
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thia- and selena-diazole containing polymers for near-infrared light-emitting diodes†
Giulia
Tregnago
a,
Timothy T.
Steckler
b,
Oliver
Fenwick
c,
Mats R.
Andersson
bd and
Franco
Cacialli
*a
aDepartment of Physics and Astronomy and London Centre for Nanotechnology, University College London, London WC1E 6BT, UK. E-mail: f.cacialli@ucl.ac.uk
bChalmers University of Technology, Gothenburg, SE-412 96, Sweden
cISIS & icFRC, Université de Strasbourg & CNRS, 8, allée Gaspard Monge, 67000 Strasbourg, France
dIan Wark Research Institute, University of South Australia, Mawson Lakes, South Australia 5095, Australia
First published on 17th February 2015
Abstract
We report the optical characterization of near-infrared (NIR) emitters for polymer light-emitting diode (PLEDs) applications based on the copolymerization of a phthalimide-thiophene host polymer with a low-gap emitter containing the bisthienyl(benzotriazolothiadiazole) unit. We investigate different loadings of the low-gap emitter (in the range 1–3% by weight) and the substitution of a sulphur atom with a selenium atom to further extend the emission in the NIR up to 1000 nm. PLEDs based on copolymers with 1% loading give the best efficiency (0.09%) and show an almost pure NIR EL (95% in the NIR) peaking at 895 nm.
Over the last few years near-infrared (NIR) organic light-emitting diodes (OLEDs) have generated considerable interest for their potential application in the medical, telecommunication and defence fields.1,2 Interestingly, the peculiarity of light emission in the NIR region (700–2500 nm) can be combined with the major advantages of OLEDs such as the solution processing, the low-cost fabrication and the possibility of using flexible, conformable or even stretchable substrates.3,4 Given the importance of NIR optoelectronics, different organic compounds have been explored as red and NIR emitters, such as small molecules,5–7 metal–organic complexes8,9 and conjugated polymers,10,11 whilst inorganic nanoparticles12 and materials “improperly” but commonly indicated as perovskites13 have been used as NIR emitters in solution processed LEDs with organic charge transport layers. Furthermore, different strategies have been employed to reduce emission quenching and aggregation effects of low-gap emitters and/or promote energy transfer to NIR moieties, e.g. by blending the NIR emitters with wider gap polymers,14,15 by diluting them in a matrix by co-evaporation,16,17 by exploiting cyclic or linear molecular π-systems with appropriate ligands to inhibit aggregation18 or via charge-transfer excitons at the organic semiconductors heterojunction with a proper gap between the energy levels for the emission to fall in the NIR.19 Among these strategies, inclusion of donor–acceptor–donor (DAD) low-gap units in wider gap host polymers via copolymerization has been demonstrated as a valid approach.20,21 In particular, benzothiadiazole and both its homo- and hetero-annulated derivatives have been extensively investigated22–24 in conjunction with different host polymers for efficient NIR emission.21,25–27 A promising route to extend further the emission in the near-infrared region is the replacement of the sulphur atom in the benzothiadiazole unit with selenium. In fact, benzoselenodiazole units have been reported to reduce the polymer energy gap and lead to a red-shift of the absorption and emission spectra compared to the sulphur-containing unit.28–33 The efficiency of NIR emitters is generally lower than that of visible emitters, owing to more efficient non-radiative quenching of the excited states which follows from the smaller number of vibrational quanta needed to dump the energy of excited states in vibrational deactivation processes. Polymers for light-emitting diodes (PLEDs) with emission beyond 850 nm have been reported with external quantum efficiencies (EQEs) of only 0.02–0.05%,26,34 although recently we reported an EQE of 0.27% for a NIR PLED emitting at 885 nm.25 In this and earlier reports we found that using ambipolar host polymers such as phthalimide-thiophene25 and F8BT14,18,35 can yield high EQEs for NIR emission. Following from the success of using phthalimide-thiophene host polymers, in this study we look at modifications to the host polymer structure and the NIR emitting moiety to shift the emission further into the NIR whilst maintaining high EQEs.
We present NIR emitters (see Fig. 1a) based on a phthalimide-thiophene host polymer (P1) copolymerised with a low-gap DAD moiety based on the bisthienyl(benzotriazolothiadiazole) unit at different loadings, 1% and 3% (P2 and P3, respectively) calculated with respect to the host polymer portion for the initial ratio of reactants. To lower the energy gap, we also exchanged a sulphur atom in the thiadiazole for a selenium atom (P4). Note that the branched alkyl side chains of the DAD unit for the P4 copolymer is longer than those of the P2 and P3 copolymers. It should also be noted that the phthalimide-thiophene host polymer used in this study has a straight alkyl chain (C16H33) as the solubilizing group whereas a phthalimide-thiophene host with branched side-chains (CH(C8H17)2) was used for NIR OLEDs in our previous study.25 In the past study we showed that a straight side-chain phthalimide-thiophene copolymers showed higher and more balanced ambipolar field-effect mobilities, increased order and higher photoluminescence quantum efficiencies than its branched side-chain analogue, but in this paper we report the use of these straight-chain phthalimide-thiophene copolymers as host materials for OLEDs for the first time. In particular, we found a relatively high EQE (0.09%) for P2 with an electroluminescence (EL) at 895 nm characterized by a high spectral purity (>95% in the NIR). We also show that the substitution of the sulphur atom in the thiadiazole unit of the DAD with a selenium atom (P4) red-shifts the emission to a band peaking at 990 nm, also yielding one of the most efficient PLEDs reported to date at such a long wavelength.26
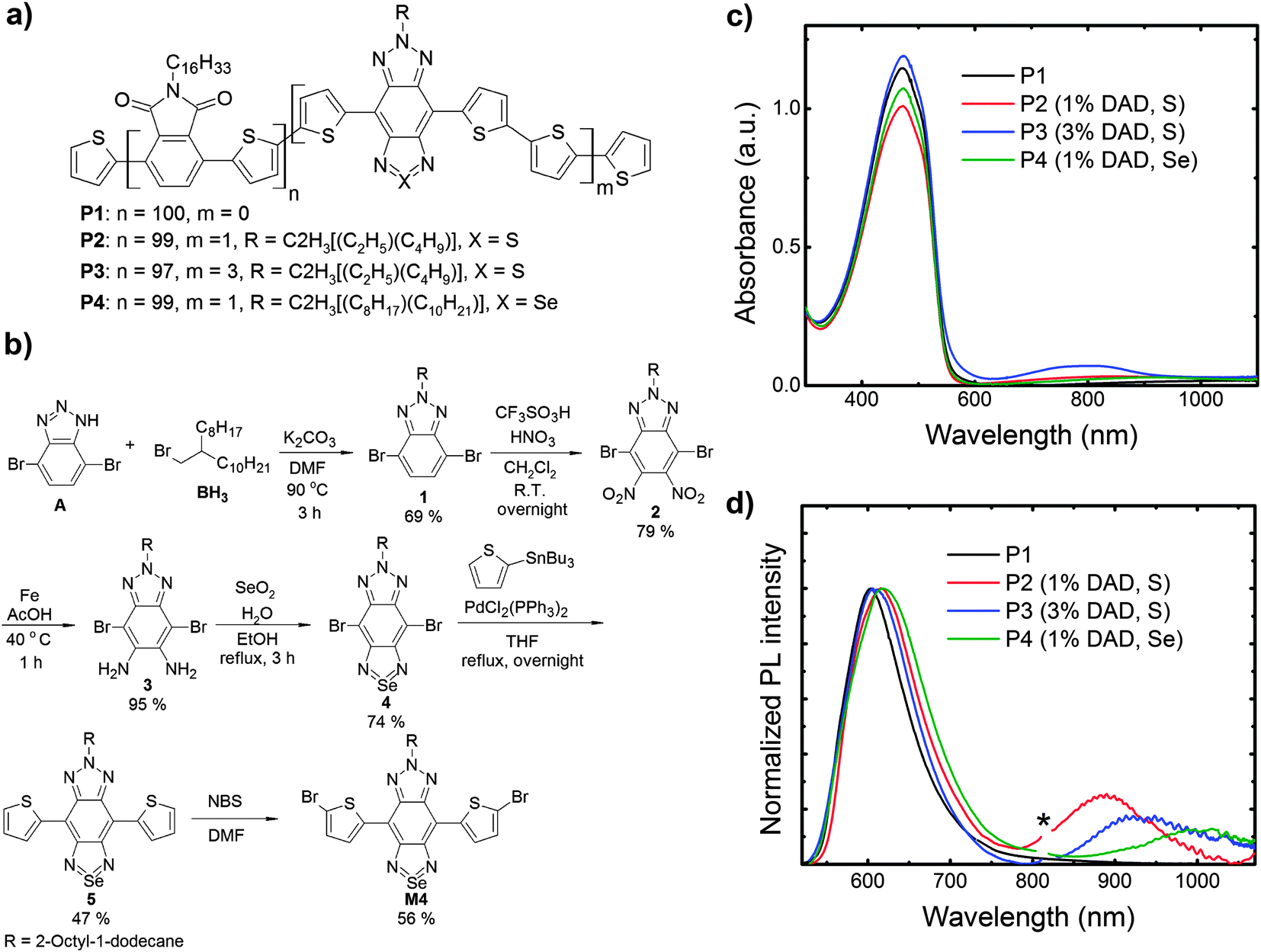 | ||
| Fig. 1 (a) Chemical structure of the wide-gap host polymer (P1) and the copolymers (P2, P3 and P4). P2 and P3 differ in the DAD loading (1% and 3% respectively). The DAD unit in P2 and P4 differs in the substitution of a sulphur atom for a selenium atom to lower the energy gap. (b) Synthesis of monomer M4 (P4 precursor). (c) Absorption spectra of the polymers thin films (100 nm) over fused silica glass. We report in the legend the percentage of DAD moieties and the type of atom (S or Se) in the DAD segment. (d) PL of polymers thin films, the “*” indicates the monochromator 2nd-order transmission of the excitation wavelength (λex = 405 nm). | ||
For polymer P4, the synthesis of the bisthienyl(benzotriazoloselenadiazole) (M4) emitter can be seen in Fig. 1b. Initially, 4,7-dibromo-1H-benzo[d][1,2,3]triazole was alkylated with 2-octyl-1-bromododecane using K2CO3 in DMF yielding 1 in 69%. Next, an improved nitration using triflic acid and fuming nitric acid in DCM resulted in the formation of 2 (79%),36 which upon reduction using iron in acetic acid gave 3 in 95%. Ring closing with SeO2 yielded 4 in 74%. Stille coupling of 4 and 2-tributylstannylthiopene resulted in 5 (47%), which was then brominated to yield M4 (56%).
Following our previous work, the synthesis of the copolymers (P1–P4) were carried out via Stille polymerization and worked up in a similar manner (see ESI†).25 Polymers P1 and P2 had number average molecular weights of 8.1 and 9.5 kg mol−1, which are similar to our previous results for these type of polymers.25 Polymers P3 and P4 had slightly higher number average molecular weights of 15.1 and 12.1 kg mol−1. All polymers were thermally stable with 1% weight loss occurring at temperatures >300 °C (under N2). For this study square-wave voltammetry was used to determine the HOMO/LUMO levels of the polymers. P1 has a HOMO of −6.05 eV and a LUMO of −3.45 eV (Fig. S1, ESI†). These values are very similar to what we reported previously for the same polymer with either a different end-capping unit or no end-capping.25 Due to the small loadings of the NIR-emitting segments, square-wave voltammetry of polymers P3–P4 showed no signal from the low gap segments as we saw previously for polymers with similar loadings.25 Likewise, based on previous studies of similarly structured compounds in the literature, we estimated the HOMO/LUMO levels of the NIR-emitting segments to be ∼−5.1/−4.0 ± 0.1 eV for segment M323 and a slightly narrower HOMO/LUMO gap for segment M4. Since the HOMO/LUMO levels of these segments lie within the HOMO/LUMO levels of the host polymer P1, this ought to allow for energy transfer to the NIR-emitting segments.37–39
We report the absorption spectra of the polymer films in Fig. 1c, in which we note that the host polymer (P1) absorption band is centred at 470 nm. As expected, all the copolymers display the host polymer absorption peak. In addition, P3 (the copolymer with 3% b.w. content of DAD) clearly shows a band at 790 nm that is not visible in the host polymer, and that we thus assign to the DAD moiety. The DAD absorption is not clearly distinguishable in the other copolymers (P2 and P4) due to the lower content (1% b.w.) of such a moiety. However the solution spectra show the absorption feature of the DAD unit peaking at 756 nm, 767 nm and 859 nm for P2, P3, and P4 respectively (see Fig. S2, ESI†). We report the PL spectra of the polymers in Fig. 1d. Emission from the host polymer (P1) features a band at 605 nm, and as intended, the copolymers also show an emission band in the NIR region, peaking at 895 nm (P2), 927 nm (P3) and 1000 nm (P4), respectively. We attribute such bands to states delocalised over the DAD segments. Emission from the host polymer is still visible in the PL spectra of the copolymers, thereby suggesting that energy transfer (ET) from the host segment to the DAD is not complete. This is consistent with the fact that the spectral overlap between the host polymer emission and the DAD unit absorption is not optimal as the emission of P1 is not centred on the DAD unit absorption (this is detailed by the shaded area in Fig. 2a). We also expect a red-shift of the lower energy band when increasing the DAD loading21 as a result of aggregation. Similarly, we expect an even more significant red-shift when substituting S with Se, owing to a lower LUMO energy level,40,41 as also suggested from the solution spectra in Fig. S2 (ESI†). Whereas this is appealing for the purpose of achieving an as pure as possible NIR emission, any red-shift should also lead to a less efficient energy transfer from P1, which is undesired. Both expectations (red-shift and less-efficiency ET, leading to lower NIR intensity) are confirmed by the trends observed in Fig. 1d. In fact, we observe that the percentage of the PL in the NIR (i.e. taken for wavelengths >700 nm) are 31% (P2), 24% (P3) and 21% (P4), respectively. Upon increasing the DAD loading from 1% (P2) to 3% (P3), we notice a red-shift of the DAD unit PL emission from 895 nm (P2) to 927 nm (P3). We attribute such a red-shift to aggregation of the DAD moieties.21 The exchange of sulphur with selenium in the benzothiadiazole also shifts the DAD unit emission further from 895 nm (P2) to 1000 nm (P4). Indeed, the introduction of benzoselenadiazole moieties is thus confirmed as a successful approach to lower the polymer energy gap and enable emission up to 1000 nm.
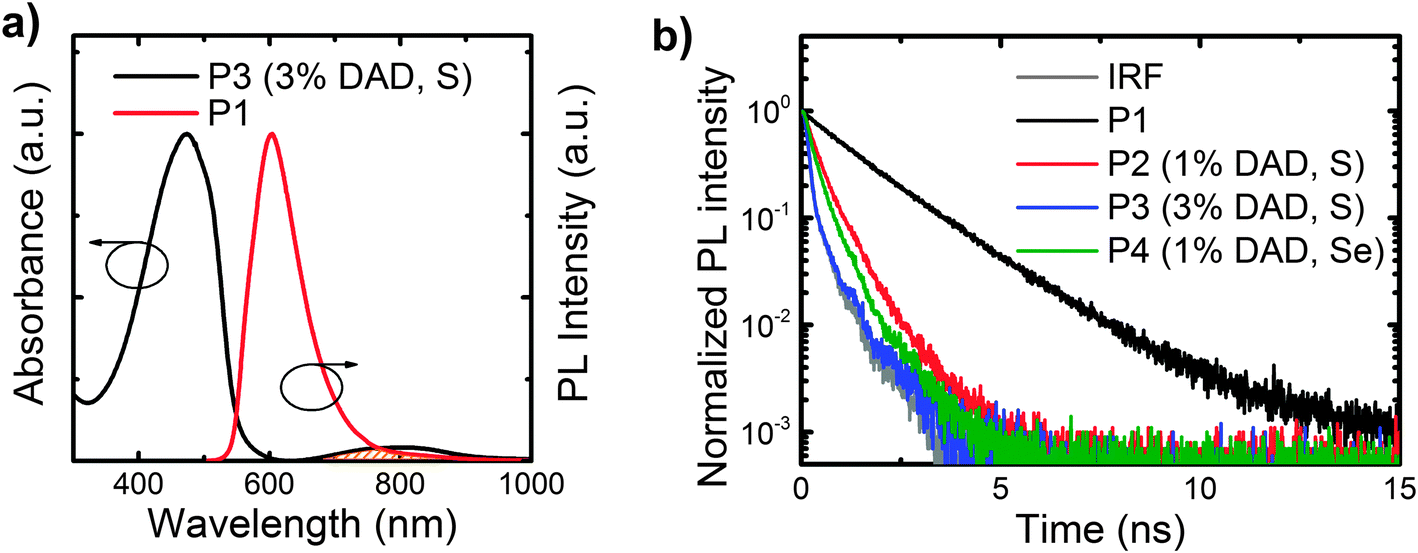 | ||
| Fig. 2 (a) Normalized absorption spectrum of P3 superimposed on the normalized emission spectrum of P1. The overlap between the absorption of the DAD unit and the P1 emission is highlighted in orange. (b) PL time decay for thin films (100 nm thick on fused silica glass) of polymers P1, P2, P3 and P4 taken at 610 nm, following excitation at 371 nm. The instrument response function (IRF) is also reported. | ||
We also found the films PLQE efficiency (in the 500–1100 nm range) to drop from 14.8% for the host polymer P1, to 1.4% for P2, 1.0% for P4, and below our 1% sensitivity limit for P3. As mentioned, such a reduction is entirely expected as a result of both the reduction of the energy gap, and because of quenching/aggregation (e.g. in the comparison P2vs.P3). To investigate further the energy transfer between the host polymer and the DAD unit, we measured the PL lifetime decays of the host polymer emission (at 610 nm) for all the samples, and we report the results in Fig. 2b. The lifetimes of the host polymer P1 can be fitted with a mono-exponential that returns a 1.65 ns time constant. The copolymers show a drop in the lifetime to 0.50 ns for P2 and to 0.32 ns for P4 whereas the lifetime of P3 is below the detection limit of the apparatus.42 We note that such reduction in the lifetime follows the PL quenching of the host polymer by the presence of the DAD unit. The quenching is stronger when using Se instead of S and when increasing the concentration of the DAD unit.
We also incorporated the copolymers into PLEDs with ITO/PEDOT:PSS anodes and Ca/Al cathodes. We report the EL spectra of the devices above in Fig. 3a. Whereas the host polymer shows essentially a single band (albeit with a main peak at 560 nm and a shoulder at 605 nm), the EL from the copolymers is predominantly in the NIR and peaking at 895 nm for P2, 939 nm for P3 and ∼990 nm for P4. Although relatively noisy, it is possible to note that the emission of P4 shows a shoulder at 807 nm. The percentage of NIR (>700 nm) EL is 95% (P2), 87% (P3) and 88% (P4), respectively. Even though the host polymer emission is also present to some extent in the EL of the copolymers, it is largely suppressed in EL compared to the PL (and in fact nearly completely suppressed in P2 devices). Such suppression (compared to the PL) is due to the energy-selective injection and trapping and transport of charges via the NIR moieties, and subsequent formation of excitons at such sites (which effectively act as traps). Residual visible EL from P3 is thus easily reconciled by taking into account the relatively high voltage needed for such a spectrum (60 V) that would also enable some degree of charge injection and transport via the host. Additionally, we note that spectra in Fig. 3a are normalised, thereby amplifying the visible spectral region for those devices that are less efficient in the NIR (as suggested by the higher noise levels of P3 and P4 spectra compared to that of P2).
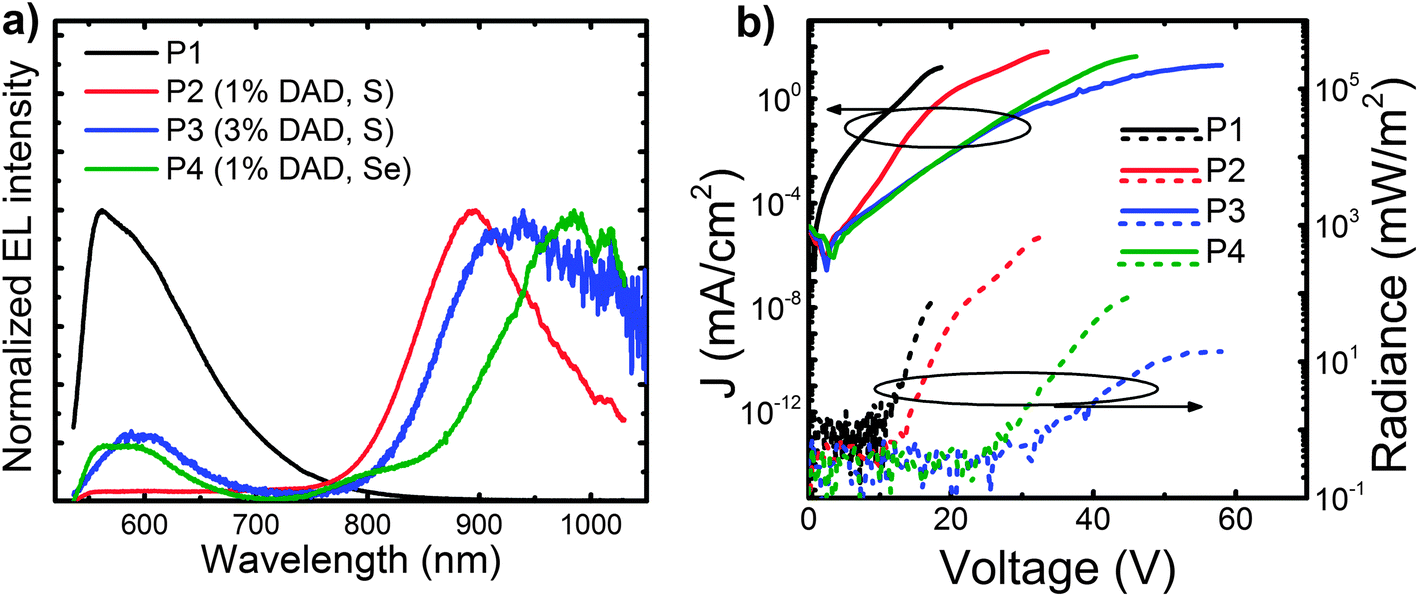 | ||
| Fig. 3 (a) EL of the polymers taken at 20 V (P1), 33 V (P2), 60 V (P3) and 42 V (P4). (b) PLEDs characteristics: current density and radiance versus voltage. The active layer thickness is ∼100 nm and the device area is 3.5 mm2. | ||
Finally, we report a summary of the PLEDs characteristics in Table 1, and the current–radiance–voltage characteristics in Fig. 3b. We find that the best results are obtained for the devices incorporating the low-content S-based DAD units (1%, P2), for which we achieve a maximum EQE of 0.091%, an irradiance of 291 mW m−2 (measured at 20 mA cm−2), and most importantly, with nearly pure NIR emission peaking at 895 nm (95% > 700 nm). These results are among the best reported in the literature for a single active layer NIR PLED at such a long wavelength.14,18,21,25,26 Regrettably, although perhaps not surprisingly, at 3% DAD loading the external quantum efficiency decreases significantly (from 0.09 to 0.006%), whereas the turn-on voltage (Von) increases from 14.3 V for P2 (1% DAD) to 28.1 V for P3 (3% DAD). The increased driving voltage is easily attributed to an increased number of traps, related to the higher concentration of DAD units, which have a lower energy gap compared to P1. In addition we attribute the EQE reduction to the expected aggregation of the DAD units, which is also corroborated further by the previously-discussed PL red-shifts. A similar trend has also been reported by some of us for other low-gap polymers copolymerized with a wide-gap host.21,35 Interestingly by comparing P2 and P4, we are also able to get an insight into the influence of the exchange of a sulphur atom for selenium as a strategy to achieve NIR emission at longer wavelengths. We notice again that such a substitution leads to a decreased EQE and an increased Von. However we point out that despite a disappointing Von, not only is Se substitution a better approach for increasing the wavelength of the emission than increasing DAD loading, but it also gives one of the most efficient devices at ∼1 μm reported to date for a polymer.
| Polymer | Max EQE (%) | V on (V) | Radianceb (mW m−2) | NIR PL peak (nm) | % PL in NIRc | NIR EL peak (nm) | % EL in NIRc |
|---|---|---|---|---|---|---|---|
| a Intercept of the I–V curve with the x-axis in a semi-log plot. b Measured at 20 mA cm−2. c Defined as λ > 700 nm. | |||||||
| P1 | 0.037 ± 0.008 | 9.0 ± 0.8 | 98 ± 13 | — | — | — | — |
| P2 | 0.091 ± 0.004 | 14.3 ± 2.9 | 291 ± 9 | 890 | 31 | 895 | 95 |
| P3 | 0.006 ± 0.002 | 28.1 ± 1.1 | 16 ± 2 | 927 | 24 | 939 | 87 |
| P4 | 0.018 ± 0.004 | 23.5 ± 1.5 | 58 ± 10 | 1000 | 21 | 990 | 88 |
We can also compare P2 to our previously reported polymer, where the only difference is that in the previous study a branched alkyl chain (–CH(C8H17)2) was used on the host polymer where as in this study we used the straight alkyl chain (C16H33).25 We can see that in this study, NIR PLEDs constructed from P2 suffer from slightly higher turn-on voltages, and a factor of 3 lower EQE (0.09% vs. 0.27%). Thus, in comparing these two host polymers, it is surprising that the increased order, luminescence and higher (and more balanced) mobility provided by the straight alkyl chain (C16H33) on the host polymer, as characterized previously,25 did not result in the best host polymer for NIR PLEDs. Interestingly, the emission for the PLED based on P2 is at 895 nm, which is red-shifted 10 nm compared to the previous study using the branched alkyl chain (885 nm). This supports the idea that lower order in the previous system likely prevented some aggregation of the NIR-emitting segments, resulting in higher performance. Even though the lower and unbalanced mobilities of the branched alkyl side-chains would have suggested higher resistance in the devices, we observe no significant differences in the operating voltages of devices when compared to the devices using linear side-chain host polymers.25 This suggests that charge trapping on the low gap DAD segments is the dominant source of resistance in these devices.
Conclusions
In summary, we have characterised the NIR emission of low-gap DAD units copolymerized with a wider gap phthalimide-thiophene polymer. PLEDs based on copolymers with 1% DAD loading give the best efficiency (∼0.09%) and EL peaking at 895 nm. We show that the copolymerization is a successful strategy to obtain almost pure NIR EL (up to 95% of the overall emission) for benzothiadiazole-based polymers. By varying the loading of the DAD moieties it is possible to shift the emission further into the NIR however at a cost of lowered PLED efficiency because of increased aggregation. As an alternative and effective approach to shift the emission into the NIR (up to 1000 nm) we report the use of Se containing materials, that produce a lower impact on PLEDs EQE and driving voltage when compared to S-based copolymers with a higher DAD loading. We have also demonstrated that in using the phthalimide-thiophene copolymer as a host polymer for NIR PLEDs, the more disordered system using the branched alkyl chain (CH(C8H17) vs. C16H33) results in better performing NIR PLEDs. In addition, we consider that there should be significant margins of improvements for the spectral purity by further engineering the chemical design of these copolymers so as to provide lower-energy-gap host units, and thus ensure better spectral overlap, and more efficient energy transfer to the NIR moieties.Acknowledgements
We thank the EC Seventh Framework Programme (FP7/2007-2013) under Grant Agreement No. 264694 (GENIUS), No. 607585 (OSNIRO), the EU Horizon 2020 Research and Innovation Programme under Grant Agreement No. 643238 (SYNCHRONICS), the Royal Society and the EPSRC. FC is a Royal Society Wolfson Research Merit Award holder.Notes and references
- T. Karu, in Biomedical Photonics Handbook, ed. T. Vo-Dinh, CRC Press, 2003 Search PubMed.
- H. Suzuki, J. Photochem. Photobiol., A, 2004, 166, 155–161 CrossRef CAS PubMed.
- M. S. White, M. Kaltenbrunner, E. D. Glowacki, K. Gutnichenko, G. Kettlgruber, I. Graz, S. Aazou, C. Ulbricht, D. A. M. Egbe, M. C. Miron, Z. Major, M. C. Scharber, T. Sekitani, T. Someya, S. Bauer and N. S. Sariciftci, Nat. Photonics, 2013, 7, 811–816 CrossRef CAS.
- T. Sekitani and T. Someya, Adv. Mater., 2010, 22, 2228–2246 CrossRef CAS PubMed.
- X. Du, J. Qi, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2012, 24, 2178–2185 CrossRef CAS.
- Y. X. Yang, R. T. Farley, T. T. Steckler, S. H. Eom, J. R. Reynolds, K. S. Schanze and J. G. Xue, J. Appl. Phys., 2009, 106, 044509 CrossRef PubMed.
- Y. Sun, C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, J. Brooks, J. J. Brown and S. R. Forrest, Appl. Phys. Lett., 2007, 90, 213503 CrossRef PubMed.
- T. V. Duncan, K. Susumu, L. E. Sinks and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 9000–9001 CrossRef CAS PubMed.
- E. L. Williams, J. Li and G. E. Jabbour, Appl. Phys. Lett., 2006, 89, 083506 CrossRef PubMed.
- G. Tzamalis, V. Lemaur, F. Karlsson, P. O. Holtz, M. Andersson, X. Crispin, J. Cornil and M. Berggren, Chem. Phys. Lett., 2010, 489, 92–95 CrossRef CAS PubMed.
- E. Perzon, F. Zhang, M. Andersson, W. Mammo, O. Inganäs and M. R. Andersson, Adv. Mater., 2007, 19, 3308–3311 CrossRef CAS.
- K. R. Choudhury, D. W. Song and F. So, Org. Electron., 2010, 11, 23–28 CrossRef CAS PubMed.
- Z. K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- P. Li, O. Fenwick, S. Yilmaz, D. Breusov, D. J. Caruana, S. Allard, U. Scherf and F. Cacialli, Chem. Commun., 2011, 47, 8820–8822 RSC.
- E. L. Williams, J. Li and G. E. Jabbour, Appl. Phys. Lett., 2006, 89, 083506, DOI:10.1063/1.2335275.
- C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, K. Aznavour, R. Bau, Y. Sun, S. R. Forrest, J. Brooks, L. Michalski and J. Brown, Angew. Chem., 2007, 46, 1109–1112 CrossRef CAS PubMed.
- R. J. Curry and W. P. Gillin, Appl. Phys. Lett., 1999, 75, 1380–1382 CrossRef CAS PubMed.
- O. Fenwick, J. K. Sprafke, J. Binas, D. V. Kondratuk, F. Di Stasio, H. L. Anderson and F. Cacialli, Nano Lett., 2011, 11, 2451–2456 CrossRef CAS PubMed.
- G. Tregnago, C. Flechon, S. Choudhary, C. Gozalves, M. Mateo-Alonso and F. Cacialli, Appl. Phys. Lett., 2014, 105, 143304, DOI:10.1063/1.4898135.
- B. C. Thompson, L. G. Madrigal, M. R. Pinto, T.-S. Kang, K. S. Schanze and J. R. Reynolds, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1417–1431 CrossRef CAS.
- T. T. Steckler, O. Fenwick, T. Lockwood, M. R. Andersson and F. Cacialli, Macromol. Rapid Commun., 2013, 34, 990–996 CrossRef CAS PubMed.
- T. L. Tam, H. Li, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 4612–4615 CrossRef CAS PubMed.
- D. G. Patel, F. Feng, Y. Y. Ohnishi, K. A. Abboud, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 2599–2612 CrossRef CAS PubMed.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J.-L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2014, 2, 22–36 RSC.
- T. T. Steckler, M. J. Lee, Z. Chen, O. Fenwick, M. R. Andersson, F. Cacialli and H. Sirringhaus, J. Mater. Chem. C, 2014, 2, 5133–5141 RSC.
- M. X. Chen, E. Perzon, M. R. Andersson, S. Marcinkevicius, S. K. M. Jonsson, M. Fahlman and M. Berggren, Appl. Phys. Lett., 2004, 84, 3570–3572 CrossRef CAS PubMed.
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage, S. P. Tiwari, S. Coppee, S. Ellinger, S. Barlow, J. L. Bredas, B. Kippelen, J. R. Reynolds and S. R. Marder, J. Mater. Chem., 2010, 20, 123–134 RSC.
- R. Q. Yang, R. Y. Tian, Q. Hou, W. Yang and Y. Cao, Macromolecules, 2003, 36, 7453–7460 CrossRef CAS.
- I. H. Jung, H. Kim, M. J. Park, B. Kim, J. H. Park, E. Jeong, H. Y. Woo, S. Yoo and H. K. Shim, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1423–1432 CrossRef CAS.
- J. H. Hou, T. L. Chen, S. Q. Zhang, H. Y. Chen and Y. Yang, J. Phys. Chem. C, 2009, 113, 1601–1605 CAS.
- L. Yang, J. K. Feng and A. M. Ren, THEOCHEM, 2007, 816, 161–170 CrossRef CAS PubMed.
- C. M. MacNeill, R. C. Coffin, D. L. Carroll and N. H. Levi-Polyachenko, Macromol. Biosci., 2013, 13, 28–34 CrossRef CAS PubMed.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539–547 CrossRef CAS PubMed.
- G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111–116 CrossRef CAS.
- O. Fenwick, S. Fusco, T. N. Baig, F. Di Stasio, T. T. Steckler, P. Henriksson, C. Flechon, M. R. Andersson and F. Cacialli, APL Mater., 2013, 1, 032108, DOI:10.1063/1.4820433.
- C. L. Coon, W. G. Blucher and M. E. Hill, J. Org. Chem., 1973, 38, 4243–4248 CrossRef CAS.
- X. Gong, J. C. Ostrowski, D. Moses, G. C. Bazan and A. J. Heeger, Adv. Funct. Mater., 2003, 13, 439–444 CrossRef CAS.
- V. Cleave, G. Yahioglu, P. Le Barny, D. H. Hwang, A. B. Holmes, R. H. Friend and N. Tessler, Adv. Mater., 2001, 13, 44 CrossRef CAS.
- C. C. Wu, J. C. Sturm, R. A. Register, J. Tian, E. P. Dana and M. E. Thompson, IEEE Trans. Electron Devices, 1997, 44, 1269–1281 CrossRef CAS.
- H. Padhy, J. H. Huang, D. Sahu, D. Patra, D. Kekuda, C. W. Chu and H. C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4823–4834 CrossRef CAS.
- H. Y. Chen, S. C. Yeh, C. T. Chen and C. T. Chen, J. Mater. Chem., 2012, 22, 21549–21559 RSC.
- Please note that the decays reported in Fig. 2b have not been deconvolved to remove the instrumental response, therefore do not look mono-exponential, but we have used an algorithm taking into account such a response to fit the data with a mono-exponential, which returns fits with a χ2 < 1.3.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and materials characterization. See DOI: 10.1039/c5tc00118h |
| This journal is © The Royal Society of Chemistry 2015 |