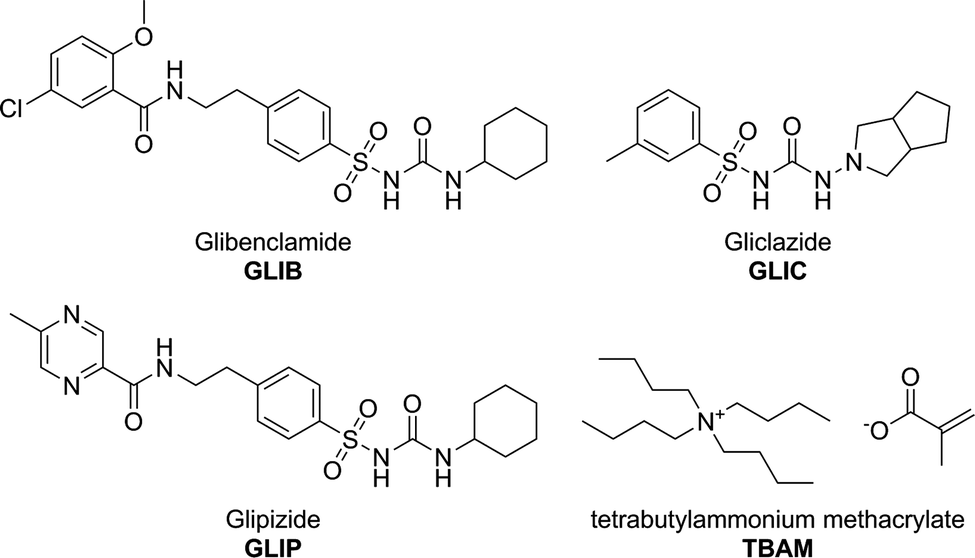
Tetrabutylammonium methacrylate as a novel receptor for selective extraction of sulphonylurea drugs from biological fluids using molecular imprinting
Received
27th July 2015
, Accepted 25th September 2015
First published on 25th September 2015
Abstract
Glibenclamide (GLIB), an oral antidiabetic medication of the sulphonylurea drug family, was stoichiometrically imprinted using tetrabutylammonium methacrylate as the functional monomer, for the first time in molecular imprinting, and utilising the sulphonylurea affinity for carboxylate anions. Solution association between the drug and the novel functional monomer was studied by 1H-NMR titrations, whereby evidence of sulphonylurea deprotonation followed by the formation of “narcissistic” GLIB dimers was found when tested in CDCl3, while an affinity constant in excess of 105 L mol−1 was measured in DMSO-d6. Detailed analysis of GLIB binding on the subsequently prepared imprinted and non-imprinted polymers confirmed the deactivation of binding sites by exchange of a proton between GLIB and methacrylate, followed by extraction of the tetrabutylammonium counterion from the polymer matrix, resulting in overall reduced binding capacities and affinities by the imprinted material under equilibrium conditions. An optimised MI-SPE protocol, which included a binding site re-activation step, was developed for the extraction of GLIB from blood serum, whereby recoveries of up to 92.4% were obtained with an exceptional sample cleanup.
Introduction
Glibenclamide (GLIB), also known as glyburide, is an oral antidiabetic medication and belongs to the second generation sulphonylurea drug family, used in the treatment of diabetes mellitus. It acts by stimulating the release of insulin from pancreatic β-cells and peripheral tissue sensitivity to insulin.1 Glibenclamide is more potent than the first generation sulphonylurea drugs and is a medication of choice for initiating treatment for non-insulin dependent diabetes mellitus when diet fails. Controlling the therapeutic concentration of glibenclamide in blood as well as the study of its pharmacokinetic profile requires rapid and sensitive analytical methods. The most commonly employed method for the analysis of glibenclamide is by HPLC, with UV or mass spectrometry detection, GC, and MEKC using non-ionic surfactants.2 All of the above methods reported in the literature require a liquid–liquid extraction step for blood sample pre-treatment, which is a labour intensive, time consuming process and is often associated with sample loss. Solid phase extraction (SPE) methods for glibenclamide extraction from human plasma have been reported using C8 or C18 cartridges coupled in line with HPLC and capillary electrophoresis.3
Herein, we wish to report on the development of novel Molecularly Imprinted Solid-Phase Extraction (MI-SPE) materials for the rapid pre-concentration and cleanup of glibenclamide in biological fluid samples prior to HPLC analysis. Molecular imprinting is a technique that enables the generation of selective binding sites within the matrix of a synthetic polymer by co-polymerisation of selected functional and cross-liking monomers in the presence of a target substance.4 Thus, solution phase complexes between the so-called template and functional monomers are locked in place upon cross-linking of the growing polymer chains. The resulting binding sites are revealed by removal of the template by simple solvent wash and are thus capable of selective rebinding of the template or closely related substances.5 Such materials have already shown promise in bioanalytical applications combining high selectivity and sample cleanup prior to subsequent analysis.6
Our approach was inspired by our previous work in the development of custom functional monomers for the recognition of anionic species.7 In particular, we have demonstrated that urea-based functional monomers are powerful receptors for carboxylate anions and have employed them in the recognition of folic acid and the analogous anticancer drug methotrexate.8 In the present work we report on the first use of tetrabutylammonium methacrylate (TBAM) in molecular imprinting, as a receptor for the sulphonylurea moiety of glibenclamide. We verified the validity of our hypothesis by 1H-NMR titrations and our results showed that TBAM greatly outperformed commercially available monomers commonly used in molecular imprinting, such as methacrylic acid (MAA) and acrylamide (AM). Molecularly imprinted polymers synthesised using TBAM as the functional monomer exhibited high selectivity for glibenclamide over similar sulphonylureas, and we were able to achieve recoveries of the drug from blood serum up to 92.4% using an optimised MI-SPE protocol.
Experimental
Materials and methods
Methacrylic acid (MAA), acrylamide (AM), tetrabutylammonium hydroxide (TBAOH), ethyleneglycol dimethacrylate (EDMA), 2,2′-azobis(2-methylpropionitrile) (AIBN), trifluoroacetic acid (TFA), and deuterated solvent for NMR were purchased from Sigma Aldrich. Polymerisation inhibitors were removed from MAA and EDMA by filtration through a basic alumina column and AIBN was recrystallised from acetone prior to use. Tetrabutylammonium methacrylate (TBAM) was synthesised by mixing equimolar amounts of tetrabutylammonium hydroxide (1.0 mol L−1 solution in methanol) and methacrylic acid in methanol. Following evaporation of the solvent under reduced pressure, TBAM was obtained as a viscous liquid and was stored in the fridge for further use. HPLC grade solvents methanol and acetonitrile were purchased from JT Baker. Pro analysis grade solvents acetone and acetic acid were purchased from Merck. Glibenclamide (GLIB) was provided by Hexpharm Pharmaceuticals Industry. Glipizide (GLIP) was purchased from Indonesia National Agency of Drug and Food Control. Gliclazide (GLIC) was provided by Dexa Medica Pharmaceuticals Industry (Fig. 1). Blood samples were provided by the Indonesian Red Cross. 1H NMR spectra were collected on a Bruker ECX 400 MHz NMR spectrometer. FT-IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR spectrometer equipped with an ATR attachment. An Agilent 1100 HPLC instrument equipped with a photodiode array detector was used for all chromatographic separations. Analyses were performed by isocratic elution using a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 water/acetonitrile mixture containing 0.01% TFA as the mobile phase and a Kinetex C18 column (5 μm, 150 mm × 4.6 mm i.d.). The flow rate was 1 mL min−1 and the detection wavelength was set at 230 nm. Using these conditions the LOD and LOQ for GLIB were 0.2 and 0.6 μg mL−1 respectively.
:60 water/acetonitrile mixture containing 0.01% TFA as the mobile phase and a Kinetex C18 column (5 μm, 150 mm × 4.6 mm i.d.). The flow rate was 1 mL min−1 and the detection wavelength was set at 230 nm. Using these conditions the LOD and LOQ for GLIB were 0.2 and 0.6 μg mL−1 respectively.
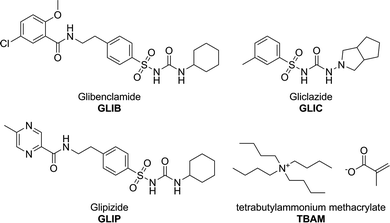 |
| | Fig. 1 Chemical structures of the analytes and the functional monomer used in this study. | |
1H NMR titration experiments
Monomer–template complexation was studied prior to polymer synthesis using 1H NMR titrations in order to establish the type and strength of interactions present in the pre-polymerisation solution. Thus, to a solution of GLIB (0.001 mol L−1) in CDCl3 or DMSO-d6 increasing amounts of each tested functional monomer were added, until at least a 10-fold excess was reached. The monomers studied were acrylamide (AM), methacrylic acid (MAA) and tetrabutylammonium methacrylate (TBAM). The complexation-induced shifts (CIS) of the GLIB urea and amide protons were followed and titration curves were then constructed. The stoichiometry of the each monomer–template complex was then confirmed using Job's method of continuous variation. Hence, equimolar solutions (0.001 mol L−1) of GLIB and each monomer were mixed in different ratios and a plot of Δδ against the molar fraction of the monomer multiplied by the CIS (Xi × Δδ) was constructed.9
Preparation of imprinted polymers
The imprinted (PGLIB) and corresponding non-imprinted (NP) polymers used in this study were prepared by thermally initiated free radical polymerisation. Briefly, GLIB (0.25 g, 0.5 mmol), TBAM (0.33 g, 1 mmol) and MAA (0.085 mL, 1 mmol) were transferred into a glass vial and mixed with 2.8 mL of acetonitrile. Upon complete dissolution, 1.9 mL (10 mmol) of EDMA were added followed by 0.02 g of AIBN. The pre-polymerisation solutions were ultra-sonicated for 5 min, purged with N2 and then hermetically sealed. The vials were placed in a water-bath thermostated at 60 °C for 24 h. The resulting rigid monoliths were smashed and washed with methanol in a Soxhlet apparatus in order to remove the template and any unreacted monomers. The coarse polymer particles were ground using a mortar and pestle, wet-sieved with acetone and the 25–50 μm fraction was collected, dried and stored at room temperature for further experiments. The corresponding non-imprinted polymer was prepared in a similar fashion but without addition of the template in the pre-polymerisation mixture.
Equilibrium rebinding experiments
Polymer affinity and capacity for the template were estimated using equilibrium rebinding experiments performed in chloroform or acetonitrile. Thus, 10 mg of each polymer were transferred in 2 mL glass vials and equilibrated with 1.5 mL of glibenclamide solution of increasing concentrations (0–3 mmol L−1) for 24 hours. The supernatants were then analysed by HPLC using the method described above. The amount of glibenclamide bound to the polymer was calculated by subtracting the amount determined after the experiment from the starting amount of the drug. The results are plotted as the concentration of free analyte in solution versus the amount of GLIB bound to the polymer to produce binding isotherms that were fitted using the appropriate binding model.
Solid phase extractions (ΜΙ-SPE)
50 mg of PGLIB and NP particles were dry packed in 3 mL SPE cartridges using 20 μm porous polyethylene frits. SPE protocol optimisation was performed by analysis of aqueous solutions of GLIB prior to analysis of blood serum samples. The optimised SPE protocol consisted of an initial conditioning step with 0.1 mmol L−1 TBAOH, loading of 2 mL of spiked blood sample, followed by an aqueous wash (1 mL) and final elution of the cartridges with 99:1 methanol/acetic acid (0.5 mL). Full vacuum was applied to the cartridges between each step. Blood serum samples were prepared by centrifugation of the collected blood at 8000 rpm for 5 minutes at 14 °C and careful collection of the clear top layer. This was spiked with 5 μg mL−1 of GLIB in 95:5 water/acetonitrile. In order to test the selectivity of the phases an equimolar mixture of GLIB, GLIC and GLIP (5 μg mL−1 each) in 95/5 water/acetonitrile was spiked into blood serum samples and applied onto the SPE cartridges. The collected fractions were analysed by HPLC using the method described above.
Results and discussion
Template–monomer association in solution
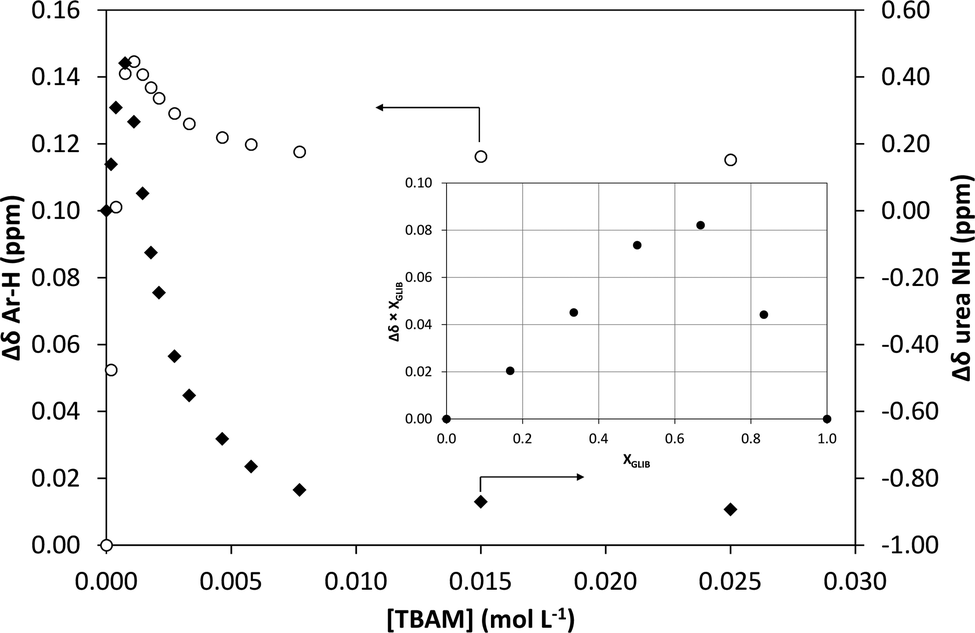
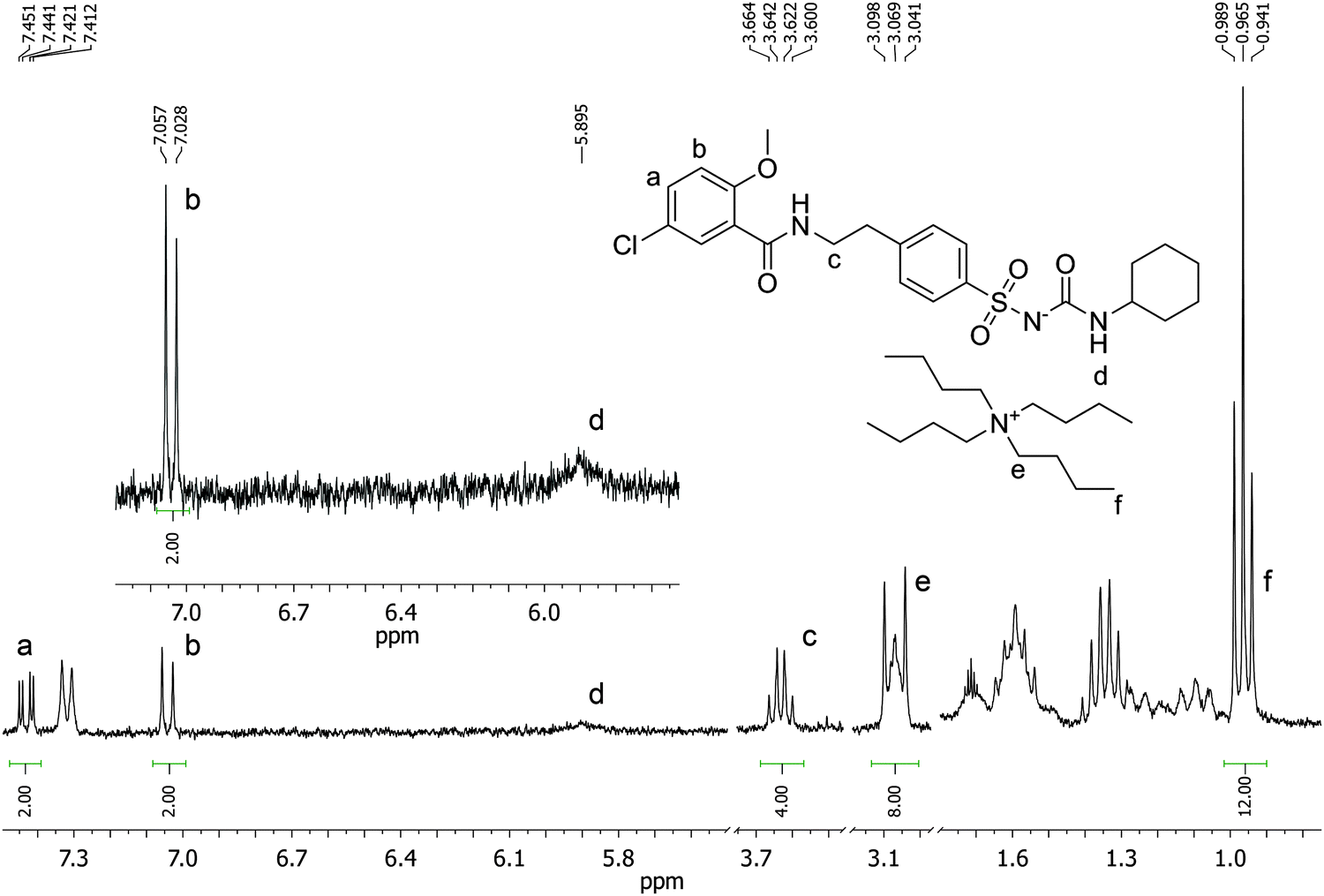
1H NMR titrations were used in order to ascertain the type and strength of interactions between GLIB (Host) and a series of functional monomers, namely AAM, MAA and TBAM (Guest). A number of signals were monitored during the titration experiments, with the urea proton situated next to the cyclohexyl group and the aromatic protons of the p-substituted ring adjacent to the sulphonyl group being the most useful, as they are directly affected by the interaction with the selected monomers. It should be noted that the NH adjacent to the sulphonyl group could not be observed in CDCl3. AAM is a neutral functional monomer, hence its interaction with GLIB was very weak and an association constant <20 L mol−1 was calculated in CDCl3. The acidic monomer MAA interacted more strongly with GLIB and an association constant of 145 L mol−1 was calculated. A rather unexpected behaviour was observed when GLIB was titrated with TBAM. Thus, in the first part of the titration, up to 1.0 equivalent of the added monomer, the urea proton shifted downfield as expected however, as soon as an excess of TBAM was added, the urea NH signal shifted up-field, and reached a negative plateau at 5.55 ppm, 0.9 ppm below the chemical shift observed at the start of the titration. A similar, albeit not as dramatic behaviour, was observed for the aromatic protons adjacent to the sulphonyl group (Fig. 2). It is noteworthy that although the irregular shape of the isotherms constructed in this case did not allow the calculation of an affinity constant, the rapid establishment of equilibrium and the large chemical shifts compared to the AAM and MMA experiments suggested a very strong interaction. Indeed a similar effect was previously reported when a series of acidic sulphonamides were titrated against basic anions, such as acetate and hydroxide.10 In this study, the authors attributed the irregular shapes of the obtained isotherms to deprotonation of the host receptor by the basic anion in the low-polarity solvent used (CD3CN). In order to further investigate the GLIB–TBAM interaction, we performed a Job plot experiment, where it was found that a 2:1 complex was formed, instead of the usual 1:1 stoichiometry found in urea–carboxylate systems. A dilution study of GLIB in CDCl3 did not reveal evidence of dimerisation, hence it was postulated that the methacrylate anion was responsible for the formation of a higher order complex. In an earlier study Gale et al. have shown that highly acidic diamidopyrrole receptors formed “narcissistic” complexes in the presence of basic anions, such as fluoride, via the formation of amide NH⋯N− (pyrrole) hydrogen bonds.11 These findings support our hypothesis that upon addition of TBAM to a solution of GLIB, the latter is deprotonated and GLIB⋯[GLIB-H+]− complexes are formed, hence observation of the interaction of GLIB with TBAM suggests the formation of a 2:1 complex, as one equivalent of TBAM is required to deprotonate an equivalent amount of GLIB, which subsequently forms H-bonds with the neutral drug to yield a “narcissistic” dimer.
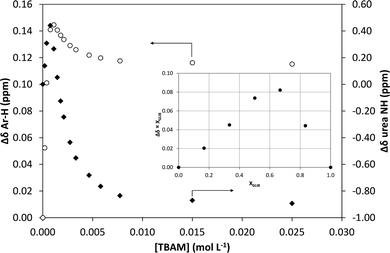 |
| | Fig. 2 Binding isotherms obtained during 1H NMR titration of GLIB with TBAM in CDCl3. Open circles correspond to the shift of the aromatic protons and closed diamonds to the urea proton. Inset: Job plot for the association of GLIB with TBAM in CDCl3 where the formation of 2:1 complexes is verified. | |
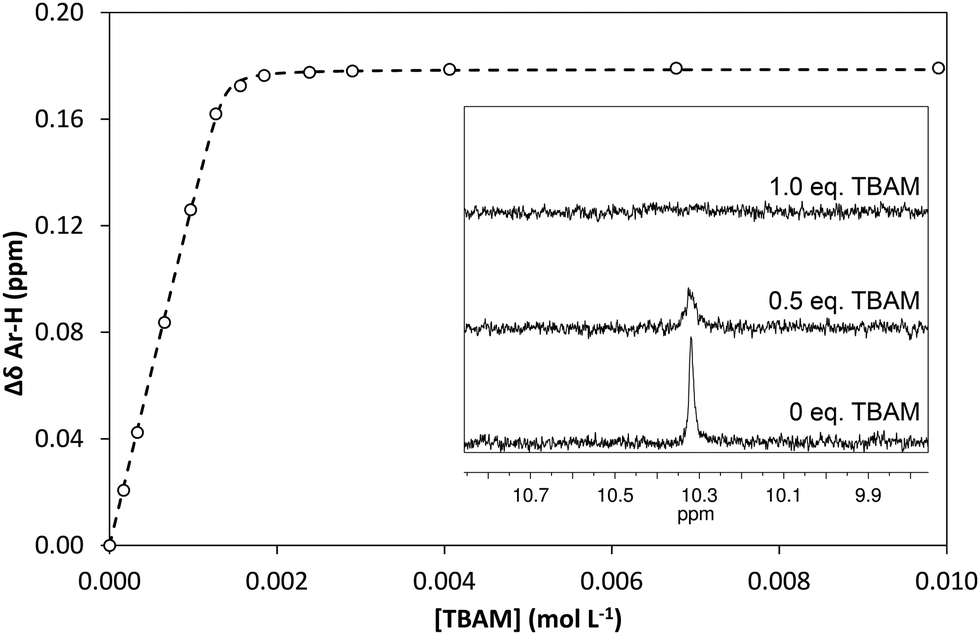
In order to quantify the strength of the GLIB–TBAM interaction, we repeated the titration experiment in DMSO-d6. In this case, the obtained 1H NMR titration isotherm suggested the formation of 1:1 complexes, verified by an additional Job plot experiment also performed in DMSO-d6, and an affinity constant >105 L mol−1 was calculated (Fig. 3). However, as seen in the Fig. 3 inset, deprotonation of the acidic urea NH also takes place in DMSO-d6, with the corresponding signal at 10.3 ppm broadening and eventually disappearing when >1.0 equivalent of TBAM is added.
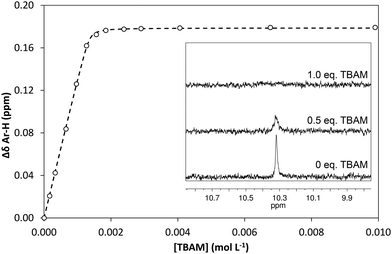 |
| | Fig. 3 Binding isotherms obtained during 1H NMR titration of GLIB with TBAM in DMSO-d6. Inset: Decrease of sulphonyl urea 1H NMR signals in the presence of increasing amounts of TBAM supporting the proposed deprotonation mechanism. | |
Polymer synthesis and characterisation
Based on the findings of the 1H NMR titration experiments, it was originally decided to prepare an imprinted polymer with a 1:1 ratio of GLIB:TBAM in acetonitrile. Nonetheless, early rebinding results indicated that although significant binding of GLIB was observed, there was virtually no imprinting effect, and both imprinted and non-imprinted polymers bound the same amount of drug (data not shown). Thus, an imprinted polymer with a 1:2:2 ratio of GLIB:MAA:TBAM was prepared, aiming to primarily target the sulphonylurea H-bond donor moiety with the anionic monomer, and also introduce secondary points of interaction, such as sulphonyl or the amide groups present in the molecule. Furthermore, the presence of an acidic monomer could counteract the deprotonation of the acidic sulphonylurea and prevent dimerisation of GLIB in the pre-polymerisation solution.
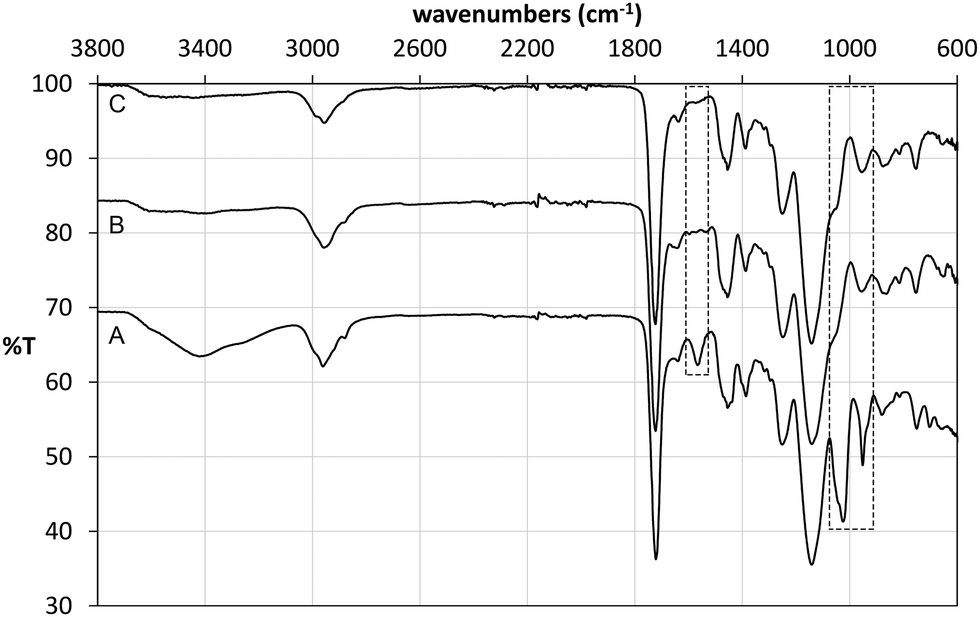
Following polymerisation, the resulting materials were washed with methanol in order to remove the template and any unreacted monomers. 1H NMR analysis of the washing solutions revealed that a significant proportion of the tetrabutylammonium cation (TBA+) was also removed from the polymer matrix, from both imprinted and non-imprinted polymers. Furthermore, signals corresponding to the vinyl protons of methacrylic acid were also detected, indicating that the monomer was not completely incorporated in the non-imprinted polymer. These findings were verified by FT-IR analysis of the materials before and after Soxhlet extraction (Fig. 4). Indeed it was found that the broad band at 3418 cm−1 (carboxylic acid O–H stretch) as well as bands at 1025 cm−1 (C–O stretch) and 952 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch) decreased significantly after washing of NP, indicating loss of methacrylate. Although we were not able to unambiguously assign any of the signals corresponding to TBAM, bands in the region of 700 cm−1 and 1450 cm−1, indicative of methylene rocking and bending respectively, appear with reduced intensity following polymer washing. An interesting observation was made based on the band at 1566 cm−1, which is attributed to carboxylate CO stretching. Hence, although this band appears in the FT-IR spectrum of NP (Fig. 4A), this is present neither in the spectrum of PGLIB before (Fig. 4B) nor after washing of the polymer (Fig. 4C). This is in agreement with the findings of solution evaluation of the GLIB–TBAM complexation and suggests that GLIB is deprotonated upon interaction with TBAM leaving neutral carboxylic acid moieties in the final polymer.
C stretch) decreased significantly after washing of NP, indicating loss of methacrylate. Although we were not able to unambiguously assign any of the signals corresponding to TBAM, bands in the region of 700 cm−1 and 1450 cm−1, indicative of methylene rocking and bending respectively, appear with reduced intensity following polymer washing. An interesting observation was made based on the band at 1566 cm−1, which is attributed to carboxylate CO stretching. Hence, although this band appears in the FT-IR spectrum of NP (Fig. 4A), this is present neither in the spectrum of PGLIB before (Fig. 4B) nor after washing of the polymer (Fig. 4C). This is in agreement with the findings of solution evaluation of the GLIB–TBAM complexation and suggests that GLIB is deprotonated upon interaction with TBAM leaving neutral carboxylic acid moieties in the final polymer.
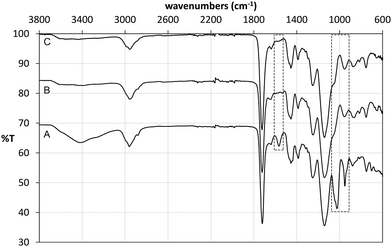 |
| | Fig. 4 ATR FT-IR spectra of NP before (A), PGLIB before (B) and PGLIB after (C) Soxhlet extraction with methanol. | |
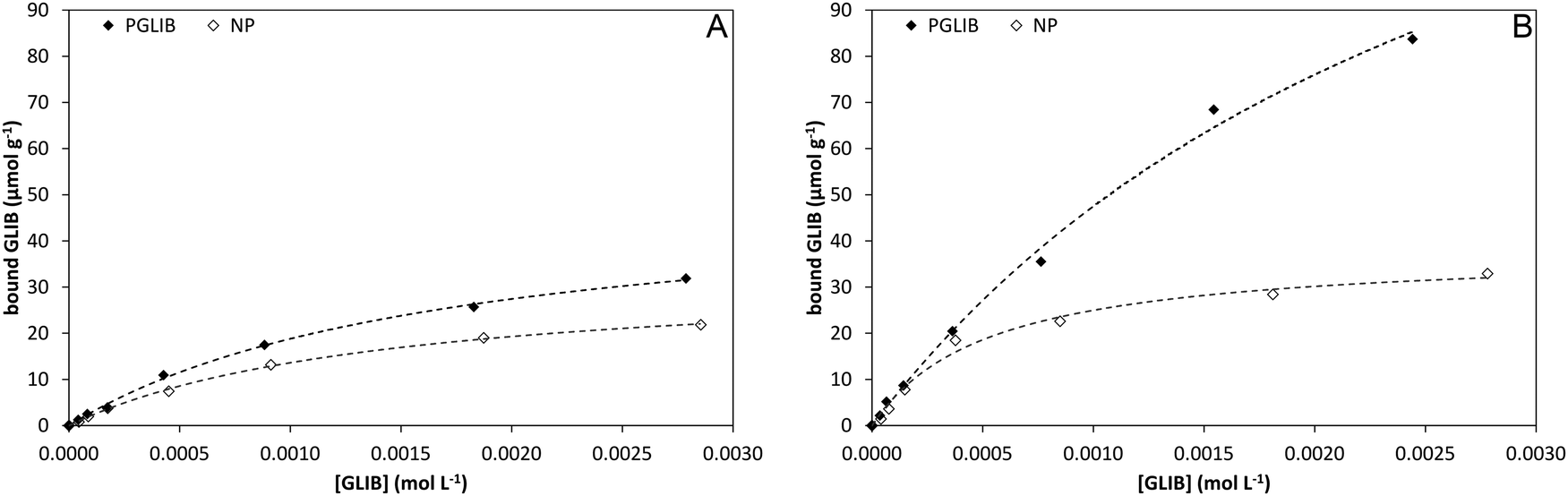
Polymer evaluation by equilibrium rebinding
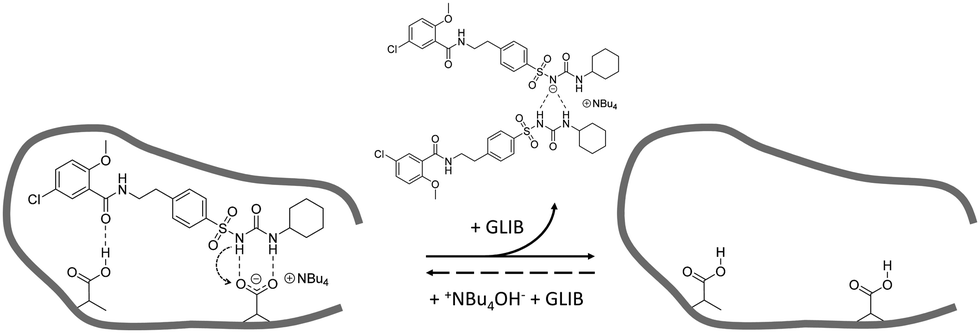
Batch rebinding experiments using PGLIB and NP were initially performed in acetonitrile, the solvent used as a porogen in the preparation of these polymers. The corresponding binding isotherms are presented in Fig. 5A, and indicate a moderate binding capacity and an imprinting effect. Fitting of the experimental results to the Langmuir binding isotherm by non-linear regression reveals similar affinity constants for the binding of GLIB on both polymers, while the imprinted polymer has a higher capacity of 50.7 μmol g−1 compared to 33.1 μmol g−1 of the non-imprinted counterpart (Table 1). The binding process was further investigated by 1H NMR spectroscopy, whereby 1 mL of a 0.002 mol L−1 solution of GLIB in CD3CN or 1 mL of pure CD3CN as control was allowed to equilibrate with 10 mg of PGLIB and NP particles. Analysis of the control supernatants showed no “bleeding” of TBA+; however, both rebinding solutions contained a 2:1 ratio of GLIB:TBA (Fig. 6). Under the conditions of this experiment the amount of TBA+ contained within the polymer (up to 5 μmol) is significantly greater than the amount of GLIB in solution (2 μmol), hence this observation suggests an interesting binding mechanism: upon addition of GLIB to each polymer containing TBAM, a proton transfer takes place from GLIB to the polymerised methacrylate anions, converting the latter to neutral carboxylic acid moieties. This process yields [GLIB-H+]− anions that form dimers and whose negative charge is balanced by the TBA+ cation, which is no longer associated with the polymer (Fig. 7).
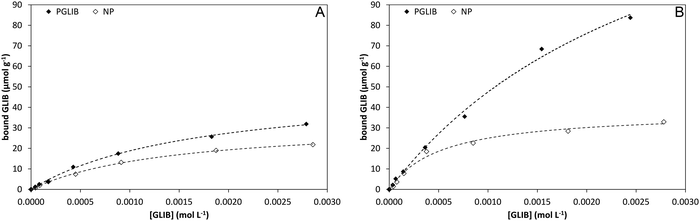 |
| | Fig. 5 Binding isotherms and corresponding Langmuir model fitted curves for PGLIB and NP in acetonitrile (A) and chloroform (B). | |
Table 1 Affinity constants (Ka, L mol−1) and number of binding sites (N, μmol g−1) calculated using the Langmuir binding model, from batch rebinding in different solvents presented in Fig. 5
| Solvent |
PGLIB |
NP |
|
K
a
|
N
|
K
a
|
N
|
| CH3CN |
0.6 × 104 |
50.7 |
0.7 × 104 |
33.1 |
| CHCl3 |
0.3 × 104 |
190.3 |
1.9 × 104 |
38.0 |
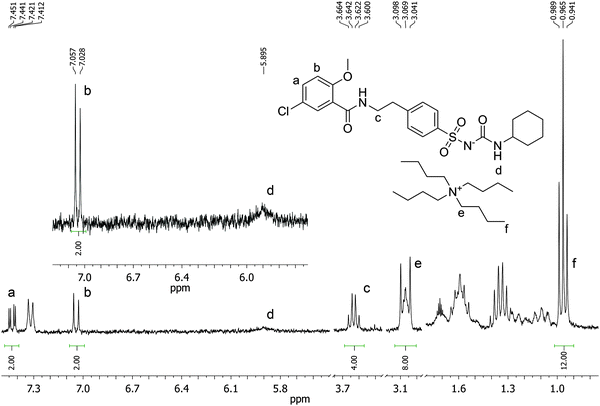 |
| | Fig. 6
1H NMR spectrum of the supernatant solution of NP (10 mg) following equilibration with a 2 mmol L−1 solution of GLIB in CD3CN showing a 2:1 ratio of GLIB:TBA signals. | |
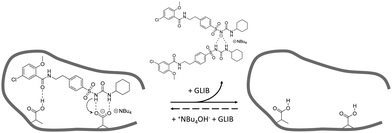 |
| | Fig. 7 Proposed binding mechanism of GLIB onto the prepared imprinted polymer and formation of the “narcissistic” GLIB dimers. | |
This mechanism was verified by the observation that following a round of equilibrium rebinding experiments and subsequent washing of PGLIB and NP particles with methanol, both polymers lost their capacity for GLIB binding. Indeed, PGLIB and NP performance after recycling was comparable to this exhibited by both polymers when they were washed with a dilute solution of HCl, whereby nearly all binding was lost, highlighting the crucial role of methacrylate anions in the binding process. Interestingly, polymer performance was fully restored following reactivation with TBAOH, indicating that the deactivation of the binding sites was reversible, a feature of great importance for materials intended to be used repeatedly in extraction processes.
In order to further investigate the binding characteristics of the synthesised polymers, the rebinding experiment was repeated in chloroform (Fig. 5B). In this case, PGLIB exhibited significantly increased binding for GLIB, with a calculated capacity of 190 μmol g−1, while NP performed only marginally better. This behaviour was attributed to the fact that chloroform caused both polymers to swell significantly, possibly revealing more binding sites on the imprinted polymer, as well as its significantly lower polarity compared to acetonitrile. The latter property results in reduced stability of charged species in solution and hence higher abundance of GLIB in its native neutral form, which is complementary to the methacrylate moieties of the imprinted polymer. On the contrary, when the rebinding experiment was performed in DMSO, no binding was observed on either PGLIB or NP, due to stronger stabilisation of [GLIB-H+]− anions, which are not compatible with the functionality present in the prepared polymers.
Extraction of glibenclamide from blood serum samples
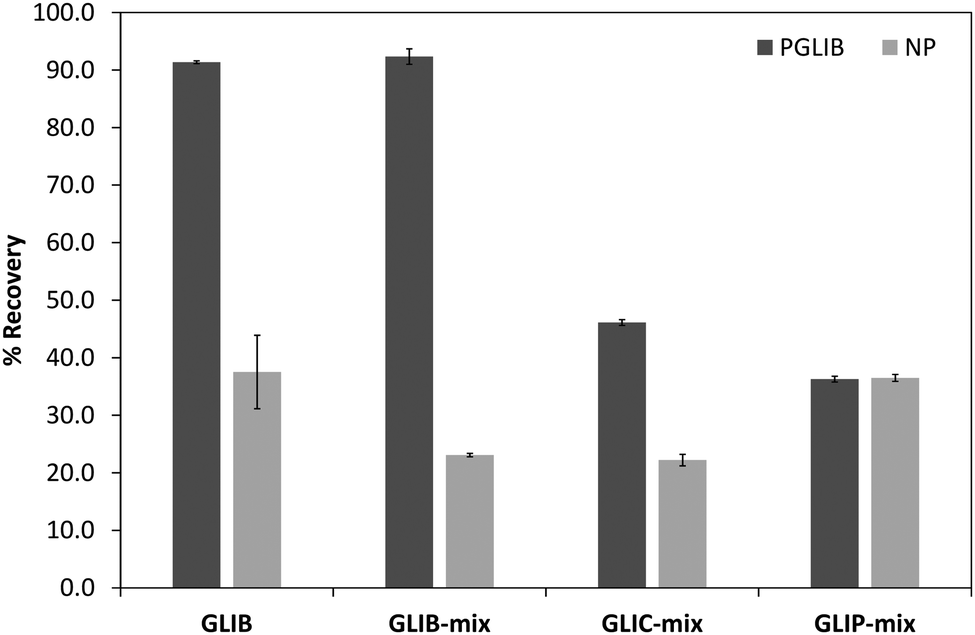
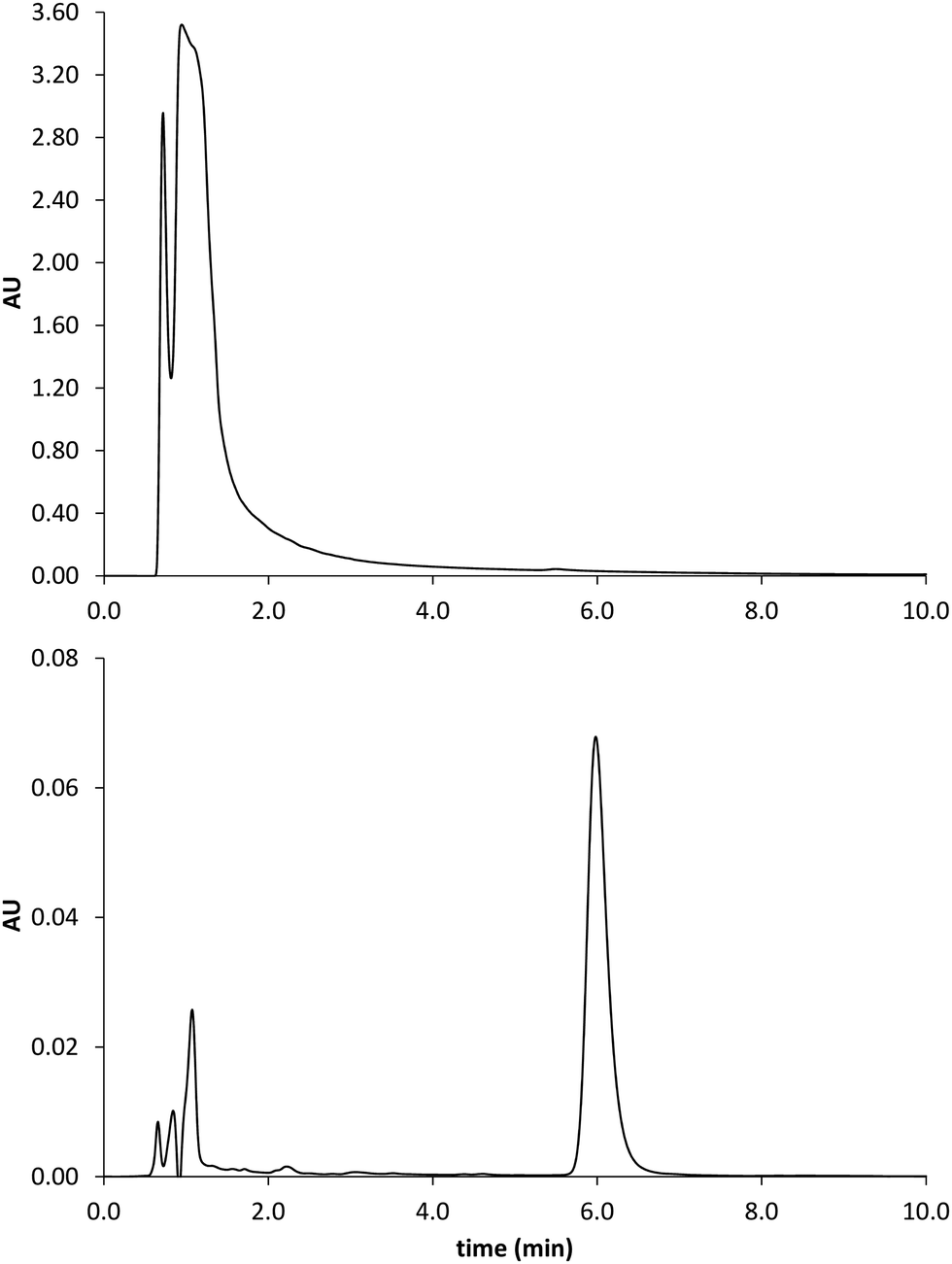
The prepared polymers were lastly evaluated as SPE sorbents for the selective extraction and pre-concentration of GLIB. Aqueous samples of GLIB were used during the initial optimisation phase where it was found that removal of TBA+ at the polymer preparation stage described previously, resulted in reduced recovery of the drug and the latter was not bound by the polymers at the loading stage. This indicated that a conditioning step with TBAOH was required in order to activate the carboxylic acid moieties present in the polymers by deprotonating them and converting them to the anionic form compatible to the sulphonylurea functional group of the target analyte. Indeed when the cartridges were conditioned with 1 mL of 0.1 mmol L−1 solution of TBAOH near quantitative removal of GLIB was achieved from the aqueous solutions. Furthermore, it was observed that template analogous compounds GLIC and GLIP were only partially retained under these conditions with 45.4% and 35.5% respective recoveries in the loading solution (Table 2). An aqueous wash to remove non-specifically bound blood serum components was then performed, during which it was observed that while no further amount of GLIB was eluted from the PGLIB cartridge, 66.7% of the drug was washed out of the NP. Significant quantities of GLIC and GLIP were also recovered from both cartridges. At this stage, several combinations of the porogen (acetonitrile) and water or chloroform were tested as washing solvents to promote molecular recognition; however, it was found that in the presence of any of the two organic solvents the majority of GLIB was eluted from the columns, as did both other competing analytes, resulting in reduced final recoveries. In view of the favourable polymer performance in purely aqueous conditions it was decided not to use any organic solvent in the washing step as a single aqueous wash was sufficient to achieve high recoveries for GLIB, up to 92.4% (Fig. 8), and exceptional sample cleanup, as seen in Fig. 9. It should be noted that the entire MI-SPE method development and optimisation, with over 60 full extraction cycles, was performed using a total of three cartridges, highlighting the robustness of the imprinted materials, as well as the efficiency of the regeneration protocol employed.
Table 2 Recoveries (%) of sulfonylurea drugs from blood serum samples using PGLIB and NP in each fraction of the optimised SPE protocol (n = 3)
| Fraction |
PGLIB |
NP |
| GLIB |
GLIC |
GLIP |
GLIB |
GLIC |
GLIP |
| Loading |
7.6 ± 0.3 |
45.4 ± 1.8 |
32.5 ± 0.7 |
10.2 ± 0.4 |
69.0 ± 0.8 |
35.1 ± 0.5 |
| Wash |
0.0 |
8.5 ± 0.2 |
31.3 ± 2.1 |
66.7 ± 8.6 |
8.9 ± 0.7 |
28.4 ± 0.6 |
| Elution |
92.4 ± 1.3 |
46.1 ± 0.5 |
36.2 ± 0.5 |
23.1 ± 0.3 |
22.1 ± 1.1 |
36.5 ± 0.6 |
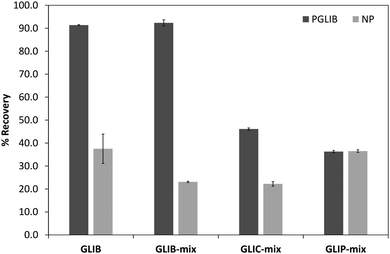 |
| | Fig. 8 Recovery (%) of the different tested analytes from spiked blood serum samples using the optimised SPE protocol (n = 3). | |
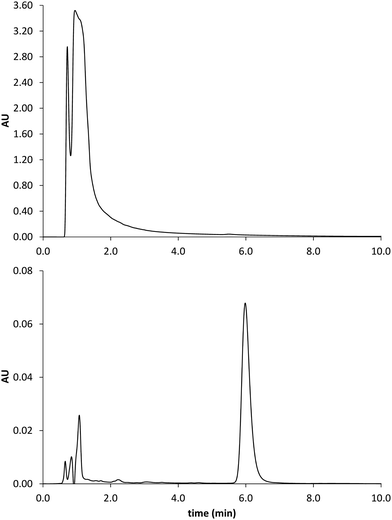 |
| | Fig. 9 Chromatograms obtained by analysis of the spiked serum sample before (upper trace) and after MISPE (lower trace). GLIB elutes at 6 min. | |
Conclusions
Stoichiometric imprinting of glibenclamide (GLIB), an oral antidiabetic medication of the sulphonylurea family, was investigated by introducing tetrabutylammonium methacrylate (TBAM) as a receptor for ureas for the first time in molecular imprinting. The affinity of GLIB for TBAM as well as acrylamide and methacrylic acid was measured by 1H NMR titrations, and the new monomer was found to be the strongest candidate with Ka > 10 L mol−1 in DMSO-d6. When the titration experiment was performed in CDCl3, strong dimerisation of the drug was observed, which was induced by deprotonation of the acidic sulphonylurea NH by methacrylate, followed by the formation of “narcissistic” glibenclamide dimers with TBA+ as the counter-cation. Subsequently prepared imprinted polymers exhibited moderate affinity and capacity for the drug in acetonitrile; however, significantly higher binding was achieved by the imprinted polymer when rebinding was performed in chloroform. In-depth analysis of the rebinding solutions revealed the presence of GLIB in its dimerised form in a 2:1 ratio with TBA+, in agreement with the observations made during solution association studies and suggested that rebinding of GLIB to the polymers resulted in “deactivation” of the binding sites by protonation of the methacrylate moieties rendering them incompatible for binding with the sulphonylurea. When applied as sorbents in the extraction of GLIB from blood serum samples, the binding sites were reactivated by introducing a dilute TBAOH wash prior to sample loading. Following SPE protocol optimisation and using a purely aqueous wash step, exceptional sample cleanup and GLIB recoveries of up to 92.4% were achieved, while recoveries for competing analytes GLIC and GLIP were 46.1% and 36.2% respectively.
This is the first report on the use of TBAM as a functional monomer in molecular imprinting and the results presented herein demonstrate the added value that can be gained by simple modification of a commercial building block to match the functionality of a target substance. We are currently working on the development of further anionic receptors with enhanced affinity for urea and sulphonylurea compounds and will report on those in a future communication.
Acknowledgements
Financial support from the Directorate General of Higher Education, Ministry of Education and Culture of Indonesia is gratefully acknowledged.
Notes and references
- A. A. Abd Elbary, H. F. Salem and M. E. Maher, Br. J. Pharmacol. Toxicol., 2011, 2, 51–62 CAS; N. Haq, F. K. Alanazi, I. A. Alsarra and F. Shakeel, Int. J. Pharma Res. Rev., 2014, 13, 863–872 Search PubMed.
- S. AbuRuz, J. Millership and J. McElnay, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 817, 277–286 CrossRef CAS PubMed; D. Gowrisankar, J. M. Rajendra-Kumar and P. V. M. Latha, Asian J. Chem., 2005, 17, 1334–1336 Search PubMed; P. Hartvig, C. Fagerlund and O. Gyllenhaal, J. Chromatogr., 1980, 181, 17–24 CrossRef; V. Maier, J. Znaleziona, D. Jirovsky, J. Skopalova, J. Petr and J. Sevcik, J. Chromatogr. A, 2009, 1216, 4492–4498 CrossRef PubMed; M. R. Sohrabi, N. Kamali and M. Khakpour, Anal. Sci., 2011, 27, 1037–1041 CrossRef.
- E. P. Lai and S. Y. Feng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 843, 94–99 CrossRef CAS PubMed; J. R. Valdes Santurio and E. Gonzalez Porto, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 1996, 682, 364–370 CrossRef.
-
B. Sellergren, Molecularly imprinted polymers. Man made mimics of antibodies and their applications in analytical chemistry, Elsevier Science B.V., Amsterdam, 2001 CrossRef CAS; J. Steinke, D. C. Sherrington and I. Dunkin, Adv. Polym. Sci., 1995, 123, 81–125 CrossRef CAS.
- L. I. Andersson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2000, 739, 163–173 CrossRef CAS; L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC; F. G. Tamayo, E. Turiel and A. Martín-Esteban, J. Chromatogr. A, 2007, 1152, 32–40 CrossRef PubMed.
- A. Krstulja, C. De Schutter, P. Favetta, P. Manesiotis and L. A. Agrofoglio, J. Chromatogr. A, 2014, 1365, 12–18 CrossRef CAS PubMed; P. Manesiotis, S. Kashani and P. McLoughlin, Anal. Methods, 2013, 5, 3122–3128 RSC.
- P. Manesiotis, A. Riley and B. Bollen, J. Mater. Chem. C, 2014, 2, 8990–8995 RSC.
- A. J. Hall, P. Manesiotis, M. Emgenbroich, M. Quaglia, E. De Lorenzi and B. Sellergren, J. Org. Chem., 2005, 70, 1732–1736 CrossRef CAS PubMed; A. J. Hall, M. Quaglia, P. Manesiotis, E. De Lorenzi and B. Sellergren, Anal. Chem., 2006, 78, 8362–8367 CrossRef PubMed.
-
K. A. Connors, Binding constants; the measurement of molecular complex stability, John Wiley & Sons, New York, 1987 CrossRef CAS; L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, L. Mosca and F. P. Schmidtchen, Chem. – Eur. J., 2011, 17, 5972–5981 CrossRef CAS PubMed; C. Caltagirone, G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2008, 61–63, 10.1039/B713431B.
- S. Camiolo, P. A. Gale, M. B. Hursthouse, M. E. Light and A. J. Shi, Chem. Commun., 2002, 758–759 RSC.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c