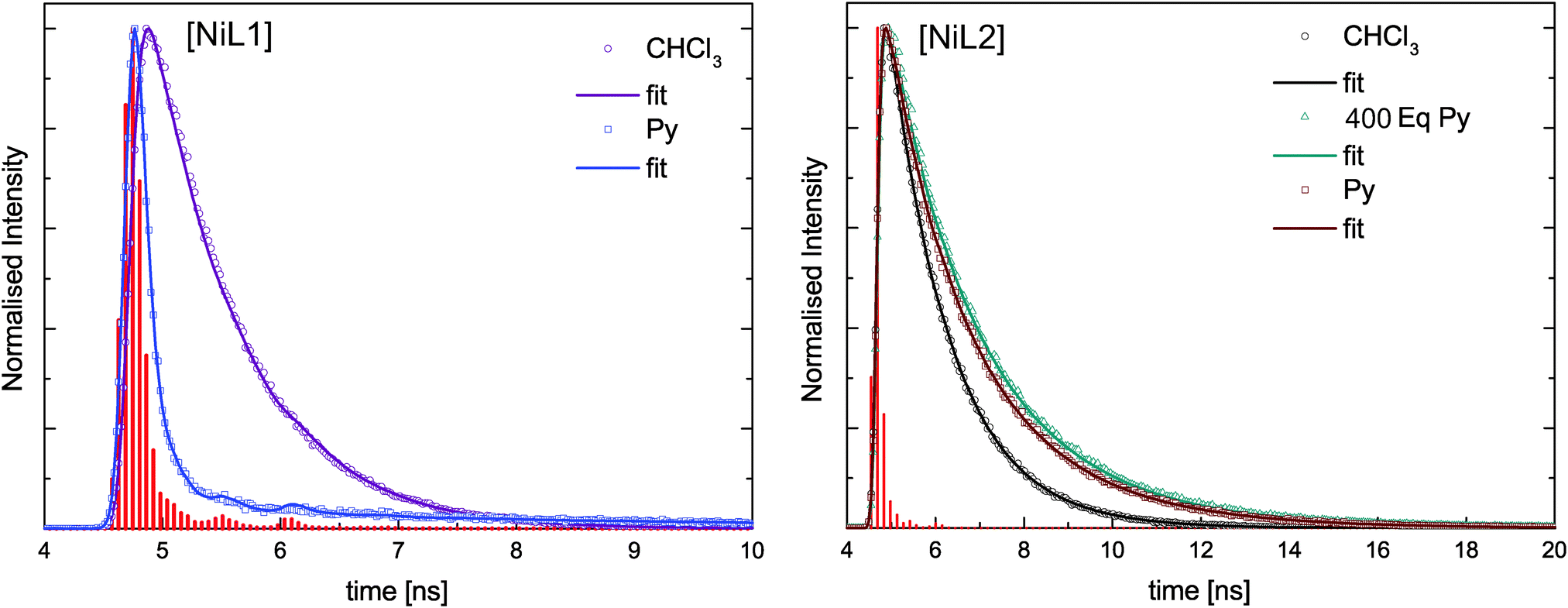 | ||
| Fig. 6 Measured fluorescence decays for the nickel complexes. The instrument response function (IRF) is represented by the red vertical bars. The solid lines are deconvolution fits of the lifetime traces. | ||
| Py equivalents | τ |
|---|---|
| [NiL1] | |
| 0 (trichloromethane solution) | 0.7 [1%] |
| 16 | 0.6 [2%] |
| 32 | 0.5 [2%] |
| 60 | 0.4 [2%] |
| 108 | 0.3 [4%] |
| 179 | <0.2 |
| 230 | <0.2 |
| 528 (pyridine solution) | <0.2 |
| [NiL2] | |
| 0 (trichloromethane solution) | 1.3 [<1%] |
| 20 | 1.3 [<1%] |
| 38 | 1.6 [<1%] |
| 73 | 1.8 [<1%] |
| 134 | 2.0 [<1%] |
| 230 | 2.0 [<1%] |
| 302 | 1.9 [<1%] |
| 403 | 1.9 [<1%] |
| 470 | 1.8 [<1%] |
| 518 | 1.7 [<1%] |
| 554 | 1.7 [<1%] |
| 644 | 1.8 [<1%] |
| 805 (pyridine solution) | 1.7 [<1%] |
For compound [NiL2], a different trend is observed upon addition of pyridine: the compound starts with a relatively longer lifetime of τ = 1.3 ns, which slightly increases upon addition of pyridine, reaching 1.8 ns in pure pyridine solution. For this complex all fluorescence decays were fitted with a monoexponential model and satisfying χ2 values were obtained.
Discussion
The complexes [ML1] and [ML2] were designed with the aim to favour possible non-radiative energy transfer between the heteroaromatic fluorophore, the donor, and the metal centre, the acceptor. Such design could give a control on the fluorescence properties of the fluorophore through changes of the spin state of the metal centre. As the distance between the donor and the acceptor plays a crucial role in the effectiveness of a non-radiative energy transfer,20 two different fluorophores with extended π-systems were used. The crystal structure of [NiL1] and [CuL1(EtOH)]·2CHCl3 were successfully analysed by X-ray diffraction. The obtained crystal structures present π–π interactions in the packing between the complexes, as well as a hydrogen bond network in case of [CuL1(EtOH)]·2CHCl3. In the latter crystal structure, it is observed that the copper complex crystallizes in a square pyramidal geometry, although it is obtained as square planar complex in the bulk material. It was already observed in corresponding phenylene-derivatives copper(II) complexes that the metal centre is always trying to break its square planar symmetry, either by coordinating solvent molecules, or by sitting in a distorted coordination sphere.17 Unfortunately no crystals suitable for X-ray diffraction were obtained for the [ML2] samples, even when using vapour–vapour or liquid–liquid slow diffusion setups.The spin state of the different complexes is usually due to their electronic configuration, with S = ½ for the 3d9 Cu(II) complexes, and S = 0 for the 3d10 Zn(II) complexes. In the case of the Ni(II) complexes, their spin state depends also on the geometry of the coordination sphere of the Ni(II) centre. In a square planar geometry, the nickel centre has a spin number of S = 0, with all its electrons paired, however, upon coordination of pyridine on the axial position(s), the changes on the splitting of the d orbitals will induce a spin state change (S = 1), as described in the literature.8,16 The magnetic moment of the complex [NiL1] was determined with the Evans method: an effective magnetic moment μeff = 0 was measured in CDCl3 solution, in contrast to a μeff = 2.78 in pyridine-d5 solution, in good agreement with a theoretical value of μSO = 2.83 for S = 1.
The investigation of the steady-state fluorescence of [ML1] compounds shows that the emission properties depend on the spin state of the metal centre. Indeed, diamagnetic metal centres, Zn(II) or Ni(II) in square planar geometry are fluorescent whereas for paramagnetic metal centres, Cu(II) or Ni(II) in square pyramidal/octahedral geometry, the fluorescence is quenched. The presence of unpaired electrons, and therefore partially filled orbitals, can give rise to an energy transfer between the fluorophore and the metal centre. A shortening of the lifetime of a given fluorophore is often observed when an energy transfer occurs, and this effect is observed for the complex [NiL1], as the emission lifetime gets shorter upon addition of pyridine, or in other words, upon coordination change induced spin state change (see Scheme 2). It has to be pointed out here that the sample [NiL1] exhibits a second contribution with a longer lifetime once a certain amount of pyridine equivalents is reached. The origin of this second component is not yet well understood and requires further investigations. The type of non-radiative energy transfer, whether it is a Förster or Dexter type cannot be pinpointed on the basis of the current investigation and requires further experiments or calculations.
The corresponding Zn(II) complex [ZnL1] displays a strong red-shift (87 nm) of the fluorescence upon coordination with pyridine. The same effect is observed for sample [ZnL2] but the red-shift is only in the order of 54 nm, indicating that the metal centre has a reduced influence on the fluorescence properties of the bigger fluorophore.
The complex [NiL2] presents a very different behaviour than [NiL1] upon addition of pyridine. Instead of being quenched, its emission properties are actually intensifying upon the addition of the first equivalents of pyridine. After reaching a maximum, the emission band intensity is decreasing; however the sample in pure pyridine solution shows stronger fluorescence than the sample in pure trichloromethane solution. The opposite trend was observed for the complex [NiL1]. The lifetime measurements also present different results for [NiL2] than for [NiL1]. The lifetime of the emission is slightly increasing from 1.3 ns to 2.0 ns upon pyridine coordination. Then as the intensity of the emission peak is decreasing, the lifetime stays approximately constant (τ = 1.7 ns in pyridine solution).
Two hypotheses can be drawn about the big difference in the luminescence properties of the two nickel complexes. A difference in the nature of the transitions involved in the fluorophore could be the reason. It is described in literature that phenanthrene, and phenanthrene-derivatives like the ligand H2L2, undergo forbidden transition, leading generally to longer lifetimes.21 Another reason could be a too long distance between the metal centre and the fluorophore, preventing any energy transfer. In this case, the effect observed on addition of pyridine would be of the same nature as solvatochromism. Finally, it is also possible that the first excited state of the fluorophore is at a lower energy than the first excited state of the metal centre, making the energy transfer impossible. Further investigations about the compound [NiL2], with varying temperature, or changing the coordinating solvent molecule i.e. for acetonitrile, are needed in order to understand the luminescence properties. Calculations of the different ground and excited states of the donor–acceptor pair would also give useful insights.
As complex [NiL1] showed an interesting coupling of the fluorescence properties with the spin state change, corresponding iron(II) complexes will be investigated in order to determine if such coupling is also obtained with a thermally induced spin crossover.
Experimental section
Synthesis
2,3-Diaminophenazine (1), 4,5-diamino-NN′-ditoluene-p-sulphonyl-o-phenylenediamine (2) and ethoxymethylene-ethylacetoacetate were synthesised as described in literature.22 Methanol, ethanol, trichloromethane and pyridine were of analytical grade and used without further purification. Nickel(II) acetate tetrahydrate (99%, Fluka), copper(II) acetate monohydrate (99%, Fluka), zinc(II) acetate dihydrate (97%, Alfa Aesar), 9,10-phenanthrenequinone (95%, Alfa Aesar) and sulphuric acid (97%, Sigma Aldrich) were used without further purification. CHN analyses were measured with a Vario El III from Elementar Analysen-Systeme. Mass spectra were recorded with a Finnigan MAT 8500 with a data system MASPEC II. NMR spectra were measured with a Varian INOVA 300. Toluene was used as reference for the Evans method.
H2L1: 2,3-diaminophenazine (1) (0.5 g) and ethoxymethyleneethylacetoacetate (0.93 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and a brownish yellow powder precipitated upon reflux. The brownish yellow powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure product H2L1 as brownish yellow crystals. Yield: 0.48 g (42%). MS (DEI-(+), 70 eV) m/z (%): 490 (100) (M+); elemental analysis calculated (found) for C34H30N4O6 (490.51 g mol−1): C 63.66 (66.30), H 5.34 (5.15), N 11.42 (11.65). 1H-NMR (DMSO, 300 MHz, ppm) δ = 12.67 (d, J = 12 Hz, –NH, 2H), 8.52 (d, J = 12 Hz, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H, 2H), 8.25 (s, Ar–H, 2H), 8.18 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 7.94 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 4.18 (qua, J = 7.5 Hz, –CH2, 4H), 2.49 (s, –CH3, 6H), 1.32 (t, J = 7.5 Hz, –CH3, 6H).
C–H, 2H), 8.25 (s, Ar–H, 2H), 8.18 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 7.94 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 4.18 (qua, J = 7.5 Hz, –CH2, 4H), 2.49 (s, –CH3, 6H), 1.32 (t, J = 7.5 Hz, –CH3, 6H).
H2L2: 6′,7′-diaminoquinoxaline-[2′,3′-d]-1,10-phenanthrene (4) (0.17 g) and ethoxymethyleneethylacetoacetate (0.26 g) were dissolved in 10 mL ethanol. The solution was refluxed during 1 hour, and a yellow powder precipitated upon reflux. The yellow powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure product H2L2 as yellow crystals. Yield: 0.24 g (74%). MS (DEI-(+), 70 eV) m/z (%): 590 (100) (M+); elemental analysis calculated (found) for C34H30N4O6 (590.63 g mol−1): C 69.14 (69.30), H 5.12 (5.15), N 9.49 (9.65). 1H-NMR (DMSO, 300 MHz, ppm) δ = 12.59 (d, J = 12 Hz, –NH, 2H), 9.13 (m, Ar–H, 2H), 8.69 (m, Ar–H, 2H), 8.65 (d, J = 12 Hz, C–H, 2H), 7.68 (m, Ar–H, 4H), 7.10 (s, Ar–H, 2H), 4.17 (qua, J = 7.5 Hz, –CH2, 4H), 2.47 (s, –CH3, 6H), 1.33 (t, J = 7.5 Hz, –CH3, 6H).
[NiL1]: H2L1 (0.2 g) and nickel(II) acetate tetrahydrate (0.12 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and an orange powder precipitated upon reflux. The orange powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure complex as orange powder. Yield: 0.20 g (88%). MS (DEI-(+), 70 eV) m/z (%): 546 (100) (M+); elemental analysis calculated (found) for C26H24N4NiO6 (547.19 g mol−1): C 57.07 (56.98), H 4.42 (4.15), N 10.24 (10.42).
[CuL1]: H2L1 (0.2 g) and copper(II) acetate monohydrate (0.10 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and a brown powder precipitated upon reflux. The brown powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure complex as brown powder. Yield: 0.21 g (91%). MS (DEI-(+), 70 eV) m/z (%): 551 (100) (M+); elemental analysis calculated (found) for C26H24N4CuO6 (552.04 g mol−1): C 56.57 (56.48), H 4.38 (4.23), N 10.15 (10.05).
[ZnL1]: H2L1 (0.2 g) and zinc(II) acetate dihydrate (0.11 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and a red powder precipitated upon reflux. The red powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure complex as red powder. Yield: 0.18 g (79%). MS (DEI-(+), 70 eV) m/z (%): 552 (100) (M+); elemental analysis calculated (found) for C26H24N4O6Zn (553.87 g mol−1): C 56.38 (56.29), H 4.37 (4.43), N 10.12 (10.13).
[NiL2]: H2L2 (0.2 g) and nickel(II) acetate tetrahydrate (0.10 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and an yellow powder precipitated upon reflux. The yellow powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure complex as yellow powder. Yield: 0.17 g (76%). MS (DEI-(+), 70 eV) m/z (%): 646 (100) (M+); elemental analysis calculated (found) for C34H28N4NiO6 (647.30 g mol−1): C 63.09 (63.16), H 4.36 (4.45), N 9.07 (9.05).
[CuL2]: H2L2 (0.2 g) and copper(II) acetate monohydrate (0.08 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and a brown powder precipitated upon reflux. The brown powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure complex as brown powder. Yield: 0.19 g (86%). MS (DEI-(+), 70 eV) m/z (%): 651 (100) (M+); elemental analysis calculated (found) for C34H28N4CuO6 (652.16 g mol−1): C 62.62 (62.58), H 4.33 (4.18), N 8.59 (8.65).
[ZnL2]: H2L2 (0.2 g) and zinc(II) acetate dihydrate (0.06 g) were dissolved in 20 mL ethanol. The solution was refluxed during 1 hour, and an orange powder precipitated upon reflux. The orange powder was filtered, washed with 5 mL cold ethanol. Recrystallization from ethanol gives the pure complex as orange powder. Yield: 0.12 g (82%). MS (DEI-(+), 70 eV) m/z (%): 652 (100) (M+); elemental analysis calculated (found) for C26H24N4O6Zn (653.99 g mol−1): C 62.44 (62.32), H 4.32 (4.27), N 8.57 (8.51).
X-ray structure analysis
The X-ray analysis of the [NiL1] and [CuL1(EtOH)]·2CHCl3 complexes was performed with a Stoe StadiVari diffractometer using graphite-monochromated MoKα radiation. The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods (SIR-97)23 and refined by full-matrix least-square techniques against Fo2–Fc2 (SHELXL-97).24 All hydrogen atoms were calculated in idealised positions with fixed displacement parameters. ORTEP-III25 was used for the structure representation, SCHAKAL-9926 to illustrate molecule packing. CCDC 1055605 and 1055606.Powder diffractograms were measured with a STOE StadiP Powder Diffractometer (STOE, Darmstadt) using Cu[Ka1] radiation with a Ge Monochromator, and a Mythen 1K Stripdetector in transmission geometry.
Optical measurements
Absorbance spectra were obtained using a Agilent UV-vis spectrophotometer 8453 (Agilent Technologies, USA) operating in a spectral range of 190–1100 nm. The spectra were measured at 298 K in quartz cells with 1 cm lightpath (Hellma, Germany). Fluorescence measurements were performed with a FLS980 (Edinburgh Instruments, UK) fluorescence spectrometer equipped with double monochromators (1800 grooves per mm) in excitation and emission. A Xe lamp (360 W) was used as excitation source for steady-state fluorescence measurements. Time-resolved measurements were performed with a pulsed supercontinuum fiber laser (WhiteLase SC-400, Fianium, UK) with a pulse width <10 ps and a repetition rate of 5 MHz. Emission from the sample was collected at an angle of 90° with respect to the excitation path. The emitted beam was focused onto a single photon counting photomultiplier (R928P, Hamamatsu, Japan) or a microchannel plate photomultiplier (MCP-PMT) for steady-state and lifetime measurements respectively. All spectra were measured at 298 K in quartz cells (Hellma, Germany) with 1 cm lightpath. The excitation wavelength and time range for lifetime measurements were selected according to the sample requirements. The time-resolved fluorescence decays were analysed by deconvolution fits accounting for the instrument response function (IRF) obtained from a Rayleigh scatterer.Conclusion
Two new Schiff base-like ligands bearing a fluorophore were synthesised and successfully used for complexation of Ni(II), Cu(II) and Zn(II). While for the Cu(II) complexes the fluorescence is quenched, the Zn(II) and Ni(II) complexes retain the photoluminescent properties. We have shown that the addition of pyridine induces a change in the geometry of the complex, and consequently a change of the spin state. While the Zn(II) complexes only display a red-shift of the emission upon coordination, the nickel(II) complexes exhibit a more complex behaviour. The sample [NiL1] showed a quenching of its fluorescence correlated to a coordination change-induced spin state change, with a dramatic decrease of the intensity and lifetime of the emission band. This is in agreement with the appearance of non-radiative energy transfer between the fluorophore and the Ni(II) centre in its S = 1 paramagnetic state. The sample [NiL2], bearing a more extended phenanthrene-based fluorophore, displays a more complex behaviour upon coordination of pyridine which requires further investigation. This will be addressed in a future work. Corresponding iron(II) complexes will also be investigated with regard to possible correlation effects between fluorescence and thermally induced spin crossover.Acknowledgements
Financial supports from the German Science foundation (SFB840) and the University of Bayreuth are acknowledged. Mathias Karg and Kristina G. Wagner acknowledge financial support from the German Research Foundation (DFG) via the Emmy-Noether Programme. We thank F. Puchtler (University of Bayreuth) for the collection of powder diffraction data.Notes and references
-
(a)
Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013 Search PubMed
; (b) Spin Crossover in Transition Metal Compounds I-III, ed. P. Gütlich and H. Goodwin, Springer, Berlin/Heidelberg, 2004, pp. 233–235 Search PubMed
-
(a) J. Linares, E. Codjovi and Y. Garcia, Sensors, 2012, 12, 4479–4492 CrossRef CAS PubMed
-
P. N. Martinho, C. Rajnak and M. Ruben, in Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013, pp. 375–404 Search PubMed
- B. Weber, W. Bauer and J. Obel, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed
-
(a) J. Larionova, L. Salmon, Y. Guari, A. Tokarev, K. Molvinger, G. Molnár and A. Bousseksou, Angew. Chem., Int. Ed., 2008, 47, 8236–8240 CrossRef CAS PubMed
-
(a) J.-F. Letard, J. Mater. Chem., 2006, 16, 2550–2559 RSC
-
B. Weber, in Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013, pp. 55–76 Search PubMed
-
(a) S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sonnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed
-
(a) M. Matsuda, H. Isozaki and H. Tajima, Chem. Lett., 2008, 37, 374–375 CrossRef CAS
- L. Salmon, G. Molnár, D. Zitouni, C. Quintero, C. Bergaud, J.-C. Micheau and A. Bousseksou, J. Mater. Chem., 2010, 20, 5499 RSC
- C.-F. Wang, R.-F. Li, X.-Y. Chen, R.-J. Wei, L.-S. Zheng and J. Tao, Angew. Chem., Int. Ed., 2015, 54, 1574–1577 CrossRef CAS PubMed
- H. Matsukizono, K. Kuroiwa and N. Kimizuka, Chem. Lett., 2008, 37, 446–447 CrossRef CAS
- Y. Garcia, F. Robert, A. D. Naik, G. Zhou, B. Tinant, K. Robeyns, S. Michotte and L. Piraux, J. Am. Chem. Soc., 2011, 133, 15850–15853 CrossRef CAS PubMed
-
(a) A. Santoro, L. J. Kershaw Cook, R. Kulmaczewski, S. A. Barrett, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2015, 54, 682–693 CrossRef CAS PubMed
- M. Engeser, L. Fabbrizzi, M. Licchelli and D. Sacchi, Chem. Commun., 1999, 1191–1192 RSC
- L. Wolf and E.-G. Jäger, Z. Anorg. Allg. Chem., 1966, 346, 76–91 CrossRef CAS
- C. Lochenie, S. Schlamp, A. P. Railliet, K. Robeyns, B. Weber and Y. Garcia, CrystEngComm, 2014, 16, 6213 RSC
- W. Bauer, T. Ossiander and B. Weber, Z. Naturforsch., B: J. Chem. Sci., 2010, 323–328 CAS
-
(a) N. Chopin, M. Médebielle, O. Maury, G. Novitchi and G. Pilet, Eur. J. Inorg. Chem., 2014, 6185–6195 CrossRef CAS
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC
-
B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence. Principles and applications, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed
-
(a) N. P. Loveless, K. C. Brown and R. H. Horrocks, J. Org. Chem., 1981, 46, 1182–1185 CrossRef CAS
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
(a)
C. K. Johnson and M. N. Burnett, ORTEP-III, Oak-Ridge National Laboratory, Oak-Ridge, TN, 1996 Search PubMed
-
E. Keller, Schakal-99, University of Freiburg, Freiburg, Germany, 1999 Search PubMed
Footnotes |
| † Dedicated to Prof. Manfred Scheer on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1055605 and 1055606. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc00837a |
| This journal is © The Royal Society of Chemistry 2015 |