
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiphoton microfabrication of conducting polymer-based biomaterials†
John. G.
Hardy‡
abc,
Derek S.
Hernandez‡
d,
Damian M.
Cummings
c,
Frances A.
Edwards
*c,
Jason B.
Shear
*d and
Christine E.
Schmidt
*ab
aDepartment of Biomedical Engineering, The University of Texas at Austin, Austin, TX 78712, USA
bJ. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Biomedical Sciences Building JG-53, P.O. Box 116131, Gainesville, FL 32611-6131, USA. E-mail: schmidt@bme.ufl.edu
cDepartment of Neuroscience, Physiology and Pharmacology, University College London, London, WC1E 6BT, UK. E-mail: f.a.edwards@ucl.ac.uk
dDepartment of Chemistry, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: jshear@cm.utexas.edu
First published on 10th March 2015
Abstract
We report the application of multiphoton microfabrication to prepare conducting polymer (CP)-based biomaterials that were capable of drug delivery and interacting with brain tissue ex vivo, thereby highlighting the potential of multiphoton lithography to prepare electroactive biomaterials which may function as implantable neural biointerfaces (e.g. electrodes).
Electrical fields are known to interact with biological tissues (including cardiac, muscle, nerve and skin tissues), and have been shown to play roles in a variety of biological processes (e.g. cell signalling).1,2 Electroactive biomaterials capable of acting as electrical interfaces with the body (including cardiac pacemakers and electrodes for stimulation of the brain) have been approved for clinical application.3–7
The tuneable properties of CPs (e.g. derivatives of polyaniline, polypyrrole, polythiophene) make them attractive components of electroactive biomaterials for drug delivery devices, electrodes or tissue scaffolds.8–19 CPs have been processed into a variety of materials morphologies including films (via electropolymerization,20 vapour deposition,21 solution casting,22 spin coating23 or dip coating24), fibers (via electrospinning25 or wet spinning26), foams (via sacrificial templating27) and hydrogels (by crosslinking solutions28). While such approaches are effective routes to functional electroactive materials, the ability to prepare biomaterials with de-novo designed architectures,29 particularly CP-based materials30–32 is highly appealing for biomedical applications because the material must interface with biological tissues which tend to be topologically complex and feature anisotropic and asymmetric features.
Researchers have demonstrated it is possible to print CP-based materials using screen printing,30 rotary printing30 (potentially in a roll-to-roll fashion), inkjet printing (potentially in 3D)30 and nozzle extrusion,30 as described in detail in an insightful review from Wallace and co-workers.30
Multiphoton fabrication is an approach that potentially allows the fabrication of de-novo designed architectures with features on biologically relevant length scales (i.e. μm-scale), and, has previously been used to manufacture biodegradable biopolymer-based biomaterials with well-defined topographies capable of interacting with both bacterial and mammalian cells.33–36 It is possible to render polymer structures fabricated via multiphoton lithography electroactive through the growth of a sample spanning network of metal nanoparticles within the polymer matrix,37 or by fabricating electroactive polymer-based structures (e.g. polypyrrole) using the requisite lasers to initiate polymerization of the monomer and prepare electroactive materials on glass or in Nafion® sheets.38–40
With a view to the preparation of functional CP-based biomaterials we used multiphoton lithography to fabricate arrays of polypyrrole wires. The arrays were capable of electrically-triggered drug delivery from drug loaded wires in vitro and of interfacing with a slice of mouse brain ex vivo.
Arrays of polypyrrole wires were fabricated between silver contacts on glass via multiphoton lithography (an example of the wire fabrication process is shown in a video in the supplementary information). A titanium-doped sapphire laser with a wavelength of 740 nm and beam diameter of 1 μm was used to initiate the polymerization of pyrrole (100 μL) in chloroform (900 μL), enabling the fabrication of polypyrrole wires (Fig. 2A and B and Fig. S2, ESI†). Once the fabrication process was complete, the substrate was washed with ethanol to remove traces of pyrrole and dried under high vacuum. The polypyrrole could be doped with camphorsulfonic acid (CSA, Fig. 1) during polymerization at a CSA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pyrrole molar ratio of 1:4, followed by washing with ethanol and vacuum drying. Alternatively, the polypyrrole could be doped post-polymerization by reduction of the polypyrrole with sodium borohydride,41 immersion in a solution of carboxyfluorescein (Fluor-CO2H, Fig. 1) in hexafluoroisopropanol (1 mg mL−1), re-oxidation during air drying, rinsing with water and vacuum drying.
:pyrrole molar ratio of 1:4, followed by washing with ethanol and vacuum drying. Alternatively, the polypyrrole could be doped post-polymerization by reduction of the polypyrrole with sodium borohydride,41 immersion in a solution of carboxyfluorescein (Fluor-CO2H, Fig. 1) in hexafluoroisopropanol (1 mg mL−1), re-oxidation during air drying, rinsing with water and vacuum drying.
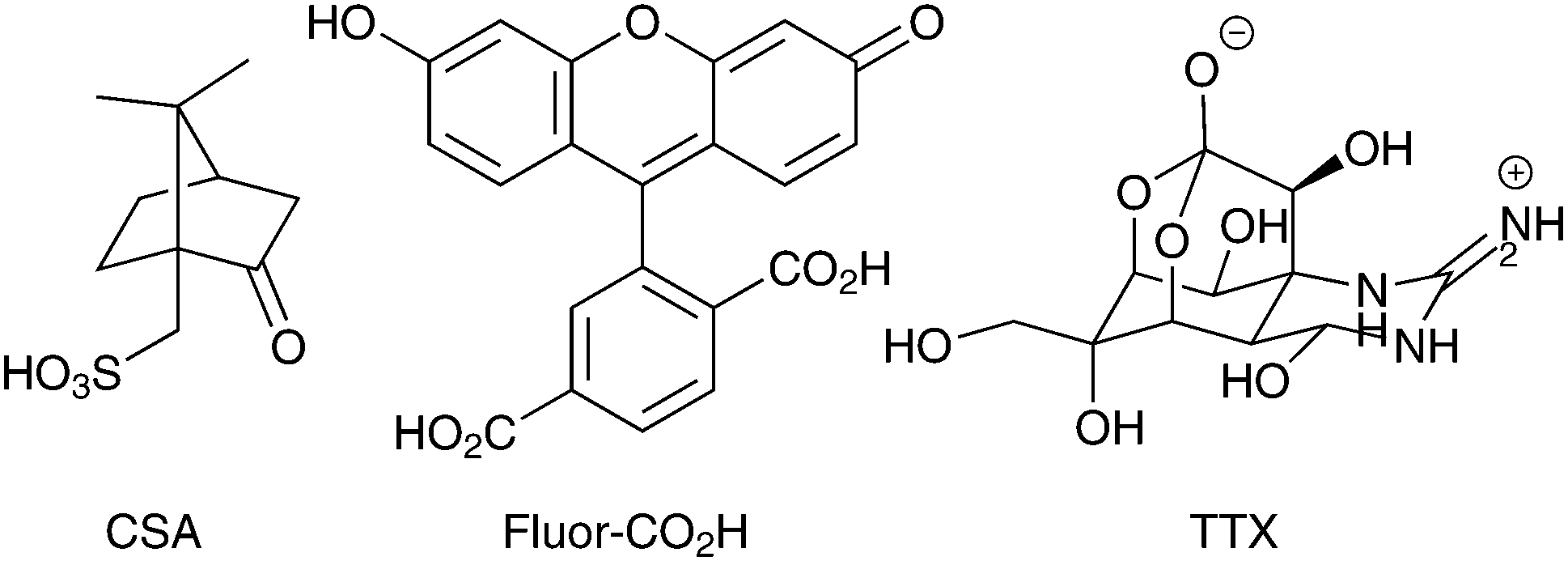 | ||
| Fig. 1 Camphorsulfonic acid (CSA), carboxyfluorescein (Fluor-CO2H), and tetrodotoxin (TTX). | ||
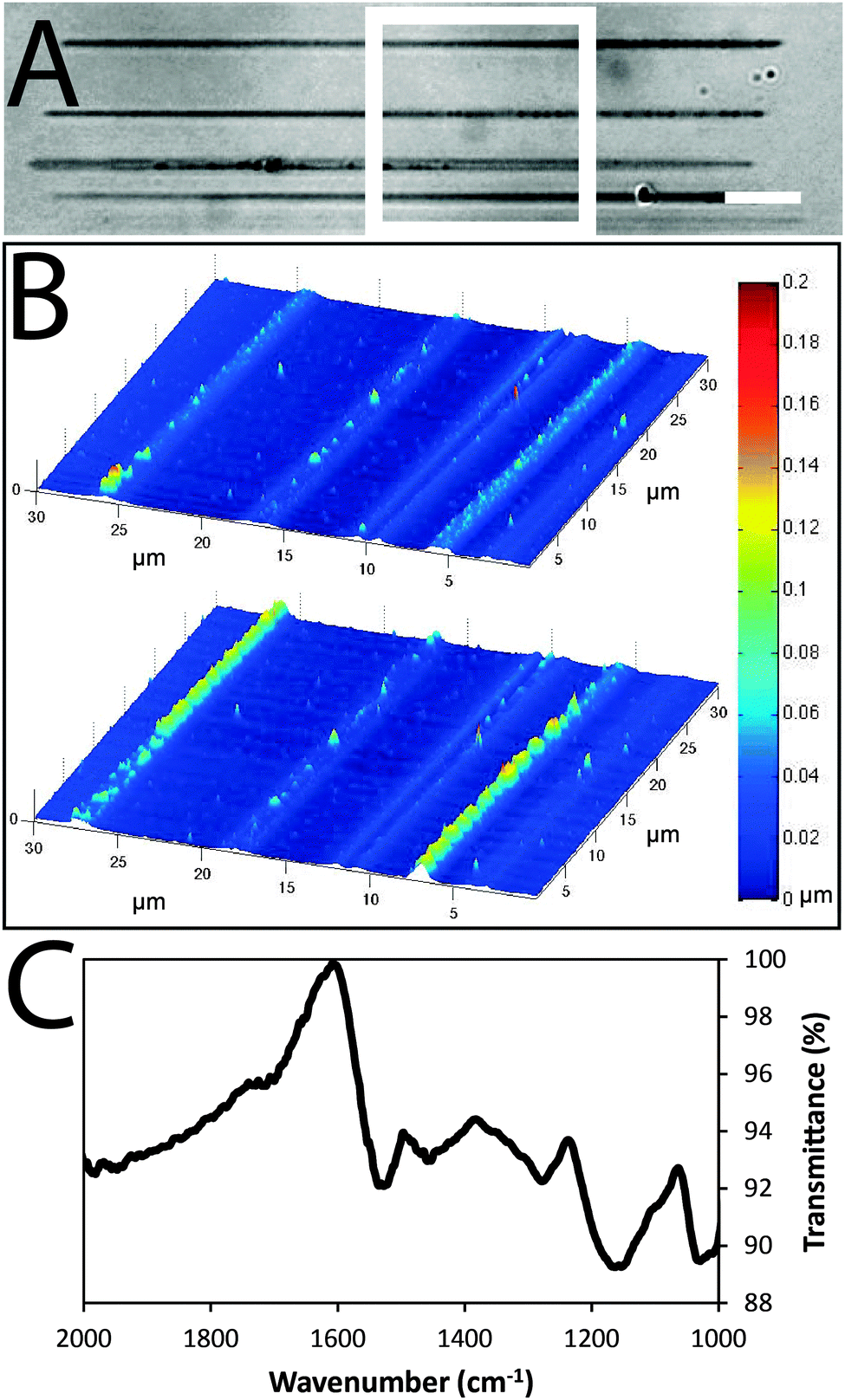 | ||
| Fig. 2 (A) Optical microscope image of black polypyrrole wires. The dotted white outline represents the scan area for AFM images, and the white scale bar represents 10 μm. (B) AFM image of wires in dry (top) and hydrated (bottom) states. (C) FTIR spectrum of CSA-doped polypyrrole. | ||
Images of the wires obtained via atomic force microscopy (Fig. 2B) suggest their widths are approximately 1.5 μm and thicknesses are approximately 0.1 μm in the dry state (confirmed by SEM, Fig. S1, ESI†) which swell to thicknesses of approximately 0.2 μm when hydrated in phosphate buffered saline (PBS). Energy-dispersive X-ray spectroscopy maps confirm the polypyrrole wires to be carbon-rich (Fig. S1, ESI†). Analysis of larger pieces of analogously synthesized polypyrrole with Fourier transform infrared (FTIR) spectroscopy recorded in attenuated total reflectance (ATR) mode (Fig. 2C) showed characteristic vibrational bands associated with C–C and C–N stretching at 1527 and 1423 cm−1, respectively. The band at 1278 cm−1 is attributed to C–H or C–N in-plane deformations, the bands at 1154 and 1029 cm−1 are attributed to C-H bending, and the shoulder at 1714 cm−1 indicates partial overoxidation during the polymerization process.
The conductance of the polypyrrole wires was measured in accordance with protocol IPC-TM-650, number 2.5.17.2 described by the Institute for Interconnecting and Packaging Electronic Circuits. We found that the CSA-doped polypyrrole wires had conductivities of 3.4 (±0.8) × 10−6 S cm−1, which is similar to that reported in the literature for CSA-doped bulk polypyrrole.42
We studied the capability of the polypyrrole wires to deliver a model drug (carboxyfluorescein, Fig. 1) into PBS. The electrical stimulation paradigm for electrically-triggered delivery was a simple potential step of 0.6 V applied for 30 seconds across the array of wires (Fig. 3A). Data is presented as ng released from arrays of 30 wires because of the difficulties associated with precisely determining the loading efficiency of such low quantities of drug (ng regime), and the potential for sodium borohydride-mediated degradation of polypyrrole.41 Passive release of drug (a common problem for most drug delivery systems) from the unstimulated polypyrrole matrix was observed (Fig. 3B), which is likely to be poorly entrapped drug because of inefficiencies in the loading process which is not as simple as for our previously reported solution processable degradable electroactive polymers.22 Electrical stimulation of the wires initiates ion transport within the polymer matrix and alters the oxidation state and conformation of the polypyrrole,43,44 thereby enhancing carboxyfluorescein release compared to the unstimulated control with approximately double the quantity released during the short experiment (Fig. 3B). While passive release is notable, and the quantity of drug released upon electrical stimulation is low (ng regime), it should be possible to address this by printing structures with different dimensions (particularly surface area to volume ratio), and investigating alternative doping methodologies; nevertheless, our results highlight the potential of such multiphoton fabricated structures to deliver small molecules or drugs (e.g. anti-inflammatories) upon the application of an electrical stimulus.
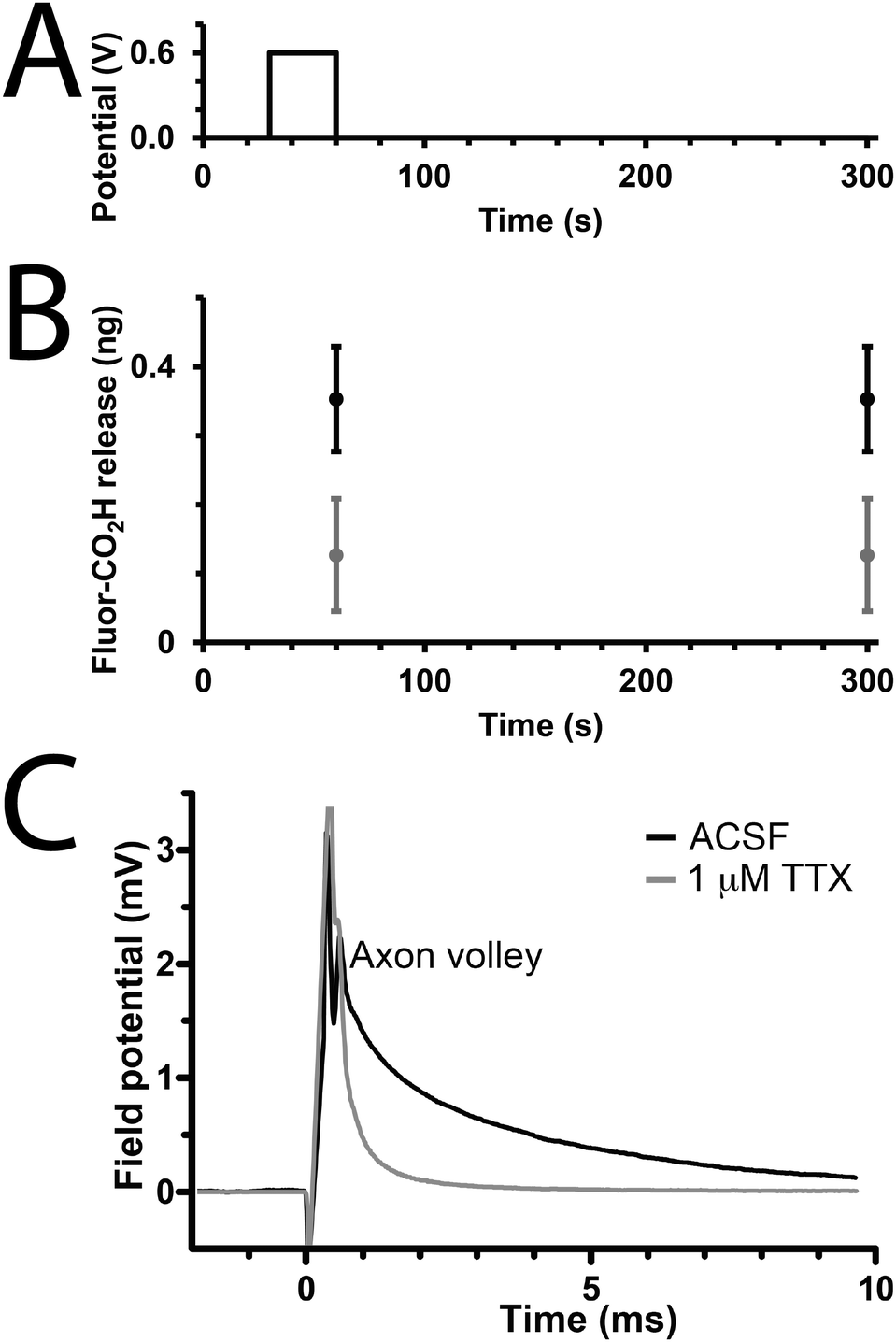 | ||
| Fig. 3 (A and B) Electrically-triggered “drug” delivery. (A) Electrical stimulation paradigm, rest 30 seconds, application of a potential step of 0.6 V for 30 seconds, rest for 240 seconds. (B) Electrically-triggered delivery of Fluor-CO2H as quantified by fluorimetry. Grey circles represent unstimulated control samples, black circles represent electrically stimulated samples; error bars represent standard deviation (n = 5). (C) Electrical stimulation of a slice of mouse brain in the absence (black line) or presence of tetrodotoxin (TTX, grey line). Stimulation paradigm: application of a potential step of 2 V for 100 μs. | ||
To demonstrate that de novo designed CP-based materials produced via multiphoton lithography have the potential for future development as implantable neural interfaces, we used arrays of polypyrrole wires as an electrode to interact with a slice of mouse brain ex vivo. The arrays were positioned to stimulate the Schaffer collaterals in the stratum radiatum and a recording electrode was positioned in the CA1 region of the stratum radiatum to record from CA3–CA1 synapses. A potential step of 2 V was applied for 100 μs, and a corresponding response was recorded by the electrode in the CA1 region (Fig. 3B, black line). This result indicates that the CP-based materials interact with the nervous system. The addition of the sodium channel inhibitor tetrodotoxin (TTX, Fig. 1, 1 μM) to the medium resulted in the disappearance of the axon volley after stimulation (Fig. 3B, grey line), thereby confirming that, while we could not discern a postsynaptic response, the response recorded was indeed physiological, and caused by the firing of action potentials in response to stimulation provided by the polypyrrole wires.
The electrochemical stability of the polypyrrole wires was preserved to acceptable levels over the course of the experiments (Fig. S3, ESI†), yet it is known to decrease over long periods of time.45 Other CPs46 such as poly(3,4-ethylenedioxythiophene) have been demonstrated to have improved electrochemical stabilities over analogous time periods,45 and it is noteworthy that arrays of poly(3,4-ethylenedioxythiophene) can also be fabricated using multiphoton lithography (Fig. S4, ESI†). Preclinical trials have shown CPs such as polypyrrole to be relatively non-immunogenic, with histological analyses revealing no significant inflammation in the vicinity of polypyrrole-based electrodes implanted in Sprague-Dawley rat brains after 3 or 6 weeks.47 By comparison the results reported for poly(3,4-ethylenedioxythiophene)-based neural electrodes in rats have been somewhat mixed with Kipke, Martin and coworkers reporting modest levels of global tissue reaction of approximately the same magnitude as for silicon probes after 6 weeks,48 whereas Malliaras and coworkers reported lower levels of tissue reaction than for silicon probes after 4 weeks;49 however, these conflicting results are likely to be ascribed to differences in the dopant anions (perchlorate or polystyrenesulfonate, respectively), the mechanical properties between the underlying substrates (iridium or parylene, respectively), or indeed differences in species of rats used in the studies (Sprague-Dawley or Wistar, respectively). Clearly, all of these issues would need to be addressed before clinical translation is possible.
Conclusions
Herein we report the preparation of electroactive polymer (CP)-based biomaterials via multiphoton lithography. Polypyrrole-based biomaterials were synthesized in one step from commercially available starting materials. The physicochemical properties of the polypyrrole wires were characterized. The ability of the polypyrrole-based materials to deliver small quantities of a model drug (carboxyfluorescein) was demonstrated in vitro, as was their ability to interact with a slice of mouse brain (ex vivo) on the application of an electrochemical stimulus. Such CP-based materials have prospects for the development of de novo designed implantable electrodes for interaction with the nervous system, either when inserted in the central nervous system as highlighted,3 or the peripheral nervous system (inserted into peripheral nerve fascicles to trigger the activity of specific axons), and may also be useful to deliver small molecules or drugs in a highly localized fashion.43,50–52Acknowledgements
We thank Joshua Bolinger of the Texas Materials Institute for supplying glass substrates with strips of silver deposited on them. We thank the University of Florida for financial support in the form of start-up resources. We thank the MRC for grant ref: MR/J011851/1 for financial support for Damian Cummings.Notes and references
- R. H. Funk, T. Monsees and N. Ozkucur, Prog. Histochem. Cytochem., 2009, 43, 177 CrossRef PubMed.
- C. D. McCaig, A. M. Rajnicek, B. Song and M. Zhao, Physiol. Rev., 2005, 85, 943 CrossRef PubMed.
- M. L. Kringelbach, N. Jenkinson, S. L. Owen and T. Z. Aziz, Nat. Rev. Neurosci., 2007, 8, 623 CrossRef CAS PubMed.
- D. R. Merrill, M. Bikson and J. G. R. Jefferys, J. Neurosci. Methods, 2005, 141, 171 CrossRef PubMed.
- G. G. Wallace, S. E. Moulton and G. M. Clark, Science, 2009, 324, 185 CrossRef CAS PubMed.
- G. G. Wallace, M. J. Higgins, S. E. Moulton and C. Wang, Nanoscale, 2012, 4, 4327 RSC.
- D. M. Thompson, A. N. Koppes, J. G. Hardy and C. E. Schmidt, Annu. Rev. Biomed. Eng., 2014, 16, 397 CrossRef CAS PubMed.
- M. Berggren and A. Richter-Dahlfors, Adv. Mater., 2007, 19, 3201 CrossRef CAS.
- T. F. Otero, J. G. Martinez and J. Arias-Pardilla, Electrochim. Acta, 2012, 84, 112 CrossRef CAS PubMed.
- T. F. Otero, Polym. Rev., 2013, 53, 311 CrossRef CAS.
- S. E. Moulton, M. J. Higgins, R. M. I. Kapsa and G. G. Wallace, Adv. Funct. Mater., 2012, 22, 2003 CrossRef CAS.
- W. K. Oh, O. S. Kwon and J. Jang, Polym. Rev., 2013, 53, 407 CrossRef CAS.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679 CrossRef CAS.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Biomaterials, 2008, 29, 3393 CrossRef CAS PubMed.
- M. Muskovich and C. J. Bettinger, Adv. Healthcare Mater., 2012, 1, 248 CrossRef CAS PubMed.
- M. Irimia-Vladu, Chem. Soc. Rev., 2014, 43, 588 RSC.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341 CrossRef CAS PubMed.
- B. Guo, L. Glavas and A. C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263 CrossRef CAS PubMed.
- J. G. Hardy, J. Y. Lee and C. E. Schmidt, Curr. Opin. Biotechnol., 2013, 24, 847 CrossRef CAS PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724 CrossRef CAS PubMed.
- E. M. Stewart, M. Fabretto, M. Mueller, P. J. Molino, H. J. Griesser, R. D. Short and G. G. Wallace, Biomater. Sci., 2013, 1, 368 RSC.
- J. G. Hardy, D. J. Mouser, N. Arroyo-Currás, S. Geissler, J. K. Chow, L. Nguy, J. M. Kim and C. E. Schmidt, J. Mater. Chem. B, 2014, 2, 6809 RSC.
- L. Pettersson, S. Ghosh and O. Inganas, Org. Electron., 2002, 3, 143 CrossRef CAS.
- D. Mawad, K. Gilmore, P. Molino, K. Wagner, P. Wagner, D. L. Officer and G. G. Wallace, J. Mater. Chem., 2011, 21, 5555 RSC.
- L. Ghasemi-Mobarakeh, M. P. Prabhakaran, M. Morshed, M. H. Nasr-Esfahani, H. Baharvand, S. Kiani, S. S. Al-Deyab and S. Ramakrishna, J. Tissue Eng. Regener. Med., 2011, 5, e17 CrossRef CAS PubMed.
- S. J. Pomfret, P. N. Adams, N. P. Comfort and A. P. Monkman, Polymer, 2000, 41, 2265 CrossRef CAS.
- J. G. Hardy, R. C. Cornelison, R. C. Sukhavasi, R. J. Saballos, P. Vu, D. L. Kaplan and C. E. Schmidt, Bioengineering, 2015, 2, 15 CrossRef PubMed.
- D. Mawad, E. Stewart, D. L. Officer, T. Romeo, P. Wagner, K. Wagner and G. G. Wallace, Adv. Funct. Mater., 2012, 22, 2692 CrossRef CAS.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773 CrossRef CAS PubMed.
- B. Weng, R. L. Shepherd, K. Crowley, A. J. Killard and G. G. Wallace, Analyst, 2010, 135, 2779 RSC.
- S. E. Moulton and G. G. Wallace, J. Controlled Release, 2014, 193, 27 CrossRef CAS PubMed.
- L. Sasso, P. Vazquez, I. Vedarethinam, J. Castillo-León, J. Emnéus and W. E. Svendsen, Sensors, 2010, 10, 10986 CrossRef CAS PubMed.
- C. N. LaFratta, J. T. Fourkas, T. Baldacchini and R. A. Farrer, Angew. Chem., Int. Ed., 2007, 46, 6238 CrossRef CAS PubMed.
- A. I. Ciuciu and P. J. Cywinski, RSC Adv., 2014, 4, 45504 RSC.
- S. Maruo and J. T. Fourkas, Laser Photon. Rev., 2008, 2, 101 CrossRef.
- E. C. Spivey, E. T. Ritschdorff, J. L. Connell, C. A. McLennon, C. E. Schmidt and J. B. Shear, Adv. Funct. Mater., 2013, 23, 333 CrossRef CAS.
- R. T. Hill, J. Lyon, R. Allen, K. Stevenson and J. Shear, J. Am. Chem. Soc., 2005, 127, 10707–10711 CrossRef CAS PubMed.
- K. Yamada, Y. Kimura, S. Suzuki, J. Sone, J. Chen and S. Urabe, Chem. Lett., 2006, 35, 908 CrossRef CAS.
- K. Yamada, A. Kyoya, J. Sone and J. Chen, Opt. Rev., 2009, 16, 208 CrossRef CAS.
- K. Yamada, A. Kyoya, J. Sone and J. Chen, Opt. Rev., 2011, 18, 162 CrossRef CAS PubMed.
- M. Bengoechea, I. Boyano, O. Miguel, I. Cantero, E. Ochoteco, J. Pomposoa and H. Grande, J. Power Sources, 2006, 160, 585 CrossRef CAS PubMed.
- M. A. Chougule, G. D. Khuspe, S. Sen and V. B. Patil, Appl. Nanosci., 2013, 3, 423 CrossRef CAS.
- V. Pillay, T. S. Tsai, Y. E. Choonara, L. C. du Toit, P. Kumar, G. Modi, D. Naidoo, L. K. Tomar, C. Tyagi and V. M. Ndesendo, J. Biomed. Mater. Res., Part B, 2014, 102, 2039 CrossRef PubMed.
- P. Meredith, C. J. Bettinger, M. Irimia-Vladu, A. B. Mostert and P. E. Schwenn, Rep. Prog. Phys., 2013, 76, 034501 CrossRef CAS PubMed.
- A. Kros, N. A. J. M. Sommerdijk and R. J. M. Nolte, Sens. Actuators, B, 2005, 106, 289 CrossRef CAS PubMed.
- C. Vallejo-Giraldo, A. Kelly and M. J. Biggs, Drug Discovery Today, 2014, 19, 88 CrossRef CAS PubMed.
- P. M. George, A. W. Lyckman, D. A. LaVan, A. Hegde, Y. Leung, R. Avasare, C. Testa, P. M. Alexander, R. Langer and M. Sur, Biomaterials, 2005, 26, 3511 CrossRef CAS PubMed.
- K. A. Ludwig, J. D. Uram, J. Yang, D. C. Martin and D. R. Kipke, J. Neural Eng., 2006, 3, 59 CrossRef PubMed.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Garfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Senaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575 CrossRef PubMed.
- D. T. Simon, S. Kurup, K. C. Larsson, R. Hori, K. Tybrandt, M. Goiny, E. W. Jager, M. Berggren, B. Canlon and A. Richter-Dahlfors, Nat. Mater., 2009, 8, 742 CrossRef CAS PubMed.
- D. Svirskis, J. Travas-Sejdic, A. Rodgers and S. Garg, J. Controlled Release, 2010, 146, 6 CrossRef CAS PubMed.
- Z. Yue, S. E. Moulton, M. Cook, S. O'Leary and G. G. Wallace, Adv. Drug Delivery Rev., 2013, 65, 559 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, SEM and SEM-EDX images. See DOI: 10.1039/c5tb00104h |
| ‡ Indicates these authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |