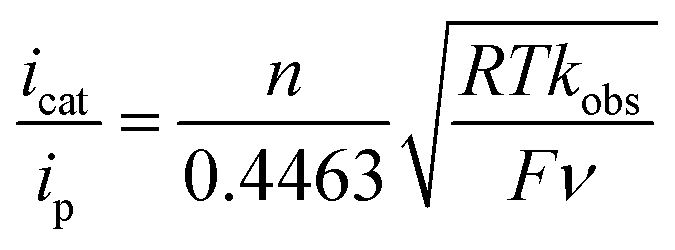DOI:
10.1039/C5SC00398A
(Edge Article)
Chem. Sci., 2015,
6, 2737-2745
Increasing the rate of hydrogen oxidation without increasing the overpotential: a bio-inspired iron molecular electrocatalyst with an outer coordination sphere proton relay†
Received
2nd February 2015
, Accepted 4th March 2015
First published on 5th March 2015
Abstract
Oxidation of hydrogen (H2) to protons and electrons for energy production in fuel cells is currently catalyzed by platinum, but its low abundance and high cost present drawbacks to widespread adoption. Precisely controlled proton removal from the active site is critical in hydrogenase enzymes in nature that catalyze H2 oxidation using earth-abundant metals (iron and nickel). Here we report a synthetic iron complex, (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl), that serves as a precatalyst for the oxidation of H2, with turnover frequencies of 290 s−1 in fluorobenzene, under 1 atm of H2 using 1,4-diazabicyclo[2.2.2]octane (DABCO) as the exogenous base. The inclusion of a properly tuned outer coordination sphere proton relay results in a cooperative effect between the primary, secondary and outer coordination spheres for moving protons, increasing the rate of H2 oxidation without increasing the overpotential when compared with the analogous complex featuring a single pendant base. This finding emphasizes the key role of pendant amines in mimicking the functionality of the proton pathway in the hydrogenase enzymes.
Introduction
Nature has developed the ability to perform catalytic chemical transformations at amazing rates and efficiencies. For example, hydrogenase enzymes are able to both produce and oxidize H2 at rates exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 and at low overpotentials (high efficiencies).1–3 Such remarkable performance has only been achieved through active sites that have evolved to minimize free energy differences between the reactive intermediates of the catalytic process, with the outer coordination sphere organized to shuttle protons between the surrounding matrix and the metal center.4 Understanding and mimicking the function of hydrogenase enzymes is a particularly compelling challenge, as increasing efforts worldwide are devoted to utilizing clean, renewable resources for power. Solar and wind have the potential to eliminate our dependence on fossil fuels; however, their irregular geographic and temporal availability underlies the necessity for efficient energy storage technologies. One promising strategy is to electrochemically convert energy into chemical bonds in the form of a fuel such as H2, which can be stored, transported and used when needed.5 Current H2 fuel cell technologies rely on high-cost, low abundant metals, such as platinum, to accomplish the oxidation of H2, limiting their widespread use.6 Hydrogenase enzymes, which use earth-abundant Ni and Fe metals, serve as inspiration to develop non-precious metal molecular catalysts for H2 production and oxidation with comparable rates and efficiencies.7–10
000 s−1 and at low overpotentials (high efficiencies).1–3 Such remarkable performance has only been achieved through active sites that have evolved to minimize free energy differences between the reactive intermediates of the catalytic process, with the outer coordination sphere organized to shuttle protons between the surrounding matrix and the metal center.4 Understanding and mimicking the function of hydrogenase enzymes is a particularly compelling challenge, as increasing efforts worldwide are devoted to utilizing clean, renewable resources for power. Solar and wind have the potential to eliminate our dependence on fossil fuels; however, their irregular geographic and temporal availability underlies the necessity for efficient energy storage technologies. One promising strategy is to electrochemically convert energy into chemical bonds in the form of a fuel such as H2, which can be stored, transported and used when needed.5 Current H2 fuel cell technologies rely on high-cost, low abundant metals, such as platinum, to accomplish the oxidation of H2, limiting their widespread use.6 Hydrogenase enzymes, which use earth-abundant Ni and Fe metals, serve as inspiration to develop non-precious metal molecular catalysts for H2 production and oxidation with comparable rates and efficiencies.7–10
Development of functional mimics of the [FeFe]-hydrogenase has focused on optimizing the first and second coordination spheres of the active catalytic site.11–15 Less attention has been paid towards the development and utility of the outer coordination sphere in synthetic catalysts to improve catalytic performance.16 Although outer coordination sphere interactions may seem remote from the active site, recent work by Shaw has demonstrated that these effects can play a significant role in catalytic transformations.16 Synthetic iron complexes that heterolytically cleave H2 have appeared in the literature including those that catalyze the oxidation of H2 using a chemical oxidant.17–19 Our Center reported (CpC6F5)Fe(PtBu2NBn2)H (PR2NR′2 = RP(CH2N(R′)CH2)2PR), a molecular iron electrocatalyst for the oxidation of H2 with an active site that has been thermodynamically tuned through modification of the first coordination sphere, and a pendant amine in the second coordination sphere to heterolytically cleave H2.20 Very recently, we reported the related (CpC5F4N)Fe(PRNR′PR)Cl family of complexes (PRNR′PR = R2PCH2N(R′)CH2PR2) for molecular electrocatalytic oxidation of H2 that included a detailed mechanistic study of the catalytic process.21 In that study, deprotonation of the Fe(III) hydride was identified as having a significant thermodynamic barrier due to the mismatch between the pKa values of the metal hydride and that of the protonated pendant base, limiting the rate of electrocatalytic H2 oxidation. We now report the synthesis of a modified iron complex that incorporates an additional proton relay in the outer coordination sphere to facilitate intramolecular proton movement. This new iron complex, (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl), 1-Cl, electrocatalytically oxidizes H2 (1 atm) at a rate more than two orders of magnitude faster than the first iron-based, homogeneous H2 oxidation electrocatalyst, (CpC6F5)Fe(PtBu2NBn2)(H) and one order of magnitude faster than the second generation (CpC5F4N)Fe(PRNR′PR)(H) catalysts.20,21 Equally remarkable, the addition of an outer sphere proton relay to a (CpC5F4N)Fe(PRNR′PR)(H) catalyst increased the rate without an increase in the overpotential, illustrating the power of controlling proton movement for improved catalytic performance. Our study clearly demonstrates that outer coordination sphere effects can significantly enhance the performance of bio-inspired synthetic molecular catalysts.
Results
Synthesis and characterization of (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl) (1-Cl) and electrocatalytic intermediates
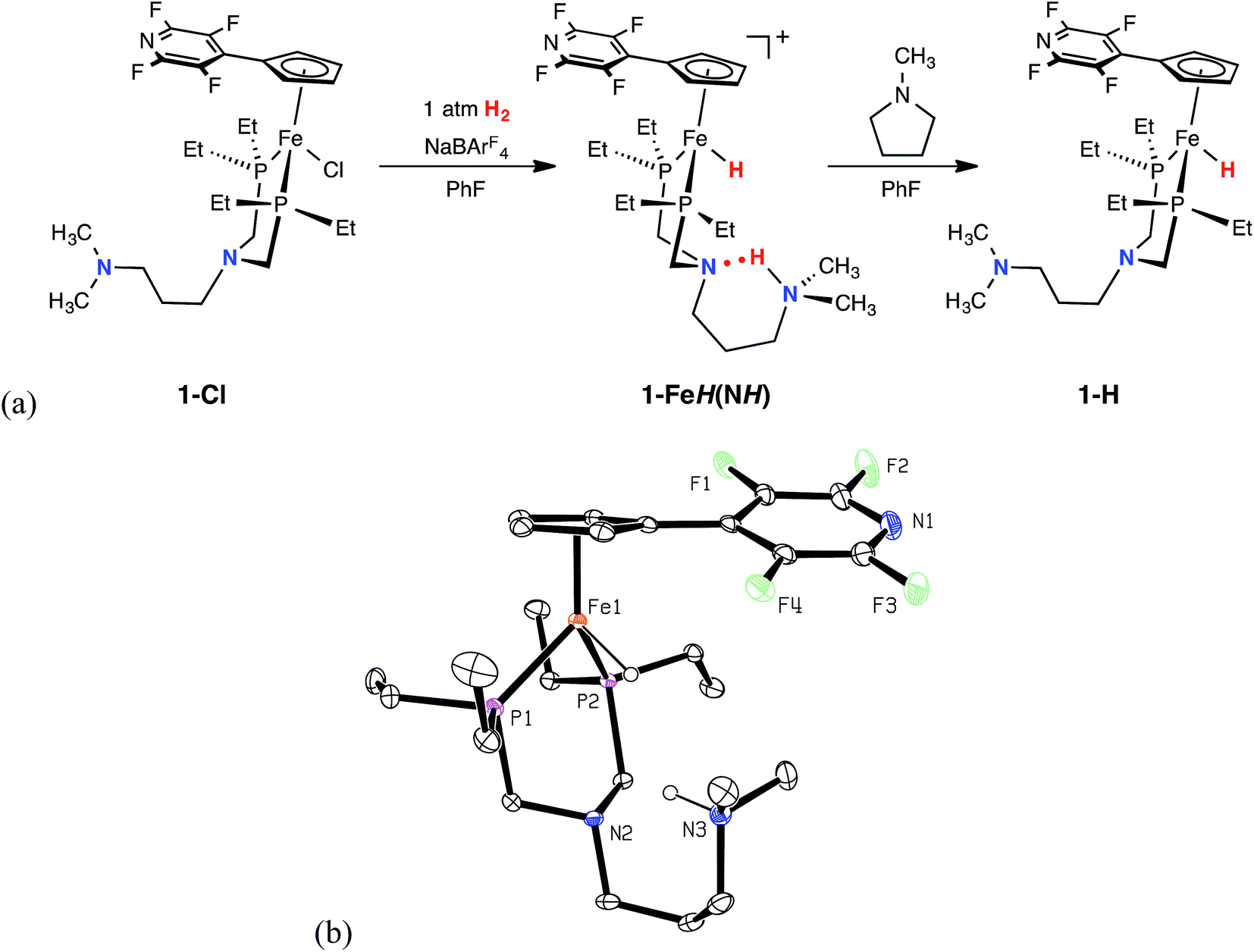
The green iron chloride complex, 1-Cl, was prepared by treating (PEtN(CH2)3NMe2PEt)FeCl2 with NaCpC5F4N, and isolated in 92% yield. Reaction of 1-Cl with NaBArF4 (ArF = 3,5-bis(trifluoromethyl)phenyl) under an atmosphere of H2 generated the protonated iron hydride species, 1-FeH(NH), which has been characterized by heteronuclear NMR spectroscopy and single crystal X-ray diffraction (Fig. 1, and ESI Table S1†). Spectroscopic NMR studies of 1-FeH(NH) in solution were diagnostic of dynamic behavior involving the H2 ligand (see below); however, at −40 °C, the 1H NMR spectrum exhibits iron hydride (−17.9 ppm) and protonated amine (10.4 ppm) resonances, indicative of heterolytically cleaved H2. In the solid state (Fig. 1), 1-FeH(NH) adopts a three-legged piano-stool geometry, where the six-membered ring comprised of the PNP backbone and the iron center is in a boat conformation. The structure illustrates that heterolytic cleavage of H2 has occurred, as was also observed in the recently reported neutron diffraction structure of (CpC5F4N)Fe(H)(PtBu2NtBu2H).22 The proton located on the outer coordination sphere amine forms a six-membered ring with the second coordination sphere pendant base through hydrogen bonding. Addition of five equivalents of N-methylpyrrolidine deprotonated complex 1-FeH(NH) and furnished the iron hydride species, 1-H, as a viscous, red oil in 51% yield.
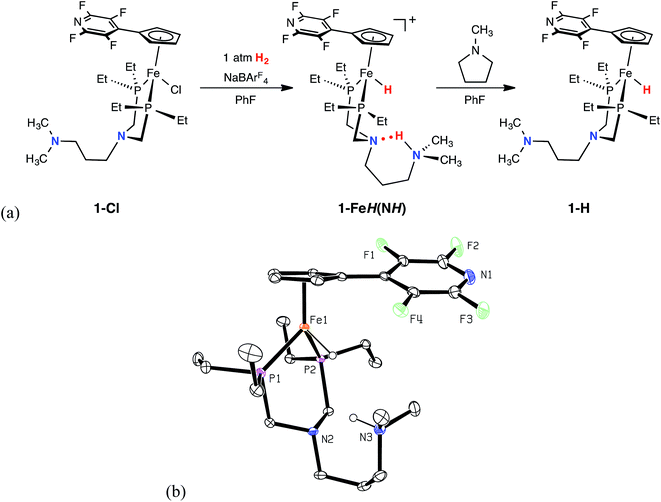 |
| | Fig. 1 (a) Observable catalytic intermediates from the reaction of 1-Cl with NaBAr4F under 1 atm H2. (b) Representation of the solid-state structure of 1-FeH(NH) with 30% probability ellipsoids. The BArF4 anion and all hydrogen atoms with the exception of the proton and the hydride are omitted for clarity. | |
Electrochemical characterization of the iron complexes and electrocatalytic oxidation of H2
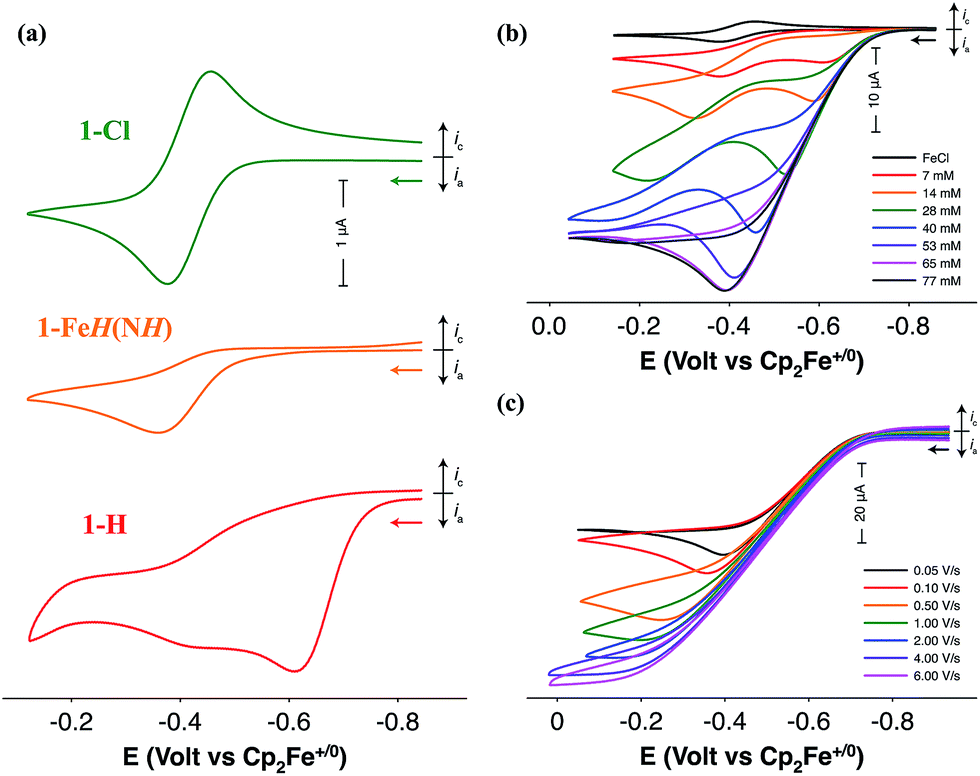
Electrochemical characterization of the iron complexes were carried out in fluorobenzene solutions to avoid catalyst inhibition from solvent binding to the 16-electron complex.21 The cyclic voltammogram (CV) of 1-Cl exhibits a reversible one-electron oxidation wave assigned to the FeII/III couple at −0.42 V versus the ferrocenium/ferrocene couple (Fig. 2a). Irreversible oxidation waves for the protonated iron hydride, 1-FeH(NH) (Ep = −0.36 V) and the iron hydride, 1-H (Ep = −0.61 V) are observed in their CVs (Fig. 2a). Electrocatalytic oxidation studies of H2 are carried out after addition of NaBArF4 to 1-Cl under 1 atm H2 in fluorobenzene. Cyclic voltammograms recorded upon subsequent additions of either 1,4-diazabicyclo[2.2.2]octane (Fig. 2b, DABCO, pKa = 18.2 for H-DABCO+ in CH3CN) or N-methylpyrrolidine (Fig. S1,† pKa = 18.5 for N-methylpyrrolidinium in CH3CN) result in a current enhancement consistent with electrocatalytic H2 oxidation.23,24 Base concentration and scan rate independence is achieved at 0.1 M DABCO and 6 V s−1 (Fig. 2c and S2†), indicating that steady state conditions were achieved, yielding a reliable catalytic current (icat) for calculation of the turnover frequency (TOF). Similar results were obtained when using N-methylpyrrolidine as exogenous base (Fig. S1†). Bulk electrolysis of a 0.5 mM solution of 1 in the presence of 1 atm of H2 and 77 mM N-methylpyrrolidine confirmed H2 oxidation through the quantitative analysis of the protons via NMR spectroscopy, resulting in an average current efficiency of 95% and a minimum turnover number of 6.| | 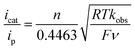 | (1) |
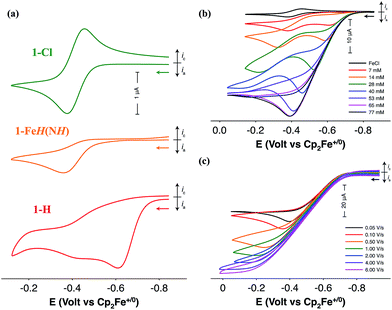 |
| | Fig. 2 (a) CVs of 1-Cl (green), 1-FeH(NH) (orange), and 1-H (red) in fluorobenzene solution at 50 mV s−1. (b) CVs of a fluorobenzene solution of 1-FeH(NH) upon addition of DABCO under 1 atm H2 at 50 mV s−1. The initial CV (black) corresponds to 1-Cl prior to activation with NaBAr4F. (c) CVs of a fluorobenzene solution of 1-H in the presence of 77 mM DABCO under 1 atm H2 as a function of scan rate. Conditions for all experiments: 1 mM [Fe], 0.1 M [nBu4N][B(C6F5)4], 22 °C. | |
The TOF for catalytic H2 oxidation with 1-Cl was calculated using eqn (1).25,26 In this equation, ip is the current under non-catalytic conditions, icat is the catalytic current, n is the number of electrons (2) involved in the catalytic reaction, kobs is the observed first-order rate constant (TOF), R is the universal gas constant, T is the temperature in Kelvin, F is Faraday's constant, ν is the scan rate, and 0.4463 is a constant determined by numerical solution of the diffusion equations.27 The TOF for the oxidation of H2 catalyzed by 1-Cl was determined to be 290 s−1 in the presence of ≥77 mM DABCO under 1 atm of H2 at a half-wave potential (Ecat/2) of −0.46 V. The TOF for the oxidation of H2 catalyzed by 1-Cl was determined to be 180 s−1 in the presence of ≥65 mM N-methylpyrrolidine, the base used in previously reported catalytic studies with (CpC5F4N)Fe(PEtNMePEt)(Cl) (Fig. S1†). The thermodynamic potential for the oxidation of H2 using a 10:1 (base to acid) buffered solution of DABCO was determined by open circuit potential measurement and was found to be 1.22 V, resulting in a 0.65 V overpotential at Ecat/2 (Fig. S3–S5†).28,29 A buffered solution was used for the determination of the overpotential to minimize the experimental uncertainty in the measurement, in accord with recent recommendations.29
Discussion
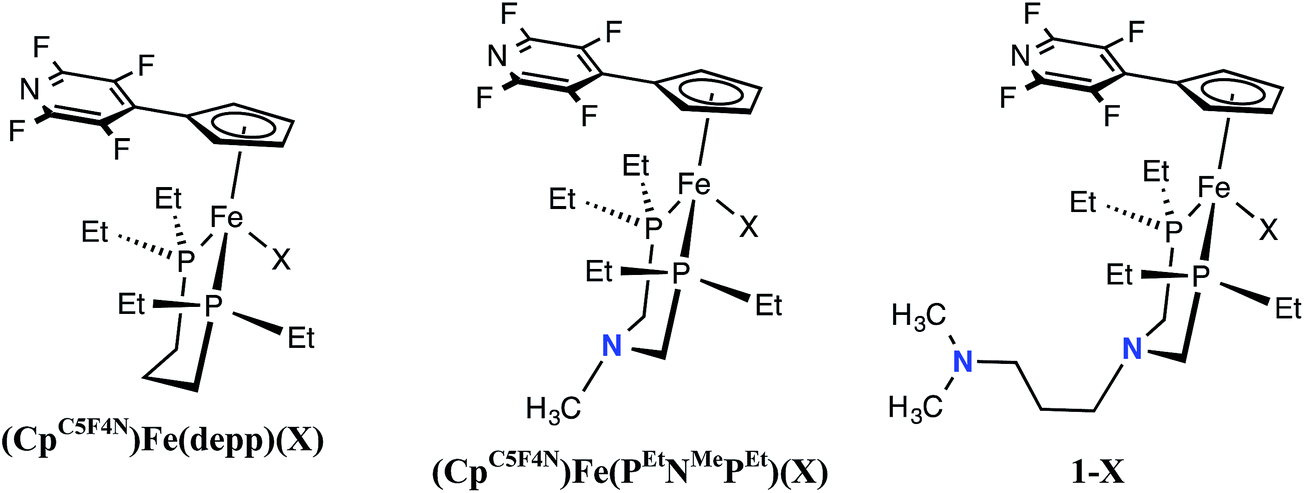
Comparison of (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(X), (CpC5F4N)Fe(PEtNMePEt)(X) and (CpC5F4N)Fe(Et2P(CH2)3PEt2)(X) (X = Cl, H)
To illustrate the impact of the number of proton relays on the rate of H2 oxidation, electrochemical and electrocatalytic studies using iron complexes without pendant amines ((CpC5F4N)Fe(depp)(X) (X = Cl, H), Scheme 1) and with one pendant amine ((CpC5F4N)Fe(PEtNMePEt)(X), Scheme 1) were conducted under identical conditions to those of 1-X. For the three Fe complexes, the reversible Fe(III/II) couple of the Cl− complexes differ by only 40 mV (Table 1 and Fig. S6†). Similarly, the peak of the waves of the Fe(II) hydride complexes range from −0.56 to 0.61 V (Table 1 and Fig. S7†). The (CpC5F4N)Fe(depp)(H)(III/II) couple is reversible, while the (CpC5F4N)Fe(PEtNMePEt)(H)(III/II) wave is irreversible, but becomes reversible at scan rates ≥0.5 V s−1. The Fe(III/II) couple for 1-H is irreversible up to scan rates as fast as 50 V s−1 (Fig. S8†). The irreversible nature of the Fe(II) hydride oxidation of (CpC5F4N)Fe(PEtNMePEt)(H) and 1-H can be attributed to the intramolecular deprotonation of the hydride by the pendant amines.21 In the presence of exogenous base, the reversibility of the Fe(II) hydride oxidation waves as a function of scan rate can be used to estimate pseudo-first-order rate constants for intermolecular deprotonation. For (CpC5F4N)Fe(depp)(H) this rate was estimated to be <0.1 s−1 and >25 s−1 for (CpC5F4N)Fe(PEtNMePEt)(H).21 Similar studies of 1-H show no reversibility, indicating the rate of intermolecular deprotonation is likely ≫25 s−1 (Fig. S9†). This data indicates that providing a pathway to move the proton further away from the metal center results in more facile deprotonation by the exogenous base.
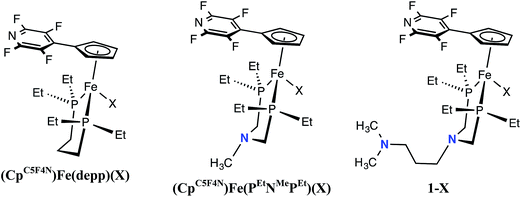 |
| | Scheme 1 Complexes compared to examine the role of the number of pendant amines (X = Cl, or H). | |
Table 1 Electrochemical and electrocatalytic data for complexes with increasing number of proton relays
| Compound |
Fe(III/II) couplea (V) X = Cl |
Fe(III/II) coupleb (V) X = H |
TOFc (s−1) |
Overpotentiald (V) |
|
Conditions: fluorobenzene solution, 1 mM [Fe], 0.1 M [nBu4N][B(C6F5)4], 50 mV s−1, 22 °C.
Values reported are for the oxidative peak potential.
Reported values were determined upon addition of a DABCO solution, and are given under base- and scan rate-independent conditions (see above and the ESI† for additional information).
Reported values were determined upon addition of a 10:1 DABCO to DABCO-H+ solution as the exogenous base, and are given under base- and scan rate-independent conditions (see above and the ESI† for additional information).
Not Catalytic (NC) – no significant current enhancement was observed, indicating this complex is not a catalyst for H2 oxidation.
|
| (CpC5F4N)Fe(depp)(X) |
−0.45 |
−0.60 |
NCe |
— |
| (CpC5F4N)Fe(PEtNMePEt)(X) |
−0.41 |
−0.56 |
34 |
0.64 |
|
1-X
|
−0.42 |
−0.61 |
290 |
0.65 |
Electrocatalytic H2 oxidation with 1-Cl resulted in a TOF of 180 s−1 using N-methylpyrrolidine as the exogenous base and a TOF of 290 s−1 when using DABCO. Although N-methylpyrrolidine and DABCO have similar pKa values, the less sterically demanding nature of DABCO yields faster rates of H2 oxidation, indicating deprotonation by exogenous base is likely a rate-limiting factor. In order to study the effect of the outer coordination sphere pendant amine on catalysis, H2 oxidation TOFs and overpotentials with Fe(depp)(X) and Fe(PEtNMePEt)(X) and DABCO were determined (Table 1 and Fig. S10 and 11†). The rates of H2 oxidation by the three compounds increase as the number of pendant amines increases, with no significant change (<10 mV) in the overpotential. This data is again consistent with the hypothesis that increasing the rate of deprotonation of the Fe(III) hydride increases the rate of catalysis, rather than an increase attributable to the driving force (overpotential) of catalysis.
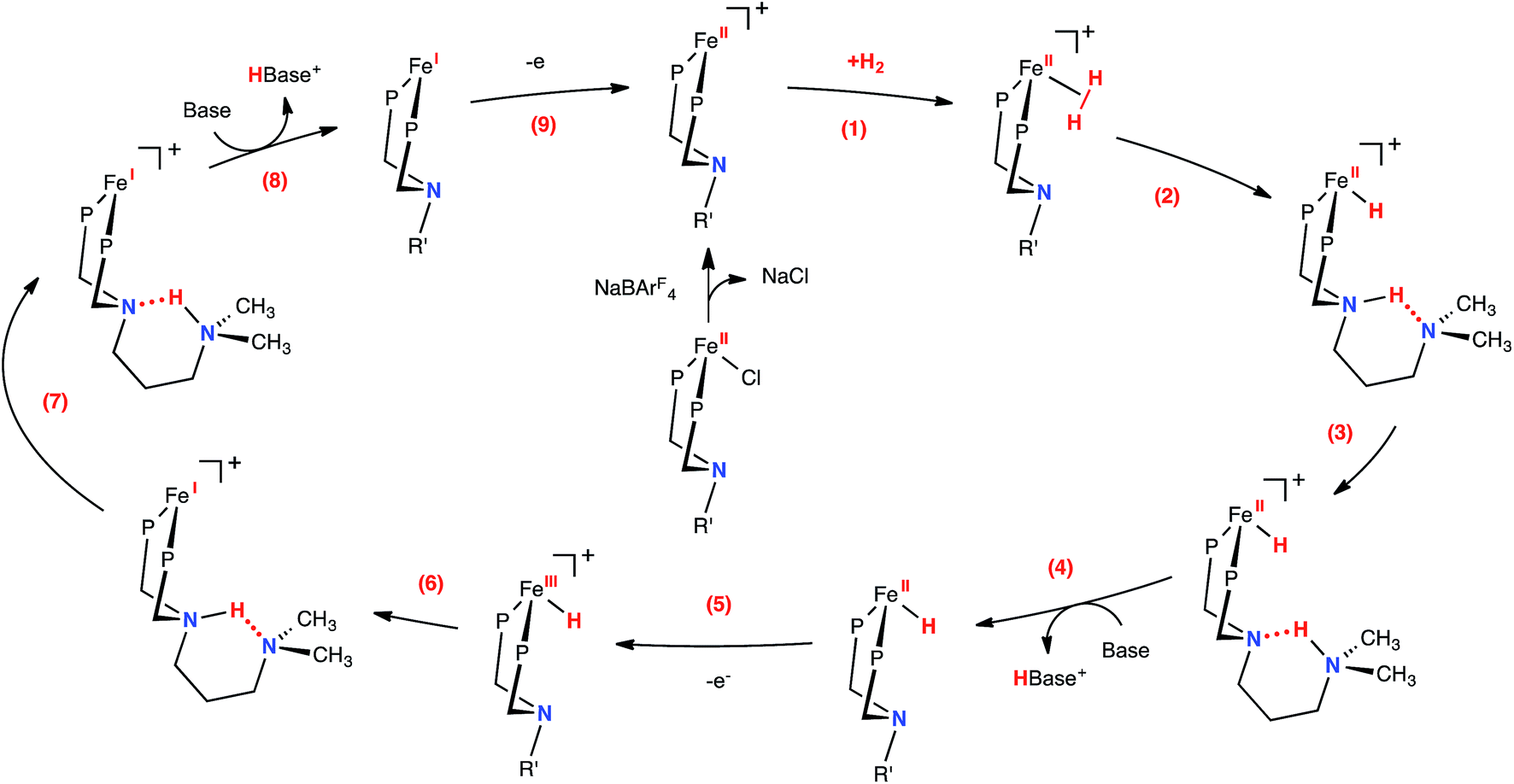
Proposed mechanism of electrocatalytic H2 oxidation
The proposed mechanism for H2 oxidation by 1-Cl is shown in Fig. 3 (some intermediates have been omitted for clarity; see Fig. S12–14† for complete mechanistic details). Addition of H2 to the unsaturated, 16-electron Fe(II) complex is followed by heterolytic cleavage to form the protonated iron hydride 1-FeH(NH). A dihydrogen bonding30 interaction between the proton and hydride in this complex is likely, as previously observed in the analogous [(CpC5F4N)Fe(H)(PtBu2NtBu2H)]+ complex.22 Computed thermodynamic and kinetic parameters for these steps (Fig. 3, steps 1–3) suggest that the resulting species are close in free energy and the barriers are less than 9 kcal mol−1 (Fig. S13†), and therefore do not limit the observed rate of oxidation of H2. These results are further supported by the dynamic behavior observed by 1H NMR spectroscopy (Fig. S15†). At −40 °C (at 500 MHz), the proton and hydride environments of 1-FeH(NH) are resolved, and the FeH (−17.93 ppm) and NH (10.39 ppm) resonances are independently observed. This peak separation corresponds to a proton hydride exchange of ∼6.4 × 103 s−1 (ΔG‡ of 12 kcal mol−1 according to simple transition state theory arguments) at the coalescence temperature of ∼20 °C. The coalesced proton-hydride resonance is observed at −6.1 ppm (65 °C). This deviates from the expected value of about −3.8 ppm, and is consistent with contributions from other energetically-matched species (i.e. Fe(η2-H2) complex). In addition to the fluxional behavior of the proton and hydride resonances, the ethyl and methylene resonances of PEtN(CH2)3NMe2PEt also coalesce at elevated temperatures (Fig. S15†). At −40 °C (500 MHz), the two ethyl environments are resolved, and the two resonances are independently observed (0.71 and 0.81 ppm). This peak separation corresponds to a Fe–PNP inversion (Fig. S16†) rate of 25 s−1 at the coalescence temperature of ∼40 °C (ΔG‡ of 16 kcal mol−1). This behavior is consistent with loss of H2 and formation of the pseudosymmetric, fluxional 16-electron species, and is consistent with the computational results. Given the calculated energies and this experimentally determined rate for heterolytic cleavage, it is apparent that these steps are not rate-limiting for catalysis in this system.
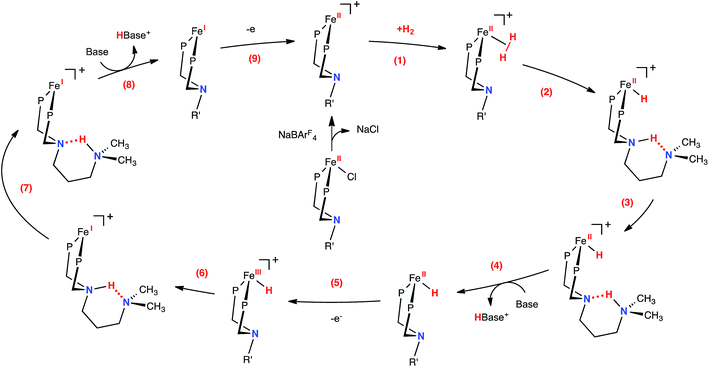 |
| | Fig. 3 Proposed mechanism for the electrocatalytic oxidation of H2 by [(CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)]+. Steps (4) and (8) include opening of the six-membered ring (not shown), prior to deprotonation by exogenous base (see Fig. 4, S13 and S14†). The CpC5F4N and ethyl substituents on the phosphorus atoms have been omitted for clarity (R′ = CH2CH2CH2N(CH3)2). | |
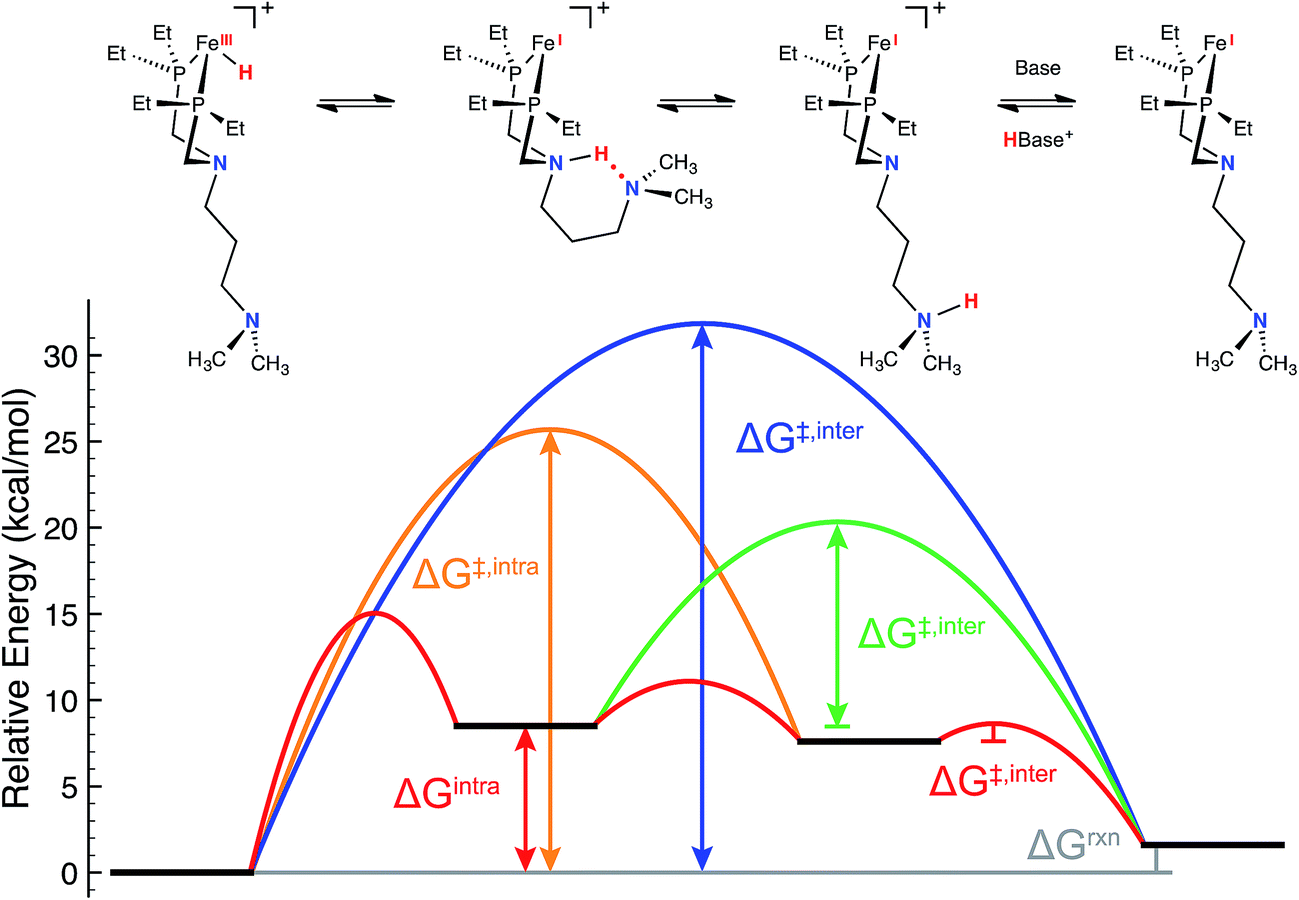
Consistent with our current study, previous experimental and computational work describing the mechanism of H2 oxidation by the Fe–PRNR′PR family of complexes indicated that H2 addition and cleavage (i.e., Fig. 3, steps 1 and 2) and the subsequent deprotonation of the pendant amine of the Fe(II) hydride (i.e., Fig. 3, steps 3–4) do not significantly contribute to the overall catalytic rate. In contrast, the intramolecular followed by intermolecular deprotonation steps of the Fe(III) hydride are the steps that limit the overall observed rate of catalysis (Fig. 3, steps 6–8, Fig. 4).21 The large difference in acidity between the Fe(III) hydride and the protonated, secondary sphere pendant amine (9 pKa units) makes the intramolecular proton transfer thermodynamically unfavorable. Additionally, the poor steric accessibility of the protonated pendant amine introduces a further penalty for removal of the proton by an exogenous base (e.g.Fig. 4, green ΔG‡inter). A detailed computational analysis of H2 oxidation by [(CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)]+ using N-methylpyrrolidine as the exogenous base was carried out, allowing for direct comparison to the previously published [(CpC5F4N)Fe(PEtNMePEt)]+ complex.21 The calculations revealed that introduction of an outer coordination sphere amine results in a more suitably energy-matched pathway for the proton from the Fe(III) center to the periphery of the catalyst (Fig. 4, red trace). The presence of the outer coordination sphere amine stabilizes the aminium in the second coordination sphere through intramolecular hydrogen bonding, which results in a reduced pKa difference between the Fe(III) hydride intermediate and protonated amine by about 3 pKa units with respect to the [(CpC5F4N)Fe(PEtNMePEt)]+ complex (Fig. S9†).21 Proton transfer between the two pendant amines is very facile, and the intramolecular hydrogen bond between them easily breaks, exposing the protonated outer sphere amine to the exogenous base, diminishing the steric penalty for deprotonation. It is important to point out that direct deprotonation of the Fe(III) hydride either by the outer sphere amine (Fig. 4, orange ΔG‡intra) or by an exogenous base (Fig. 4, blue ΔG‡inter) is kinetically disfavored because of the scarce accessibility of the base to the metal hydride. A detailed mechanistic discussion of the various pathways for the proton movement is provided in the ESI.† As observed in the remarkable TOF exhibited by this catalyst, the low energy pathway provided by the addition of the outer coordination sphere pendant amine to move the proton away from the Fe(III) active site mimics the functionality of hydrogenase enzymes, in which protons shuttle through different amino acids for rapid proton movement.
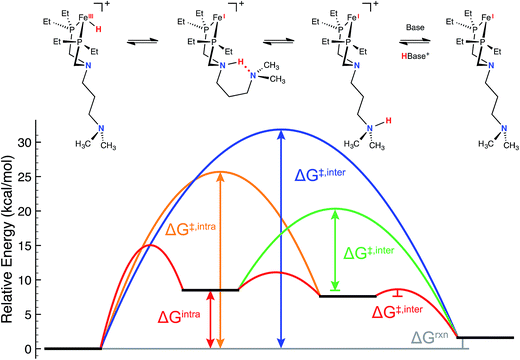 |
| | Fig. 4 Free energy diagram for the key intra- and intermolecular deprotonation steps of [(CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(H)]+ by N-methylpyrrolidine as obtained from density functional theory calculations (see ESI† for additional details). Red lines: lowest-free energy deprotonation pathway (upper reaction scheme); blue line: direct deprotonation of the iron(III) hydride by the exogenous base; orange line: deprotonation of the iron(III) hydride by the outer coordination sphere pendant amine; green line: deprotonation of the second coordination sphere pendant amine by exogenous base. The CpC5F4N substituent has been omitted for clarity. | |
Conclusions
These results conclusively demonstrate that controlled movement of protons from the primary, to the secondary and outer coordination spheres dramatically improves the rate of H2 oxidation in an iron-based molecular electrocatalyst without increasing the overpotential. Although this report focuses on a single catalyst, the comprehensive thermodynamic analysis highlights the broader implications of this approach, specifically how mimicking enzyme functionality to minimize free energy differences between catalytic intermediates can be used to improve catalyst performance. One of the most exciting results from this work is the demonstrated use of rational catalyst design to control proton movement and lower the barrier of the rate-limiting step in catalysis by the previously reported (CpC5F4N)Fe(PRNR′PR)(H) family. As electrocatalytic rates of H2 oxidation escalate, an increased focus on lowering the overpotential of these catalysts will be crucial.
Experimental
All manipulations of free phosphine ligands and metal reagents were carried out under argon using standard vacuum line, Schlenk, and inert atmosphere glovebox techniques. Solvents were purified by passage through neutral alumina using an Innovative Technology, Inc., PureSolv™ solvent purification system. THF-d8 and bromobenzene-d5 were vacuum transferred from NaK and CaH2, respectively after stirring for a minimum of 24 h under an inert atmosphere prior to use. DABCO was dried under vacuum prior to use. N-Methylpyrrolidine was dried over KOH, degassed, distilled under vacuum, and stored in an argon-filled glovebox. Elemental analyses were performed by Atlantic Microlab, Inc. in Norcross, GA. The following compounds were prepared according to literature procedures: NaCpC5F4N,22 and [(Et2O)2H][B(C6F5)4].31 Additional experimental details can be found in the ESI, pages S3–S7.†
Instrumentation
NMR.
Spectra were recorded on a Varian Inova spectrometer (500 MHz for 1H) at 22 °C unless otherwise noted. All 1H chemical shifts have been internally calibrated using the monoprotio impurity of the deuterated solvent. The 31P{1H} NMR spectra were referenced to external phosphoric acid at 0 ppm. The 19F NMR spectra were referenced to external fluorobenzene at −113.15 ppm. A fluorobenzene solution of tetramethylsilane (3.0 mM) was used as the solvent for variable temperature NMR experiments.
Electrochemistry.
All experimental procedures were conducted at ambient temperature, 22–23 °C, under argon in a vacuum atmospheres drybox. A standard three-electrode configuration was employed in conjunction with a CH Instruments potentiostat interfaced to a computer with CH Instruments 700 D software. All voltammetric scans were recorded using glassy-carbon working electrode disks of 1 mm diameter (Cypress Systems EE040). A glassy-carbon rod (Structure Probe, Inc.) and platinum wire (Alfa-Aesar) were used as auxiliary electrodes and quasi-reference electrodes, respectively. All glassware for electrochemical experiments was oven dried overnight and allowed to cool to room temperature before use. [Cp2Co][PF6] and (η6-C6H6)2Cr were used as an internal standard, and all potentials reported within this work are measured vs. the Cp2Co+/0 couple at −1.33 V or the (η6-C6H6)2Cr+/0 couple at −1.22 V and are reported vs. Cp2Fe+/0. Bases were measured and transferred to electrochemical solutions via gas-tight syringes. Controlled potential electrolyses were performed using a high-power BASi potentiostat (Fig. S17†).
UV-vis.
UV-vis data were collected under argon with an Ocean Optics USB2000+ spectrometer (Fig. S18†).
X-ray diffraction.
The crystal selected for diffraction studies was immersed in Paratone-N oil, placed on a Nylon loop, and transferred to a precooled cold stream of N2. A Bruker KAPPA APEX II CCD diffractometer with 0.71073 Å Mo Kα radiation was used. The space group was determined on the basis of systematic absences and intensity statistics. The structure was solved by direct methods and refined by full-matrix least squares on F2. All non-hydrogen atoms were refined anisotropically unless otherwise stated. Hydrogen atoms were placed at idealized positions and refined using the riding model unless otherwise stated. Data collection and cell refinement were performed using Bruker APEX2 software. Data reduction and absorption correction was performed using Bruker's SAINT and SADABS programs, respectively.32,33 Structural solution and refinements were completed using SHELXS-97 and SHELXL-97 (ref. 34) using the OLEX2 software package35 as a front-end.
Computational details.
All possible reaction intermediates involved in the catalytic cycle and the transitions states between them were optimized via density function theory (DFT) calculations. The hybrid B3P86 functional36,37 was used along with the Stuttgart–Dresden relativistic effective core potential for Fe and its associated basis set (SDD)38 and the 6-31G* basis set for all the atoms with one additional p polarization function39 for the protic hydrogens. This level of theory has previously shown great accuracy for thermodynamic and kinetics parameters of first row transition metal based catalytic systems.40-42 Harmonic vibrational frequencies were calculated at the optimized geometries using the same level of theory to estimate the zero-point energy (ZPE) and the thermal contributions (298 K and 1 atm) to the free energy. Transition states were verified by intrinsic reaction coordinate (IRC) calculations. All the molecular structures were optimized in fluorobenzene using the SMD continuum model43 of solvation. Calculations were performed using Gaussian 09 suite of programs.44
Preparation of Et2PCH2N((CH2)3NMe2)CH2PEt2
A 50 mL Schlenk flask was loaded with 0.67 g (22 mmol) of paraformaldehyde, 15 mL of absolute ethanol, and a stir bar. Cycling between vacuum and nitrogen degassed the vessel, and 2.54 mL (22 mmol) of diethylphosphine was added by syringe with rapid stirring. After 0.5 h, 1.40 mL (11 mmol) of 3-(dimethylamino)-1-propylamine was added by syringe. The suspension rapidly became a clear, colorless solution over the course of 1 h. After 24 h, the solvent was removed under vacuum, and 3.20 g (94% yield) of a colorless oil identified as Et2PCH2N((CH2)3NMe2)CH2PEt2 was collected. 1H{31P} NMR (THF-d8, 22 °C): δ 1.06 (t, J = 7.6 Hz, 12H, P(CH2CH3)2), 1.28–1.48 (m, 8H, P(CH2CH3)2), 1.58 (dt, J = 14.2, 7.1 Hz, 2H, NCH2CH2CH2NMe2), 2.13 (s, 6H, N(CH3)2), 2.22 (app. t, J = 7.1 Hz, 2H, NCH2CH2CH2NMe2), 2.67 (app. t, J = 7.1 Hz, 2H, NCH2CH2CH2NMe2), 2.74 (s, 4H, PCH2N). 31P{1H} NMR (THF-d8, 22 °C): δ −32.9 (s). 13C{1H} NMR (THF-d8, 22 °C): δ 15.5 (d, JCP = 14 Hz, P(CH2CH3)2), 19.6 (d, JCP = 13 Hz, P(CH2CH3)2), 26.5 (s, NCH2CH2CH2NMe2), 46.0 (s, N(CH3)2), 55.9 (t, JCP = 8 Hz, NCH2CH2CH2NMe2), 58.4 (dd, JCP = 5, 8 Hz, PCH2N), 58.7 (s, NCH2CH2CH2NMe2).
Preparation of (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl) (1-Cl)
A 20 mL scintillation vial was charged with 0.083 g (0.65 mmol) of FeCl2, 2 mL THF and a stir bar. A solution of Et2PCH2N((CH2)3NMe2)CH2PEt2 (0.20 g, 0.65 mmol) in 3 mL of THF was added to the suspension with rapid stirring, and the mixture rapidly became golden yellow. After stirring for 1 h, the resulting solution was stored at −35 °C for 30 minutes. A chilled (10 mL, −35 °C for 20 minutes) THF solution of NaCpC5F4N (0.16 g, 0.65 mmol) was added in a dropwise fashion with rapid stirring, causing an immediate color change to an intense green. After 24 h, the solvent was removed under vacuum. The residue was extracted with diethyl ether, and the resulting mixture was filtered through a pad of Celite. The solvent was removed under vacuum to afford 0.37 g (92% yield) of a dark green solid identified as (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl). The product was recrystallized from a saturated solution of diethyl ether at −35 °C to afford analytically pure material. Anal. calcd for C25H40ClF4FeN3P2: C, 49.08; H, 6.59; N, 6.87. Found: C, 49.34; H, 6.64; N, 6.89. 1H{31P} NMR (THF-d8, 22 °C): δ 1.19 (t, J = 7.7 Hz, 6H, P(CH2CH3)2), 1.23 (t, J = 7.7 Hz, 6H, P(CH2CH3)2), 1.55 (p, J = 7.0 Hz, 2H, NCH2CH2CH2NMe2), 1.94–2.08 (m, 4H, P(CH2CH3)2), 2.11 (s, 6H, N(CH3)2), 2.14–2.38 (m, 6H, P(CH2CH3)2 and NCH2CH2CH2NMe2), 2.26 (d, J = 12.6 Hz, 2H, NCH2P), 2.42 (t, J = 7.0 Hz, 2H, NCH2CH2CH2NMe2), 2.77 (d, J = 12.6 Hz, 2H, NCH2P), 3.77 (app. t, J = 2.1 Hz, 2H, C5H4C5F4N), 5.43 (app. p, J = 1.8 Hz, 2H, C5H4C5F4N). 31P NMR (THF-d8, 22 °C): δ 44.5 (s). 19F NMR (THF-d8, 22 °C): δ −141.0 (m), −96.6 (m).
Characterization of [(CpC5F4N)Fe(PEtN(CH2)3N(H)Me2PEt)(H)][BAr4F] (1-FeH(NH))
A 5 mL scintillation vial was charged with 1 (0.015 g, 24 μmol), NaBArF4 (0.022 g, 24 μmol), and a stir bar. Bromobenzene-d5 (1 mL) was added with rapid stirring under an atmosphere of H2, resulting in a rapid color change from intensely green to a transparent red-orange. The mixture was filtered through a pad of Celite, and the resulting solution was characterized by NMR spectroscopy. 1H NMR (fluorobenzene, 25 °C): δ 0.75 (br m, 6H, P(CH2CH3)2), 0.86 (br m, 6H, P(CH2CH3)2), 1.18–1.37 (br m, 6H, P(CH2CH3)2 and NCH2CH2CH2NMe2), 1.48 (br m, 4H, P(CH2CH3)2), 1.67 (br d, J = 11.7 Hz, NCH2P), 2.19 (br d, J = 11.7 Hz, NCH2P), 2.27 (s, 6H, N(CH3)2), 2.31 (br t, J = 5 Hz, 2H, NCH2CH2CH2NMe2), 2.46 (br t, J = 5 Hz, 2H, NCH2CH2CH2NMe2), 4.40 (br s, 2H, C5H4C5F4N), 5.07 (br s, 2H, C5H4C5F4N), 7.63 (br s, 4H, BArF4), 8.29 (br s, 8H, BArF4). 31P NMR (bromobenzene-d5, 22 °C): δ 69.0 (br s). 19F NMR (fluorobenzene, 22 °C): δ −143.2 (br s, C5H4C5F4N), −93.7 (br s, C5H4C5F4N), −62.3 (br s, BArF4).
Preparation of (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(H) (1-H)
A 20 mL scintillation vial was charged with 1 (0.15 g, 0.24 mmol), NaBArF4 (0.22 g, 0.24 mmol), and a stir bar. Fluorobenzene (10 mL) was added with rapid stirring under an atmosphere of H2, resulting in a rapid color change from intensely green to a transparent red-orange. After 1 h, N-methylpyrrolidine (0.13 mL, 1.2 mmol) was added by microsyringe and the solution immediately became purple-red in color. After stirring for 24 h, the solvent was removed under vacuum. The residue was extracted with toluene, and filtered through a pad of Celite. Removal of solvent afforded a viscous red oil (0.072 g, 51% yield) identified as 1-H. 1H{31P} NMR (THF-d8, 22 °C): δ −17.0 (s, 1H, FeH), 0.93 (t, J = 7.6 Hz, 6H, P(CH2CH3)2), 1.12 (t, J = 7.6 Hz, 6H, P(CH2CH3)2), 1.29–1.70 (m, 8H, P(CH2CH3)2), 1.69–1.87 (m, 4H, NCH2CH2CH2NMe2 and PCH2N), 2.12 (s, 6H, N(CH3)2), 2.19 (t, J = 6.9, 2H, NCH2CH2CH2NMe2 or NCH2CH2CH2NMe2), 2.34 (t, J = 7.0, 2H, NCH2CH2CH2NMe2 or NCH2CH2CH2NMe2), 2.88 (d, J = 12.0, 2H, PCH2N), 4.54 (app. t, J = 2.0 Hz, 2H, C5H4C5F4N), 5.04 (app. p, J = 2.1 Hz, 2H, C5H4C5F4N). 31P NMR (THF-d8, 22 °C): δ 65.2 (d, JPH = 71.9 Hz). 19F NMR (THF-d8, 22 °C): δ −144.7 (m), −96.8 (m).
Preparation of [(DABCO)2H][B(C6F5)4]
A 20 mL scintillation vial was charged with DABCO (0.50 g, 4.4 mmol), 5 mL of fluorobenzene, and a stir bar. A fluorobenzene solution (10 mL) of [(Et2O)2H][B(C6F5)4 (1.85 g, 2.23 mmol) was added dropwise, and resulted in the precipitation of a white solid. After 4 h, the solvent was removed, and a white solid (1.88 g, 93% yield) identified as [(DABCO)2H][B(C6F5)4] was isolated. 1H NMR (dichloromethane-d2, 22 °C): δ 2.91 (s, 24H, CH2), 13.81 (s, 1H, N–H). 13C{1H} NMR (dichloromethane-d2, 22 °C): δ 46.0 (s). 19F NMR (dichloromethane-d2, 22 °C): δ −166.7 (t, J = 18 Hz, B(C6F5)4), −162.7 (t, J = 20 Hz, B(C6F5)4), −132.4 (d, J = 10 Hz, B(C6F5)4).
Electrochemical H2 oxidation catalyzed by [(CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)]+
A 1.0 mL fluorobenzene solution containing (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl) (1.0 mM) and [Bu4N][B(C6F5)4] (0.1 M) was prepared. Cyclic voltammograms were recorded at varying scan rates to determine the non-catalytic current (ip). NaBArF4 (0.9 mg) was added with rapid stirring under an atmosphere of H2 resulting in a color change from green to orange. Aliquots of a stock solution of DABCO (0.48 M) in fluorobenzene or a freshly prepared buffered solution of DABCO (see below) were added by microsyringe and cyclic voltammograms were collected after each addition.
Bulk electrolysis
Bulk electrolysis was performed using a high-power BASi potentiostat with a four-necked flask. One neck was sealed with a rubber septum through which a copper wire was fed that pierced through a cylinder of reticulated vitreous carbon, and functioned as the working electrode. The second and third necks were equipped with a reference electrode and a counter electrode suspended from copper wire that was fed through rubber septa. A AgCl-coated silver wire suspended in a 0.1 M fluorobenzene solution of [nBu4N][B(C6F5)4] in a glass tube with a Vycor frit served as the reference electrode. The counter electrode consisted of a Ni–Cr coiled wire and a 0.1 M fluorobenzene solution of [nBu4N][B(C6F5)4] in a glass tube with a glass frit. The fourth neck was fitted with a septum through which a copper wire attached to a second working electrode (1 mm PEEK-encased glassy carbon, Cpress Systems EE040) was fed. The second working electrode was used to record cyclic voltammograms. A needle was placed through this septum to introduce H2 into the cell. The flask contained 10 mL of a 0.1 M [nBu4N][B(C6F5)4] solution in fluorobenzene as well as 1, (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(Cl) (0.5 mM) and a small amount of Cp2CoPF6 (∼0.9 mM) as an internal reference. Before and after adding NaBArF4 (10 mg, 11 μmol) and N-methylpyrrolidine (80 μL, 77 mM), two cyclic voltammograms were recorded to ensure that the solution was under catalytic conditions and to determining the applied potential for bulk electrolysis. Controlled-potential electrolysis was performed at approximately 0.1 V positive of the peak potential for (CpC5F4N)Fe(PEtN(CH2)3NMe2PEt)(H). After the application of 4.01 C of charge (corresponding to 4.2 calculated turnovers) a 200 μL aliquot of the solution was removed and mixed with 400 μL of a 20 mM solution of Verkade's base (2,8,9-triisopropyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]undecane, 30 mg dissolved in 5 mL tetrahydrofuran). Verkade's base (pKa of the conjugate acid = 33.6 in CH3CN)45 sequestered all protons from the protonated N-methylpyrrolidine (pKa of the conjugate acid = 18.5 in CH3CN) produced from bulk electrolysis, forming protonated Verkade's base. The amount of Verkade's base (δ 118.0) and protonated Verkade's base (δ −11.9) can be quantified by 31P{1H} NMR spectroscopy, based on the known concentration of the base. The amount of protons generated (3.6 mM in a 10 mL solution, 0.036 mmol, corresponding to 3.6 turnovers) from the oxidation of H2 can be determined. A second sample was taken after passing 5.90 C of charge (corresponding to 6.2 calculated turnovers), giving the amount of protons as 6.5 mM in a 9.8 mL solution (0.064 mmol, corresponding to 6.6 turnovers). An average Faradic efficiency of 95% was calculated for H2 oxidation.
Open circuit potential measurement of a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 DABCO to DABCO-H+ solution
:1 DABCO to DABCO-H+ solution
A stock solution of DABCO (0.030 g, 0.27 mmol), [(DABCO)2H][B(C6F5)4] (0.027 g, 30 μmol) and [Bu4N][B(C6F5)4] (0.276 g, 0.30 mmol) in fluorobenzene was prepared by transferring the compounds to a 3 mL volumetric flask and diluting with solvent. A 1 mL aliquot of the solution was transferred to the electrochemical cell that was prepared according the method reported by Roberts and Bullock.28 The solution was continuously sparged with H2, and after 1 min, the open circuit potential was measured. Measurements were repeated until a consistent value was obtained, which signaled saturation of H2. Following one more repeated open circuit potential measurement, an average open circuit potential of 1.222 V was determined. Data for these experiments appears in Fig. S4.†
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This research was supported as part of the Center for Molecular Electrocatalysis, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. Pacific Northwest National Laboratory is operated by Battelle for the U.S. Department of Energy.
Notes and references
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- Y. Nicolet and J. C. Fontecilla-Camps, J. Biol. Chem., 2012, 287, 13532–13540 CrossRef CAS PubMed.
- C. Madden, M. D. Vaughn, I. Díez-Pérez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 CrossRef CAS PubMed.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2013, 26, 407–414 CrossRef.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- W. J. Shaw, M. L. Helm and D. L. DuBois, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1123–1139 CrossRef CAS PubMed.
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- F. Gloaguen and T. B. Rauchfuss, Chem. Soc. Rev., 2009, 38, 100–108 RSC.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682–684 CrossRef CAS PubMed.
- M. Y. Darensbourg and R. D. Bethel, Nat. Chem., 2012, 4, 11–13 CrossRef CAS PubMed.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS PubMed.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS PubMed.
- N. Wang, M. Wang, J. Liu, K. Jin, L. Chen and L. Sun, Inorg. Chem., 2009, 48, 11551–11558 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- N. Wang, M. Wang, Y. Wang, D. Zheng, H. Han, M. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2013, 135, 13688–13691 CrossRef CAS PubMed.
- T. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS PubMed.
- J. M. Darmon, S. Raugei, T. Liu, E. B. Hulley, C. J. Weiss, R. M. Bullock and M. L. Helm, ACS Catal., 2014, 4, 1246–1260 CrossRef CAS.
- T. Liu, X. Wang, C. Hoffmann, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2014, 53, 5300–5304 CrossRef CAS PubMed.
- K. T. Leffek, P. Pruszynski and K. Thanapaalasingham, Can. J. Chem., 1989, 67, 590–595 CrossRef CAS.
- J. F. Coetzee and G. R. Padmanabhan, J. Am. Chem. Soc., 1965, 87, 5005–5010 CrossRef CAS.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- J. M. Savéant, Acc. Chem. Res., 1980, 13, 323–329 CrossRef.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2000 Search PubMed.
- J. A. S. Roberts and R. M. Bullock, Inorg. Chem., 2013, 52, 3823–3835 CrossRef CAS PubMed.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS PubMed.
- Z. M. Heiden, S. Chen, M. T. Mock, W. G. Dougherty, W. S. Kassel, R. J. Rousseau and R. M. Bullock, Inorg. Chem., 2013, 52, 4026–4039 CrossRef CAS PubMed.
-
Bruker, SAINT Program for Data Reduction, Bruker AXS Inc., Madison, WI, USA, 2007 Search PubMed.
-
Bruker, SADABS Program for Absorption Correction of Area Detector Frames, Bruker ASX Inc., Madison, WI, USA, 2007 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed.
- S. Chen, S. Raugei, R. Rousseau, M. Dupuis and R. M. Bullock, J. Phys. Chem. A, 2010, 114, 12716–12724 CrossRef CAS PubMed.
- N. Kumar, D. M. Camaioni, M. Dupuis, S. Raugei and A. M. Appel, Dalton Trans., 2014, 43, 11803–11806 RSC.
- S. Raugei, D. L. DuBois, R. Rousseau, S. Chen, M.-H. Ho, R. M. Bullock and M. Dupuis, Acc. Chem. Res., 2015, 48, 248–255 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 2447–2464 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B01, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- P. B. Kisanga, J. G. Verkade and R. Schwesinger, J. Org. Chem., 2000, 65, 5431–5432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, syntheses, X-ray crystallographic data (CIF), and detailed theoretical discussion of presented and alternative mechanisms. CCDC 1046291. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00398a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence