
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocycles: lessons from the distant past, recent developments, and future directions
Andrei K.
Yudin
*
Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: ayudin@chem.utoronto.ca; Blog: http://www.amphoteros.com
First published on 3rd November 2014
Abstract
A noticeable increase in molecular complexity of drug targets has created an unmet need in the therapeutic agents that are larger than traditional small molecules. Macrocycles, which are cyclic compounds comprising 12 atoms or more, are now recognized as molecules that “are up to the task” to interrogate extended protein interfaces. However, because macrocycles (particularly the ones based on peptides) are equipped with large polar surface areas, achieving cellular permeability and bioavailability is anything but straightforward. While one might consider this to be the Achilles' heel of this class of compounds, the synthetic community continues to develop creative approaches toward the synthesis of macrocycles and their site-selective modification. This perspective provides an overview of both mechanistic and structural issues that bear on macrocycles as a unique class of molecules. The reader is offered a historical foray into some of the classic studies that have resulted in the current renaissance of macrocycles. In addition, an attempt is made to overview the more recent developments that give hope that macrocycles might indeed turn into a useful therapeutic modality.
1. Introduction
A macrocycle is a molecule that contains a cyclic framework of at least twelve atoms. Although the size of naturally occurring macrocycles can reach 50+ atoms in the largest ring, a recent analysis of natural products suggested that 14-, 16-, and 18-membered frameworks are the most common naturally occurring macrocyclic scaffolds.1 Over the years, many different classes of macrocyclic compounds have been synthesized and/or isolated from natural sources.2Fig. 1 depicts a selection of just three molecules of this class of compounds. In the history of organic chemistry, crown ethers emerged as the first subclass of synthetic macrocycles that offered a clear relationship between structure and function. Shown in Fig. 1 is 18-crown-6, which possesses micromolar affinity for potassium ions in methanol (Fig. 1A).3 To date, crown ethers exemplify an intuitively clear consequence of molecular-level organization of electron-donating oxygen atoms on a useful property – metal ion recognition. Also included in Fig. 1 are octreotide, a cyclic octapeptide that mimics the natural hormone somatostatin and is used for the treatment of growth hormone producing tumors, and erythromycin, a macrolide that is widely used as an antibiotic to treat bacterial infections. Similar to crown ethers, octreotide and erythromycin benefit from the organization of their function-defining elements.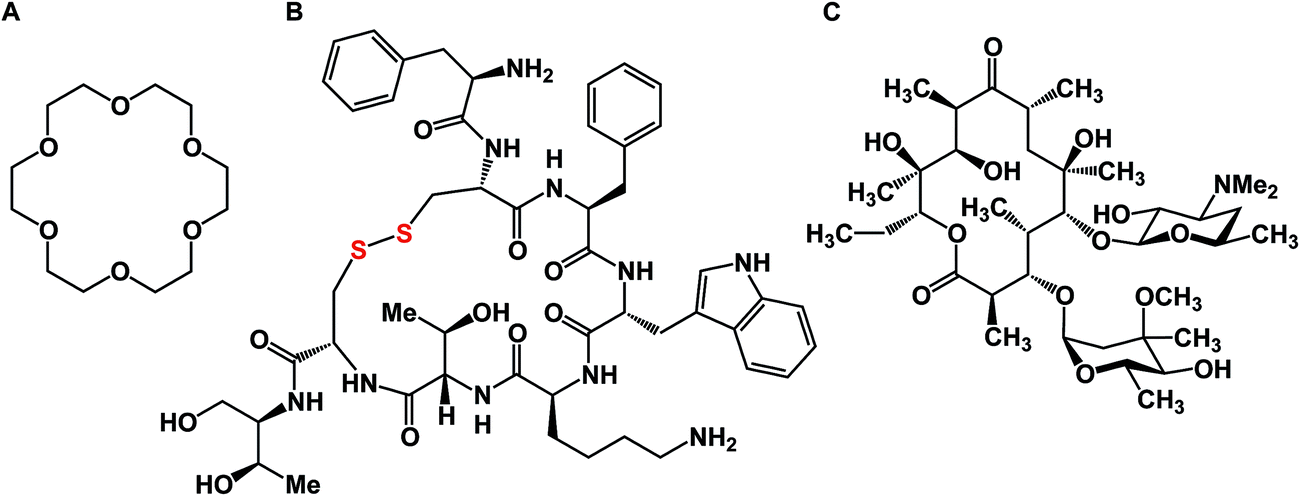 | ||
| Fig. 1 Representative macrocycles. (A): [18]-Crown-6; (B): octreotide; and (C): erythromycin. | ||
While macrocycles have found use in many different areas of chemical science, it is the preorganization of binding elements in the course of a biologically relevant protein/ligand interaction that has been the topic of particularly intense efforts over the past decade. The sustained interest in macrocycles has coincided with a noticeable increase in molecular complexity of therapeutic targets. A growing appreciation of complex protein–protein interactions, which are not easily addressed using small molecules, calls for the development of inhibitors that are more sophisticated than traditional small molecules. In this regard, one might contemplate a difference in how researchers intuit about small molecules in comparison to macrocycles. The small molecule “frame of mind” is about maximizing enthalpic interactions such as hydrogen bonds and salt bridges. On the other hand, bioactive macrocycles are commonly designed with the goal of preorganizing an unstructured linear fragment into a well-defined conformation. The underlying reasoning is to diminish entropic penalties in the course of a protein/ligand interaction. This is not to say that small molecules are somehow exclusively geared to address the enthalpic term of the free energy of binding, whereas macrocycles – its entropy component. In reality, these two energy contributions are exquisitely intertwined.
Among different classes of macrocycles, cyclic peptides and peptidomimetics have received the major share of attention in drug discovery, which is easily explained by the existence of powerful synthetic and biological methods to rapidly put together the amino acid building blocks these molecules consist of. Chemical synthesis of cyclic peptides benefits from the availability of relatively inexpensive orthogonally protected amino acids. In the realm of biological synthesis, DNA and RNA-templated approaches provide a mechanism for translation of nucleotide codons into amino acid-containing oligomers. Besides a straightforward relationship with amino acid building blocks, there is another reason that peptide-based molecules have received a disproportional amount of attention: cyclic amino acid-containing molecules offer a measurable correlation with the behavior of their linear counterparts. This relationship can serve as a validation tool to establish the merits of converting a linear amino acid sequence into its cyclized form. Beyond preorganization, there is a significant practical consequence of constraining a peptide sequence into a macrocycle, namely an opportunity to address the central liability of linear peptides – their propensity to undergo rapid proteolytic degradation in cells. Indeed, the main reason cyclic peptides resist proteolysis is that their structures do not fit into endopeptidase active sites. Endopeptidases (both intracellular and the ones in the plasma compartment) are known for their propensity to recognize β-sheets in segments that typically involve 4–5 amino acid residues (Fig. 2).4 This is not easily achievable with medium-sized macrocycles, which is why they are characterized by reduced rates of proteolysis.
 | ||
| Fig. 2 Peptidases prefer extended conformations: a substrate-derived aminomethylene inhibitor (A) and its complex with the Rous sarcoma virus (RSV) protease S9 (B) (pdb id: 1a94). | ||
The aforementioned properties might paint an erroneous picture that cyclic peptides and other macrocycles in and of themselves belong to some “privileged” class of molecules for therapeutic intervention, the kind that holds clear answers to the challenges facing modern drug discovery, which is inundated with complex protein targets. Unfortunately, this is far from the truth because the large polar surface areas that accompany high amide content come typically at the expense of cellular permeability, which significantly limits the bioavailability of peptide macrocycles. Indeed, one can consider this to be the Achilles' heel of all large therapeutics designed for intracellular targets. P-glycoprotein (P-gp), a membrane protein, whose job is to remove foreign molecules from cells, presents an additional obstacle to macrocycles.5 As a result, the greatest conceptual challenge that faces this area of inquiry is how to devise effective synthetic tools that simultaneously bear on the favorable drug-like properties of macrocycles and ensure their target engagement. These two properties are not correlated in drug discovery, which is less of an issue when it comes to small molecules, but can turn into a major consideration in the case of macrocycles. Fig. 3 illustrates this conundrum using the example of cyclosporine A. This molecule is known for its relatively good passive membrane permeability that arises from the network of intramolecular hydrogen bonds that are presumed to form while cyclosporine enters and passes through the lipid bilayer.6 A molecular-level analysis of the interaction between cyclosporine A and its cellular target cyclophilin paints a different conformational picture, in which the amide groups are involved in target recognition and thus relinquish the intramolecular interactions that contributed to cyclosporine A’s entry into the cell. Many researchers have been taking notes from this “cyclosporine lesson” on cellular permeability of large therapeutic agents. The goal of these endeavors has been to design molecules with favorable drug-like properties, such as enhanced cellular permeability, by minimizing their effective polar surface areas. Unfortunately, this quest is not likely to simultaneously deliver optimal target engagement that operates on principles which are related, but seek to align hydrogen bonds donors and acceptors in an “outward” orientation. Without a doubt, this dichotomy of properties is the biggest challenge in this vibrant research area, and successful molecules are most likely going to be outliers, rather than a group that obeys certain rules.
 | ||
| Fig. 3 Cyclosporine A: membrane conformation (A) and conformation during target engagement (B, pdb id: 2z6w). | ||
2. Synthesis of macrocycles
At its core, the challenge of macrocycle construction is about solving the dreaded “ring/chain” equilibrium, well familiar to polymer chemists. A reasonable strategy to minimize the probability of oligomerization and improve cyclization efficiency is to use high dilution or resort to solid-phase synthesis, which affords pseudo-high dilution conditions.7 A clever solution that bypasses the need for slow-addition/high-dilution involves phase separation of the macrocyclization catalyst from its substrate.8 An exhaustive treatment of the known methods of cyclization is not the goal of this perspective as it will be almost impossible to deliver an adequate discussion of all available methodologies. Most of these methods are merely performed with the goal of inducing intramolecular reactivity and are not conceptually different from linear molecule synthesis. However, there are technologies that tackle the challenge of ring-chain equilibrium, and bias reactions away from generating oligomers and polymers without relying on high-dilution.The differentiation between cyclization and polymerization modes of reactivity is formally captured in the concept of effective molarity, which allows one to parameterize the entropic consequences of ring/chain equilibrium.9 A recent paper by James and co-workers introduced another useful parameter – the so-called Emac (Efficiency of macrocyclization), which is derived from experimentally determined reaction yield and concentration and is useful in comparing macrocyclization technologies.10 Synthetic methods that provide good isolated yields of cyclic products often capitalize on the inherent propensity of linear precursors to fold into conformations that are conducive to cyclization. This section discusses the effects of preorganization on reaction efficiency, with a focus on macrocycle modification as a means to produce analogs, and touches upon ways to increase the accessible diversity of structures through the use of biological methods.
2.1 Preorganization of linear precursors for cyclization
Effective preorganization of linear precursors is the function of non-covalent interactions that may operate prior to or during the cyclization. In this regard, peptide preorganization through hydrogen bonding and ion pairing bears resemblance to protein folding. It is well known that about one half of the single domain proteins in the Protein Data Bank have their N- and C-terminal residues in close proximity.11 This amount is significantly higher than expected on a random probability basis. While the exact reasons for this phenomenon are still debated, similar trends were observed in substantially shorter molecules. In their study of end-to-end loop closure kinetics in polypeptide chains, Daidone and Smith concluded that the loop-closure kinetics in longer peptides are determined by the formation of intra-peptide hydrogen bonds and transient β-sheet structure, which accelerates the search for contacts among residues distant in sequence.12 Significantly, intramolecular hydrogen bonds were found to lower the free energy of loop closure for longer peptides. The observation of a rollover to slower kinetics and the absence of intra-peptide hydrogen bonds for shorter peptides provided evidence of intrinsic stiffness of the polypeptide chains. Ion-pair interactions, which are inversely proportional to the square of the distance between the charges, were shown by Daidone and Smith to be effective in bringing the ends of linear chains together in fairly short sequences. A kinetic study of this process in solvents of low dielectric constants revealed that pre-cyclization architectures are maintained with two chain ends in close contact through Coulombic interactions. Imaginative approaches to study this kind of behavior are found in the work of Schmuck and co-workers. These authors evaluated the effect of electrostatic attraction between peptide chain termini on the formation of zwitterionic peptide cyclodimers (Fig. 4A).13 This study further stresses the significance of hydrogen bond-enforced ion pair formation and its effect on peptide conformation.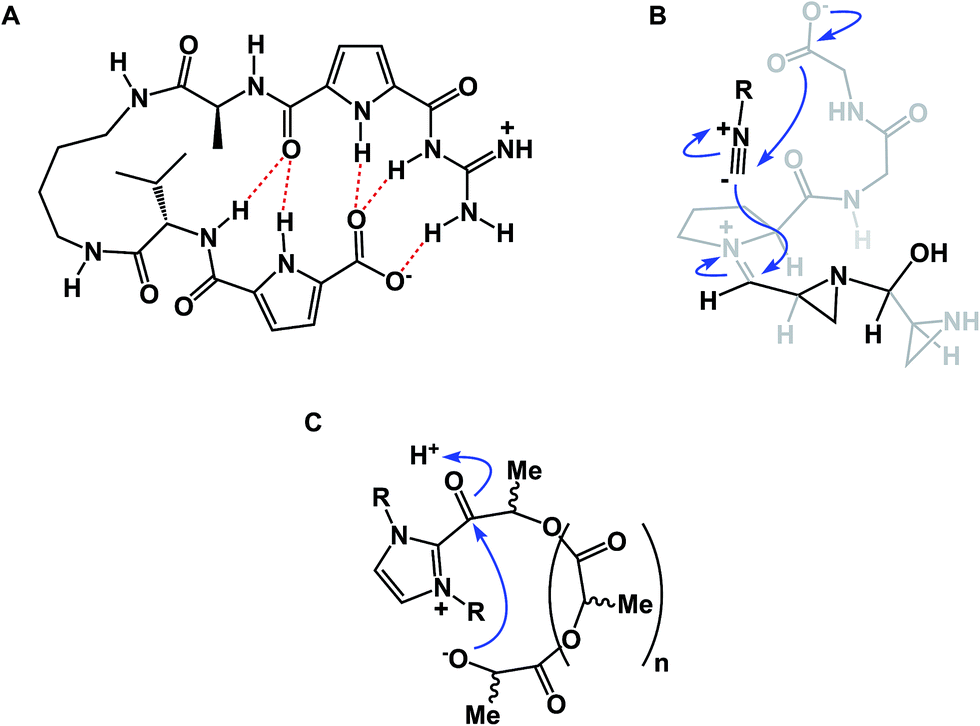 | ||
| Fig. 4 Zwitterionic control over conformation of linear peptide precursors. (A): Zwitterion-driven formation of circular conformations in solution; (B): zwitterionic control of multicomponent peptide cyclization; and (C): zwitterion-driven formation of cyclic polymers. | ||
Our lab's work in the area of aziridine aldehyde-driven macrocyclization of linear peptides14 has identified electrostatic attraction between the chain termini as one of the decisive factors responsible for the attainment of favorable pre-cyclization conformations and the absence of cyclodimerization and oligomerization by-products at relatively high concentrations. Aziridine aldehydes, which readily dimerize to a fused oxazolidine-containing ring system in N-unprotected form, were central to the success of this chemistry.15 These molecules show solvent-dependent dissociation into an open dimer form that controls the facial selectivity of isocyanide attack at the incipient iminium ion (Fig. 4B). This process is believed to guide the peptide chain toward formation of an intermediate mixed anhydride. The resulting macrocycles are equipped with N-acyl aziridine rings that allow for site-selective structural modification. Building on the idea of zwitterionic control over pre-cyclization conformation, Londregan and colleagues at Pfizer have developed a pyridine N-oxide-driven macrocyclization.16 The electrostatic factors involved in this work also echo Waymouth's studies in the area of cyclic polymer synthesis, where zwitterionic control assisted in attaining cyclic conformations prior to ring closure (Fig. 4C).17 In another thought-provoking study that benefits from zwitterionic control over conformation, Zheng and co-workers explored N-heterocyclic carbene-mediated zwitterionic polymerization toward the synthesis of cyclic peptoids.18
2.2 Post-cyclization reactions and side chain reactivity
A substantial proportion of effort expended in the area of bioactive macrocycles is aimed at making structural analogs. Late stage modification of macrocycles is a promising area of research because, once a scaffold with favorable properties has been identified, its folding pattern is likely to be retained in close analogs. Kessler and co-workers found that the presentation of pharmacophoric groups in sterically restricted peptides is predominantly controlled by the amino acid chirality and is less related to the cycle size. The χ1 angle, which defines the conformation about the C(α)–C(β) bond, is the main determinant of the side chain presentation. Kessler's “spatial screening” method allows for systematic interrogation of the geometric parameters of cyclic peptides.19Due to the decisive role of backbone chirality on the macrocycle conformation, methods that allow one to site-selectively modify a macrocyclic compound are expected to be particularly useful in efforts to generate analogs. In their classic work, Seebach and colleagues demonstrated that C-alkylation of sarcosine residues in cyclic tetrapeptides occurs with remarkably high levels of site-selectivity (Fig. 5).20 Curiously, even a molecule as complex as cyclosporine A could be selectively modified by deprotonation/electrophile trapping. The nucleophilic substitution reaction with alkyl halides at low temperature was shown to result in the introduction of side chains at the sarcosine’s methylene moiety of cyclosporine A, whereas N-alkylation became a competing process only at elevated temperature.21
 | ||
| Fig. 5 Seebach's C-selective alkylation of cyclic peptides. | ||
While some macrocycles demonstrate substantial rigidity of structure, many of them are rather flexible, inviting a possibility for transannular interactions. These interactions can play an important organizational role. However, unwanted and irreversible transannular reactivity can also take place, particularly in smaller rings. The origins of this effect in cyclic peptides can be traced back to the environment-sensitive amide nucleophilicity. In this regard, an interesting historical analogy to cyclol hypothesis of protein folding might be drawn. A misguided foray into explaining protein folding by D. Winch was based on cyclol formation between adjacent amides. While Pauling quickly showed this proposal to be fundamentally incorrect as the driving force in protein folding,22 cyclol formation is a well-recognized process that is behind green fluorescent protein (GFP)'s chromophore maturation (Fig. 6A).23 Due to the proximity of functional groups, similar types of reactions can be expected to operate within the structures of cyclic peptides. Intramolecular collapse has indeed been observed even in relatively simple cyclic amide-containing molecules.24 This process is also responsible for the formation of cyclols in the course of spontaneous cyclization of tripeptide sequences.25 Cyclol-mediated transannular ring closure and condensation in cyclic peptides was also documented by Heimgartner and colleagues (Fig. 6B).26
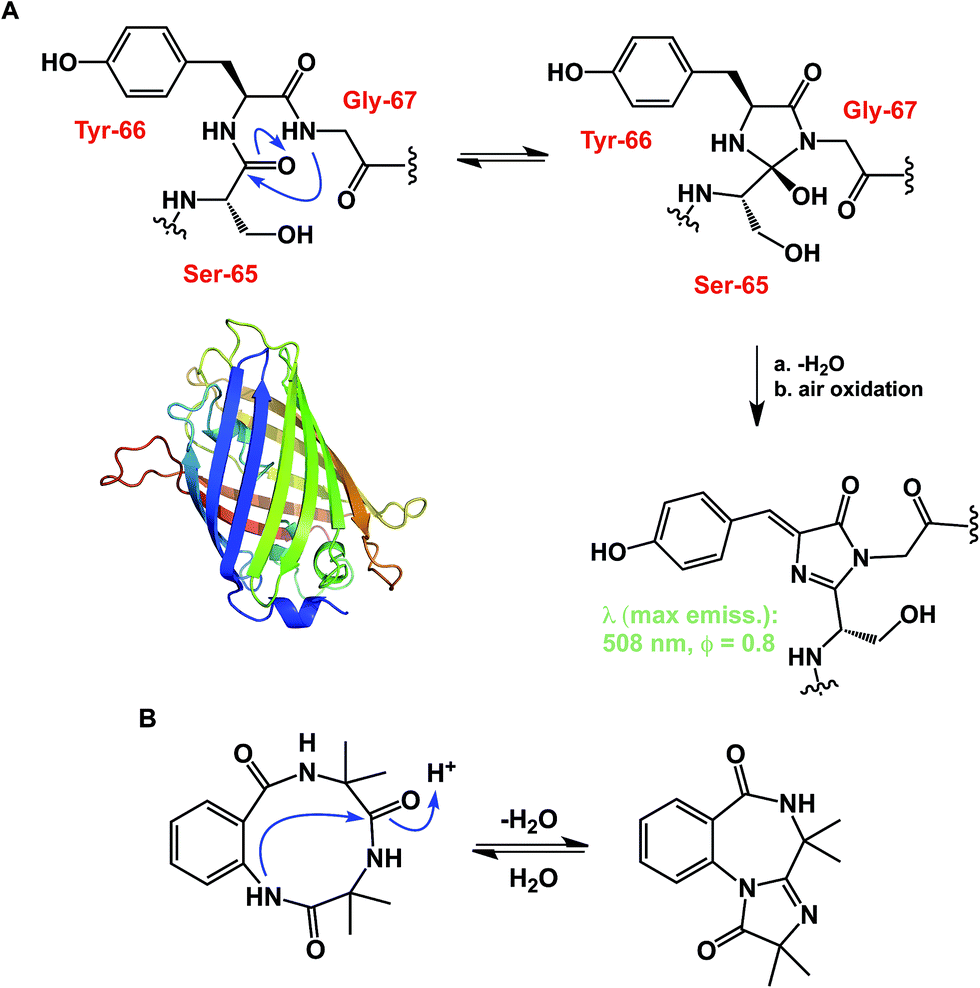 | ||
| Fig. 6 (A): Cyclol-driven formation of the GFP chromophore; and (B): transannular collapse in cyclic peptides. | ||
An interesting case showing intramolecular collapse in medium-sized rings served as the key step in Baran's palau'amine synthesis (Fig. 7A).27 In this example, the desired transannular cyclization proceeded through the guanidine tautomer, delivering the hallmark trans-5,5 ring system of the natural product. Impressive levels of control over transannular reactivity can be exercised through the use of enantioselective catalysis, which can be seen in Jacobsen's asymmetric transannular Diels–Alder reactions (Fig. 7B).28 Interesting transannular reactions in peptide macrocycles have been documented by Porco and colleagues (Fig. 7C).29 In this work, base-mediated transannular cyclizations of macrocyclic bis-lactams driven by olefin isomerization/intramolecular conjugate addition have allowed the authors to access both bicyclic and tricyclic frameworks. While these cases exemplify targeted transannular reactions, they also suggest that caution needs to be exercised when macrocyclic molecules are being synthesized and stored for prolonged periods of time. It is particularly significant in the library mode of synthesis, where LC/MS is routinely used as the main element of quality control, making it difficult to elucidate any rearranged products that may have the same mass as the target molecule.
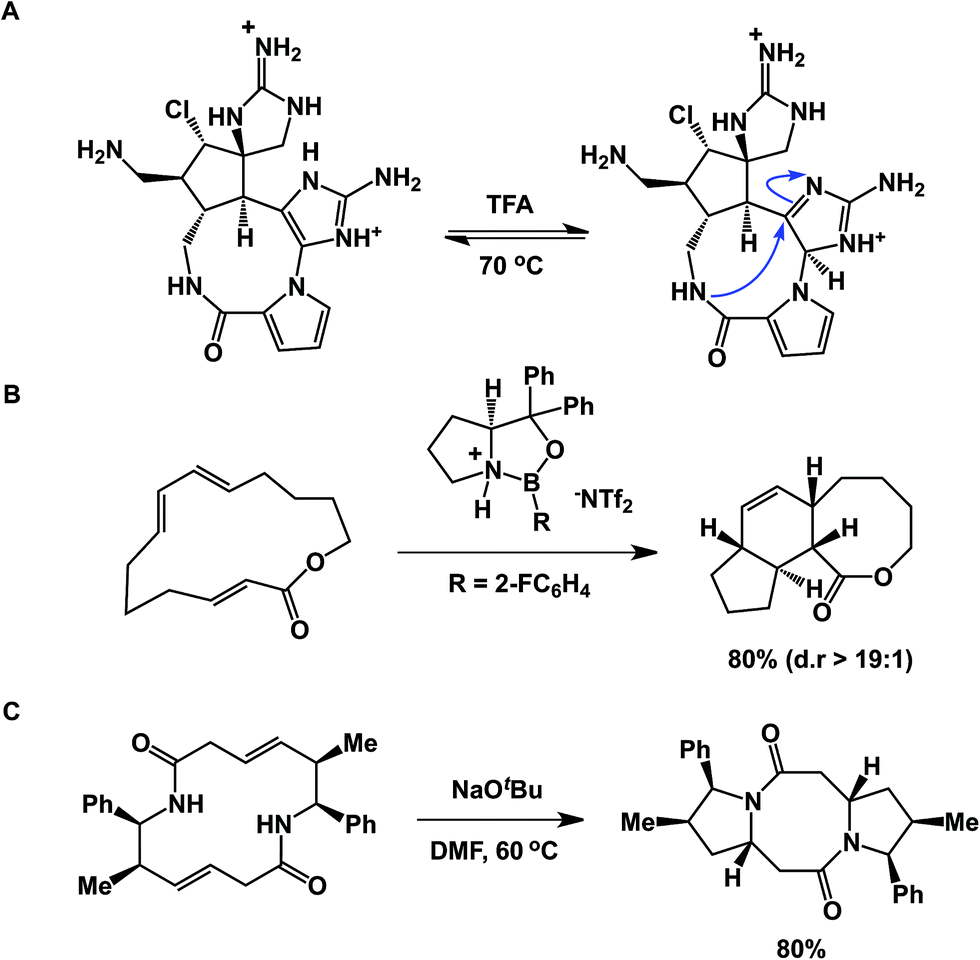 | ||
| Fig. 7 (A): Transannular attack during palau'amine synthesis; (B): asymmetric control over transannular Diels–Alder cycloaddition; and (C): transannular collapse driven by olefin isomerization-conjugate addition. | ||
While amide collapse in large rings is rather rare, the formation of disulfide bonds is not only well-documented, but also serves a significant organizational role in a number of functionally important molecules, including natural products such as romidepsin (Fig. 8A). Romidepsin is a histone deacetylase (HDAC) inhibitor, whose three-dimensional structure is maintained through a single disulfide unit. Defensins constitute much larger ring macrocycles whose structures are “stitched together” by way of a network of disulfide bonds. Molecules such as θ-defensin (Fig. 8B) form the basis of human innate immunity. They are believed to disable susceptible organisms through disruption of the target cell membrane's structural elements.30 Disulfide bonds in macrocycles serve other important functional roles. Alewood and colleagues demonstrated the pivotal role of vicinal disulfides in affording rigidification of cyclized amino acid sequences.31 Some peptide macrocycles contain knotted arrangements of multiple disulfide linkages. Given the reversibility of disulfide formation, disulfide scrambling might present an intractable refolding problem. However, complex disulfide-rich molecules often display remarkable fidelity with regard to “returning” to the original state under redox cycling.32
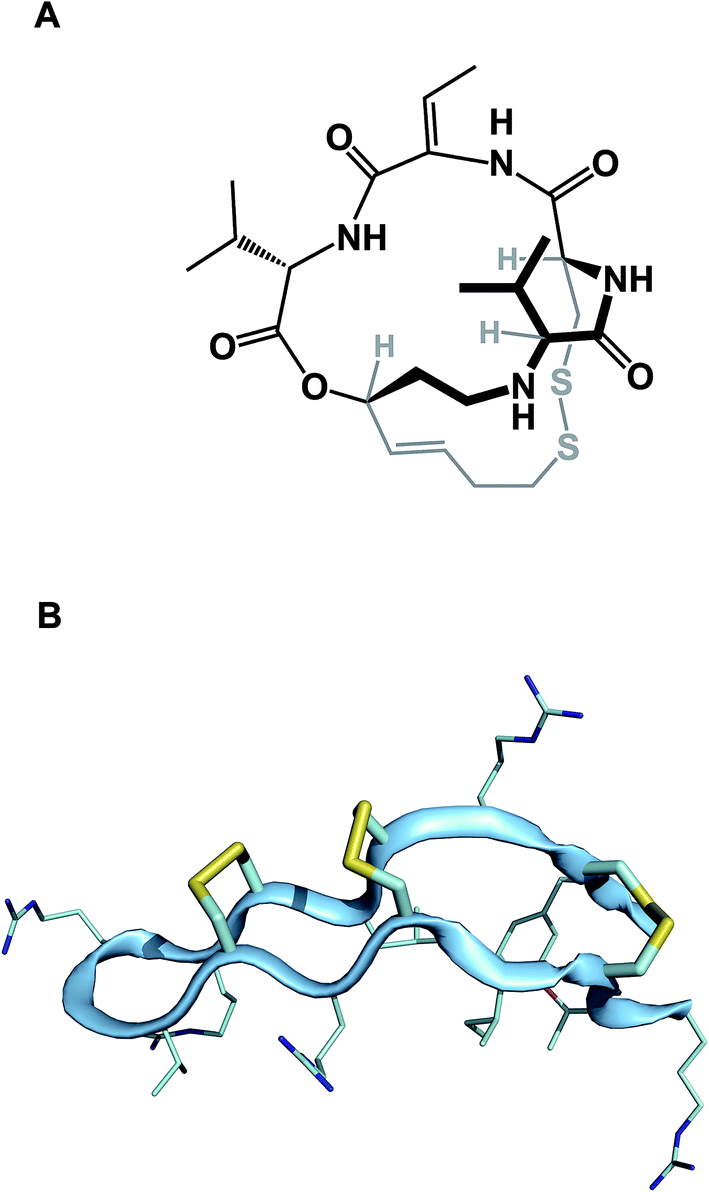 | ||
| Fig. 8 (A): Romidepsin – disulfide-containing natural product; and (B): θ-defensin and its network of disulfide bonds (shown in yellow). | ||
The idea of using side chain-to-side chain reactivity to enhance the desired presentation of a particular secondary structure has been around for some time. Early efforts by Schultz and co-workers centered around the use of intramolecular disulfide bonds between cysteines in order to increase the helical character of small peptides.33 Grubbs later employed ring-closing metathesis (RCM) to address this challenge.34 However, it was not until Schafmeister and Verdine's insight that α-aminoisobutyric acid increases the relative amount of the α-helical form in peptides, that significant improvement in the relative proportion of α-helicity became possible (Fig. 9).35 The combination of alpha, alpha-disubstituted amino acids and RCM furnished “stapled” peptides that have since been applied in a variety of disciplines.36 Interestingly, alkane-based linkers that hold these molecules in their α-helical forms are not innocent by-standers: crystallographic evidence suggests that they can engage in stabilizing hydrophobic interactions at the stapled peptide/MCL-1 binding interface.37
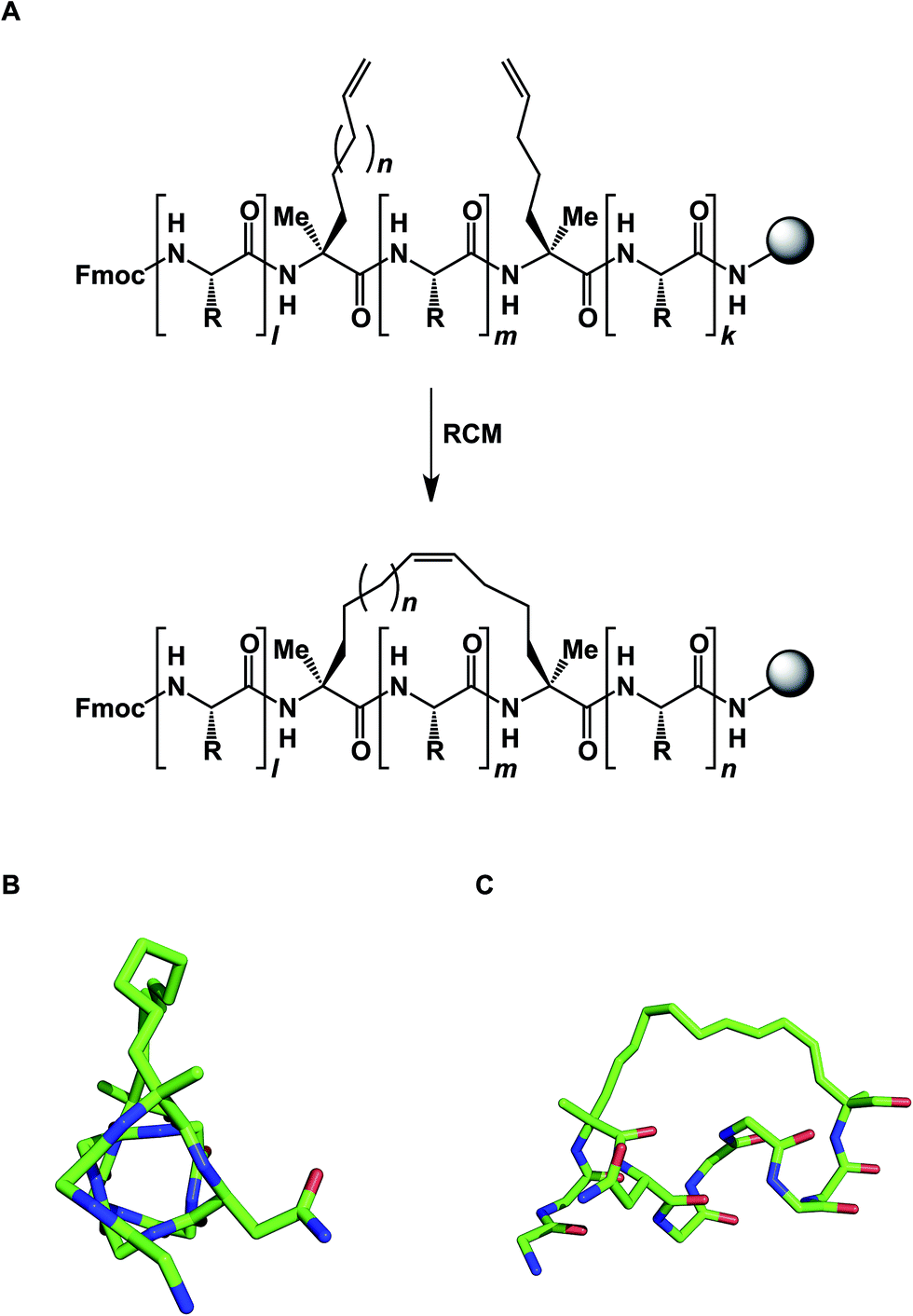 | ||
| Fig. 9 (A): Stapled peptide synthesis; (B and C): two views of the stapled peptide bound to MDM2 (side chains are omitted for clarity, pdb id: 3v3b). | ||
The proximity of functional groups in the molecules of macrocycles can give rise to interesting structural rearrangements. Building on their earlier studies, Takeya and co-workers discovered a thioamide-driven, site-selective epimerization that takes place in the structures of certain bicyclic peptide natural products.38 In a similar vein, researchers at Novartis showed that site-selective epimerization of the unmethylated leucine residue of a fungal cyclodepsipeptide is possible by way of O-alkylation followed by oxazole formation and hydrolysis (Fig. 10A).39 This example further teaches that local environments can exert profound influence over site-selective reactivity in macrocycles. Interestingly, the crystal structure reported for this molecule explains why the non-methylated leucine residue is the most nucleophilic one. This finding also echoes earlier teachings of Seebach and co-workers, who developed conditions for site-selective deprotonation of macrocycles.20 Yet another intriguing manifestation of intramolecular reactivity comes courtesy of Smythe and Meutermans, who demonstrated that challenging macrocycles can be accessed through the use of a benzylamine-derived auxiliary via a ring-contraction strategy (shown in red in Fig. 10B).40 In this example, the intramolecular collapse is driven by the relative strength of the product amide bond.
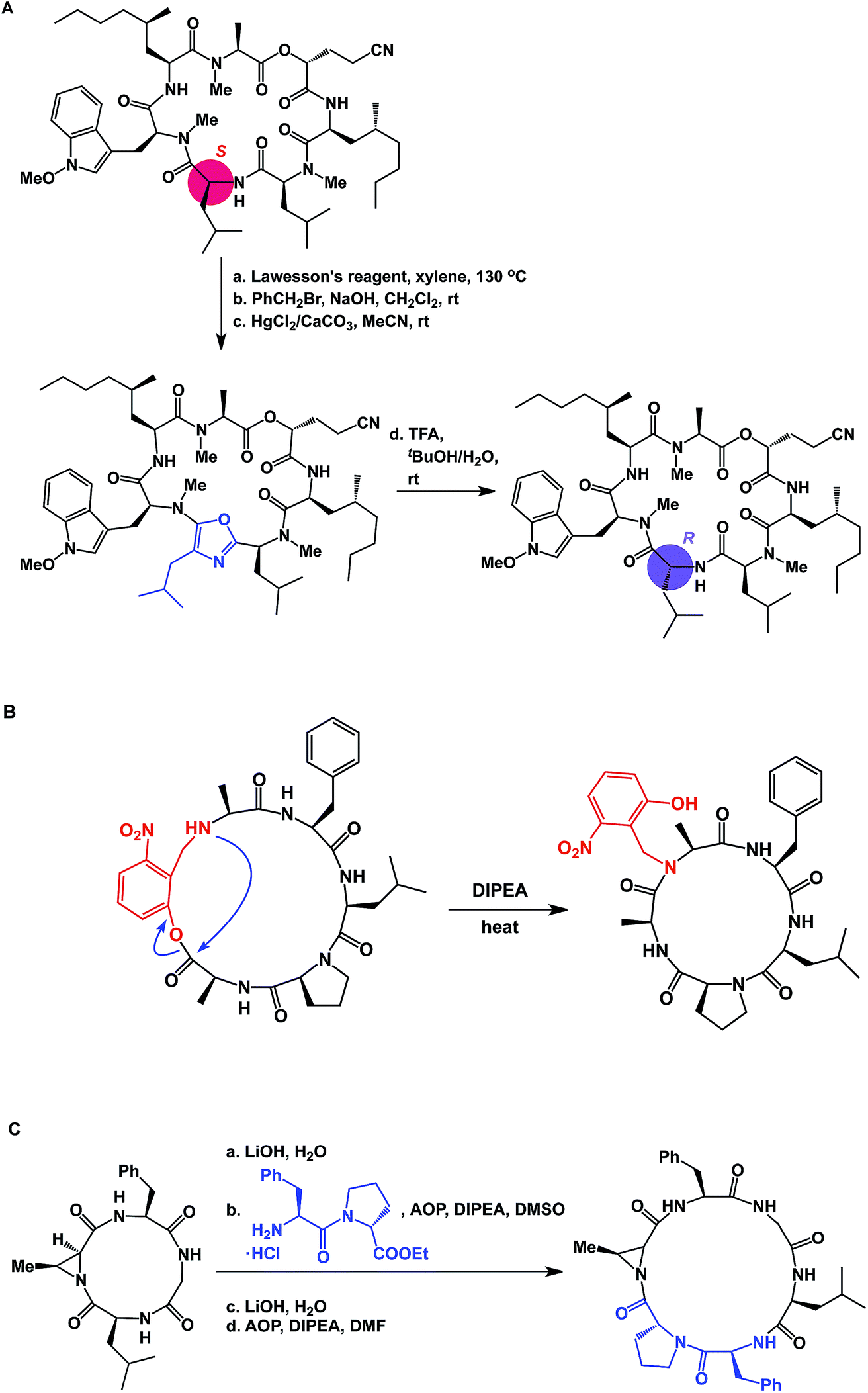 | ||
| Fig. 10 (A): Site-selective epimerization of a cyclic peptide; (B): ring contraction strategy for macrocycle synthesis; and (C): integrative approach to macrocycles. | ||
While the cases discussed above illustrate rearrangements of cyclic peptides or their side chain reactivity, until recently there have been no examples wherein site-selective ring scission of macrocycles was shown to be synthetically useful. By focusing on the relative weakness of aziridine amides compared to the regular amides, our lab has advanced the concept of site-specific integration of amino acid fragments into the structures of cyclic peptides using a synthetic sequence of hydrolysis/ligation/re-cyclization (Fig. 10C).41
Reagent- and catalyst-controlled modification of macrocycles is another vibrant area of research. Miller and co-workers reported on their use of peptide-based catalysts toward site-selective modification of erythromycin A (shown in Fig. 1C).42 Burke and colleagues resorted to fundamental principles of physical organic chemistry in order to electronically tune reagents toward useful site-selective functionalization reactions and applied this method to the chemoselective functionalization of the complex natural product amphotericin B.43
It is appropriate to mention the way in which many cyclic peptides and other macrocycles are biosynthesized because the logic of this chemistry is mirrored in some solid-phase synthesis approaches. A number of naturally occurring macrocyclic peptides are the products of non-ribosomal biosynthesis, during which synthetases tether activated linear intermediates through thioester linkages. Interestingly, isolated thioesterases were shown to promote macrocyclization of linear peptides immobilized on synthetic solid supports.44 This reaction involves transacylation to the active-site serine followed by deacylation upon intramolecular attack by the amino-terminal nucleophile. A practical example that cleverly emulates the biosynthetic assembly of cyclic peptides has emerged from Tranzyme. Over the years, this company amassed a large library of macrocycles using a solid-phase procedure in which the linear precursor had been connected to the solid support through a thioester linkage.45 In this chemistry, the macrocyclization event coincides with the N-terminus of the linear chain attacking the thioester group, resulting in traceless product release. Cyclative cleavage to generate macrocycles was also cleverly employed by Rademann and co-workers in their triazole ligation studies.46
2.3 Molecular diversity via biosynthetic approaches
One of the long-standing goals of drug discovery is to increase the diversity of accessible structures to allow for broader sampling of the structural space. Combinatorial biochemical methods such as phage display, aptamer SELEX, and mRNA display discussed in this section have shown tremendous promise for the discovery of biopolymer-derived ligands to biological targets. These methods utilize large populations of species such as bacteriophage, and then apply environmental pressure in order to focus the populations by selection. Binding to a biological target of interest is the most commonly used selection pressure. In phage display, each species carries an oligonucleotide tag that not only serves to encode chemical structure, but also provides a template for amplification. Libraries of up to 1013 members have been successfully generated and used in efforts to discover disulfide-based macrocyclic ligands to protein targets. Incidentally, the RGD (arginine–glycine–aspartic acid) sequence, widely used in cell adhesion studies, emerged from this effort.47Several innovative approaches at the intersection of biology and synthetic chemistry have appeared in recent years. The presence of nucleophilic thiol functional groups in peptides has been explored in imaginative methods that target constrained macrocyclic scaffolds. Fast and quantitative cyclization of linear peptides with unprotected side chains and multiple free cysteines is possible through the use of simple bromomethyl-functionalized aromatic scaffolds. This chemistry, developed by Timmerman and colleagues, runs fast and clean with linear peptides that are 2–30 amino acids long and provides a means to immobilize multiple peptide loops onto a synthetic scaffold.48 Using mesitylene scaffolds reported by Timmerman, Heinis and colleagues developed a phage display strategy for the selection of bicyclic peptides (Fig. 11). The linear precursors were designed as repertoires with three reactive cysteines that were separated in sequence by several random amino acid residues. The resulting constructs were then fused to the phage gene-3-protein. Subsequent conjugation with tris-(bromomethyl)benzene generated phage-based peptide conjugates and affinity selections led to the discovery of a specific human plasma kallikrein inhibitor, among other applications.49
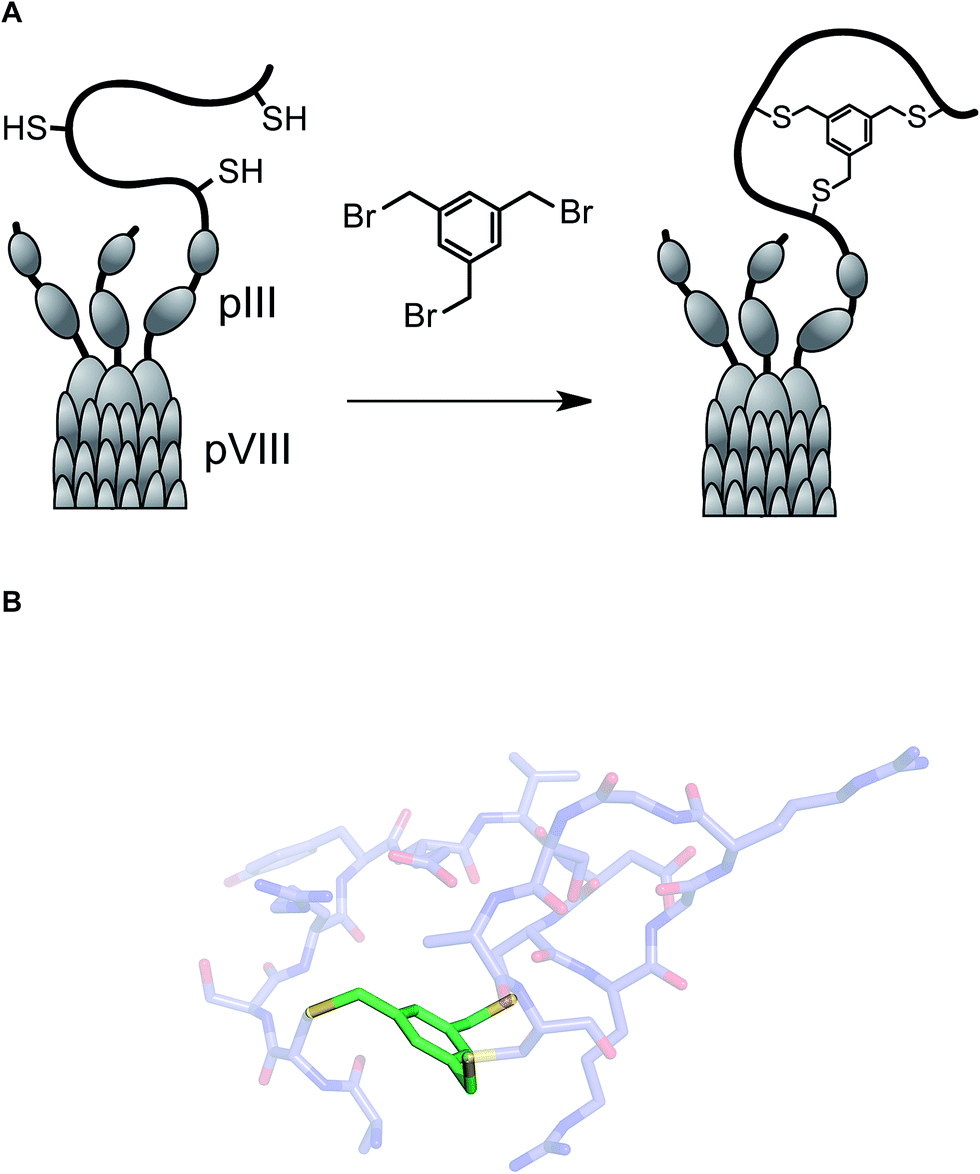 | ||
| Fig. 11 (A): Integration of tris(bromomethyl)xylene into the phage display workflow; and (B): a pose of a crystallographically characterized xylene-constrained inhibitor of human urokinase-type plasminogen activator (pdb id: 3qn7). | ||
Liu and colleagues used multistep DNA-templated organic synthesis to translate libraries of DNA sequences into libraries of sequence-programmed macrocycles and subjected the resulting DNA-macrocycle conjugates to in vitro selections for protein target affinity. The sequence of the amplified DNA template allowed the authors to de-convolute the results of their binding experiments.50 Meanwhile, using wPURE and flexizyme-mediated in vitro translation systems, Suga and co-workers genetically reprogrammed the initiation event during translation. It was shown that the translation apparatus tolerated not only proteinogenic amino acids, but also accepted N-acyl groups equipped with various functionalities. This list included electrophilic chloromethyl group on the N-terminus or a lysine side chain, which led to the ribosomal synthesis of cyclic peptides containing thioether bonds.51 This genetic reprogramming approach, in conjunction with mRNA display, has resulted in the discovery of high affinity binders for a number of relevant targets.
Szostak and colleagues have developed cysteine-mediated approaches for macrocyclization via lanthionine bridge formation or alkylation with dibromo xylene linkers. These approaches have been combined with mRNA display to rapidly prepare and screen macrocyclic peptide libraries.52 In another approach, Fasan and co-workers explored a clever chemo-biosynthetic strategy toward the generation of macrocyclic organo-peptide hybrids (MOrPHs). This feat was accomplished using a catalyst-free dual oxime-/intein-mediated ligation between ribosomally synthetized precursor proteins containing two orthogonal ligation points and a panel of bifunctional small molecules.53
Most of the biological methods of synthesis mentioned above enable the preparation of astounding numbers of diverse macrocycles that are accessible for biological screens. In addition, a direct link between genotype and phenotype allows for the rapid screening and deconvolution of these large and diverse libraries. However, the disadvantage of these methods is that they deliver relatively high molecular weight compounds with large polar surface areas, which are unlikely to be cell-permeable. A certain analogy with the state of the art in combinatorial chemistry of the 1990's might be appropriate. This comparison suggests that emphasis on the numbers is not related to the real bottleneck of drug discovery. History has shown that it is the molecular properties such as microsomal stability and cellular permeability, rather than target engagement, that are the most challenging steps in drug discovery. A synergistic relationship between biology and chemical synthesis undoubtedly needs to be nurtured if macrocycles are to become a useful modality in drug discovery.
3. Properties of macrocycles
When compared to small molecules, a macrocycle/protein interaction is inherently more complex. In some cases, the induced fit model was shown to adequately describe this interaction.54 Akin to the structural biologists' regard for induced fit, synthetic chemists view the Curtin–Hammett principle as one of the pillars of not only chemical reactivity, but many other molecular properties. By stressing the importance of kinetics, the Curtin–Hammett principle reveals an interesting analogy to macrocycle/protein target interaction. This principle states that the ground state conformation is not necessarily the reactive (or relevant) one. Significantly, this dictum applies only to systems in which a rapid inter-conversion between two or more accessible conformations can be established. The possibility shown in Fig. 12A describes two accessible macrocycle conformations that can interconvert. If the barrier to interconversion is low and the active conformation is readily sampled, it is immaterial to be concerned about conformational analysis of macrocycles because the system is unlikely to “miss” the desired state. On the other hand, if the barrier is high, the free energy of binding may not be sufficient to induce the desired conformation. This is analogous to the so-called “non-Curtin–Hammett” scenario that exists in chemical reactivity. While this is a rare phenomenon, some macrocycles with complex conformational energy landscapes are likely to undergo slow conformational interconversion, which is expected to influence their properties. For instance, the crystallographically characterized binding conformation of an SH2 domain inhibitor shown in Fig. 12B was readily attained after incubating the macrocycle in the presence of the SH2 protein at 50 °C for 10 minutes before allowing the system to crystallize at room temperature.55 The results of crystallization experiments at room temperature alone were inferior to those obtained by heating at 50 °C first, followed by crystallization. The challenges inherent to understanding macrocycle conformations are further mirrored in the difficulties that exist during computational docking of macrocycle ligands.56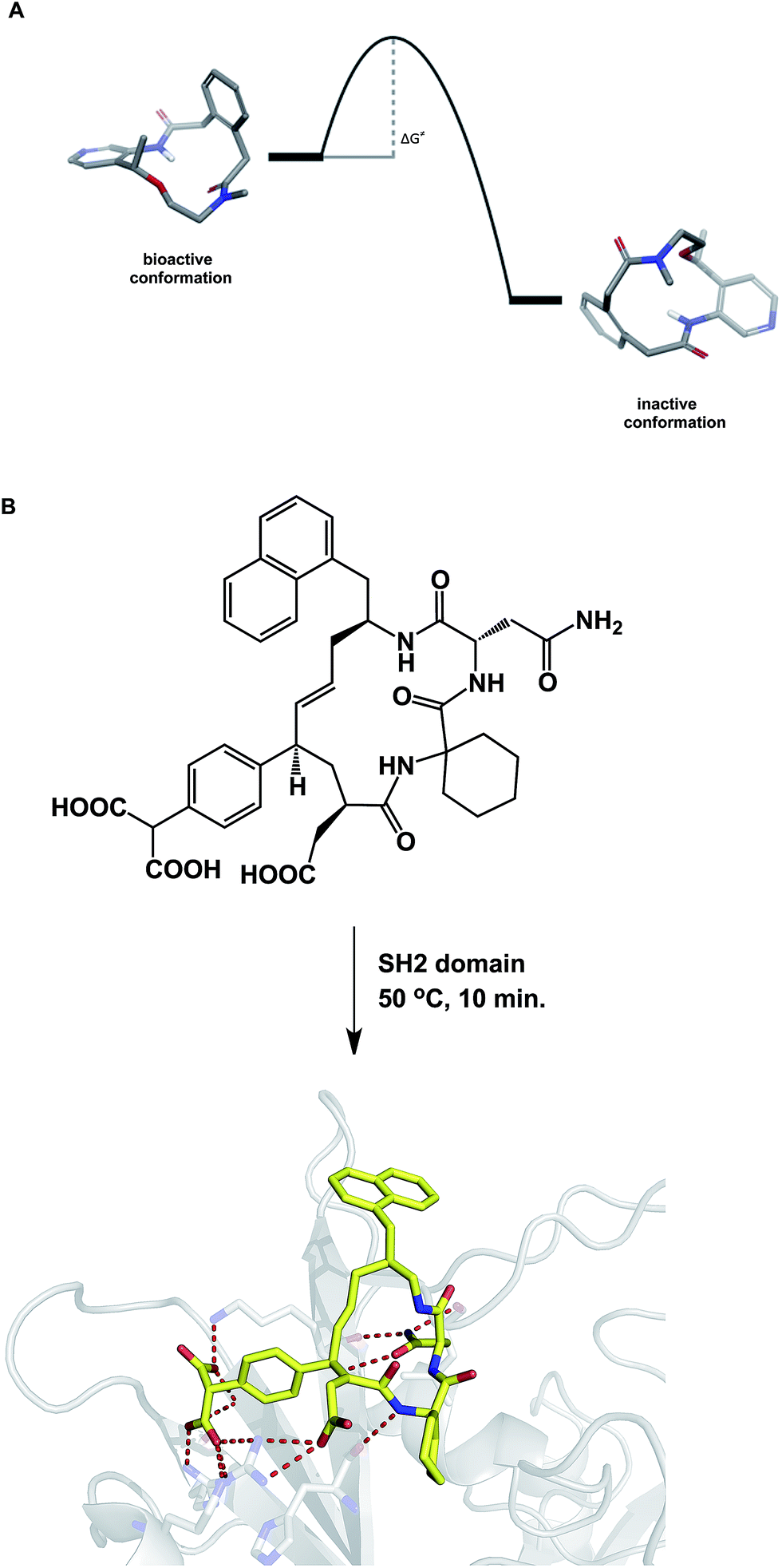 | ||
| Fig. 12 (A): A hypothetical potential energy diagram linking two macrocycle conformations; and (B): co-crystallization with macrocycles may require energy to overcome the conformational barrier between different states (pdb id: 3aob). | ||
It follows that, while the induced fit is a well-recognized phenomenon, attention must be paid to the inherent capacity of a given macrocycle scaffold to be amenable to the reorganization that is needed in such a process. Given the established correlation between cellular permeability and intramolecular hydrogen bonds (vide infra), one can easily appreciate the challenge: either target engagement or cellular permeability may correspond to a conformation that is not readily accessible. One of the main challenges that faces the field of macrocycles lies in having to simultaneously address these two disparate goals.
To further complicate matters, there are some thought-provoking studies that challenge the dogma that constrained molecule/target interactions are primarily entropy-driven. In a series of elegant experiments, Martin and co-workers questioned the prevailing view that preorganization must have a favorable entropic component (Fig. 13A).57 The authors demonstrated that entropies of binding of preorganized ligands to their targets could be disfavored when compared to the less potent, yet flexible, controls. It was shown that the enhanced enthalpy of binding could arise from an unexpected involvement of protein/ligand polar contacts. Using PDZ domains as model biological receptors, Spaller and co-workers came up with similar conclusions during their evaluation of macrocycles and stressed the significance of proper linear controls when analyzing a series of macrocycle binders (Fig. 13B).58
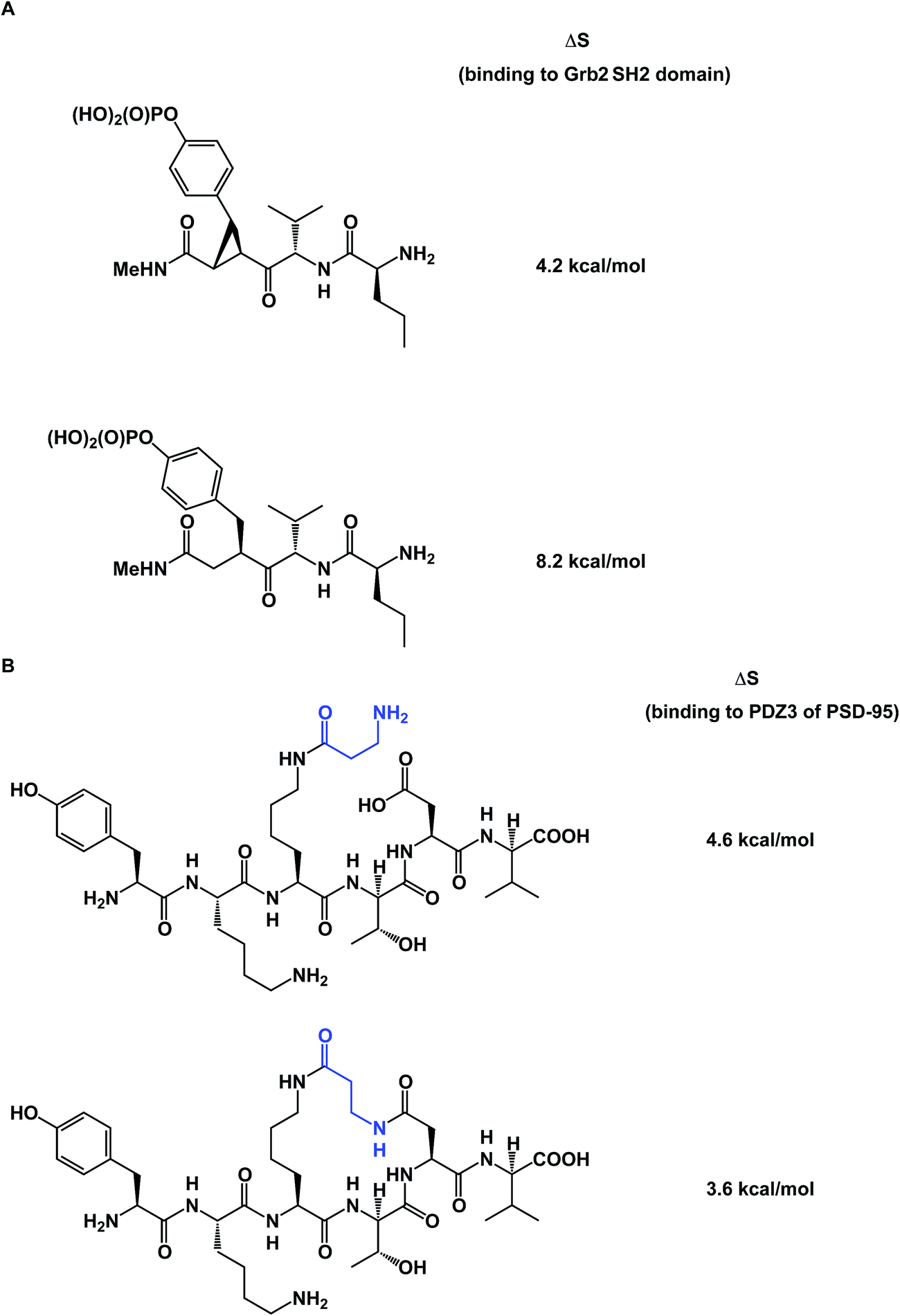 | ||
| Fig. 13 (A): Constrained small molecules do not always result in more favorable entropy of binding; and (B): macrocycles can show unfavorable binding entropy compared to linear controls. | ||
3.1 Conformational analysis
The discussion above leads to the conclusion that it is important to study and understand the conformational preferences in a given macrocycle series. Fig. 14 presents a condensed version of some powerful methods that are now routinely available to chemists interested in studying cyclic peptides and other types of macrocycles. For instance, recording the 1H NMR chemical shift changes of NH groups as a function of temperature helps identify the slowly exchanging protons that are likely to be tied up in intramolecular hydrogen bonds. In the course of their study of cyclophane macrocycles, which represent a particularly under-populated region of chemical space, James and co-workers resorted to EXSY spectroscopy in order to elucidate chemical exchange between conformations sampled by the macrocycles under scrutiny.59 The presence of cross-peaks in the EXSY spectra suggested an exchange between different conformations, which were found to slowly interconvert. The exchange rates for the transformation of detected conformations can be established from the experimentally determined parameters during EXSY experiments. X-ray crystallography is another valuable tool in the conformational analysis of macrocycles, although caution is advised with regard to the relevance of this analysis as solvent-dependent behavior in solution can lead to completely different conclusions.60 Circular dichroism can be indispensable in the studies of protein secondary structure fragments embedded in macrocycles. Here too, one has to be aware of the danger to over-interpret the data. For instance, in an intriguing case, Fairlie demonstrated how a macrocycle with α-helical sub-structure displays β-sheet-like spectrum.61 The computational prediction of macrocyclic conformations in both free and bound states is significantly more challenging compared to small molecules.56 In contrast to small molecules, the degrees of freedom in macrocycles are not mutually exclusive and perturbations are “coupled” in such a way that changing one dihedral angle in the backbone affects other dihedral angles, which causes computational conversion to take significantly longer.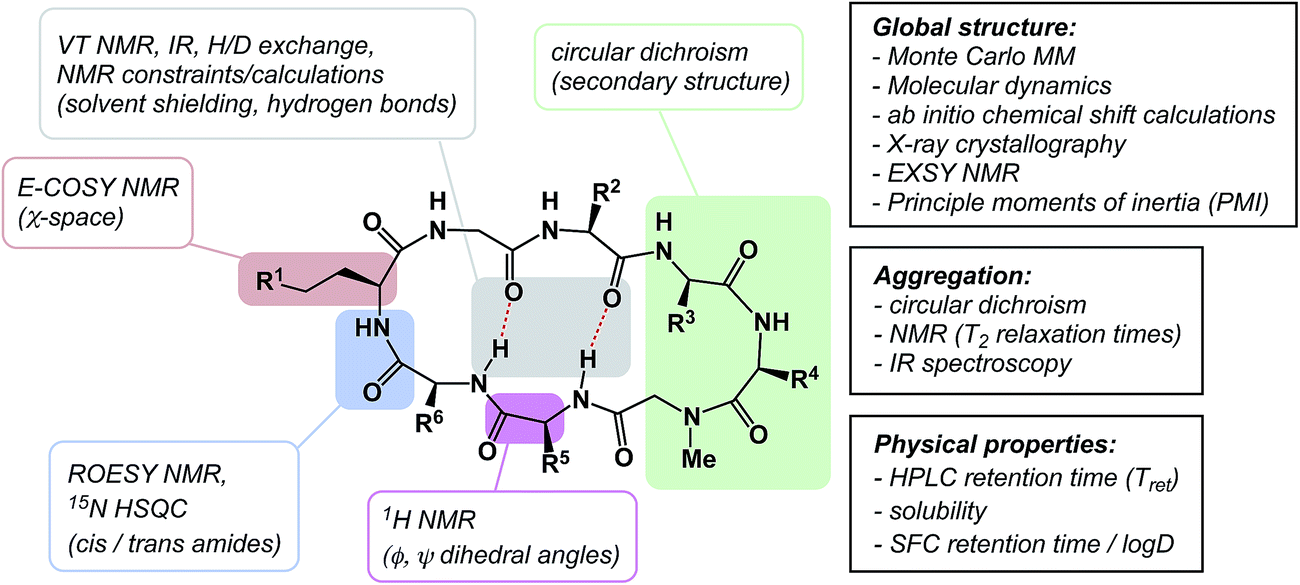 | ||
| Fig. 14 From measurement to computation: assessing the properties of macrocycles. | ||
NMR can be a useful tool for the rational design of conformationally rigidified macrocyclic scaffolds. Fairlie and colleagues were able to show that the 13-membered rings obtained by homologation of a cyclic tetrapeptide are characterized by a marked improvement in the conformational rigidity of the ring.62 Due to their rigidity, the resulting α3β architectures of cyclic tetrapeptides are especially well-suited for the interrogation of HDAC enzymes.63 While main-chain chirality is believed to be the major factor influencing macrocycle conformation, a recent report by Hunter and colleagues provides a rare and exciting possibility that diastereotopically-dependent structural modification of the cyclic peptide core with fluorine atoms can play an important role in structural rigidification (Fig. 15A).64
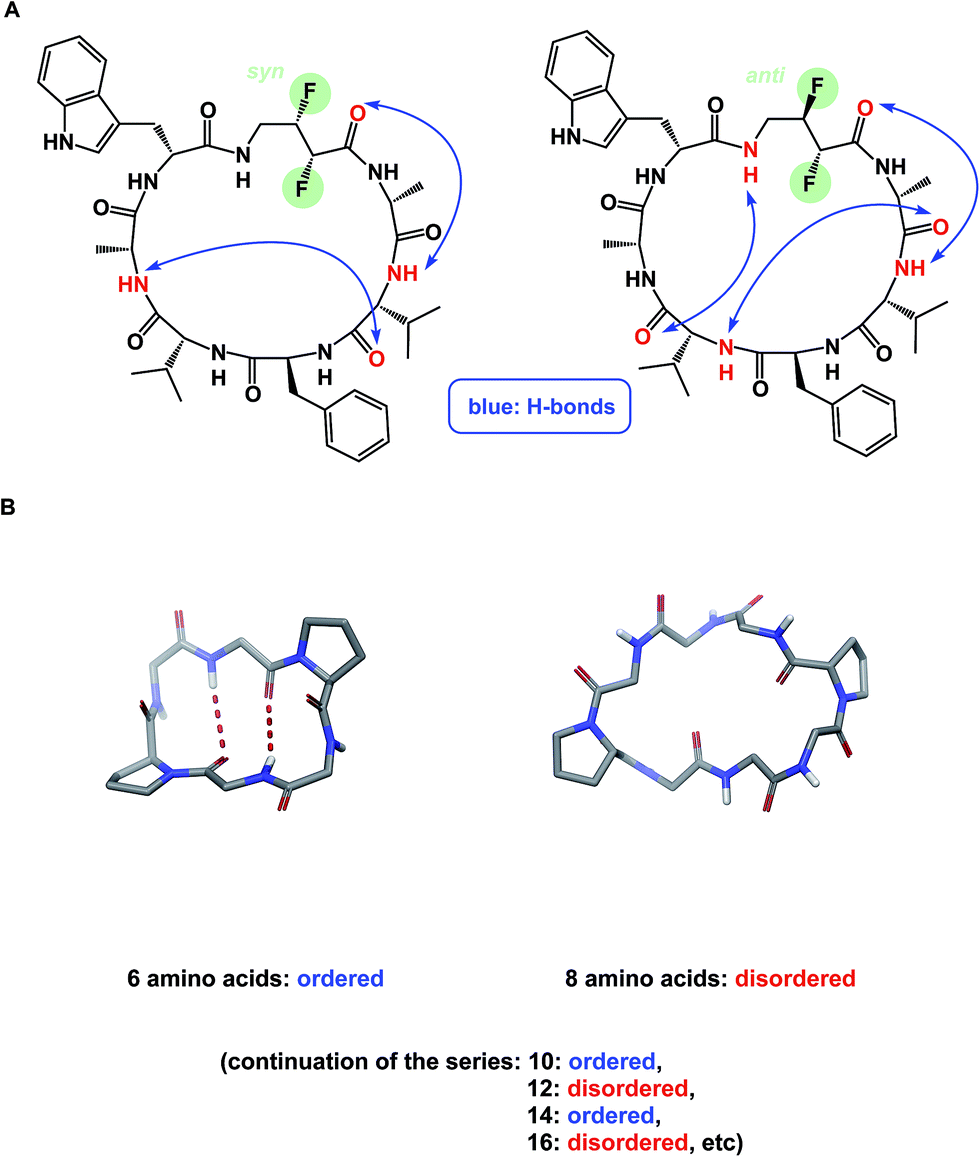 | ||
| Fig. 15 (A): Control over macrocycle conformation using fluorination; and (B): periodicity of β-sheet formation in cyclic peptides. | ||
The solution conformation deduced by NMR can also afford insights needed for the rational design of therapeutic lead structures. In an earlier study, Dinsmore and colleagues at Merck showed how the solution structure of a conformationally flexible inhibitor of FTase suggested a macrocyclic analogue with a substantially improved inhibition profile.65 Recently, a hybrid sequential molecular mechanics/quantum mechanical approach to modeling cyclic peptides has resulted in an effective method for predicting their 1H and 13C NMR chemical shift values. The utility of this method was tested in a blind fashion and excellent agreement with the experimental NMR chemical shifts was observed.66
α-Helix, β-sheet, and β-turn are the most common types of protein secondary structure. These structural types are prevalent in both solution- and solid-phase structures of proteins and are known to mediate the vast majority of protein–protein interactions. Unfortunately, these secondary structures are rarely observed in small linear peptides. Cyclization provides an opportunity to “freeze” these motifs, making them amenable to conformational analysis and structure/function studies. An interesting consequence of constraining amino acid motifs was noted by Wishart and co-workers, who observed size-dependent periodicity of β-sheet content in cyclic peptides (Fig. 15B).67 This study proved the long-standing hypothesis that cyclic peptides containing 6, 10, and 14 α-amino acid residues in length exhibit high β-sheet content, whereas macrocycles of 8, 12, and 16 residues exist as random coils. Robinson and colleagues further established (D)-Pro–(L)-Pro linker as an effective means of creating turn structures with high β-sheet content,68 while Nowick introduced ornithine-derived turn elements as a means to generate and stabilize modular β-sheet motifs.69
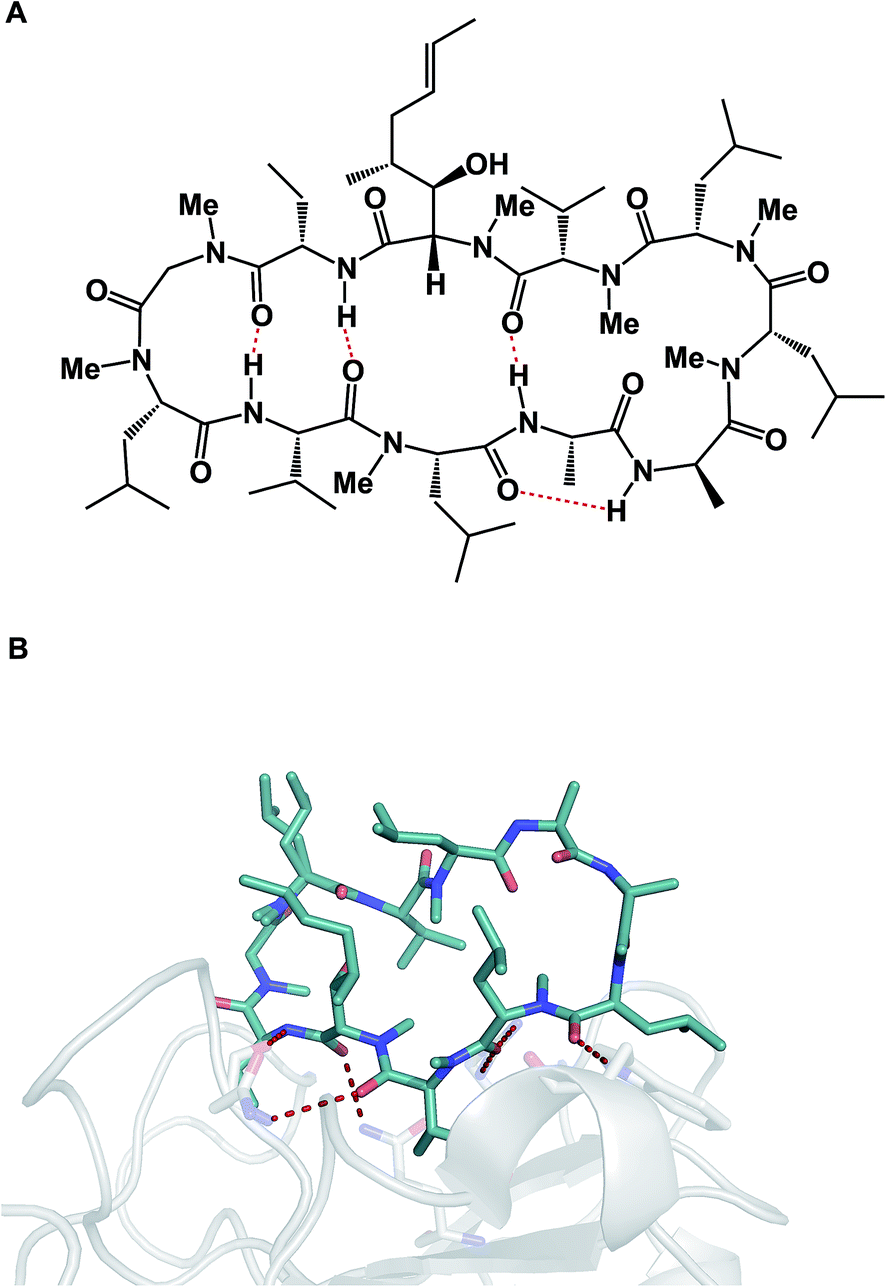
α-Helices are known to dominate protein–protein interfaces,70 which is why stapled peptides (macrocyclic peptides covalently constrained with hydrocarbon linkers that stabilize α-helices), have found a range of applications in chemical biology and drug discovery.36 Recently, Pentelute and co-workers resorted to cysteine-mediated SNAr reactions between peptides and perfluorinated aromatics in order to force linear peptides into stapled versions with constrained presentation of α-helical motifs.71 Unnatural amino acid turn elements can induce unusual turn structures in the molecules of well-known natural products. Thus, Overhand and co-workers resorted to rigid furanoid sugar amino acids to interrogate the β-hairpin structural elements of gramicidin S.72 Our lab recently resorted to strategically placed exocyclic amides to ensure conformational homogeneity of macrocycles.60 Ulrich and Komarov employed UV light to control the conformation of cyclic peptides equipped with photochemically sensitive groups (Fig. 16A).73
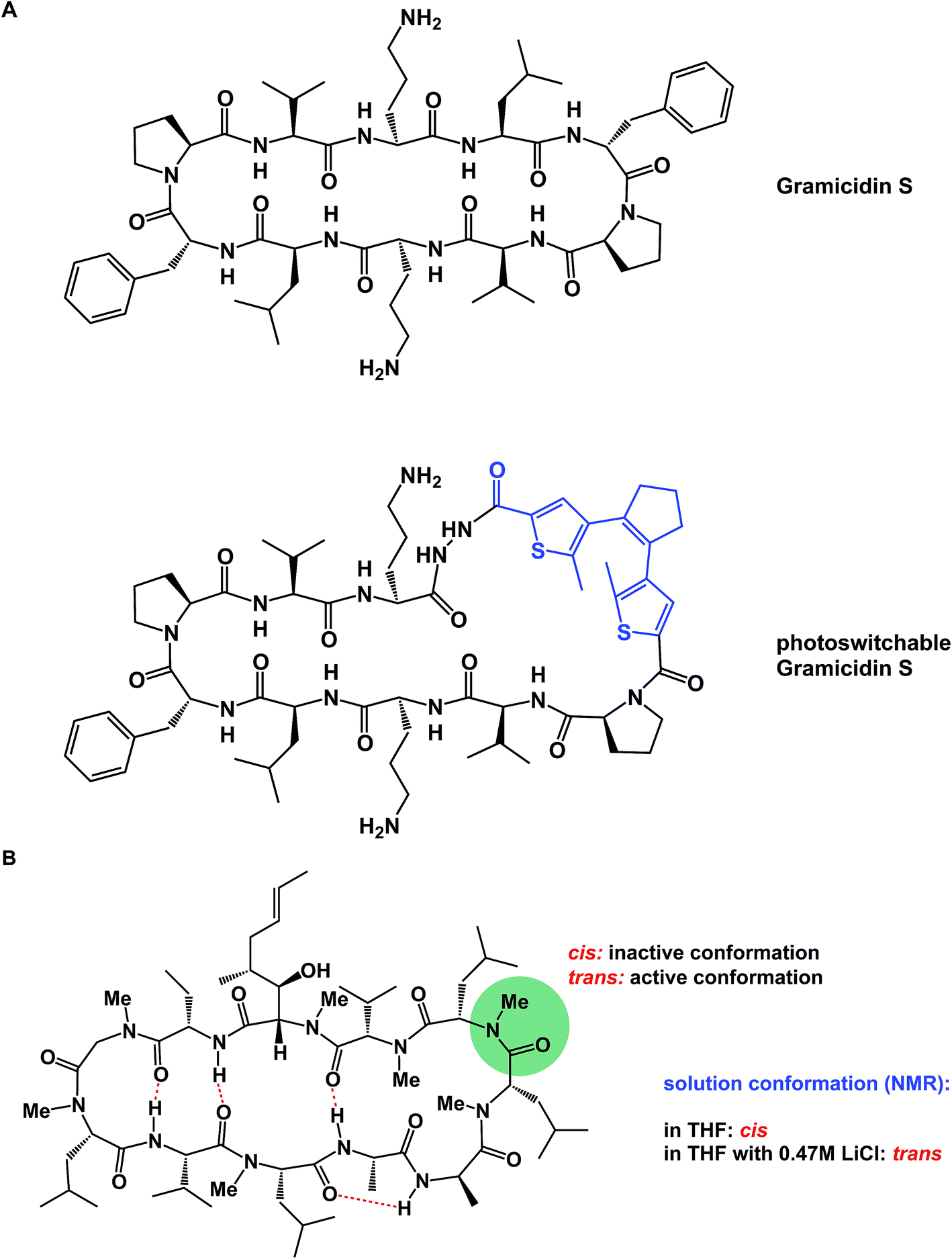 | ||
| Fig. 16 (A): Light-induced control of gramicidin S; and (B): inducing conformational changes in cyclosporine A. | ||
Various additives can exert a profound influence on the solution conformation of macrocycles. In their classic study, Rich and co-workers demonstrated that the addition of lithium chloride to the solution of cyclosporine A in THF influences the geometry of the Leu–Leu bond. It was established that if the Leu–Leu amide linkage in cyclosporine A is in its cis state, the molecule is biologically inactive against its cyclophilin target. Rich and co-workers further demonstrated that the addition of LiCl to cyclosporine A in THF shifts the cis/trans equilibrium toward the bioactive trans form (Fig. 16B). Remarkably, the authors were able to show that the LiCl perturbation method works to alter even the biological properties of cyclosporine A. It was found that a significantly more potent inhibition of cyclophilin takes place in the presence of LiCl.74
The conformational properties of macrocycles can have a direct influence on their chemical reactivity. Our lab recently demonstrated macrocycle-dependent regioselectivity during site-selective modification of macrocyclic electrophiles containing N-acyl aziridines. In this chemistry, the peptide structure dictated the adoption of different reactive conformations of the N-acyl aziridine embedded in the ring (Fig. 17). When the aziridine amide adopts a cis conformation, azide anion attacks the α-carbon of Azy (aziridine carboxylic acid) exclusively, leading to the formation of a trans amide-like transition state, wherein allylic strain is minimized. Alternatively, attack at the β-carbon of the Azy residue leads to the development of a less favorable cis-like amide bond.41 The fact that this regiochemistry is different from that observed in the case of linear Azy-containing peptides attests to the influence of macrocycle conformation over chemical reactivity. This idea is substantiated by the fact that attack at the α-carbon of the Azy residue is preferred in linear peptides.
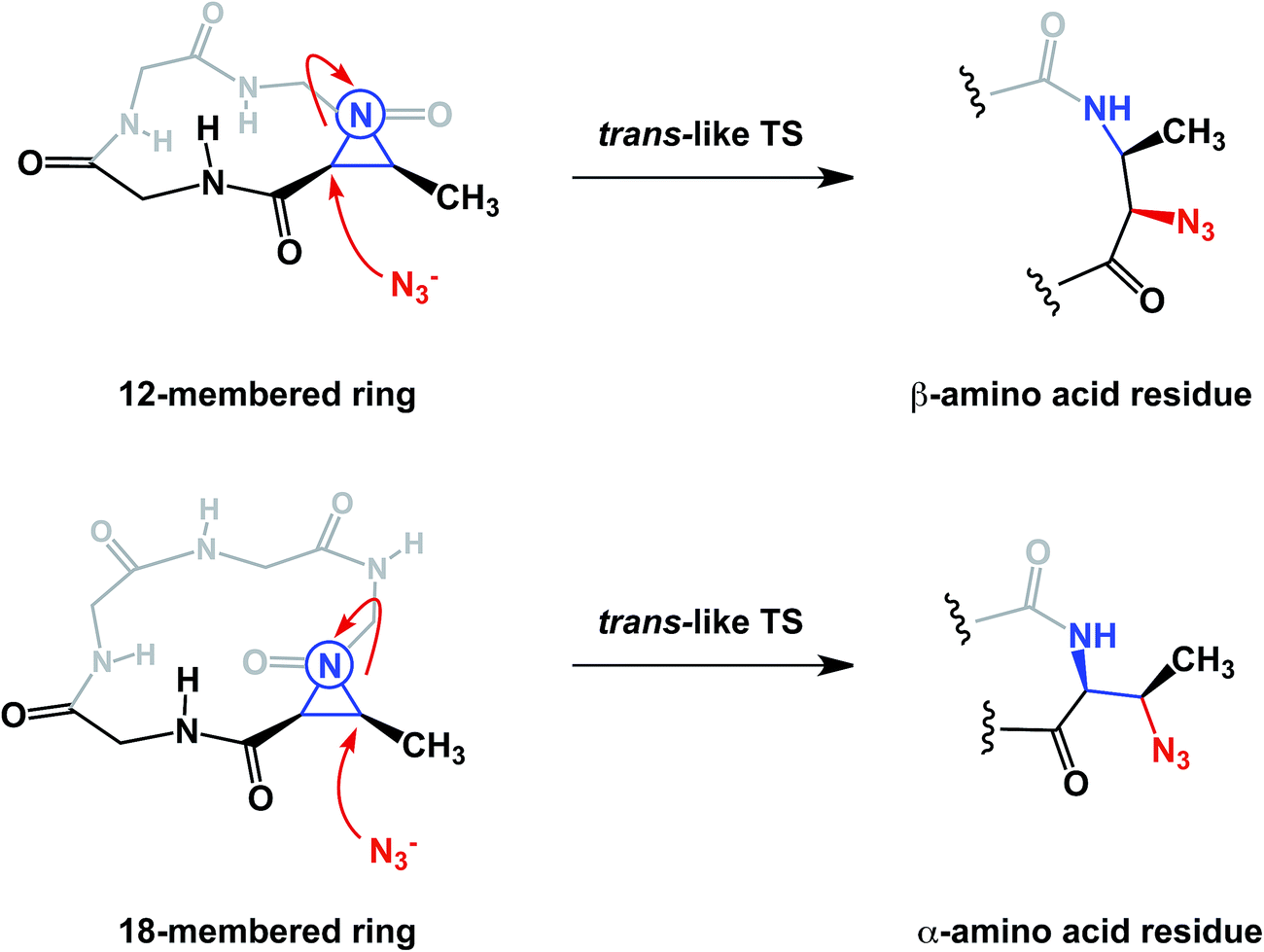 | ||
| Fig. 17 Kinetic control over cis- and trans- like transition states in macrocycles (amino acid side chains are omitted for clarity). | ||
The knowledge of the three-dimensional properties of constrained amino acid sequences can lead to impressive levels of control over macrocycle self-assembly in solution. Hydrogen bond-driven formation of peptide nanotubes is perhaps the best-known example of this sort of control. Ghadiri and co-workers demonstrated that, when constrained in macrocycles, alternating D- and L-amino acid sequences give rise to the formation of well-defined nanotubes that have found application as antibacterial agents.75 This example demonstrates the possibility of controlling aggregation of cyclic peptides in solution using relative stereochemistry of amino acid residues (Fig. 18).
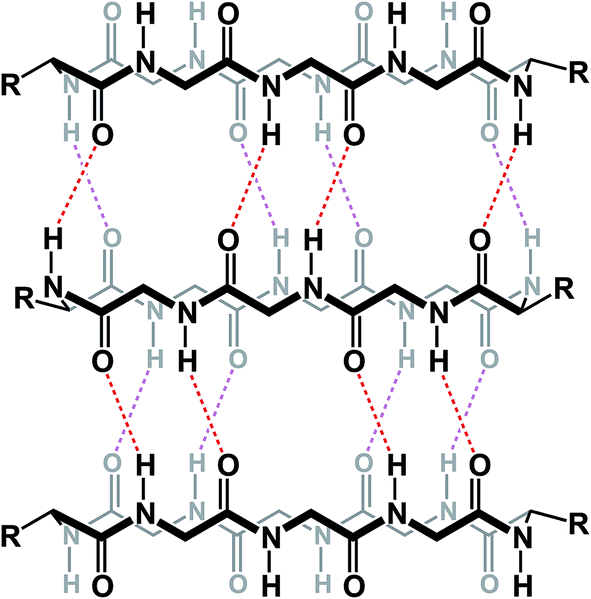 | ||
| Fig. 18 Formation of nanotubes from cyclic peptides (alternating stereochemistry not included for clarity). | ||
Lastly, the possibility of attaining well-defined conformations in cyclic peptides can have a direct link to their performance in catalysis. Herrmann and co-workers recently introduced a metallopeptide design based on a stable cyclic peptide scaffold that was maintained by an intramolecular disulfide linkage. The authors applied an alanine scanning technique to optimize the catalytic performance of their cyclic peptide catalyst system in Friedel–Crafts and Diels–Alder reactions.76
3.2 Structural biology of macrocycles
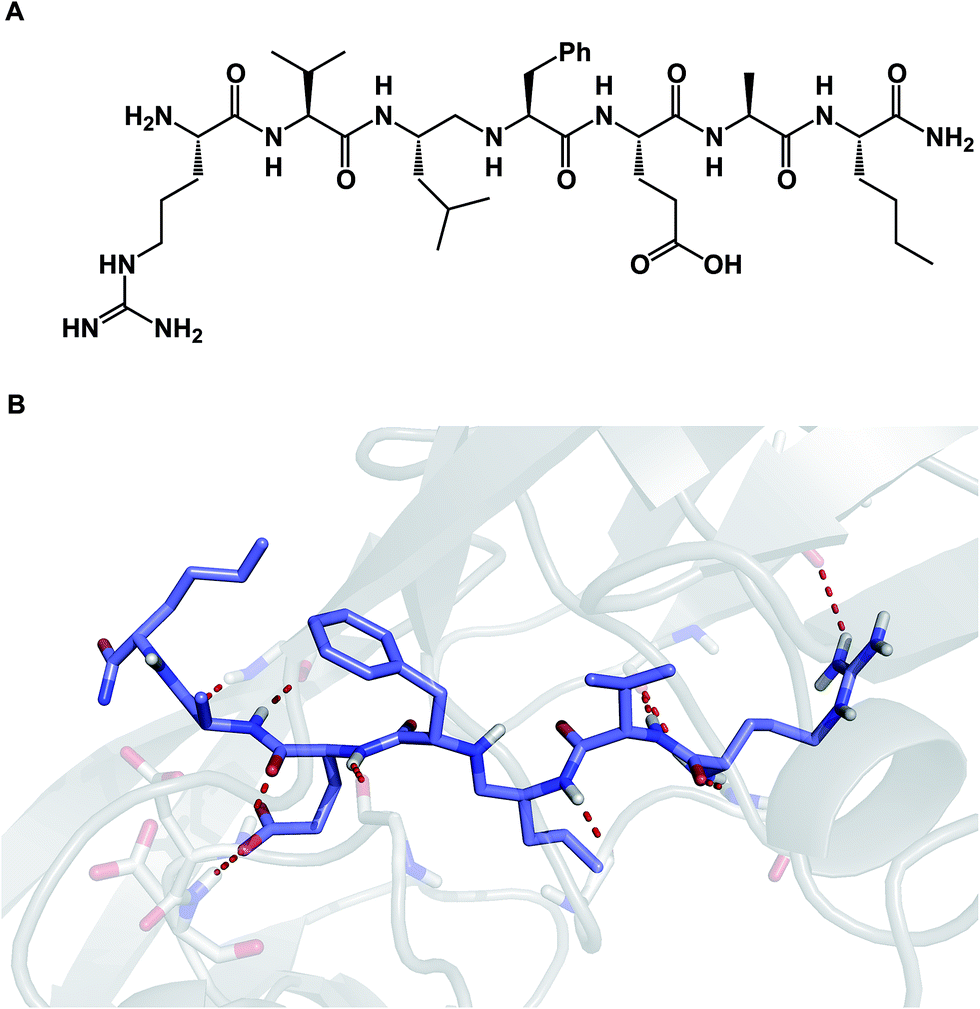
Due to their relatively large size, macrocycles display unique features upon interaction with their protein targets. Topology of the protein surface may dictate if a macrocycle adopts an “edge-on” or a “face-on” binding mode.77 This analysis by Whitty and co-workers further suggests that a preferred collection of macrocycles aimed at general-purpose drug discovery would benefit from molecules equipped with large and small substituents distributed around the ring. This is expected to increase the chances of finding useful compounds that can interrogate a wide range of protein binding site topologies. Whitty's analysis also suggests that at least five strong binding energy “hot spots” need to be present in a macrocycle, which is more than what is required for the binding a conventional small molecule drug. Interestingly, the hot spots can be set further apart from what is acceptable for conventional binding sites that are considered druggable. A recent report on the comprehensive analysis of protein surface loops suggests a mechanism by which “hot loops” can be identified and turned into constrained peptide inhibitors.78In cyclic peptides, the correct conformational recapitulation of the amino acid sequence involved in a biological interaction is required for effective agonism or antagonism of a protein target. Incorrect positioning of the binding determinants can easily lead to the wrong conformation. Even a fairly small deviation from the optimal geometry can bring about dramatic consequences. For example, the RGD sequence of amino acids is known to mediate interactions between cell surface integrin receptors and matrix proteins such as vitronectin, laminin, fibrinogen, and fibronectin. That this tripeptide sequence mediates multiple protein-dependent pathways implies distinct conformations of the RGD sequence in different matrix proteins. Kessler and colleagues showed that, when placed into the framework of a 15-membered ring, RGD correctly represents the binding epitope of fibronectin.79 This study illuminates the importance of choosing the geometrically correct turn motif. Even a minor deviation from the optimal arrangement of dihedral angles was found to result in a significant decline of potency. A constrained RGD/integrin complex was later crystallographically characterized, revealing that the RGD motif inserts into the crevice between the propeller and βA domains and makes contacts with both (Fig. 19A).80
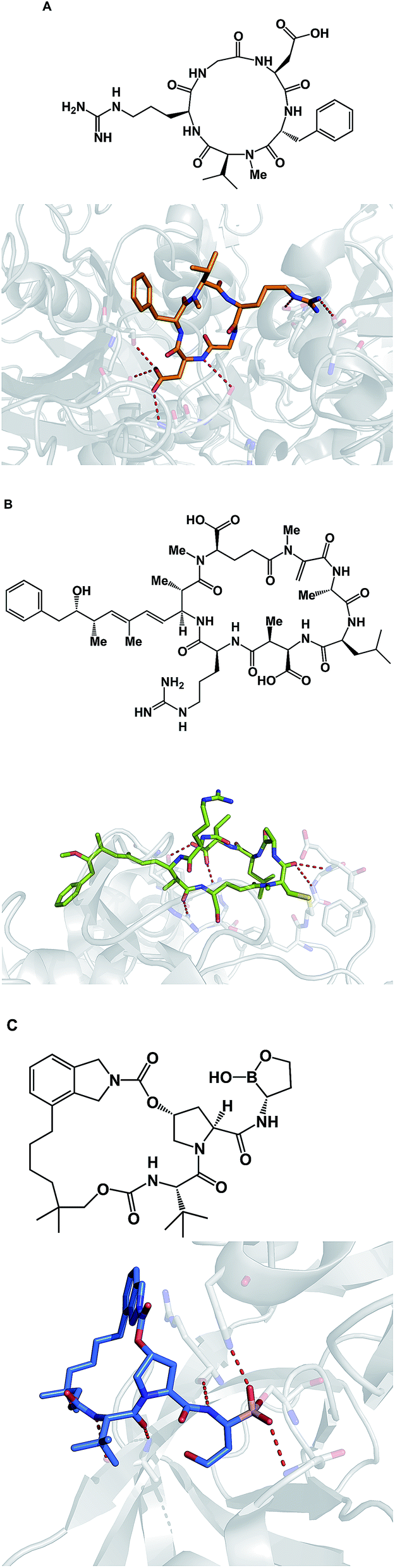 | ||
| Fig. 19 Selected crystal structures of macrocycles and their targets. (A): An RGD-based macrocycle complexed with the extracellular segment of integrin α5β3 (pdb id: 1lfg); (B): microcystin – a covalent phosphatase inhibitor (pdb id: 1fjm); and (C): Anacor's boron-containing macrocyclic HCV inhibitor (pdb id: 2xni). | ||
Macrocycles are also capable of covalent inhibition of their protein targets, which is notable given how much attention has been devoted to the development of covalent inhibitors. It is quite possible that combining the benefits that accrue as a result of increasing an inhibitor's polar surface with covalent bond formation can result in significant gains in potency. Given the possibility of rapid target ID, synthetic cyclic peptide electrophiles are likely to emerge as important tool compounds, particularly in phenotypic screens. In this regard, cyclic peptide natural products such as microcystin provide an inspiration. Thus, microcystin forms a covalent bond with the surface-exposed nucleophilic Cys-273 residue of PP1 phosphatase through Michael addition to the dehydroalanine moiety (Fig. 19B).81 Synthetic macrocycles that interact with oxygen-based active site residues include boron-containing molecules that were designed at Anacor.82 In this study, a new series of HCV NS3 serine protease inhibitors equipped with a cyclic boronate moiety at the P1 position of an HCV inhibitor scaffold, were developed and characterized by X-ray crystallography (Fig. 19C).
3.3 Cellular permeability and oral bioavailability
While the aforementioned structural features of macrocycles are significant from the standpoint of target engagement, they are unfortunately not related to properties that ensure bioavailability and other therapeutically relevant characteristics. Large polar surface area is the biggest obstacle preventing cyclic peptides and other macrocycles from being taken up by cells. The lion's share of current efforts goes into identifying macrocycles with drug-like properties.There are two main ways in which chemicals are thought to permeate cellular membranes: passive and active transport. The passive type of cellular entry is characterized by molecular diffusion driven by a concentration gradient, whereas the active entry type is energy-driven and involves molecular transporters. The most common assays that are used in comparative studies of a macrocycle's capacity to traverse cells are PAMPA (Parallel Artificial Membrane Permeability Assay) and Caco-2 cellular permeability assays.83 In this regard, it is exciting to note some surprisingly simple permeability surrogates that have appeared in the literature. One of them is based on supercritical fluid chromatography, which has enabled improved permeability design.84
The correlation between three-dimensional structure and cellular permeability is a fascinating area of contemporary research. Fernandez introduced the concept of under-wrapped hydrogen bonds (under-wrap = expose to solvent) and applied a variety of metrics to rank peptide ligands in terms of their cellular permeability, arguing that manipulation of intramolecularly under-wrapped electrostatic interactions in proteins can be exploited as a strategy to create molecules with enhanced ability to penetrate biological membranes.85 Lokey's group carried out a detailed study aimed at further examining the hypothesis that intramolecular hydrogen bonds improve passive membrane permeability of cyclic peptides. This investigation confirmed that membrane permeability of cyclic peptides is likely governed by a combination of intramolecular hydrogen bonding along with the protection of amide NH groups from solvation.86 Subtle differences in structure can play a decisive role in this process. For example, diastereomeric macrocycles can display notable differences in their ability to penetrate cells, which speaks to the adoption of different patterns of hydrogen bonds in structurally similar compounds.87 In the case of cyclic peptides, many efforts are aimed at improving cellular permeability by selective N-methylation of backbone amides. This strategy brings about a reduction of the polar surface area of a given cyclic peptide and increases the probability of intramolecular hydrogen bonds between the remaining NH amides and carbonyl oxygens. Using a library of 54 cyclic peptides with different N-methylation patterns, Kessler's lab designed structures that represent highly Caco-2 permeable templates amenable for grafting applications. This has become possible due to the defining role of the macrocycle core elements, and not the side chains, on the overall conformation.88 Interestingly, complete N-methylation can be detrimental to cellular permeability, highlighting a delicate balance that exists between a given molecule's permeability and lipophilicity. Thus, Lokey and co-workers showed that a partially N-methylated derivative shown in Fig. 20A was significantly more permeable in PAMPA assays than the corresponding per-methylated version.89 This was attributed to the more solvent-exposed nature of amide carbonyl oxygens in the per-methylated molecule's conformation, hinting at its higher effective polar surface area and, hence, diminished lipophilicity. Such findings are not intuitively clear by a visual examination of structures alone. Thankfully, computational tools can be effective in efforts to rationalize and predict the pattern of hydrogen bonds that is optimal for cellular permeability.90
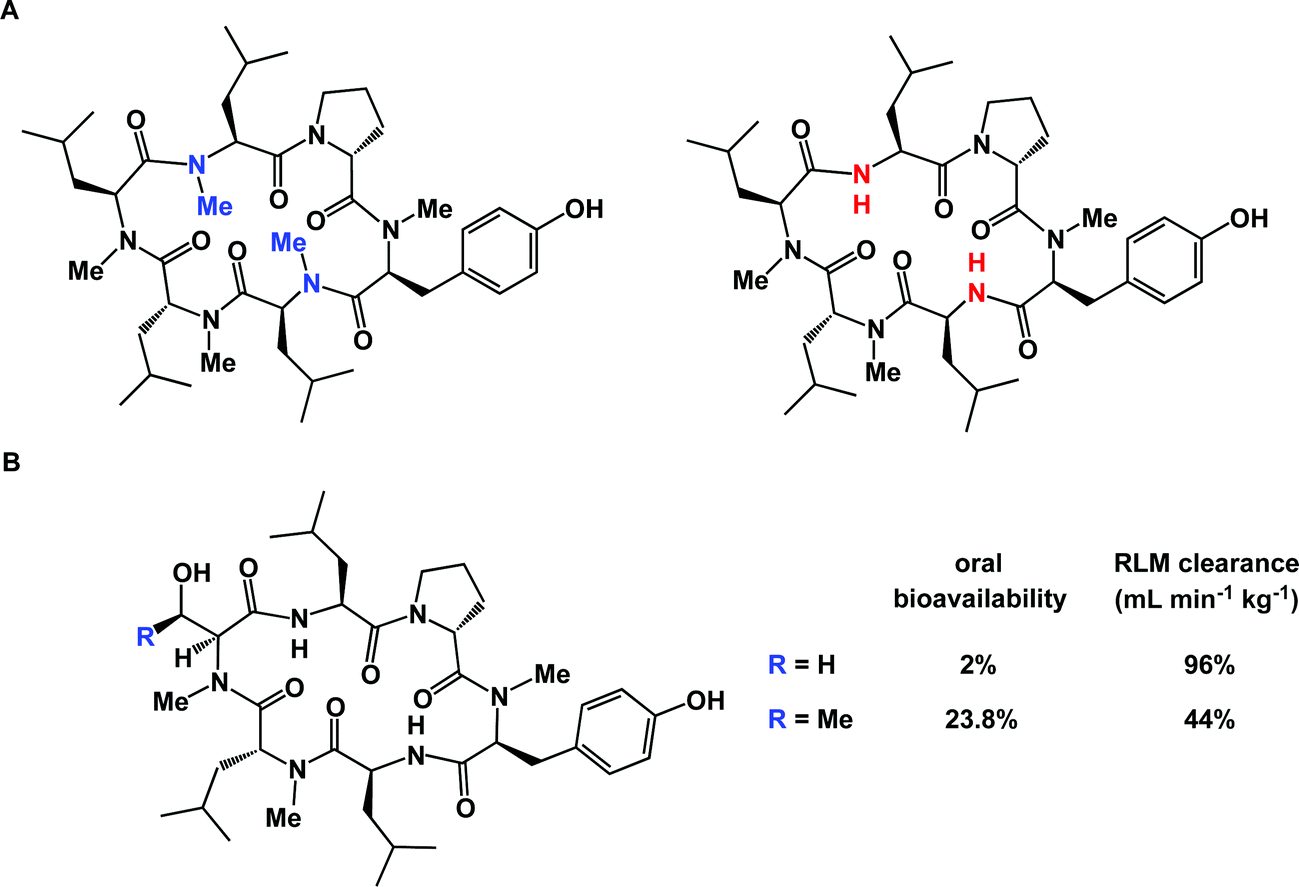 | ||
| Fig. 20 Subtle structure/properties effects in macrocycles. (A): Excessive N-methylation can be a detriment to cellular permeability; and (B): a dramatic effect of serine for threonine substitution on intrinsic clearance in rat liver microsome. | ||
It should be noted that one important factor that needs to be taken into account when considering N-methylation is the potential for chemical instability, which has been reported for excessively N-methylated peptides.91 In this regard, it is encouraging to see studies which suggest that macrocycles without N-methylation can be orally bioavailable. At present, this comes at the expense of relying on excessively hydrophobic side chains to shield polar amide groups from solvent exposure.92 It will be important to see follow-up cases where a greater variety of side chains can be accommodated in this approach.
When it comes to active membrane transport, the situation is substantially more complicated.93 Although parsing out the involvement of protein transporters at an early stage of lead generation could be extremely challenging, it is advisable to understand whether or not a given macrocycle series is subject to P-gp efflux. Structural data, pointing at the mechanisms by which P-gp can interact with macrocycles, has appeared in recent years. For instance, in an intriguing paper, Chang and colleagues showcased how the “dreaded” P-gp protein accommodated both enantiomers of a cyclic peptide molecule in its cavity (Fig. 21A).5 Nureki and co-workers reported on their structural characterization of a key multidrug and toxic compound extrusion (MATE) family transporter in complex with an in vitro selected thioether-based macrocyclic peptide (Fig. 21B).94 These studies suggest yet another parameter that awaits understanding in this area of research: how to modulate transporter-mediated removal of macrocycles from cells. This relates to both diminishing efflux in order to ensure adequate target engagement and enhancing efflux to avoid cytotoxicity due to accumulation.
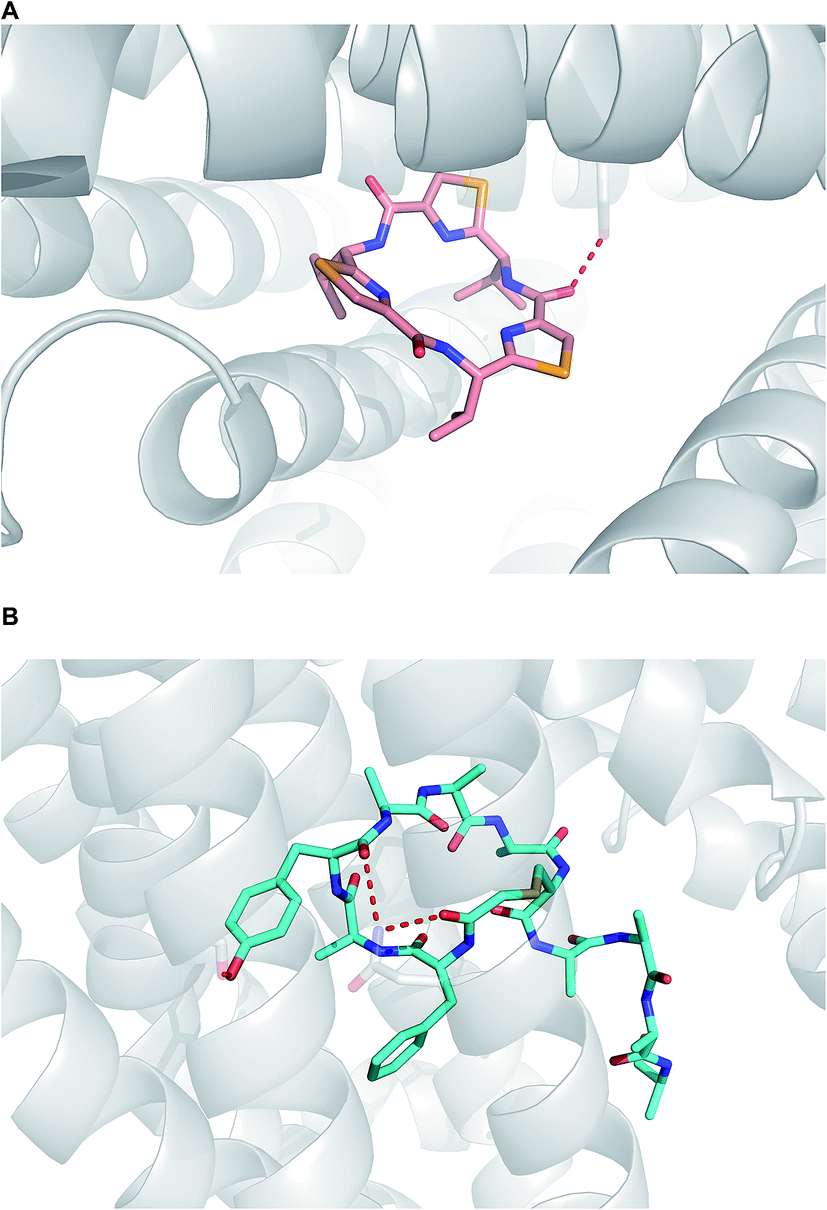 | ||
| Fig. 21 Macrocycles complexed with transporters. (A): P-gp complexed with a selenium-containing cyclic peptide (pdb id: 3g61); and (B): multidrug and toxic compound extrusion transporter complexed with a cyclic peptide (pdb id:3vvr). | ||
Oral bioavailability adds another layer of complexity to evaluating the potential of macrocycles as promising drug candidates. Orally active and/or bioavailable peptide macrocycles that are currently on the market include cyclosporine A, linaclotide, and some somatostatin analogs, such as octreotide. Clearly, all of these compounds violate the “rule of 5”, further reinforcing the significance of understanding oral bioavailability of molecules that belong to the “Beyond the rule of 5” (Bro5) class.
Microsomal stability is typically evaluated by subjecting a compound to rat liver microsomes (RLM). This kind of study can be exceptionally useful as concrete steps aimed at improving molecular profiles can be identified.95 Unfortunately, full metabolite identification (MetID) profiles of macrocycles can be very expensive and are not performed routinely, at least in the academic setting. Streamlining of these assays is expected to be enabling and will result in innovative chemical approaches to site-selective macrocycle modification. The poorly-understood balance between the three-dimensional structure and microsomal stability of macrocycles is further illuminated by comparing the dramatic difference observed when structurally similar molecules are exposed to RLM analysis (Fig. 20B).96 The serine derivative was shown to have 96 mL min−1 kg−1 RLM clearance, whereas the threonine-containing congener was substantially less stable (44 mL min−1 kg−1 RLM clearance). Oddly enough, the serine-containing peptide actually had an oral bioavailability of only 2% compared to the threonine-containing peptide which was 23.8% orally bioavailable. This example underscores the highly empirical nature of efforts to identify orally bioavailable macrocycles and suggests that finding a correlation between oral bioavailability and scaffold design is likely to be challenging. Yet, the discovery of orally bioavailable peptides continues to be the subject of intense investigations97 and oral bioavailability can be established even for fairly large molecules.98 It should also be acknowledged that, while striving for oral bioavailability is a worthy goal, peptides provide an opportunity to develop exquisitely potent compounds, which can offset their poor drug-like properties. Ultimately, reaching the desired therapeutic profile can be realized using special additives. A case in point is octreotide: in a well-known study, its oral bioavailability was reported to be only 0.3%, yet it could be dramatically improved by formulation.99 In this regard, the advent of nanotechnology and new drug delivery modalities is expected to play a pivotal role in ensuring that macrocycles reach their therapeutic targets and remain reasonably bioavailable.
4. Conclusions
Macrocycles constitute an exciting class of molecules with a tremendous upside in drug discovery and other fields of inquiry. Their complex structures invite the development of novel cyclization technologies with improved efficiency. It is equally important to have the modern tools of synthesis bear on site-selective structural modification of existing macrocycle cores. In efforts to come up with new synthetic tools aimed at macrocycles, chemists need to be aware of the propensity of macrocycles to be susceptible to transannular interactions. These interactions can deliver dividends in areas that require conformational constraint, but attention must be paid to the potentially detrimental consequences of particularly strong transannular interactions that can lead to unanticipated intramolecular reactivity and “collapse”. The availability of broadly applicable methods that address these long-standing goals will further facilitate synthesis-driven improvement of macrocyclic lead molecules in drug discovery. At the same time, detailed knowledge of the three-dimensional preferences of macrocycles, complicated by the presence of partially rotatable bonds, will serve areas in which functional outcomes are rooted in one's ability to modulate intermolecular interactions. Control over the so-called χ-space,100 that defines conformational movement of side chains, is one of the most difficult areas to address. In this regard, there is a certain irony in overemphasizing the significance of new ways to form macrocyclic ring structures: these studies offer constraints over ϕ and ψ dihedral angles, but they provide little towards control in the “χ-space” which defines the side chain orientation (Fig. 22). | ||
| Fig. 22 Peptide macrocycles and the challenge of conformational control in all areas of accessible space. | ||
When it comes to cyclic peptides, it would be useful to see a greater variability with regard to accessible synthetic building blocks. Indeed, approaches to macrocycles based on improving compound profiles using amino acid modifications tend to underemphasize the relative significance of non-amino acid building blocks – the ones that do not suffer from the limitations of peptide linkages. Chemists need to keep in mind that, just because amino acids are readily available in their protected forms and are suited for streamlined synthesis of linear precursors to cyclization, it does not mean that there is anything inherently special to them (an exception is when the goal is to rationally constrain a particular protein secondary structure motif). As a result, overemphasizing the attempts to render amino acid-derived materials “more palatable” might result in approaches that detract from rapid optimization of the function-defining characteristics of molecules. In this regard, structure-driven efforts aimed at peptidomimetic macrocycles should be encouraged.
The most difficult questions in the area of biologically active macrocycles will likely relate to reconciling the synthetic and biological approaches to synthesis. The domain of chemical synthesis is fertile with methods that enable the development of molecules with optimal pharmacological profiles. Unfortunately, the accessible molecular diversity is still a challenging proposition. In contrast, biological synthesis appears to readily provide enormous molecular diversity, albeit at the expense of offering only a limited palette of useful building blocks. In addition, the molecules that are accessible with biological methods are rarely attractive drug candidates, particularly when it comes to intracellular targets. The inherent friction between the synthetic and biological domains of synthesis is likely to result in exciting innovations in the years to come.
Acknowledgements
I would like to thank the past and present members of my research group at the University of Toronto. I would like to particularly acknowledge the efforts of Dr Conor Scully, Dr Jen Hickey, Dr Chris White, Benjamin Chung, Dr Ryan Hili, Dr Vishal Rai, and Serge Zaretsky. The tireless contributions of scientists at Encycle Therapeutics, led by Dr Andrew Roughton, further contributed to our emerging understanding of orally bioavailable macrocycles. NSERC, CIHR, and CQDM are gratefully acknowledged for their support of our research operations over the years. I would also like to thank Dr Spiros Liras of Pfizer, Dr Scott Cowen of AstraZeneca, Dr Graham Simpson of GlaxoSmithKline, and Dr Yusheng Xiong of Merck for their valuable input to our research programs. Professor Eric Marsault and his team at the Université de Sherbrooke are thanked for their important contributions to the development of our macrocycle library. Elena Dobrovetsky of SGC is thanked for fruitful discussions concerning SH2 domain/macrocycle interactions.References
- A. T. Frank, N. S. Farina, N. Sawwan, O. R. Wauchope, M. Qi, E. M. Brzostowska, W. Chan, F. W. Grasso, P. Haberfield and A. Greer, Mol. Diversity, 2007, 11, 115–118 CrossRef CAS PubMed.
- L. A. Wessjohann, E. Ruijter, D. Garcia-Rivera and W. Brandt, Mol. Diversity, 2005, 9, 171–186 CrossRef CAS.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- P. K. Madala, J. D. A. Tyndall, T. Nall and D. P. Fairlie, Chem. Rev., 2010, 110, PR1–PR31 CrossRef CAS PubMed.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Science, 2009, 323, 1718–1722 CrossRef CAS PubMed.
- J. G. Beck, J. Chatterjee, B. Laufer, M. U. Kiran, A. O. Frank, S. Neubauer, O. Ovadia, S. Greenberg, C. Gilon, A. Hoffman and H. Kessler, J. Am. Chem. Soc., 2012, 134, 12125–12133 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- A.-C. Bédard and S. K. Collins, J. Am. Chem. Soc., 2011, 133, 19976–19981 CrossRef PubMed.
- A. D. Cort, G. Ercolani, L. Mandolini and P. Mencarelli, Chem. Commun., 1993, 538 RSC.
- J. C. Collins and K. James, MedChemComm, 2012, 3, 1489 RSC.
- M. M. G. Krishna and S. W. Englander, Proc. Natl. Acad. Sci., 2005, 102, 1053–1058 CrossRef CAS PubMed.
- I. Daidone, H. Neuweiler, S. Doose, M. Sauer and J. C. Smith, PLoS Comput. Biol., 2010, 6, e10000645 Search PubMed.
- C. Schmuck, T. Rehm, F. Gröhn, K. Klein and F. Reinhold, J. Am. Chem. Soc., 2006, 128, 1430–1431 CrossRef CAS PubMed.
- R. Hili, V. Rai and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 2889–2891 CrossRef CAS PubMed.
- R. Hili and A. K. Yudin, J. Am. Chem. Soc., 2006, 128, 14772–14773 CrossRef CAS PubMed.
- A. T. Londregan, K. A. Farley, C. Limberakis, P. B. Mullins and D. W. Piotrowski, Org. Lett., 2012, 14, 2890–2893 CrossRef CAS PubMed.
- H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed.
- L. Guo, S. H. Lahasky, K. Ghale and D. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS PubMed.
- T. Weide, A. Modlinger and H. Kessler, Top. Curr. Chem., 2007, 272, 1–50 CrossRef CAS.
- S. A. Miller, S. L. Griffiths and D. Seebach, Helv. Chim. Acta, 1993, 76, 563–595 CrossRef CAS.
- D. Seebach, A. K. Beck, H. G. Bossler, C. Gerber, S. Y. Ko, C. W. Murtiashaw, R. Naef, S.-I. Shoda, A. Thaler, M. Krieger and R. Wenger, Helv. Chim. Acta, 1993, 76, 1564–1590 CrossRef CAS.
- L. Pauling and C. Niemann, J. Am. Chem. Soc., 1939, 61, 1860–1867 CrossRef CAS.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- T. Guedez, A. S. Núñez, E. Tineo and O. Núñez, J. Chem. Soc., Perkin Trans. 2, 2002, 2078–2082 RSC.
- K. A. Carpenter, G. Weltrowska, B. C. Wilkes, R. Schmidt and P. W. Schiller, J. Am. Chem. Soc., 1994, 116, 8450–8458 CrossRef CAS.
- J. M. Villalgordo and H. Heimgartner, Helv. Chim. Acta, 1997, 80, 748–766 CrossRef CAS.
- I. B. Seiple, S. Su, I. S. Young, C. A. Lewis, J. Yamaguchi and P. S. Baran, Angew. Chem., Int. Ed., 2009, 49, 1095–1098 CrossRef PubMed.
- E. P. Balskus and E. N. Jacobsen, Science, 2007, 317, 1736–1740 CrossRef CAS PubMed.
- C. Han, S. Rangarajan, A. C. Voukides, A. B. Beeler, R. Johnson and J. A. Porco, Org. Lett., 2009, 11, 413–416 CrossRef CAS PubMed.
- M. E. Selsted and A. J. Ouellette, Nat. Immunol., 2005, 6, 551–557 CrossRef CAS PubMed.
- A. Brust, C.-I. A. Wang, N. L. Daly, J. Kennerly, M. Sadeghi, M. J. Christie, R. J. Lewis, M. Mobli and P. F. Alewood, Angew. Chem., Int. Ed., 2013, 52, 12020–12023 CrossRef CAS PubMed.
- R. J. Clark, H. Fischer, L. Dempster, N. L. Daly, K. J. Rosengren, S. T. Nevin, F. A. Meunier, D. J. Adams and D. J. Craik, Proc. Natl. Acad. Sci., 2005, 102, 13767–13772 CrossRef CAS PubMed.
- D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 9391–9392 CrossRef CAS.
- H. E. Blackwell, J. D. Sadowsky, R. J. Howard, J. N. Sampson, J. A. Chao, W. E. Steinmetz, D. J. O'Leary and R. H. Grubbs, J. Org. Chem., 2001, 66, 5291–5302 CrossRef CAS PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- M. L. Stewart, E. Fire, A. E. Keating and L. D. Walensky, Nat. Chem. Biol., 2010, 6, 595–601 CrossRef CAS PubMed.
- Y. Hitotsuyanagi, Y. Matsumoto, S. I. Sasaki, K. Yamaguchi, H. Itokawa and K. Takeya, Tetrahedron Lett., 2001, 42, 1535–1537 CrossRef CAS.
- B. Oberhauser, K. Baumann, B. Grohmann and H. Sperner, Synlett, 1999, 893–896 CAS.
- W. D. F. Meutermans, S. W. Golding, G. T. Bourne, L. P. Miranda, M. J. Dooley, P. F. Alewood and M. L. Smythe, J. Am. Chem. Soc., 1999, 121, 9790–9796 CrossRef CAS.
- C. J. White, J. L. Hickey, C. C. G. Scully and A. K. Yudin, J. Am. Chem. Soc., 2014, 136, 3728–3731 CrossRef CAS PubMed.
- C. A. Lewis and S. J. Miller, Angew. Chem., Int. Ed., 2006, 45, 5616–5619 CrossRef CAS PubMed.
- B. C. Wilcock, B. E. Uno, G. L. Bromann, M. J. Clark, T. M. Anderson and M. D. Burke, Nat. Chem., 2012, 4, 996–1003 CrossRef CAS PubMed.
- R. M. Kohli, C. T. Walsh and M. D. Burkart, Nature, 2002, 418, 658–661 CrossRef CAS PubMed.
- E. Marsault, H. R. Hoveyda, R. E. Gagnon, M. L. Peterson, M. Vëzina, C. Saint-Louis, A. Landry, J.-F. C. C. O. Pinault, L. Ouellet, S. Beauchemin, S. Beaubien, A. Mathieu, K. Benakli, Z. Wang, M. Brassard, D. Lonergan, F. C. C. O. Bilodeau, M. Ramaseshan, N. Fortin, R. Lan, S. Li, F. Galaud, V. E. R. Plourde, M. Champagne, A. Doucet, P. Bhërer, M. Gauthier, G. Olsen, G. E. R. Villeneuve, S. Bhat, L. Foucher, D. Fortin, X. Peng, S. Bernard, A. Drouin, R. Dëziel, G. Berthiaume, Y. L. Dory, G. L. Fraser and P. Deslongchamps, Bioorg. Med. Chem. Lett., 2008, 18, 4731–4735 CrossRef CAS PubMed.
- Ahsanullah and J. Rademann, Angew. Chem., Int. Ed., 2010, 49, 5378–5382 CrossRef CAS PubMed.
- E. Koivunen, B. Wang and E. Ruoslahti, Nat. Biotechnol., 1995, 13, 265–270 CrossRef CAS PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- Z. J. Gartner, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- Y. Sako, Y. Goto, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 241–249 CrossRef CAS PubMed.
- K. Josephson, A. Ricardo and J. W. Szostak, Drug Discovery Today, 2014, 19, 388–399 CrossRef CAS PubMed.
- J. R. Frost, F. Vitali, N. T. Jacob, M. D. Brown and R. Fasan, ChemBioChem, 2012, 14, 147–160 CrossRef PubMed.
- M. D. Cummings, J. Lindberg, T.-I. Lin, H. de Kock, O. Lenz, E. Lilja, S. Felländer, V. Baraznenok, S. Nyström, M. Nilsson, L. Vrang, M. Edlund, Å. Rosenquist, B. Samuelsson, P. Raboisson and K. Simmen, Angew. Chem., Int. Ed., 2010, 49, 1652–1655 CrossRef CAS PubMed.
- J. Phan, Z.-D. Shi, T. R. Burke Jr and D. S. Waugh, J. Mol. Biol., 2005, 353, 104–115 CrossRef CAS PubMed.
- P. Bonnet, D. K. Agrafiotis, F. Zhu and E. Martin, J. Chem. Inf. Model., 2009, 49, 2242–2259 CrossRef CAS PubMed.
- A. P. Benfield, M. G. Teresk, H. R. Plake, J. E. DeLorbe, L. E. Millspaugh and S. F. Martin, Angew. Chem., Int. Ed., 2006, 45, 6830–6835 CrossRef CAS PubMed.
- D. G. Udugamasooriya and M. R. Spaller, Biopolymers, 2008, 89, 653–667 CrossRef CAS PubMed.
- A. R. Bogdan, S. V. Jerome, K. N. Houk and K. James, J. Am. Chem. Soc., 2012, 134, 2127–2138 CrossRef CAS PubMed.
- S. Zaretsky, C. C. G. Scully, A. J. Lough and A. K. Yudin, Chem.–Eur. J., 2013, 19, 17668–17672 CrossRef CAS PubMed.
- R. W. Driver, H. N. Hoang, G. Abbenante and D. P. Fairlie, Org. Lett., 2009, 11, 3092–3095 CrossRef CAS PubMed.
- M. P. Glenn, M. J. Kelso, J. D. A. Tyndall and D. P. Fairlie, J. Am. Chem. Soc., 2003, 125, 640–641 CrossRef CAS PubMed.
- A. Montero, J. M. Beierle, C. A. Olsen and M. R. Ghadiri, J. Am. Chem. Soc., 2009, 131, 3033–3041 CrossRef CAS PubMed.
- X.-G. Hu, D. S. Thomas, R. Griffith and L. Hunter, Angew. Chem., Int. Ed., 2014, 53, 6176–6179 CrossRef CAS PubMed.
- C. J. Dinsmore, M. J. Bogusky, J. C. Culberson, J. M. Bergman, C. F. Homnick, C. B. Zartman, S. D. Mosser, M. D. Schaber, R. G. Robinson, K. S. Koblan, H. E. Huber, S. L. Graham, G. D. Hartman, J. R. Huff and T. M. Williams, J. Am. Chem. Soc., 2001, 123, 2107–2108 CrossRef CAS.
- S. Zaretsky, J. L. Hickey, M. A. St. Denis, C. C. G. Scully, A. L. Roughton, D. J. Tantillo, M. W. Lodewyk and A. K. Yudin, Tetrahedron, 2014, 70, 7655–7663 CrossRef CAS PubMed.
- A. C. Gibbs, L. H. Kondejewki, W. Gronwald, A. M. Nip, R. S. Hodges, B. D. Sykes and D. S. Wishart, Nat. Struct. Biol., 1998, 5, 284–288 CrossRef CAS PubMed.
- J. A. Robinson, Acc. Chem. Res., 2008, 41, 1278–1288 CrossRef CAS PubMed.
- R. J. Woods, J. O. Brower, E. Castellanos, M. Hashemzadeh, O. Khakshoor, W. A. Russu and J. S. Nowick, J. Am. Chem. Soc., 2007, 129, 2548–2558 CrossRef CAS PubMed.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- G. M. Grotenbreg, M. S. M. Timmer, A. L. Llamas-Saiz, M. Verdoes, G. A. van der Marel, M. J. van Raaij, H. S. Overkleeft and M. Overhand, J. Am. Chem. Soc., 2004, 126, 3444–3446 CrossRef CAS PubMed.
- O. Babii, S. Afonin, M. Berditsch, S. Reier, P. K. Mykhailiuk, V. S. Kubyshkin, T. Steinbrecher, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2014, 53, 3392–3395 CrossRef CAS PubMed.
- J. L. Kofron, P. Kuzmič, V. Kishore, G. Gemmecker, S. W. Fesik and D. H. Rich, J. Am. Chem. Soc., 1992, 114, 2670–2675 CrossRef CAS.
- S. Fernandez-Lopez, H. S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- L. Zheng, A. Marcozzi, J. Y. Gerasimov and A. Herrmann, Angew. Chem., Int. Ed., 2014, 53, 7599–7603 CrossRef CAS PubMed.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 716–722 CrossRef PubMed.
- J. Gavenonis, B. A. Sheneman, T. R. Siegert, M. R. Eshelman and J. A. Kritzer, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef PubMed.
- R. Haubner, W. Schmitt, G. Hölzemann, S. L. Goodman, A. Jonczyk and H. Kessler, J. Am. Chem. Soc., 1996, 118, 7881–7891 CrossRef CAS.
- J. P. Xiong, Science, 2002, 296, 151–155 CrossRef CAS PubMed.
- J. Goldberg, H. Huang, Y. Kwon, P. Greengard, A. C. Nairn and J. Kuriyan, Nature, 1995, 376, 745–753 CrossRef CAS PubMed.
- X. Li, Y.-K. Zhang, Y. Liu, C. Z. Ding, Y. Zhou, Q. Li, J. J. Plattner, S. J. Baker, S. Zhang, W. M. Kazmierski, L. L. Wright, G. K. Smith, R. M. Grimes, R. M. Crosby, K. L. Creech, L. H. Carballo, M. J. Slater, R. L. Jarvest, P. Thommes, J. A. Hubbard, M. A. Convery, P. M. Nassau, W. McDowell, T. J. Skarzynski, X. Qian, D. Fan, L. Liao, Z.-J. Ni, L. E. Pennicott, W. Zou and J. Wright, Bioorg. Med. Chem. Lett., 2010, 20, 5695–5700 CrossRef CAS PubMed.
- I. J. Hidalgo, T. J. Raub and R. T. Borchardt, Gastroenterology, 1989, 96, 736–749 CAS.
- G. H. Goetz, L. Philippe and M. J. Shapiro, ACS Med. Chem. Lett., 2014, 140811082631009 Search PubMed.
- A. Fernández, Nat. Biotechnol., 2004, 22, 1081–1084 CrossRef PubMed.
- T. Rezai, J. E. Bock, M. V. Zhou, C. Kalyanaraman, R. S. Lokey and M. P. Jacobson, J. Am. Chem. Soc., 2006, 128, 14073–14080 CrossRef CAS PubMed.
- T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2006, 128, 2510–2511 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- T. R. White, C. M. Renzelman, A. C. Rand, T. Rezai, C. M. McEwen, V. M. Gelev, R. A. Turner, R. G. Linington, S. S. F. Leung, A. S. Kalgutkar, J. N. Bauman, Y. Zhang, S. Liras, D. A. Price, A. M. Mathiowetz, M. P. Jacobson and R. S. Lokey, Nat. Chem. Biol., 2011, 7, 810–817 CrossRef CAS PubMed.
- A. Alex, D. S. Millan, M. Perez, F. Wakenhut and G. A. Whitlock, MedChemComm, 2011, 669–674 RSC.
- M. Anteunis and C. Auwera, Int. J. Pept. Protein Res., 1988, 31, 301–310 CrossRef CAS PubMed.
- T. A. Hill, R.-J. J. Lohman, H. N. Hoang, D. S. Nielsen, C. C. Scully, W. M. Kok, L. Liu, A. J. Lucke, M. J. Stoermer, C. I. Schroeder, S. Chaousis, B. Colless, P. V. Bernhardt, D. J. Edmonds, D. A. Griffith, C. J. Rotter, R. B. Ruggeri, D. A. Price, S. Liras, D. J. Craik and D. P. Fairlie, ACS Med. Chem. Lett., 2014, 140804105706000 Search PubMed.
- K. Sugano, M. Kansy, P. Artursson, A. Avdeef, S. Bendels, L. Di, G. F. Ecker, B. Faller, H. Fischer, G. Gerebtzoff, H. Lennernaes and F. Senner, Nat. Rev. Drug Discovery, 2010, 9, 597–614 CrossRef CAS PubMed.
- Y. Tanaka, C. J. Hipolito, A. D. Maturana, K. Ito, T. Kuroda, T. Higuchi, T. Katoh, H. E. Kato, M. Hattori, K. Kumazaki, T. Tsukazaki, R. Ishitani, H. Suga and O. Nureki, Nature, 2013, 496, 247–251 CrossRef CAS PubMed.
- U. Christians and K. F. Sewing, Pharmacol. Ther., 1993, 57, 291–345 CrossRef CAS.
- A. C. Rand, S. S. F. Leung, H. Eng, C. J. Rotter, R. Sharma, A. S. Kalgutkar, Y. Zhang, M. V. Varma, K. A. Farley, B. Khunte, C. Limberakis, D. A. Price, S. Liras, A. M. Mathiowetz, M. P. Jacobson and R. S. Lokey, MedChemComm, 2012, 3, 1282 RSC.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2012, 81, 136–147 Search PubMed.
- R. J. Clark, J. Jensen, S. T. Nevin, B. P. Callaghan, D. J. Adams and D. J. Craik, Angew. Chem., Int. Ed., 2010, 49, 6545–6548 CrossRef CAS PubMed.
- J. Drewe, G. Fricker, J. Vonderscher and C. Beglinger, Br. J. Pharmacol., 1993, 108, 298–303 CrossRef CAS PubMed.
- V. J. Hruby, G. Li, C. Haskell-Luevano and M. Shenderovich, Biopolymers, 1997, 43, 219–266 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |