
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cell-penetrating protein designed for bimodal fluorescence and magnetic resonance imaging†
Qin
Wu‡
a,
Qinqin
Cheng‡
a,
Siming
Yuan
a,
Junchao
Qian
b,
Kai
Zhong
b,
Yinfeng
Qian
c and
Yangzhong
Liu
*a
aCAS Key Laboratory of Soft Matter Chemistry, CAS, High Magnetic Field Laboratory, Department of Chemistry, University of Science and Technology of China, Hefei, 230026, Anhui, China. E-mail: liuyz@ustc.edu.cn
bHigh Magnetic Field Laboratory, Chinese Academy of Sciences, Hefei, 230031, China
cRadiology Department, the First Affiliated Hospital of Anhui Medical University, Hefei, 230022, China
First published on 11th August 2015
Abstract
Multimodal imaging is a highly desirable biomedical application since it can provide complementary information from each imaging modality. We propose a protein engineering-based strategy for the construction of a bimodal probe for fluorescence and magnetic resonance imaging. A recombinant protein was generated by the fusion of a supercharged green fluorescence protein (GFP36+) with a lanthanide-binding tag (dLBT) that can stably bind two Gd3+ ions. The GFP36+–dLBT fusion protein showed strong fluorescence and exhibited efficient contrast enhancement in magnetic resonance imaging. This protein probe improves the MR relaxation more efficiently than Gd-DTPA (gadopentetate dimeglumine). The superior cell-penetrating activity of GFP36+ allows the efficient cellular uptake of this fusion protein and it can thus be used as a cellular imaging probe. Dual imaging was conducted in vitro and in mice. This result indicates that the fusion of different functional domains is a feasible approach for making multi-modal imaging agents.
Introduction
Over the past decades, a variety of molecular imaging techniques, such as positron emission tomography (PET), X-ray computed tomography (CT), magnetic resonance imaging (MRI) and fluorescence imaging (FI), have found numerous useful applications.1,2 Each method has its inherent advantages and limitations. Therefore, a single imaging method may not meet all of the requirements to solve a particular diagnostic problem.3 A combination of different imaging methods may achieve multi-modal imaging and facilitate bench-to-clinical applications.4,5 A typical example is a probe that combines fluorescence and magnetic resonance imaging.6,7 These two imaging modalities are highly complementary so that hybrid probes can exploit both the sensitivity of FI and the resolution and deep tissue penetration of MRI.8–10The synthesis of bimodal FI/MRI imaging probes is typically based on the chemical conjugation of two distinct functional modalities, such as organic molecules and inorganic nanoparticles.6,11,12 Although the chemical syntheses of bimodal probes have been investigated intensively, complex synthetic procedures are often required, thus limiting the general applicability of this strategy. The biological properties of these products, such as toxicity, are hard to predict. A lack of chemical stability and a property such as photobleaching are other possible complications.13–15
Although biocompatible molecules such as proteins have been used to prepare multi-modal imaging agents, chemical modification is by far the most common approach to link a particular functional group to a protein of interest.16,17 The introduction of various chemical substituents could limit further applications since only certain types of chemical modification are compatible with the clinical use of such proteins.18
To address these challenges, we developed an alternative strategy for obtaining a bimodal FI/MRI imaging probe, based solely on a protein engineering approach. Fusion proteins often retain the functional properties of the individual domains that comprise the fusion. Functional fusion proteins with MRI and FI modalities could serve as dual-imaging probes without the need for any further chemical modification. As a proof-of-concept, a GFP36+–dLBT protein was generated by the fusion of a supercharged green fluorescence protein (GFP36+) and a double-lanthanide-binding tag (dLBT), using standard protein expression procedures (Scheme 1). The dual imaging of this protein probe was tested in vitro, in cells and in mice. The results confirmed that the fusion of proteins with different functional domains is a feasible approach for making multi-modal imaging probes.
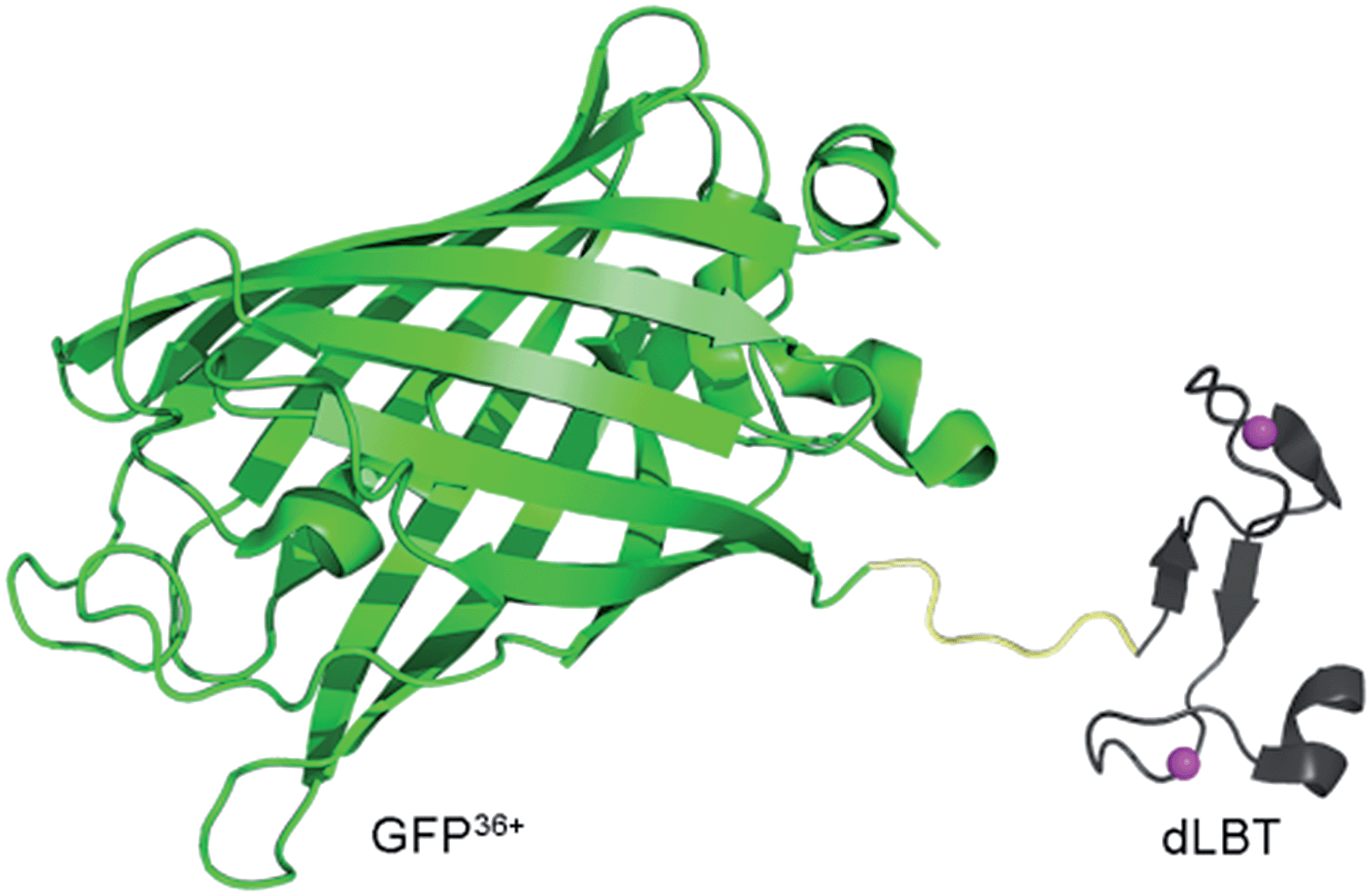 | ||
| Scheme 1 Cartoon representation of a bimodal imaging probe of a GFP36+–dLBT fusion protein. Two gadolinium ions bound to dLBT are shown as pink spheres. | ||
Results and discussion
Preparation and characterization
GFP36+ is a variant of a green fluorescence protein with 36 positive charges (also termed +36GFP).19 This supercharged GFP protein is highly aggregation-resistant and retains its fluorescence even after having been boiled or cooled.19 It shows superior cell-penetrating activity and for that reason it was used as a protein carrier for the delivery of siRNA into cells.20 dLBT is an artificially designed peptide tag with a high affinity for lanthanide ions.21 The peptide sequence of dLBT has been optimized for MRI purposes by the introduction of an H2O coordination site to Gd3+ in the Gd–dLBT complexes.22 This peptide can be fused to proteins with the retention of its Gd3+ binding ability.22 The cell-penetration property of the supercharged GFP36+ domain allows the cellular imaging of this fusion protein. To the best of our knowledge, this is the first bimodal FI/MRI agent made using an entirely genetically encoded protein.The GFP36+–dLBT fusion protein was obtained by expression in E. coli and purified by standard methods (see the Experimental section for more details). To improve its stability, a flexible glycine–serine linker was inserted between GFP36+ and dLBT (Scheme 1).23 After comparing different lengths of the linker, (GGS)9 was chosen for further investigation, as this linker results in a stable fusion protein with appropriate MR imaging properties (Fig. S2†).
In vitro characterization
To explore whether the fusion protein retains the functionality of the two individual domains, the fluorescence and metal binding properties of GFP36+–dLBT were analyzed in vitro. Inductively coupled plasma mass spectroscopy (ICP-MS) measurements showed that the fusion protein bound two Gd3+ ions (Fig. 1A, Table S1†). The number of gadolinium ions in the protein is consistent with the design of the fusion protein.22 No gadolinium was detected in the GFP36+ protein, suggesting that the presence of the (His)6 tag does not alter the binding of Gd3+ ions to the protein. UV titration showed that the binding constant of the GFP36+–dLBT fusion protein (Kd = 114 nM) is comparable to the literature data for other LBT variants (Fig. S3†).22 Fluorescence measurements showed that the spectra of the Gd(III)-bound GFP36+–dLBT fusion protein and the GFP36+ protein were nearly identical (Fig. 1B), indicating that the fluorescence quantum yield of the GFP protein is not perturbed by the fusion of dLBT and the binding of Gd(III). Thus, the fusion protein retains the functions of fluorescence and gadolinium binding, and hence is suitable for optical and MRI bimodal imaging.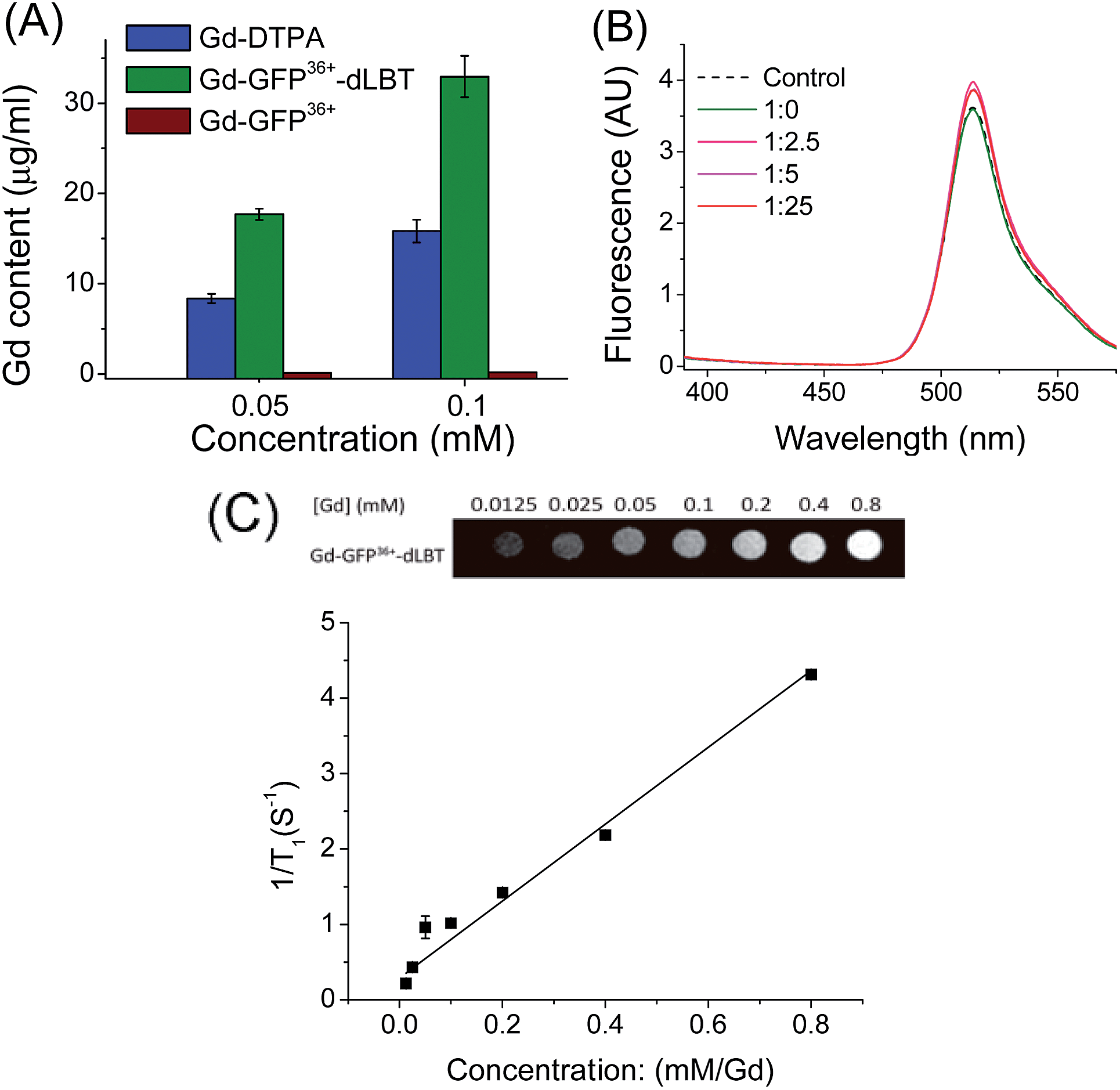 | ||
Fig. 1
In vitro characterization of the GFP36+–dLBT protein. (A) ICP-MS measurement of the Gd(III) content in Gd-DTPA (blue), Gd–GFP36+–dLBT (green) and Gd–GFP36+ (brown). Both GFP36+–dLBT and GFP36+ proteins contain a (His)6 tag. (B) Fluorescence spectra of GFP36+–dLBT with Gd3+ titration. The dashed line denotes the GFP36+ control; the solid lines indicate the 10 μM GFP36+–dLBT with the addition of Gd3+ ions. The molar ratios of [protein]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Gd3+] are given. The spectra were recorded in HEPES buffer from 400–580 nm with an excitation wavelength of 395 nm. (C) T1-weighted MR image produced with a spin echo sequence (TR 300 ms, TE 14 ms) of the GFP36+–dLBT protein at different concentrations of gadolinium. The molar relaxivity rates, r1 (1/T1), were obtained by linear fitting of the experimental data. Error bars denote the standard deviation. :[Gd3+] are given. The spectra were recorded in HEPES buffer from 400–580 nm with an excitation wavelength of 395 nm. (C) T1-weighted MR image produced with a spin echo sequence (TR 300 ms, TE 14 ms) of the GFP36+–dLBT protein at different concentrations of gadolinium. The molar relaxivity rates, r1 (1/T1), were obtained by linear fitting of the experimental data. Error bars denote the standard deviation. | ||
We next measured the relaxivity and MR contrast enhancement of the fusion protein. A contrast agent used clinically, gadopentetate dimeglumine (Gd-DTPA), was used for comparison. The Gd–GFP36+–dLBT complex clearly exhibits MR contrast enhancement, whereas no detectable signal was observed in the GFP36+ protein without the fusion of dLBT (Fig. S3†). This observation confirms the binding of Gd3+ ions to the dLBT domain. Imaging by MRI showed increasing contrast with an increase in the concentration of Gd–GFP36+–dLBT (Fig. 1C). The longitudinal relaxation rate (r1) was measured at different gadolinium concentrations. Fitting the data gave a T1 relaxivity value of 5.1 mM−1 s−1 (Fig. 1C). This value is greater than that of Gd-DTPA (3.67 mM−1 s−1) under the same conditions (Fig. S5†). The GFP36+–dLBT protein can thus serve as an efficient T1 contrast agent. The greater relaxivity enhancement of GFP36+–dLBT should in all likelihood be attributed primarily to the reduced rotational correlation of the protein in comparison to the small molecule contrast agent Gd-DTPA.24
Dual imaging in cells
Since the GFP36+ protein also possesses the unique feature of being able to penetrate cells, the dual imaging capability of GFP36+–dLBT was tested. Cells were analyzed using confocal laser microscopy after treatment with Gd–GFP36+–dLBT for four hours. Cytoplasmic fluorescence clearly shows the internalization of GFP36+–dLBT in the cells (Fig. 2A). Flow cytometry showed that the cellular uptake of GFP36+–dLBT increased rapidly with an increase in protein concentration (Fig. S6†). We also measured the uptake of Gd–GFP36+–dLBT by the quantification of Gd ions in cells. The intracellular concentration of Gd increased with an increase in the concentration of Gd–GFP36+–dLBT (Fig. 2B). This result confirmed that Gd ions are internalized together with Gd–GFP36+–dLBT, suggesting the possible application of this protein in cellular MR imaging.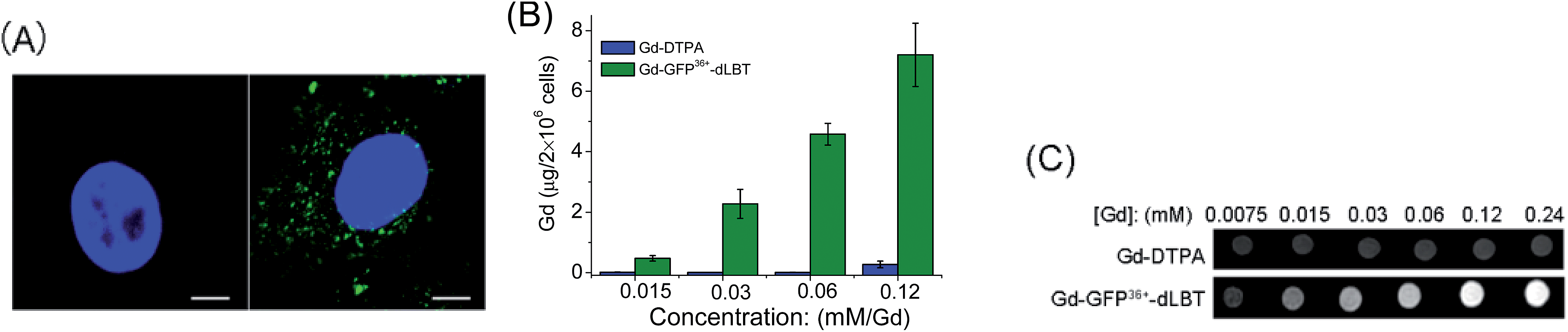 | ||
| Fig. 2 Cellular imaging of Gd–GFP36+–dLBT. (A) Confocal laser microscopy images of HeLa cells treated with PBS (left) or 1 μM Gd–GFP36+–dLBT (right) for 4 h. In blue: nuclear staining (DAPI), in green: GFP36+. The scale bar is 15 μm. (B) Gd content in HeLa cells measured using ICP-MS after incubation with Gd-DTPA or Gd–GFP36+–dLBT for 4 h. Data are shown as the mean ± SD of three independent experiments. (C) T1-weighted MR images of 2 × 106 HeLa cells treated with Gd-DTPA or Gd–GFP36+–dLBT at different concentrations. Cells were washed 5 times to remove the free protein. | ||
The T1-weighted MR image was measured on HeLa cells treated with Gd–GFP36+–dLBT. The enhanced signal strength was concentration-dependent (Fig. 2C). The application of Gd-DTPA showed no such MR enhancement, which was in line with expectations, since Gd-DTPA is an extracellular contrast agent incapable of entering cells.25 Similar results were observed for HepG2 cells (Fig. S7†). The different protein concentrations used in the fluorescence and MRI assays correspond to the sensitivity of the two imaging methods.
Dual imaging in vivo
The in vivo imaging capabilities of Gd–GFP36+–dLBT were investigated in tumor-bearing mice. The HepG2 tumor was implanted by injection into the flank above the upper left thigh. After the tumor was grown to an appropriate size, Gd–GFP36+–dLBT was injected intratumorally followed by in vivo imaging. Fluorescence images were obtained before (0 h) and after injection (3 h) using a whole body imaging system. The images show fluorescence at the site of injection (dashed circle and arrow) (Fig. 3A). After fluorescence imaging, the complementary MRI contrast efficiency was measured at the same time points using a 9.4 T MRI scanner. The T1-weighted MR images also showed contrast enhancement at the site of injection (Fig. 3B). In comparison with fluorescence imaging, MRI showed higher resolution and more accurately revealed the location and distribution of the injected Gd–GFP36+–dLBT. This difference is in line with expectations based on the distinctive features of the two imaging methods.26 Gd–GFP36+–dLBT thus possesses the necessary sensitivity for fluorescence imaging, as well as the penetration and accurate positioning for MR imaging.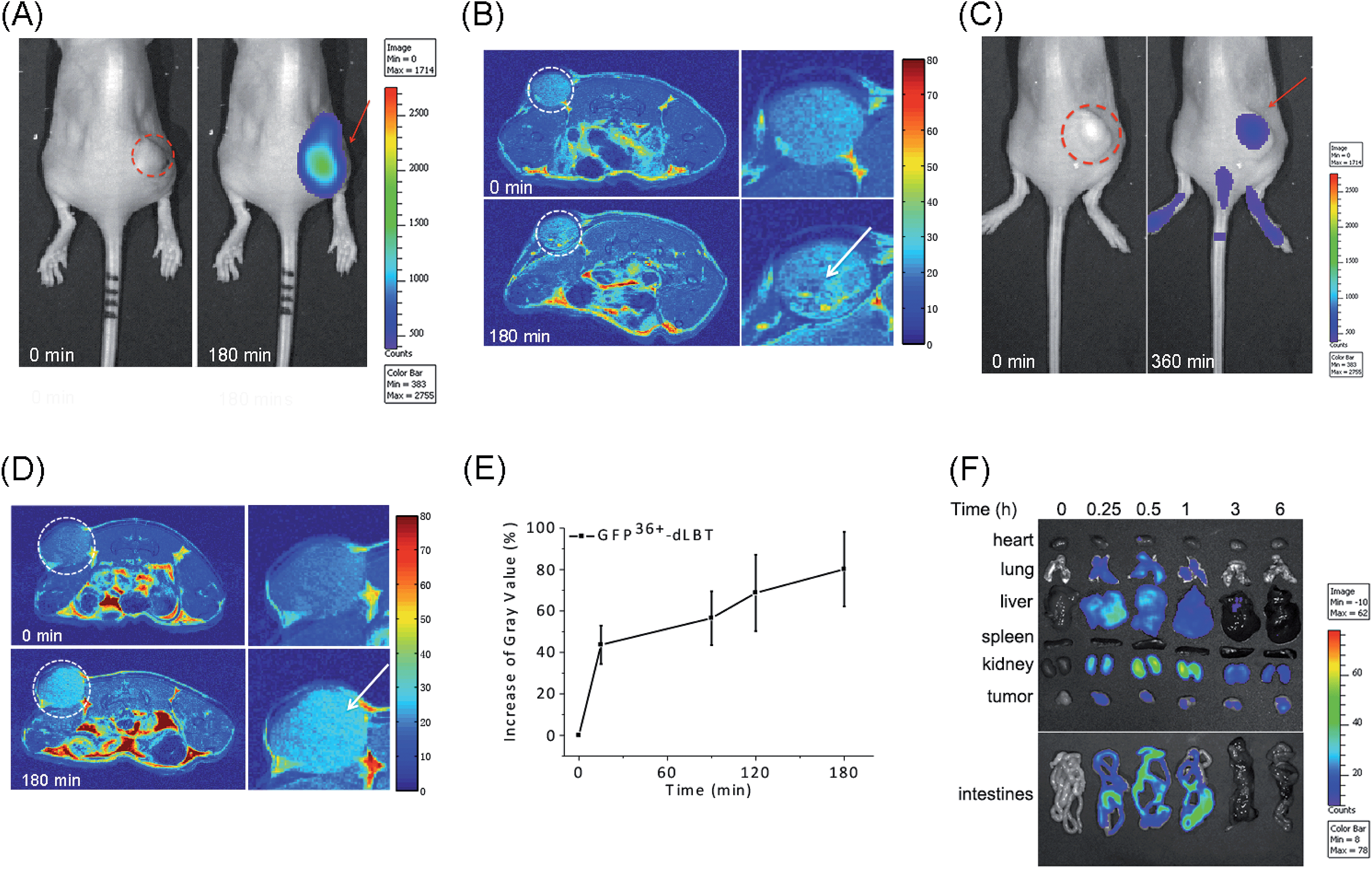 | ||
| Fig. 3 In vivo imaging of Gd–GFP36+–dLBT. The dotted circles indicate the tumor site and the arrows indicate the enhanced fluorescence signal or MR contrast. (A) Fluorescence imaging of nude mice bearing HepG2 tumors before (left) and after (right) intratumoral injection of Gd–GFP36+–dLBT at a dose of 7.5 mg kg−1. (B) T1-weighted MR images of the tumor sites before and 3 h after intratumoral injection with Gd–GFP36+–dLBT. The enlarged figures of the tumor site are shown on the right hand side. (C) Fluorescence imaging of a tumor before (left) and 6 h after (right) tail vein injection with Gd–GFP36+–dLBT (7.5 mg kg−1). (D) T1-weighted MR images of a tumor before (upper) and 3 h after (bottom) tail vein injection with Gd–GFP36+–dLBT. (E) Quantitative analysis of the tumor MR images at various times. The intensities of the MR images were determined by standard region-of-interest measurements with ImageJ. Error bars denote the standard deviation. (F) Ex vivo fluorescence imaging of the collected organs and tumors from the HepG2 xenograft nude mice at various times (0, 0.25, 0.5, 1, 3 and 6 h) after the tail vein injection of the imaging probe. | ||
To test whether Gd–GFP36+–dLBT can be used for tumor imaging, tumor-bearing mice received Gd–GFP36+–dLBT via tail vein injection. Fluorescence and T1-weighted MR images were recorded to assess the distribution of Gd–GFP36+–dLBT. Both measurements showed enhanced signals in the tumor after the injection of Gd–GFP36+–dLBT (Fig. 3C and D). The time-dependent increase of the contrast enhancement indicates the accumulation of the protein in the tumor (Fig. 3E). In addition, fluorescence imaging also showed the increased accumulation of the probe in the tumor based on ex vivo measurements at different times after the injection (Fig. 3F). Taken together, these results suggest that Gd–GFP36+–dLBT is indeed useful for in vivo imaging.
To further investigate the biodistribution of the Gd–GFP36+–dLBT probe, organs were collected at 0.25, 0.5, 1, 3, and 6 hours post-injection for ex vivo fluorescence imaging (Fig. 3F). A significant amount of the protein was observed in the liver 15 min after the injection, indicating the rapid uptake of the probe by the liver.27 This is in agreement with the literature, which indicates that cationic probes are preferentially captured by the liver.28,29 The fluorescence clearly decreased in the liver after 30 min, and reached the background level after 3 hours. A postponed accumulation and reduction were observed in the intestine. This result suggests the quick hepatic clearance of the protein. In addition, a strong fluorescence signal was also observed in the kidney, peaking at 0.5–1 h post-injection, and it had decreased after 6 hours, indicating a relatively slower renal clearance. The MR contrast enhancement in the kidney and liver was also observed (Fig. S8 and S9†). The lung showed a little protein accumulation, which was removed after 1 h. No fluorescence signal was observed in the heart and spleen. Although hepatic and renal clearance occurred, the increased accumulation of Gd–GFP36+–dLBT is still observed in the tumor in the 6 h measurements, possibly due to the enhanced permeability and retention effect in solid tumors.4 The altered vasculature and lymphatic drainage allow the accumulation of macromolecules in the tumor, resulting in the passive targeting of Gd–GFP36+–dLBT to the tumor.
The application of protein probes for multi-modal imaging has distinct advantages over chemically synthesized agents: (1) the preparation is straightforward and usually efficient. Routine protein expression and purification suffice in obtaining the probes. (2) A tandem array of protein domains can be used to avoid the interference of each functional modality, such as fluorescence quenching. (3) The function of a protein-based probe can be easily modified or further improved by adding/changing functional protein domains through standard protein engineering methods, for instance, by the fusion of an antibody to achieve specific targeting.30,31 Protein-based MRI contrast agents have been made in exactly this manner.32 Bimodal imaging has been used to monitor tumor progression and therapeutic responses.33 Cells transfected with the gene of supercharged GFP demonstrate bimodal optical imaging and an improved CEST MRI contrast in comparison to the wild type GFP.34
The cellular delivery of MRI agents is considered an important approach for the further development of contrast agents, as most currently used contrast agents are restricted to extracellular space.35 Cellular imaging agents might find application in monitoring cancer metastases and in guiding surgery for tumor removal.36 The ability to follow the metastatic process by tracking these intracellularly labeled cells at different cancer stages will provide a powerful tool for studying the mechanism of metastasis and tumor dormancy. In addition, an intracellular magnetic labeling technique has potential applications in screening antimetastasis therapies or cell-based genetic therapies.37–39 Cellular penetration agents, such as peptides, may be attached to the imaging probe to achieve this goal.6,36,38,40 In contrast, chemical modification could result in more leakage of the contrast agent from the cells and is also subject to quench relaxation.35,41 The GFP36+ protein has the unique property of being capable of cell penetration, along with displaying strong fluorescence. It is therefore an attractive building block for the construction of cell-penetrating multi-modal imaging agents. Recombinant GFP36+–dLBT possesses the dual-imaging function of both protein domains and exhibits cell penetrating activity.
Conclusions
In summary, we described a novel bimodal imaging probe generated by protein engineering. This probe is constructed by an entirely native protein sequence with a fluorescence domain and a gadolinium-binding motif. It demonstrates strong and steady fluorescence along with enhanced MRI contrast. This fusion protein also possesses cell penetration activity, so that the probe is able to enter cells efficiently. The in vitro assay indicates that this probe retains the fluorescence of the GFP protein and exhibits enhanced contrast effects upon the binding of two Gd3+ ions. The cellular assay shows that this probe is efficiently internalized, indicating its possible use for cellular imaging via fluorescence and MRI. The FI/MRI dual-imaging property of this probe can be exploited in vivo. Gd–GFP36+–dLBT accumulates in a tumor in vivo, demonstrating the utility of this approach. Thus, this protein-based dual imaging probe opens up a new way for designing functionalized imaging agents.Experimental section
Plasmid construct
The supercharged GFP (GFP36+) plasmid was generously provided from Professor Jiangyun Wang (Institute of Biophysics, Chinese Academy of Sciences). The gene for GFP36+ (236 aa) was amplified using overlap PCR with sequences 5′-GAACATATGGCTTCTAAAGGTGAACGCCTGTTC-3′ (forward primer) and 5′-GAAAAGCTTTTTGTAACGTTCGTCGCGGCC-3′ (reverse primer). The acquired PCR product was then digested with a HindIII restriction enzyme. DNA sequences encoding dLBT were flanked with a preceding (GGS)9 linker and a C-terminal (His)6 tag. The (GGS)9–dLBT–(His)6 sequence was amplified with 5′-GAAAAGCTTGGTGGCTCTGGTGGCTCTGGC-3′ and 5′-GAACTCGAGTCAGTGATGGTGGTGGTGGTGCGCCAGCAGTTCGTCACCTTC-3′. The PCR product was digested with a HindIII restriction enzyme. These two amplicons were ligated and then cloned into the NdeI and XhoI sites of a pET-21a plasmid. The construct containing the architecture of GFP36+–(GGS)9–dLBT–(His)6 was verified by DNA sequencing.Protein expression and purification
The plasmid was transformed into chemically competent BL21 (DE3) E. coli cells using a standard heat shock protocol. The cells were grown in LB media, and protein expression was induced by 0.4 mM isopropyl-thio-β-galactopyranoside (IPTG) at an OD600 of 0.6–0.8 at 16 °C for 20 h. The cells were harvested by centrifugation and resuspended in buffer A (50 mM Tris–HCl, 200 mM NaCl, 5 mM β-mercaptoethanol, 10 mM imidazole, pH 8.0). The cells were lysed by sonication on ice for 8 min. The cell lysates were centrifuged at 34500g for 30 min and the supernatant was filtered through 0.45 μm and 0.22 μm membranes. The solution was loaded onto Ni-NTA (Qiagen) columns and was washed with buffer B (50 mM Tris–HCl, 200 mM NaCl, 5 mM β-mercaptoethanol, 25 mM imidazole, pH 8.0). The GFP36+–dLBT protein was eluted with buffer C (50 mM Tris–HCl, 200 mM NaCl, 5 mM β-mercaptoethanol, 250 mM imidazole, pH 8.0). The eluate containing the GFP36+–dLBT protein was further purified by HiTrap Q XL chromatography on an AKTA Purifier. The protein was eluted using a linear gradient from 50 mM HEPES, 0.5 M NaCl to 50 mM HEPES, 1 M NaCl. The purified GFP36+–dLBT protein was analyzed by 4–25% gradient denaturing gel electrophoresis, and the gel was stained with Coomassie brilliant blue. The protein concentration was determined by UV absorption at 280 nm.
Gadolinium loading
The direct addition of GdCl3 to the protein caused protein precipitation in high concentration. The fusion protein was thus loaded with gadolinium by dialysis. A 5 molar equivalent of Gd3+ was added to the outer dialysis buffer (50 mM HEPES, 100 mM NaCl). To get rid of the excess Gd3+ in solution, the protein was subsequently dialyzed against the buffer without Gd3+ (50 mM HEPES, 100 mM NaCl) three times. After the gadolinium loading, the protein was concentrated using a centrifugal filtration device with a 3 kDa molecular weight cutoff.UV-vis titration
The UV-vis spectra were recorded on a Agilent 60 UV-vis spectrometer with a 5 mm path length quartz cuvette. 0.1 μM to 200 μM Gd3+ was titrated into 10 μM protein in 50 mM HEPES buffer (pH 7.5) containing 100 mM NaCl. The absorbance at 280 nm was recorded for data fitting.Fluorescence spectra
The fluorescence emission spectra were recorded at room temperature on a RF-5301 spectrofluorometer (Shimadzu) in a quartz cuvette with a path length of 5 mm. The excitation wavelength was set at 395 nm and the emission wavelength was recorded from 400–580 nm. Both slit widths of the excitation and emission were 5 nm.Gadolinium measurement
10 μl Gd-DTPA or Gd–GFP36+–dLBT was digested in concentrated HNO3 for 12 h and then the sample was diluted into 3 ml ultrapure water. The amount of Gd(III) was determined by ICP-MS.In vitro relaxivity measurement
The MRI contrast was evaluated on a GE 1.5 T clinical MRI scanner. An array of microcentrifuge tubes containing different concentrations of Gd–GFP36+–dLBT and Gd-DTPA were prepared for MR imaging. Phantom images were acquired with an inversion recovery turbo spin echo pulse sequence with a repetition time (TR) of 300 ms and an echo time (TE) of 14 ms. Relaxivity values (r1) were calculated from the equation r1 = 1/T1S − 1/T1C, where C is the concentration of the contrast agent in mM (Gd3+ concentrations measured by ICP-MS). T1S is the relaxation time with the contrast agent and T1C is the relaxation time without the contrast agent.Cell culture and uptake
Human Henrietta Lacks cells (HeLa) and adenocarcinomic human alveolar basal epithelial cells (A549) were incubated at 37 °C in 5% CO2. The cells were incubated with different concentrations of GFP36+–dLBT or Gd–GFP36+–dLBT for 4 hours. The cells were washed five times to remove free gadolinium ions. The uptake was measured by a flow cytometer (BD FACSCalubur™).Confocal fluorescence microscopy
The uptake of Gd–GFP36+–dLBT was analyzed by confocal microscopy. HepG2 cells were grown on coverslips placed at the bottom of wells in a tissue culture plate. After incubation, the coverslips were mounted on slides using a media containing DAPI and sealed. Imaging was performed on a Zeiss LSM 710 laser confocal scanning microscope with a 100× objective lens.Determination of the Gd3+ content in cells
The HeLa and HepG2 cells were incubated with different concentrations of Gd–GFP36+–dLBT or Gd-DTPA for 4 hours. Then the cells were trypsinized and washed several times before resuspension in PBS. The cells were adjusted to 2 × 106 cells per mL and then centrifuged at 3000 rpm for 15 min. The resulting cell pellets were digested in concentrated HNO3 overnight and diluted to 4 ml in ultrapure water for the ICP-MS measurement.T 1-weighted MR imaging
The cell suspension was acquired in the same way as described above for the ICP-MS test. T1-weighted images of the cell suspension were obtained on a 1.5 T clinical MR scanner. Phantom images were acquired with an inversion recovery turbo spin echo pulse sequence. The sequence used a repetition time (TR) of 300 ms and an echo time (TE) of 14 ms.Animals
Nude mice harboring HepG2 tumors were used for the FI and MRI experiments. All animal experiments were performed in compliance with institutional guidelines. The procedures were approved by the Animal Care and Use Committee of University of Science and Technology of China. To generate a tumor-bearing mouse model, human hepatic carcinoma (HepG2) tumors were induced into 5 week old female nude mice by the subcutaneous injection of 2.0 × 106 HepG2 cells. After the tumor grew to an appropriate size, the imaging probe was injected into the mice. Then the mice were anesthetized with an isoflurane gas mixture and positioned in the scanner for measurements.In vivo fluorescence imaging
Image acquisition was performed on a Xenogen IVIS Lumina system (Caliper Life Sciences, Hopkinton, MA, USA). The data were analyzed using Living Image 3.1 software (Caliper Life Sciences). The mice injected with Gd–GFP36+–dLBT were placed onto a warmed stage inside the IVIS Lumina light chamber and anesthesia was maintained with 2.5% isoflurane.In vivo MRI
MRI images of the HepG2 tumor-xenografted nude mice were recorded after the injection of Gd–GFP36+–dLBT at different times. The images were acquired on a 9 T MR scanner. The intensity of the MR signal was analyzed using the software ImageJ.Ex vivo analysis
The animals were sacrificed after the administration of the contrast agent. Then, the major organs and tumors were dissected and collected for fluorescence scanning. Fluorescence images of the major organs including brain, heart, liver, spleen, kidney, lung and intestines, and the tumors were obtained with a Xenogen IVIS Lumina system. The data were analyzed using Living Image 3.1 software.Acknowledgements
We thank Professor Hidde L. Ploegh (Whitehead Institute, MIT, USA.) for critical reading of the manuscript and valuable comments. This work was supported by the National Science Foundation of China (U1332210, 21171156 and 21573213), the National Basic Research Program of China (973 Program, 2012CB932502) and Collaborative Innovation Center of Suzhou Nano Science and Technology.Notes and references
- I. Y. Chen and J. C. Wu, Circulation, 2011, 123, 425–443 CrossRef PubMed.
- A. Hellebust and R. Richards-Kortum, Nanomedicine, 2012, 7, 429–445 CrossRef CAS PubMed.
- A. Y. Louie, Chem. Rev., 2010, 110, 3146–3195 CrossRef CAS PubMed.
- E. K. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv214 Search PubMed.
- Z. L. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- E. S. Olson, T. Jiang, T. A. Aguilera, Q. T. Nguyen, L. G. Ellies, M. Scadeng and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4311–4316 CrossRef CAS PubMed.
- D. Koktysh, V. Bright and W. Pham, Nanotechnology, 2011, 22, 275606 CrossRef PubMed.
- P. Verwilst, S. Park, B. Yoon and J. S. Kim, Chem. Soc. Rev., 2015, 44, 1791–1806 RSC.
- E. Debroye and T. N. Parac-Vogt, Chem. Soc. Rev., 2014, 43, 8178–8192 RSC.
- D. E. Lee, H. Koo, I. C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- J. H. Lee, Y. W. Jun, S. I. Yeon, J. S. Shin and J. Cheon, Angew. Chem., 2006, 45, 8160–8162 CrossRef CAS PubMed.
- J. Liu, K. Li, J. L. Geng, L. Zhou, P. Chandrasekharan, C. T. Yang and B. Liu, Polym. Chem., 2013, 4, 1517–1524 RSC.
- S. A. Corr, Y. P. Rakovich and Y. K. Gun'ko, Nanoscale Res. Lett., 2008, 3, 87–104 CrossRef CAS.
- J. M. Perez, T. O'Loughin, F. J. Simeone, R. Weissleder and L. Josephson, J. Am. Chem. Soc., 2002, 124, 2856–2857 CrossRef CAS PubMed.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 680–681 CrossRef CAS PubMed.
- S. Li, J. Jiang, J. Zou, J. Qiao, S. Xue, L. Wei, R. Long, L. Wang, A. Castiblanco, N. White, J. Ngo, H. Mao, Z. R. Liu and J. J. Yang, J. Inorg. Biochem., 2012, 107, 111–118 CrossRef CAS PubMed.
- H. Korkusuz, K. Ulbrich, K. Welzel, V. Koeberle, W. Watcharin, U. Bahr, V. Chernikov, T. Knobloch, S. Petersen, F. Huebner, H. Ackermann, S. Gelperina, W. Kromen, R. Hammerstingl, J. Haupenthal, F. Gruenwald, J. Fiehler, S. Zeuzem, J. Kreuter, T. J. Vogl and A. Piiper, Mol. Imag. Biol., 2013, 15, 148–154 CrossRef PubMed.
- Z. Zhao, H. Fan, G. Zhou, H. Bai, H. Liang, R. Wang, X. Zhang and W. Tan, J. Am. Chem. Soc., 2014, 136, 11220–11223 CrossRef CAS PubMed.
- M. S. Lawrence, K. J. Phillips and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 10110 CrossRef CAS PubMed.
- B. R. McNaughton, J. J. Cronican, D. B. Thompson and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6111–6116 CrossRef CAS PubMed.
- L. J. Martin, M. J. Hahnke, M. Nitz, J. Wohnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106–7113 CrossRef CAS PubMed.
- K. D. Daughtry, L. J. Martin, A. Sarraju, B. Imperiali and K. N. Allen, ChemBioChem, 2012, 13, 2567–2574 CrossRef CAS PubMed.
- D. B. Thompson, J. J. Cronican and D. R. Liu, Methods Enzymol., 2012, 503, 293–319 CAS.
- S. H. Xue, J. J. Qiao, F. Pu, M. Cameron and J. J. Yang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 163–179 CrossRef CAS PubMed.
- H. J. Weinmann, R. C. Brasch, W. R. Press and G. E. Wesbey, AJR, Am. J. Roentgenol., 1984, 142, 619–624 CrossRef CAS PubMed.
- Y. Wang, S. Song, J. Liu, D. Liu and H. Zhang, Angew. Chem., Int. Ed., 2014, 54, 536 Search PubMed.
- M. Yu and J. Zheng, Nano Lett., 2015, 10, 1021 Search PubMed.
- H. J. Lee and W. M. Pardridge, Bioconjugate Chem., 2001, 12, 995–999 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- O. Ziv, R. R. Avtalion and S. Margel, J. Biomed. Mater. Res., Part A, 2008, 85, 1011–1021 CrossRef PubMed.
- J. J. Qiao, S. Y. Li, L. X. Wei, J. Jiang, R. Long, H. Mao, L. Wei, L. Y. Wang, H. Yang, H. E. Grossniklaus, Z. R. Liu and J. J. Yang, PLoS One, 2011, 6, e18103 CAS.
- D. Grum, S. Franke, O. Kraff, D. Heider, A. Schramm, D. Hoffmann and P. Bayer, PLoS One, 2013, 8, e65346 CAS.
- A. Bhattacharya, S. G. Turowski, I. D. San Martin, A. Rajput, Y. M. Rustum, R. M. Hoffman and M. Seshadri, Anticancer Res., 2011, 31, 387–393 CAS.
- A. Bar-Shir, Y. J. Liang, K. W. Y. Chan, A. A. Gilad and J. W. M. Bulte, Chem. Commun., 2015, 51, 4869–4871 RSC.
- M. J. Allen, K. W. MacRenaris, P. N. Venkatasubramanian and T. J. Meade, Chem. Biol., 2004, 11, 301–307 CrossRef CAS PubMed.
- Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4317–4322 CrossRef CAS PubMed.
- C. Heyn, J. A. Ronald, S. S. Ramadan, J. A. Snir, A. M. Barry, L. T. MacKenzie, D. J. Mikulis, D. Palmieri, J. L. Bronder, P. S. Steeg, T. Yoneda, I. C. MacDonald, A. F. Chambers, B. K. Rutt and P. J. Foster, Magn. Reson. Med., 2006, 56, 1001–1010 CrossRef PubMed.
- L. Josephson, C. H. Tung, A. Moore and R. Weissleder, Bioconjugate Chem., 1999, 10, 186–191 CrossRef CAS PubMed.
- V. Economopoulos, Y. H. Chen, C. McFadden and P. J. Foster, Clin. Transl. Oncol., 2013, 6, 347–354 CrossRef.
- M. Lewin, N. Carlesso, C. H. Tung, X. W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- T. Yamane, K. Hanaoka, Y. Muramatsu, K. Tamura, Y. Adachi, Y. Miyashita, Y. Hirata and T. Nagano, Bioconjugate Chem., 2011, 22, 2227–2236 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01925g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |