DOI:
10.1039/C4QO00294F
(Review Article)
Org. Chem. Front., 2015,
2, 288-299
Novel strategies for catalytic asymmetric synthesis of C1-chiral 1,2,3,4-tetrahydroisoquinolines and 3,4-dihydrotetrahydroisoquinolines
Received
8th November 2014
, Accepted 14th December 2014
First published on 16th December 2014
Abstract
1,2,3,4-Tetrahydroisoquinoline is one of the most important “privileged scaffolds” present in natural products. C1-chiral tetrahydroisoquinolines have exhibited a wide variety of bioactivities and found applications as chiral scaffolds in asymmetric catalysis. This paper summarizes novel catalytic stereoselective strategies that emerged in the last ten years for synthesis of 1,2,3,4-tetrahydroisoquinoline and 3,4-dihydroisoquinoline scaffolds, beyond the traditional Pictet–Spengler and related protocols, as well as their applications in the total synthesis of alkaloid natural products.
 Wangsheng Liu | Wangsheng Liu was born in 1991. He received his B.S. degree from Tianjin Normal University in 2014, and then entered Fudan University as a graduate student. |
 Shasha Liu | Shasha Liu obtained her Ph.D. in organic chemistry at Tianjin University in 2009. She did postdoctoral studies at Syracuse University from 2010 to 2012 and worked as visiting scholar at the University of Kansas from 2012 to 2013. She has been working at School of Geological Sciences, Northeast Normal University since 2014. |
 Ruiwen Jin | Jin Ruiwen was born in 1990. She received her B.S. degree from Zhengzhou University in 2014, and then she entered Fudan University as a graduate student. |
 Hao Guo | Hao Guo was born in Tianjin, China in 1980. He received his B.S. degree in Chemistry from Nankai University in 2003, and a Ph.D. degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 2008. After postdoctoral research at Technische Universität München in Germany, he joined Fudan University as an Associate Professor in 2010. He received the Thieme Chemistry Journal Award 2011. His research interests are mainly focused on organic photochemistry. |
 Jinbo Zhao | Jinbo Zhao obtained his Ph.D. in organic chemistry at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC, CAS) in early 2010. After two consecutive postdoctoral studies in organic chemistry and medicinal chemistry respectively at Syracuse University (2010–2012) and the University of Kansas (2012–2014), United States, he joined the faculty of chemistry at Northeast Normal University as an associate professor. His research interest is focused on transition-metal catalysis and applications to natural product synthesis. |
1. Introduction
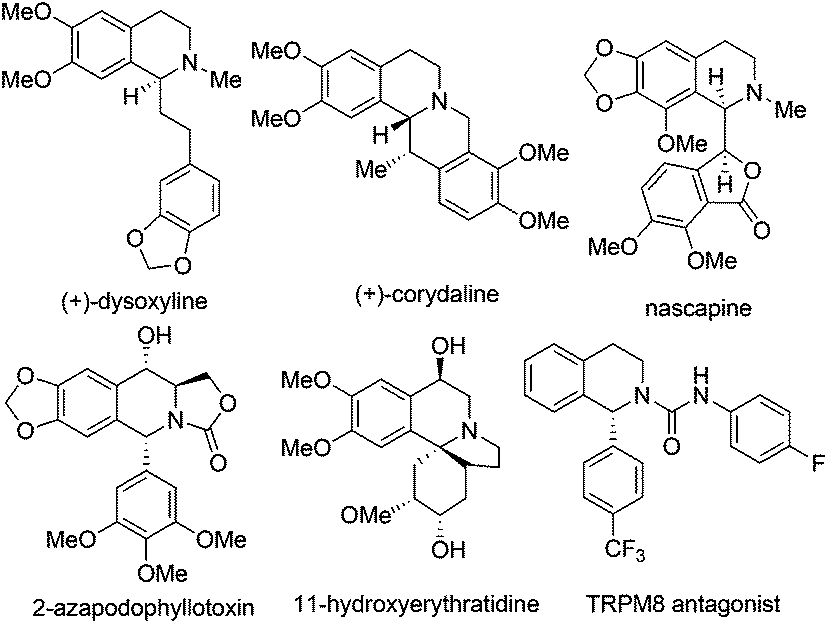
1,2,3,4-Tetrahydroisoquinoline (THIQ) represents one of the most prevalent “privileged scaffolds” created by nature,1 as exemplified by the numerous identified THIQ-containing alkaloids which demonstrate a wide range of bioactivities, including anti-tumor, anti-bacteria, anti-virus, anti-inflammation activities, etc. (Scheme 1).2 The number of bioactive small molecules bearing THIQ skeletons as potential therapeutic agents for treatment of various diseases, such as cancer, AIDS, Parkinson's disease, etc., has shown an incredible increase.3 THIQ-containing molecules have also shown increasing applications in asymmetric catalysis as chiral scaffolds.4 Not surprisingly, strategies for the assembly of THIQ-containing molecules have garnered intensive attention in the past few decades. Traditional approaches such as the Pictet–Spengler reaction and the Bischler–Napieralski cyclization/reduction sequence were successfully demonstrated and continue to show their power in the synthesis of isoquinoline and indole alkaloid frameworks. They are, however, intrinsically limited to electron-rich aromatic substrates.5 Asymmetric synthesis of C1-chiral moiety containing THIQs is of particular importance due to its presence in many THIQ type alkaloids and the usually distinct bioactivities exhibited by the two antipodes of a given structure. This paper summarizes representative recent strategies for the catalytic asymmetric synthesis of THIQ and 3,4-dihydroisoquinoline (DHIQ) scaffolds bearing the C1-chiral moiety. Updates in this field in the last ten years (2003–early 2014) are included, except for the above-mentioned classical methodologies. Asymmetric hydrogenation and transfer hydrogenation of isoquinoline derivatives are established powerful strategies that could be traced back to the 20th century and have been highlighted very recently.6 Readers are referred to elegant previous reviews for summary of non-catalytic asymmetric synthesis of THIQs.7 Reactions are discussed and classified based on their substrates and activation modes, as well as the reaction types.
 |
| | Scheme 1 Selected bioactive THIQ containing natural products and synthetic small molecules. | |
2. Recent strategies for catalytic asymmetric synthesis of C1-chiral THIQs and DHIQs
2.1 Nucleophilic addition to isoquinoline, 3,4-dihydroisoquinoline (DHIQ) and isoquinolinium salt
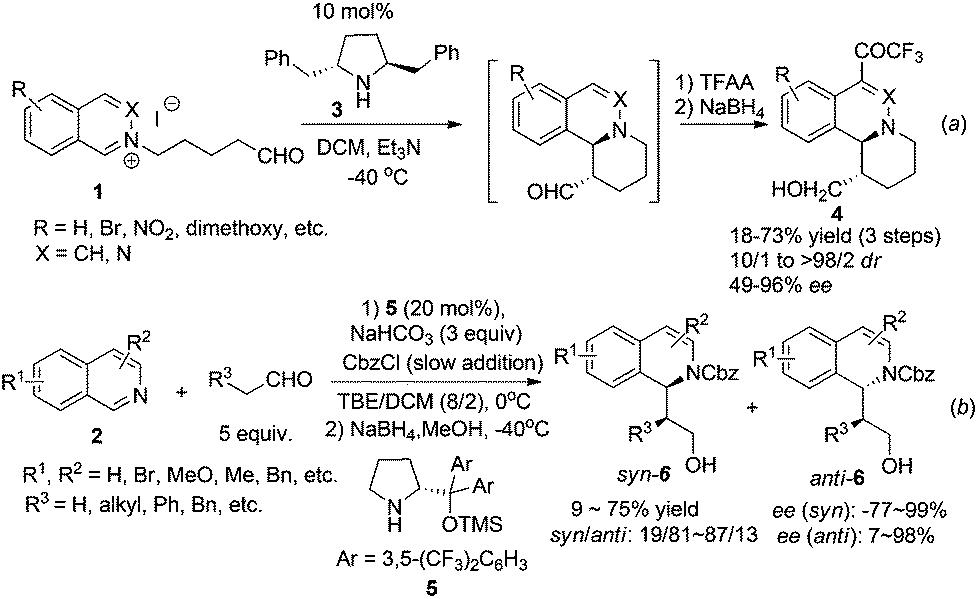
The majority of the synthetic routes for 1,2,3,4-tetrahydroisoquinolines and 3,4-dihydroisoquinolines involve asymmetric addition of nucleophiles to the readily available isoquinoline, 3,4-dihydroisoquinoline (DHIQ) or N–H tetrahydroisoquinoline (THIQ) core. This C1–Cα connection represents a straightforward approach for the synthesis of C1-chiral THIQs. Isoquinoline usually needs activation by an external alkylating or acylating reagent to form iminium species. Since the first catalytic enantioselective Reissert reaction reported by Shibasaki and coworkers in 2001,8 catalytic addition of preformed or in situ generated nucleophiles to activated iminium species has received much attention. Jørgensen et al. reported in 2005 a chiral pyrrolidine derivative catalyzed intramolecular addition to isoquinolinium ions 1 (Scheme 2a).9 High enantioselectivities and good diastereoselectivities were obtained. Mechanistic studies suggested that the reaction went through a transition state involving a crucial cation–π interaction between iminium ions and the phenyl ring in catalyst 3. The intermolecular reaction was not realized until very recently by Cozzi and coworkers starting from isoquinoline with CbzCl as the activating reagent (Scheme 2b).10 The products 6 were obtained in moderate drs and good ees, but yields were moderate. Switching the solvent to pure dichloromethane afforded anti diastereoisomers selectively in good ees. The methodology was successfully applied to the synthesis of 13-methyltetrahydroprotoberberine in seven total steps.
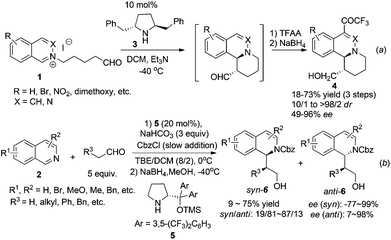 |
| | Scheme 2 Secondary amine catalyzed intra- and intermolecular asymmetric addition of aldehydes to isoquinoline. | |
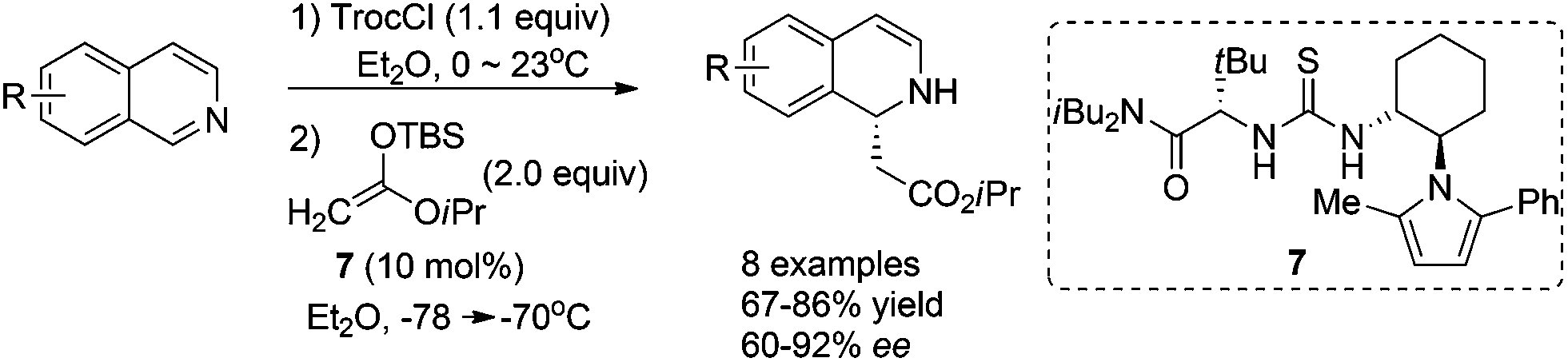
Catalyst activation of isoquinoline represents an alternative activation mode which obviates the necessity to asymmetrically activate nucleophiles. Jacobsen et al. reported in 2005 the intermolecular addition of preformed silyl ketene acetal to in situ generated N-acylisoquinolines catalyzed by chiral thiourea derivative 7, inspired by their discovery of the capability of these thiourea catalysts to activate the iminium ions in the asymmetric acylating Pictet-Spengler reaction. The reaction tolerates electronically distinct substituents on both rings and affords the corresponding DHIQs with high enantioselectivities (Scheme 3).11
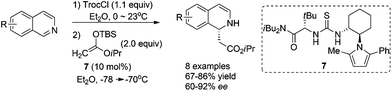 |
| | Scheme 3 Thiourea catalyzed asymmetric addition of silyl ketene acetal to isoquinoline. | |
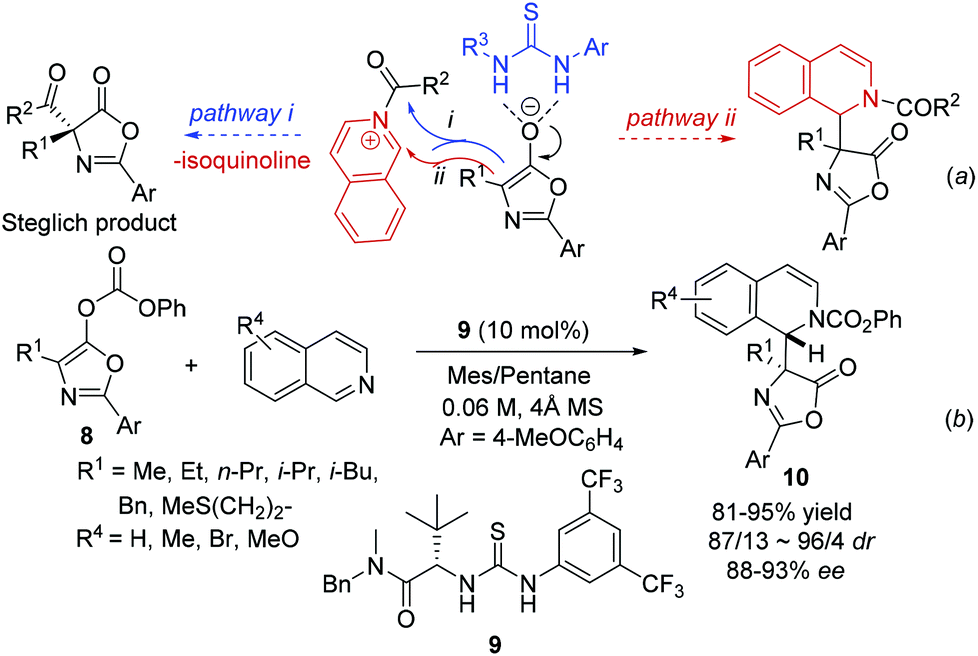
By combining an achiral nucleophilic catalyst and a chiral anion binding catalyst, Seidel and coworkers developed a novel strategy for the catalytic asymmetric Steglich rearrangement.12 When isoquinoline was used instead of DMAP, the catalyst bound anion intermediate attacks the resulting acylated isoquinoline intermediate via two possible routes leading to product formation: (i) normal attack on the carbonyl group releasing isoquinoline and the Steglich rearrangement product; and (ii) attack on the iminium carbon atom generating the THIQ product with acyl being transferred to the nitrogen in the isoquinoline (Scheme 4a). Incorporation of the isoquinoline into the final product was observed at an elevated temperature with thiourea 9via pathway ii, which delivered the products 10 in excellent yields and enantioselectivity despite the significantly lower nucleophilicity of isoquinoline than that of DMAP. Interestingly, the facial selectivity for the azlactone addition to the acylisoquinoline was opposite to that of azlactone rearrangement (Scheme 4b).
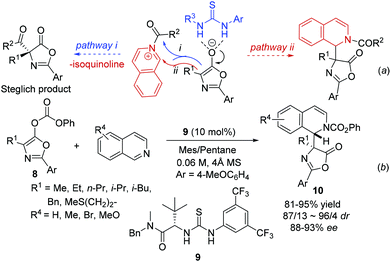 |
| | Scheme 4 Synthesis of chiral DHIQ via chiral anion binding catalysis. | |
The reactivity of 3,4-dihydroisoquinolines is quite similar to that of isoquinoline, except that the former might or might not require activation. Copper catalyzed asymmetric allylation of cyclic C,N-azomethine imine afforded C1-allylated THIQ with moderate enantioselectivities without any activating reagent. After chemical resolution the optically pure product 12 was applied to the formal total synthesis of the natural product (−)-emetine (Scheme 5a).13 Sodeoka et al. disclosed an asymmetric addition of malonate to Boc2O activated DHIQs catalyzed by chiral palladium(II) complexes (Scheme 5b).14 This reaction was applied to the facile synthesis of (R)-calycotomine in seven steps, which also defined the absolute configuration of the products 14.
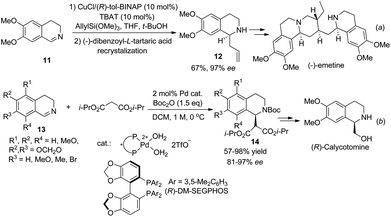 |
| | Scheme 5 Transition metal catalyzed asymmetric addition to 3,4-dihydroisoquinolines. | |
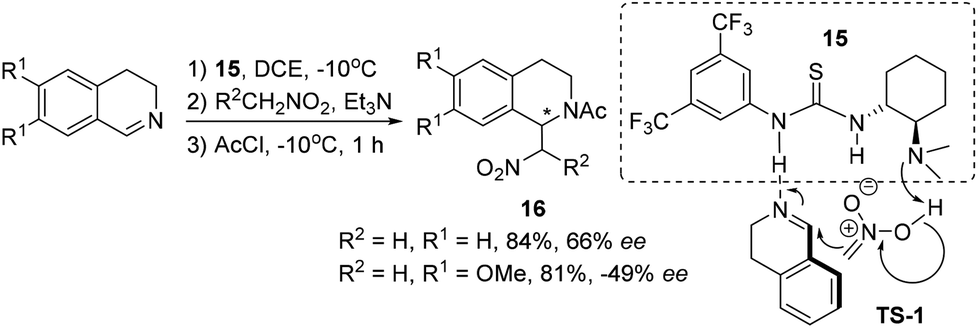
Direct catalyst activation of 3,4-DHIQ was indicated in the asymmetric aza-Henry reaction with nitroalkanes, where thiourea derivative 15 was employed as a bifunctional catalyst. However, only moderate ees were obtained for the products 16 (Scheme 6).15 This reaction works only for C1-unsubstituted cyclic imines, and the asymmetric induction is sensitive to substitutions on the DHIQ (Scheme 6, the absolute configuration not shown). Based on the information obtained from catalyst screening and mechanistic studies, a transition state (TS-1) involving a hydrogen-bonding network as depicted in Scheme 6 was proposed.
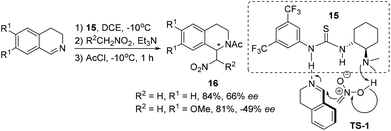 |
| | Scheme 6 Thiourea as a bifunctional organocatalyst in the aza-Henry reaction of 3,4-DHIQ with nitroalkanes. | |
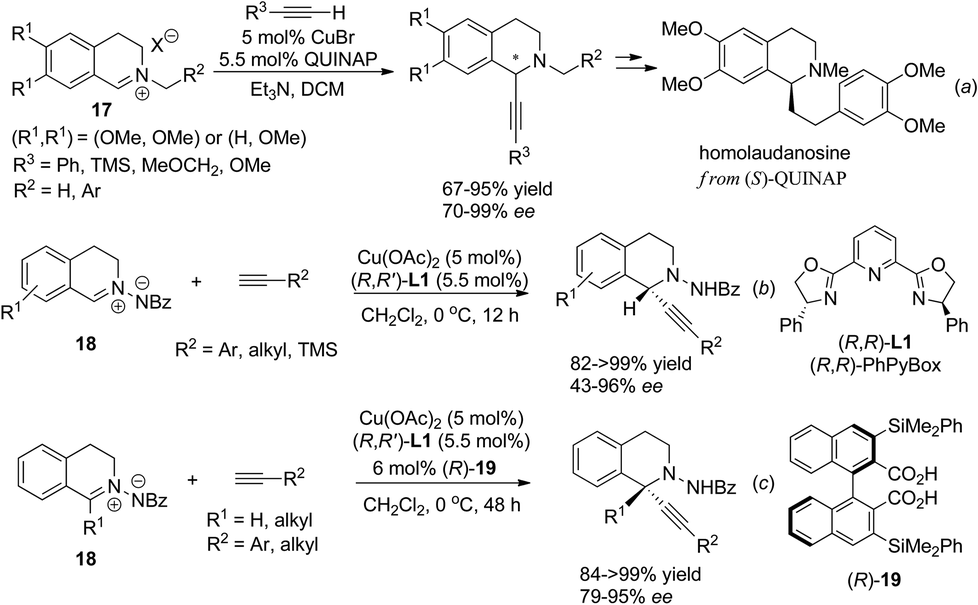
Isoquinoline iminium salts undergo facile asymmetric addition reactions with various nucleophiles. Copper catalyzed asymmetric addition of terminal alkynes was realized independently by Schreiber et al. and Li et al. in 2006, who found that under the catalysis of the CuBr/Quinap complex, a variety of alkynes reacted with alkylated iminium salt 17 with high enantioselectivity.16 This reaction was successfully applied to the concise four-step asymmetric synthesis of homolaudanosine by Schreiber and coworkers (Scheme 7a). However, it showed no optical selectivity for C1-substituted substrates. The Cu(OAc)2/(R,R)-L1 combination was shown to be viable in a similar catalytic asymmetric alkynylation of C1-unsubstituted C,N-cyclic azomethine imines 18 (Scheme 7b).17 For C1-substituted substrates, products were retrieved with moderate enantioselectivities. This problem was overcome by addition of a C2-symmetrical Brønsted acid as a co-catalyst. The 3,3′-substituents on the acid turned out to be crucial; the bulky group substituted bisacid (R)-19 led to the best enantioselectivity (Scheme 7c). The high dependence of the enantioselectivity on steric factors of the cocatalyst suggested the possible role of the acid co-catalyst being to exchange with HOAc in the activation of C,N-cyclic azomethine imines, which acted cooperatively with a copper catalyst to enhance the enantioselectivity. Besides isoquinoline iminium salts, asymmetric addition of 3,4-dihydroisoquinoline N-oxides with organozinc species was reported to give C1-aryl and alkenyl THIQs in high enantioselectivity when mediated by amino acid derived amino alcohol reagents, but reactions with catalytic amounts of chiral amino alcohol reagents resulted in moderate enantioselectivity due to competitive non-catalyzed background reactions.18,19
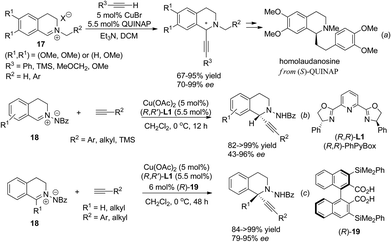 |
| | Scheme 7 Copper catalyzed asymmetric alkynylation of isoquinoline iminium salts. | |
2.2 Asymmetric cross-dehydrogenative coupling (CDC) reactions20 and merging organo-/metal catalysis and photoredox catalysis
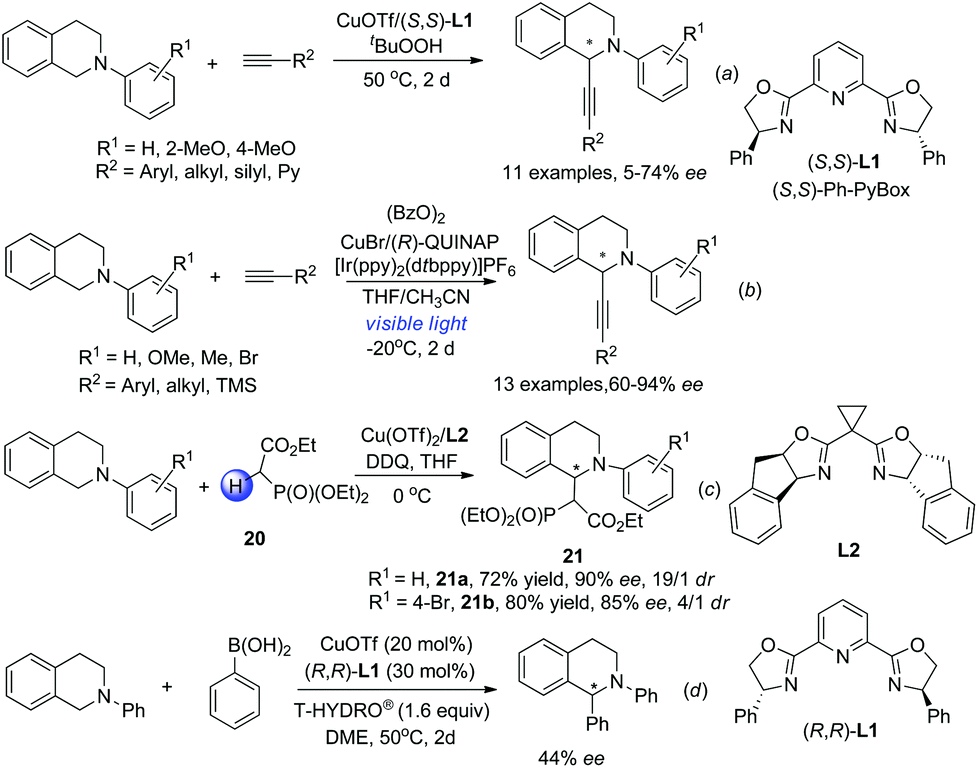
The discovery of facile oxidation of N-aryl THIQ with various oxidants opened up new avenues for the synthesis of C1-substituted THIQs. These reactions were termed “cross-dehydrogenative coupling (CDC) reactions” by Li, who, along with Murahashi, pioneered this field. The combination of N-aryl THIQ oxidation with enantioselective addition of an external nucleophile affords C1-chiral THIQs with high efficiency. The copper/bisoxazoline catalytic system has proven successful in a series of related reactions in the activation of alkynes, dicarbonyl compounds, arylboronic acids, etc. For instance, Li and coworkers discovered that in the presence of t-BuO2H, the reaction of N-aryl THIQ with different aryl substituted alkynes afforded the CDC products in moderate to good ees, while 2-pyridyl, silyl and alkyl substituted products were obtained with moderate to low ees (Scheme 8a).21 The CuOTf/QUINAP combination showed comparable selectivities in a later study with TBHP as an oxidant.16b The requirement of 50 °C or higher temperature possibly impeded the development of a highly enantioselective process. A highly enantioselective variant of this important transformation has been eventually realized very lately by Li and coworkers by merging photoredox catalysis with the CuOTf/QUINAP catalyst. This synergistic catalytic process allowed the reaction to be carried out at −20 °C under low catalyst loading, which dramatically improved the enantioselectivity versus the conventional oxidation conditions (Scheme 8b).22 The Horner–Warsworth–Emmons (HWE) reagent 20 could be activated by a Cu(OTf)2/L2 complex and react with the oxidatively generated THIQ iminium ion to deliver the CDC products 21a with high enantio- and diastereoselectivities, as reported by Wang and coworkers in 2011 (Scheme 8c).23 A related reaction with aryl boronic acids was studied by Li and coworkers in 2008. The asymmetric version of such transformation was preliminarily studied in this work with (R,R)-L1, but moderate enantioselectivity was observed (Scheme 8d).24
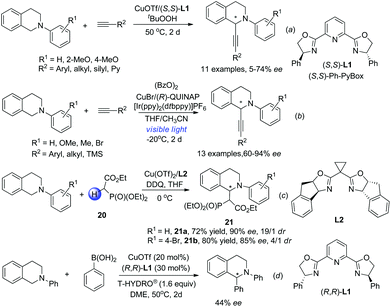 |
| | Scheme 8 Transition metal involved CDC reactions of N-aryl THIQ. | |
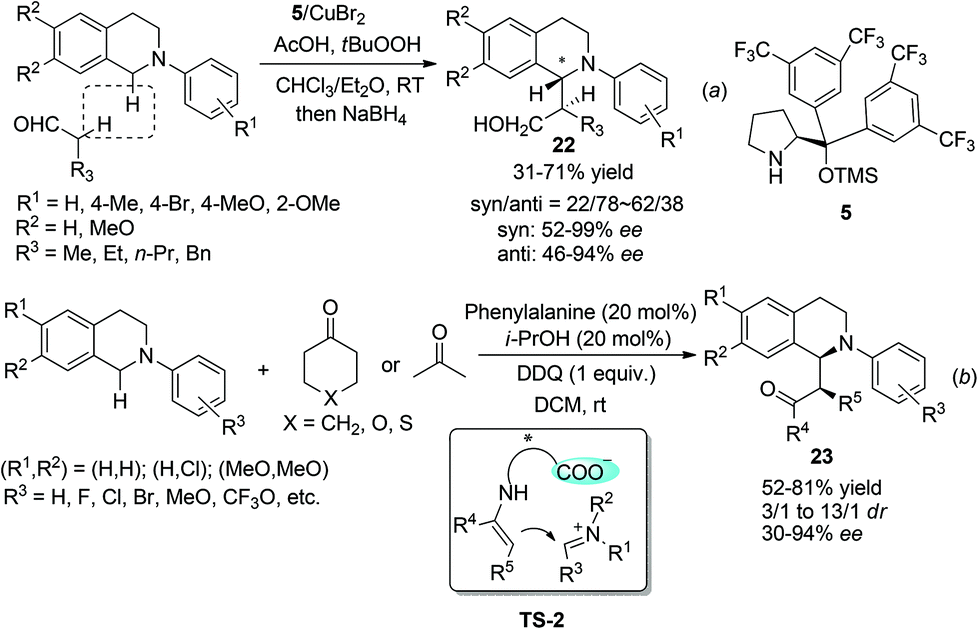
Under the catalysis of Hayashi–Jørgensen catalyst 5, aliphatic aldehydes add to the in situ generated THIQ iminium ion intermediates in an enantioselective fashion (Scheme 9a).25 The selectivity outcome turned out to be very sensitive to the reaction conditions. Under the optimized conditions the products 22 were obtained with only moderate drs but good to excellent ees. A related work with ketones by Wang and coworkers employed phenylalanine as the catalyst to form the enamine intermediate with ketones, while its pendent carboxylic acid moiety forms a chiral ion pair with the iminium ion intermediate (TS-2, Scheme 9b).26 This bifunctional catalyst brings both reacting intermediates in close proximity and is expected to improve the selectivity. Indeed, the metal free reaction between cyclic ketones and N-aryl THIQ afforded the CDC products 23 with good dr and ees, while acetone, the only reported acyclic ketone, afforded the corresponding product with only 30% ee.
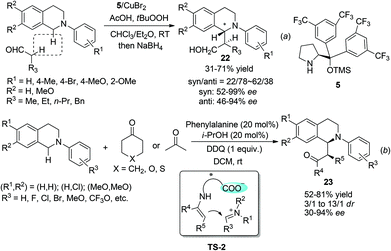 |
| | Scheme 9 Chiral amine catalyzed CDC reaction of N-aryl THIQ. | |
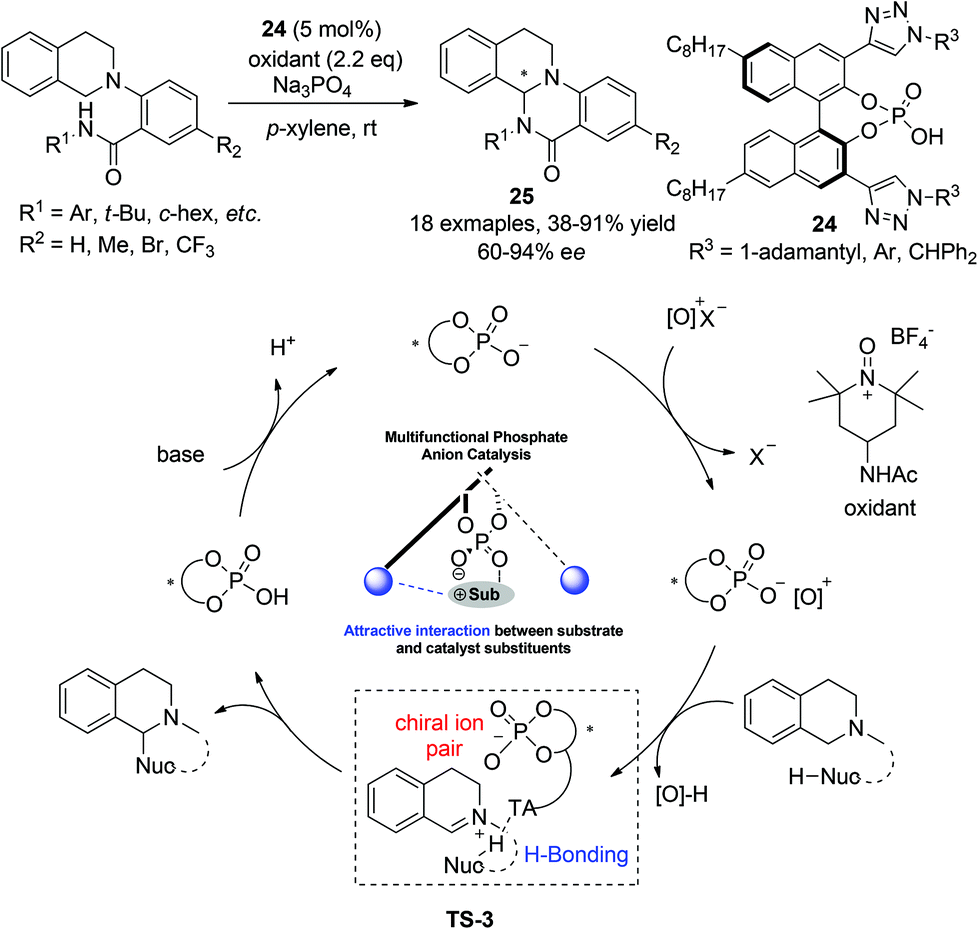
Almost at the same time, Toste and coworkers realized an enantioselective intramolecular CDC amidation reaction with multifunctional chiral phosphoric acid catalysts, featuring activation of both reaction components.27 Realization of high enantioselectivity hinges on the design of a 3,3′-(1,2,3-triazole)-containing phosphoric acid catalyst 24. The chiral phosphoric acid catalyst was proposed to form ion pairs with the cationic oxidant and remained attached to the resulting iminium intermediate after its formation. The triazole moiety in the catalysts was proposed to interact with the amide in the nucleophile, thus rendering a more rigid transition state (TS-3, Scheme 10) for asymmetric induction. Attack on the iminium intermediate by the activated amide nitrogen implements the annulation furnishing enantioenriched tricyclic THIQ products 25 (Scheme 10). This bio-inspired transition state-lowering strategy with hydrogen-bonding interactions was shown to be advantageous compared to the traditional one which relies purely on steric factors at the 3,3′-position of the phosphoric acid catalyst.
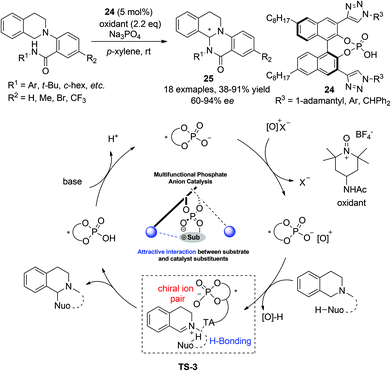 |
| | Scheme 10 Chiral phosphoric acid catalyzed intramolecular CDC-amidation of N-aryl THIQ. | |
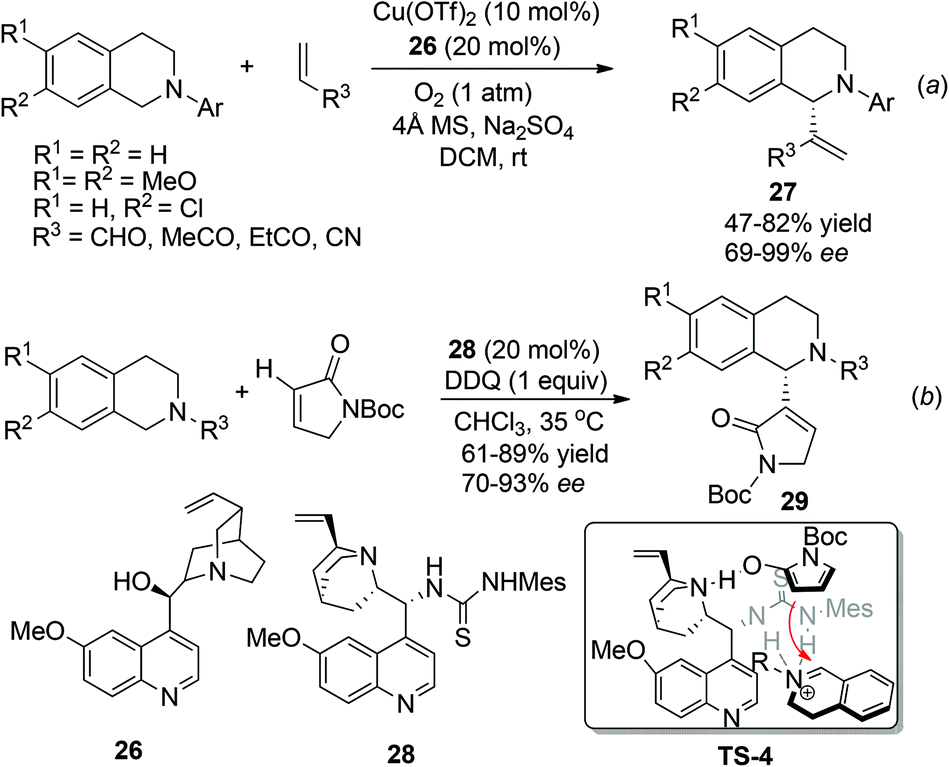
In contrast to the great success being made in the asymmetric construction of the Csp3–C1 juncture as mentioned above, examples of nucleophiles bearing sp2 hybridized carbon centers are less explored. The combined CDC-Morita–Baylis–Hillman (MBH) reaction was first reported by Li and coworkers as early as 2006,28 but its asymmetric reaction was not realized until 2012, when Wang and coworkers reported the enantioselective aerobic oxidative coupling between THIQ and electron-deficient alkenes (Scheme 11a).29 This reaction combines copper catalyzed aerobic oxidation of THIQ with asymmetric nucleophilic catalysis to produce 27 with high enantioselectivity. The reaction seemed to proceed via the classical MBH mechanism, since hydroxy tertiary amines and tertiary phosphines were also able to induce moderate enantioselectivity. Very lately, Wang and coworkers have reported a related reaction with α,β-unsaturated γ-butyrolactam catalyzed by the chiral quinine derived bifunctional thiourea catalyst 28. The reaction afforded a series of C1-heterocyclic substituted THIQs 29, featuring α-position functionalization of α,β-unsaturated γ-butyrolactam. A putative transition state TS-4 involving an activated enamine generated in situ from α,β-unsaturated γ-butyrolactam was provided to explain this unique regioselectivity (Scheme 11b).30
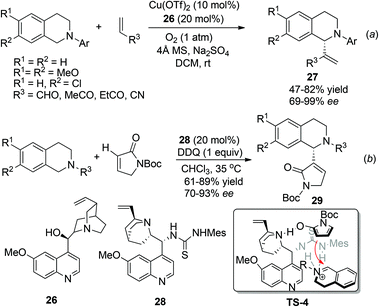 |
| | Scheme 11 Catalytic CDC-Morita–Baylis–Hillman reaction. | |
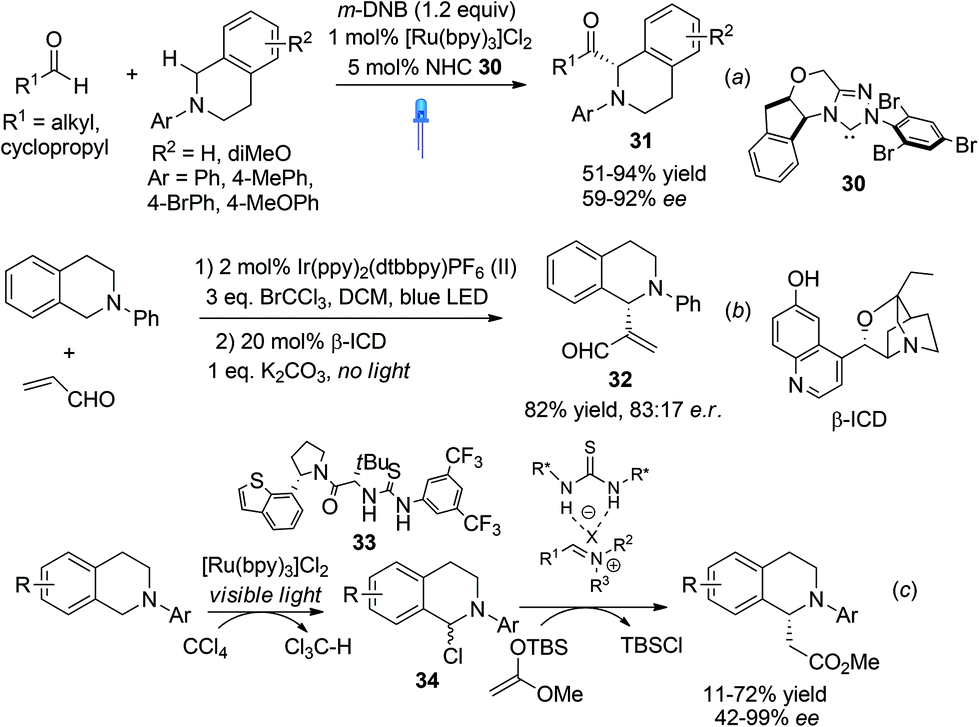
Photooxidation of N-aryl THIQ represents a greener and often more ambient alternative than traditional oxidation processes, as exemplified by the successful development of highly enantioselective asymmetric C1-alkynylation of THIQ by merging photoredox catalysis with asymmetric copper catalysis (Scheme 8b). The enantioselective functionalization of N-aryl THIQ by sequential photoredox-metal/organocatalysis achieved low enantioselectivity as observed by Tan and coworkers in the Mannich reaction of acetone with N-PMP substituted THIQ, wherein Rose Bengal, an organic dye, was used as the photocatalyst.31 Shortly after, Rovis et al. merged photoredox and NHC catalysis to realize the catalytic asymmetric α-acylation of THIQs (Scheme 12a).32 The in situ generated acyl anion via N-heterocyclic carbene (NHC) umpolung catalysis reacted with the photoredox generated iminium ion to form C1-acylated products 31 in high enantioselectivities. Notably, low catalyst loadings were required in this process.
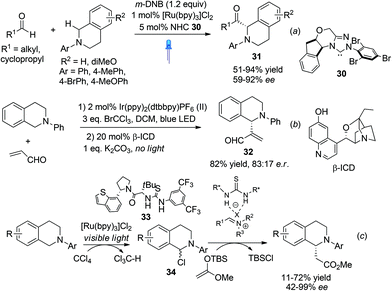 |
| | Scheme 12 CDC processes involving visible photoredox catalysis for synthesis of THIQs. | |
Xiao et al. recently described one-pot sequential photocatalysis/nucleophilic catalysis for the construction of C1-functionalized THIQs.33 Using O2 as the oxidant the reaction between N-phenyl THIQ and acrolein resulted in formation of oxidized isoquinoline. Instead, with BrCCl3 as the oxidant the desired product was obtained in moderate yield. Catalytic asymmetric reaction using β-isocupreidine (β-ICD) as the nucleophilic catalyst and an iridium complex as the photoredox catalyst retrieved product 32 with moderate 66% ee (Scheme 12b). This is reminiscent of Wang's work in 2011 which employed Cu/O2 oxidation, in which high enantioselectivities were achieved (Scheme 11a). Noticing the usually concomitant reductive generation of halide anions during light photocatalysis, Stephenson and coworkers recently combined photocatalysis with chiral anion binding catalysis to realize oxidative enantioselective C–H functionalization of THIQs with silyl ketene acetals (Scheme 12c).34 The thiourea 33 was proposed to activate the resulting intermediate α-haloamine 34 by binding to the halide ion and the resulting counterion was proposed to induce enantioselectivity. Similarly, this two-step sequential process is reminiscent of Jacobsen's chiral thiourea catalyzed addition of silyl ketene acetals to isoquinolines, which also achieved good enantioselectivity (Scheme 3). This process, however, does not require bulkier isopropyl esters to attain good enantioselectivity, albeit the yields are moderate.
2.3 1,3-Dipolar cycloaddition reactions
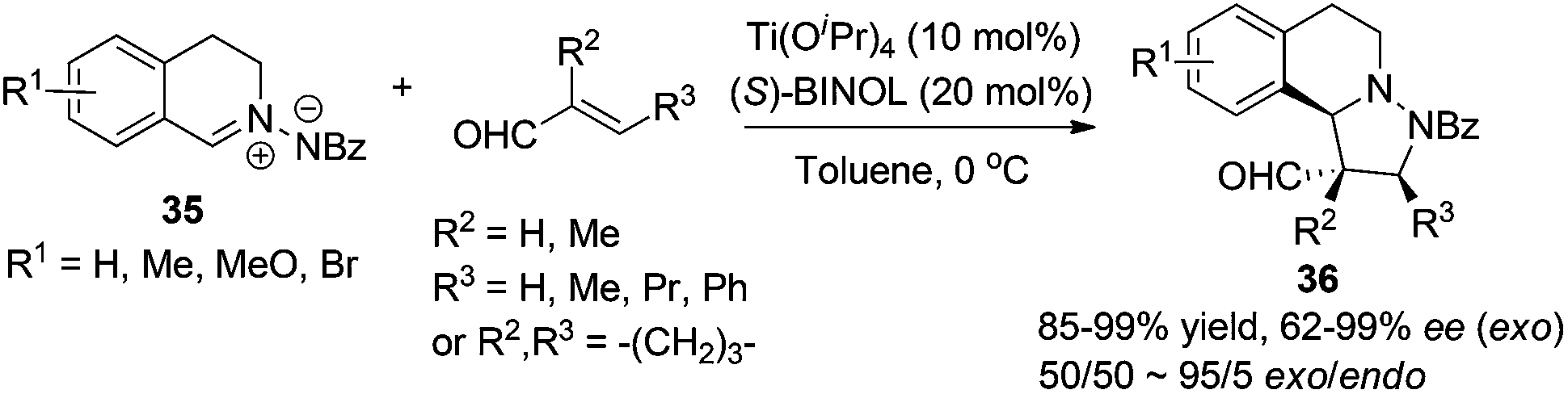
1,3-Dipolar cycloaddition reactions of C,N-cyclic azomethine imine or its analogues create ring fused THIQs bearing multiple stereogenic centers. The titanium-binolate complex catalyzed asymmetric dipolar cycloaddition of C,N-cyclic azomethine imine 35 with α,β-unsaturated aldehydes developed by Maruoka and coworkers in 2010 generates chiral THIQs 36 with the 1,3-diamine moiety (Scheme 13).35 Excellent exo/endo ratios and ees were achieved when 2/1 ratio of BINOL and Ti(OiPr)4 was employed. Substitution at the β-position (R3) on the aldehyde was crucial for the high exo selectivity, while good ees were observed in most cases with or without α- or β-substitutions.
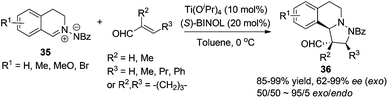 |
| | Scheme 13 Titanium Lewis acid catalyzed asymmetric 1,3-dipolar cycloaddition of C,N-cyclic methine imines with α,β-unsaturated aldehydes. | |
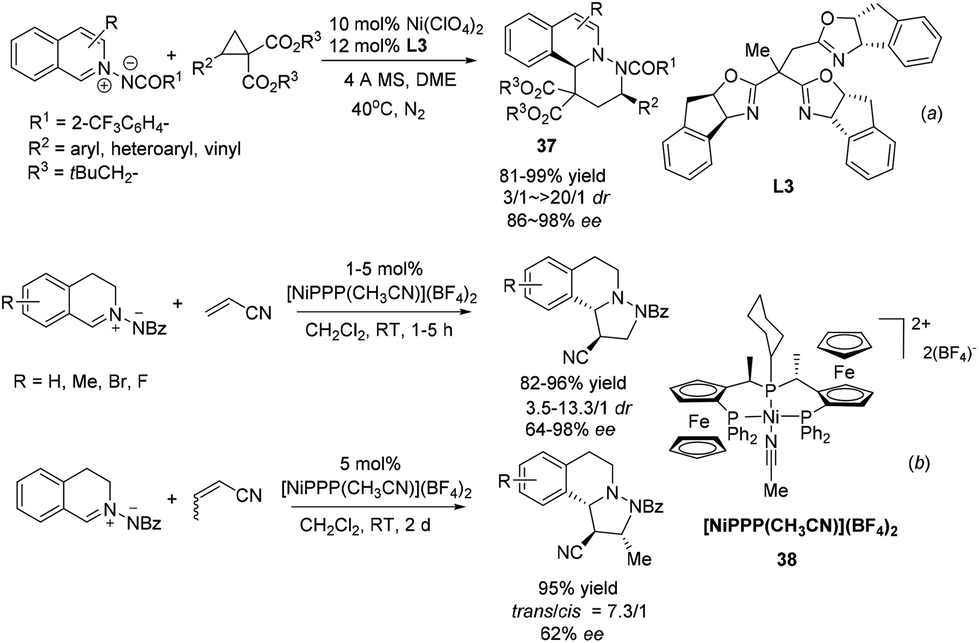
Since Maruoka's pioneering work, the 1,3-DC reaction of C,N-cyclic methine imines has received intense attention. Tang and coworkers realized a NiII/trioxazoline complex catalyzed highly enantioselective [3 + 3] cycloaddition of azomethine imines with an activated cyclopropane (Scheme 14a).36 The reaction is tolerant of a variety of different substitutions on the cyclopropane and delivers a variety of six membered heterocycle fused DHIQs 37. The reaction with R2 being H was high yielding but completely racemic. DFT calculation indicated the existence of π–π stacking interactions between the indane side arm in the ligand L3 and the phenyl group in the cyclopropane substrate, which is corroborated with experimental evidence. Another nickel based catalyst Ni[(R,Sp)-PigiPhos][BF4]238 was recently applied by Togni to the normal electron demand dipolar cycloaddition between C,N-cyclic methine imines and unsaturated nitriles in 2013 (Scheme 14b).37 The reaction with acrylonitrile afforded products with high diastereoselectivities and essentially the same enantioselectivities with 1 mol% catalyst loading within 5 hours. The reaction of crotonitrile turned out to be much slower and went to completion in 2 days to afford the product at moderate 62% ee. Mechanistic studies indicated that besides the unquestionable coordination of nitriles to the catalyst, the interaction of azomethine imines with the catalyst was also non-negligible.
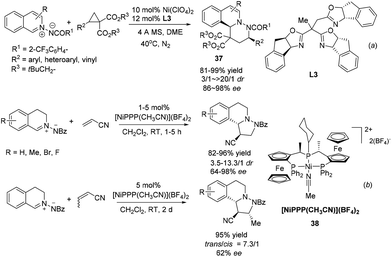 |
| | Scheme 14 Nickel Lewis acid catalyzed 1,3-dipolar cycloaddition of C,N-cyclic methine imines. | |
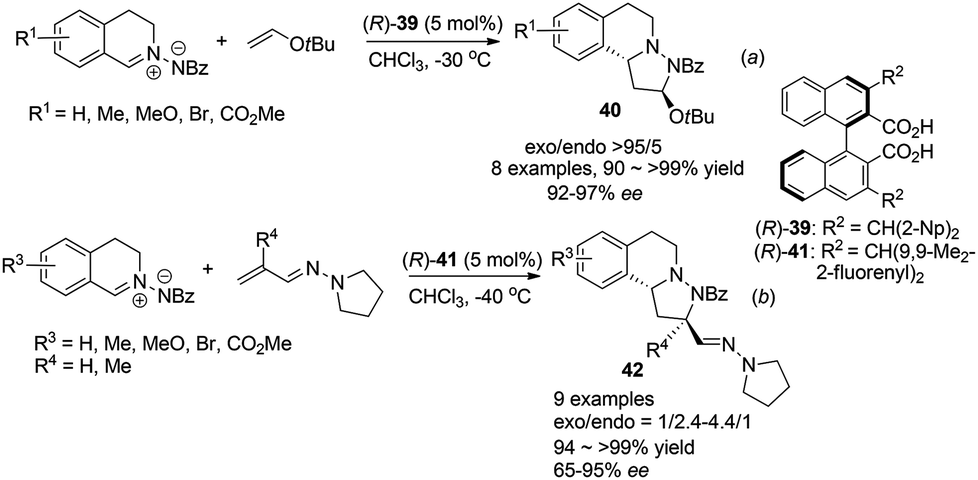
Maruoka et al. also realized inverse electron demand (IED) dipolar cycloaddition reaction of C,N-cyclic azomethine imines with electron rich t-butyl vinyl ether and vinylogous aza-enamines by activation of the 1,3-dipole, following their observation of facile protonation of such C,N-cyclic azomethine imines.38 For the reaction with vinyl ethers, high exo-selectivities and enantioselectivities were achieved with the bisacid (R)-39 (Scheme 15a). After optimization, the reaction with acrolein derived vinylogous aza-enamines afforded the corresponding products 42 with moderate exo/endo selectivities but generally high ees under the catalysis of the bulkier catalyst (R)-41 (Scheme 15b). Notably, when R′ = Me instead of H, the endo isomer became the major product (exo/endo = 1/2.4) and lower enantioselectivity of the exo isomer (68% ee) was observed. These two types of remarkable cycloaddition reactions provide tricyclic THIQ compounds with three contingent stereogenic centers efficiently in one step.
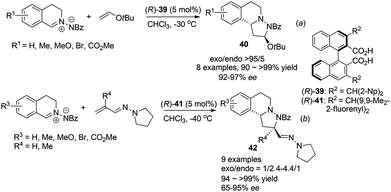 |
| | Scheme 15 Chiral Brønsted acid catalyzed asymmetric 1,3-dipolar cycloaddition reaction of C,N-cyclic methine imines. | |
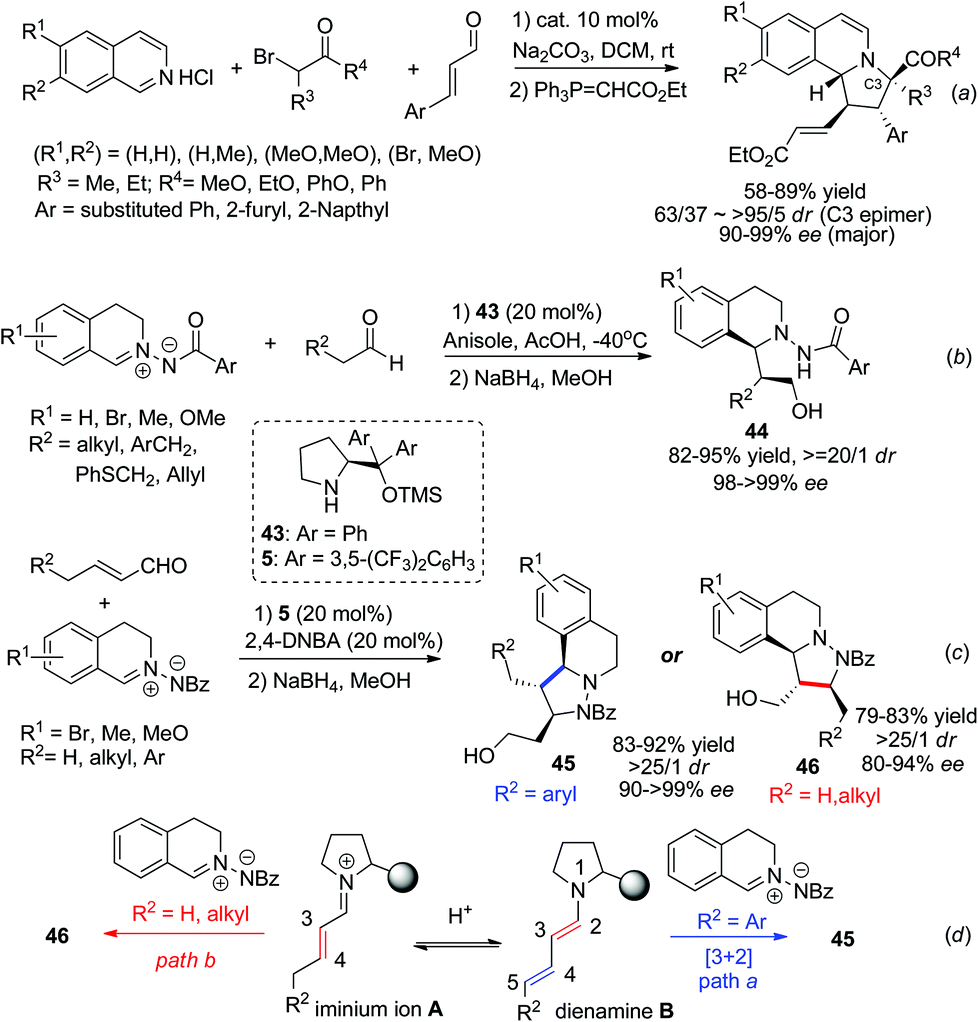
Proline derivatives have been well established in the asymmetric organocatalytic reaction of aldehydes/ketones with C,N-cyclic methine imines, as one of the strategies for nucleophile activation. Catalyst 5 was able to catalyze the three component dipolar cycloaddition featuring in situ generation of 1,3-dipoles from α-haloketones with isoquinoline salt, as reported by Jørgensen and coworkers in 2012 (Scheme 16a).39 Substituted phenyl, 2-furyl, 2-naphthyl substituted enals all reacted efficiently to afford the corresponding products with high ees. With the same strategy, Wang and coworkers reported in 2014 two examples realizing the IED cycloaddition of C,N-cyclic methine imines with enamine40 and dienamine41 (Scheme 16c) intermediates generated from aliphatic aldehydes and α,β-unsaturated aldehydes, respectively. The reaction of aldehydes proceeds via the formation of an enamine intermediate which undergoes inverse electron demand [3 + 2] cycloaddition (Scheme 16b). For the latter, the initially formed iminium intermediate A between the aldehyde and the proline silyl ether catalyst forms an equilibrium with the dienamine B. With R2 being aromatic substituents, cycloaddition occurs at the C4–C5 double bond and forms the final product 45via intermediate B. For substrates with R2 being hydrogen or alkyl groups the [3 + 2] cycloadducts occurred selectively at the C3–C4 double bond of the aldehyde, affording 46 in high enantio- and diastereoselectivities. In this case the iminium ion A was speculated to play a key role in triggering the normal electron demand cycloaddition, leading to the C3–C4 addition product via the LUMO-lowering mechanism (Scheme 16d).
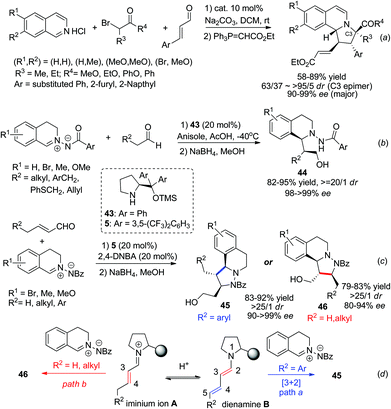 |
| | Scheme 16 Proline derivative activated HOMO-raising and LUMO-lowering strategies in the 1,3-DC of C,N-cyclic methine imines. | |
2.4
De novo construction of THIQ by asymmetric annulation
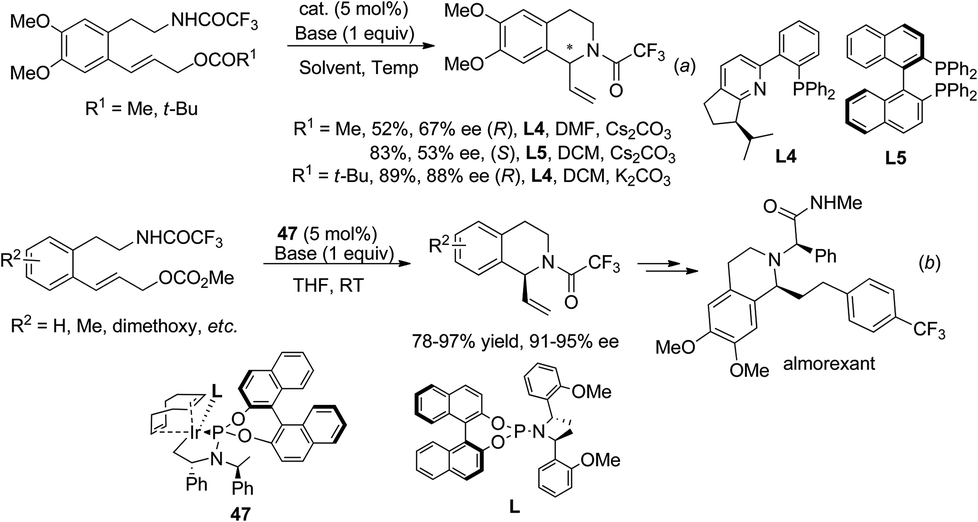
2.4.1 Asymmetric intramolecular allylic amination.
The strategy of de novo construction of THIQ via intramolecular asymmetric allylic amination was realized by Ito, Katsuki et al. in 2003 under palladium catalysis, which showed moderate to good enantioselectivities in the C1-vinylated products (Scheme 17a).42 Interestingly, the use of (R)-BINAP (L5) and (R)-Phox (L4) as ligands retrieved products with opposite configurations. A similar palladium catalyzed reaction by Ojima et al. in 2007 used monodentate phosphoramidite ligands and obtained comparable enantioselectivities.43 Feringa and coworkers developed the iridium catalyzed transformation in 2011 and achieved high enantioselectivity (Scheme 17b).44 Using this methodology the same group completed a straightforward four step total synthesis of the natural product almorexant.45
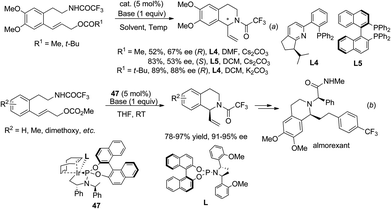 |
| | Scheme 17 Construction of C1-chiral THIQ via transition metal catalyzed asymmetric allylic amination. | |
2.4.2 Asymmetric intramolecular aza-Michael addition reaction.
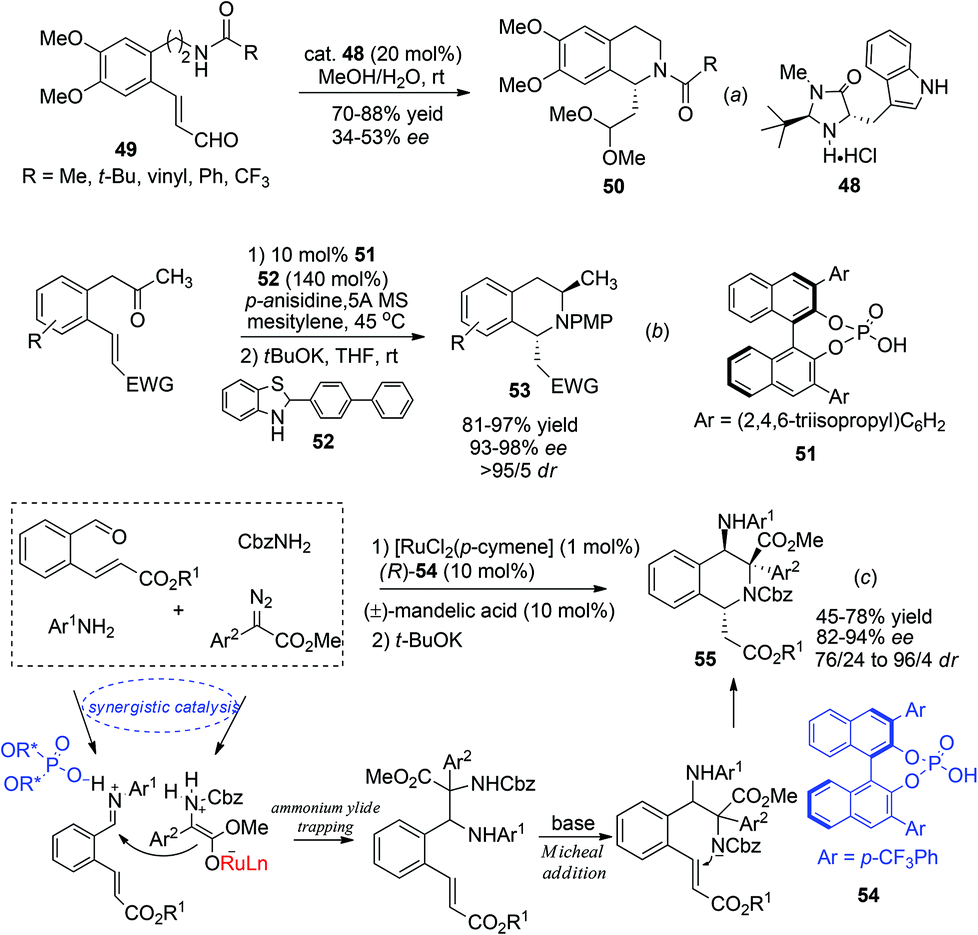
The asymmetric intramolecular aza-Michael addition reaction constitutes another important de novo synthetic approach toward construction of important nitrogen containing heterocycles, including C1-chiral THIQs. In 2003 Ihara et al. reported the use of the MacMillan catalyst 48 to trigger the aza-Michael reaction of substrate 49 affording the acetal product 50 in moderate enantioselectivities (Scheme 18a).46 This strategy, however, received more success in the synthesis of C3-chiral THIQs, wherein high enantioselectivities were achieved with the Jørgenson–Hayashi prolinol silyl ether catalyst in 2008.47
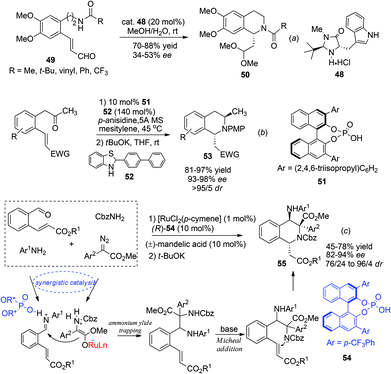 |
| | Scheme 18 Direct (a) and diastereoselectively controlled (b, c) aza-Michael addition reactions for asymmetric synthesis of C1-chiral THIQs. | |
Alternatively, by preinstallation of the C3 stereochemistry, the stereochemistry of C1 in the aza-Michael addition could be elegantly controlled in a diastereoselective manner. Enders and coworkers realized a chiral Brønsted acid catalyzed reductive amination/aza-Michael addition sequence for the synthesis of 1,3-disubstituted THIQs in 2010.48 The reductive amination afforded products with high conversion and selectivity using List's TRIP catalyst 51. Upon deprotonation, the 1,3-trans-substituted THIQ products 53 were obtained with high enantio- and diastereoselectivities (Scheme 18b). More recently, in a cascade four-component ylide trapping/aza-Michael addition reaction, 1,3,4-tetrasubstituted THIQs were produced with high enantio- and diastereoselectivities. The successful combination of ruthenium and chiral phosphoric acid catalysts 54 enabled the synergistic interaction of the ruthenium associated ammonium ylide with the chiral phosphoric acid activated iminium intermediate, creating the key C3–C4 bond with concomitant formation of two stereogenic centers.49 Subsequent treatment of the base deprotonates the aniline N–H bond and triggers a diastereoselective Michael addition to afford the final product 55 (Scheme 18c).
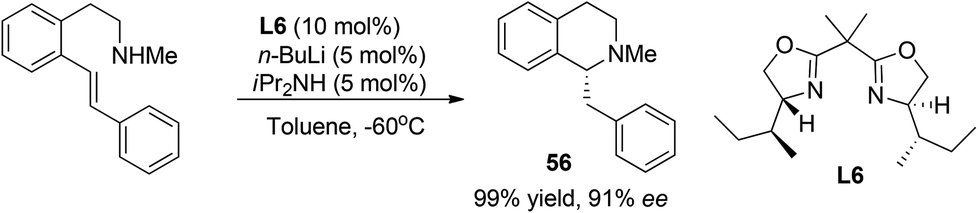
2.4.3 Asymmetric hydroamination.
Due to their high reactivity, asymmetric reactions involving organolithium reagents usually proceed with stoichiometric amounts of chiral reagents. Nevertheless, intramolecular asymmetric addition of lithium amide to styrene was realized by Tomioka et al. in a catalytic manner in 2007 (Scheme 19).50 Under optimal conditions this reaction affords N-methyl C1-benzylated THIQ 56 in high enantioselectivity with a substoichiometric amount of bisoxazoline ligand L6. However, no further substrate scope studies were performed.
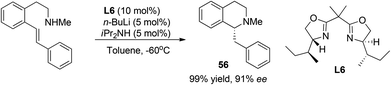 |
| | Scheme 19 Catalytic asymmetric hydroamination with an organolithium reagent. | |
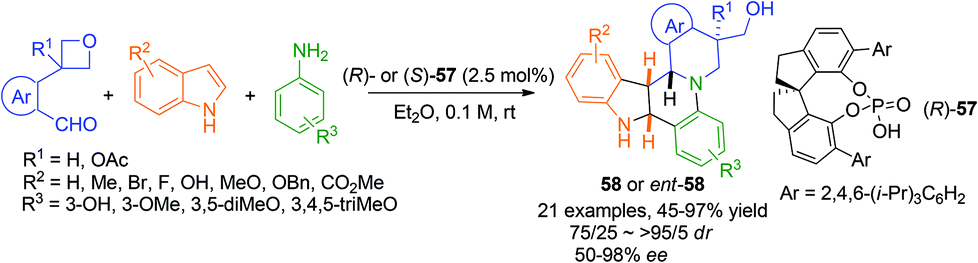
2.4.4 Asymmetric hetero Diels–Alder reaction.
A recent report by Sun and Zhu on the asymmetric three component aza-hetero Diels–Alder reaction between indoles and in situ generated N-aryl imines represents a very efficient construction of polycyclic compounds 58 (Scheme 20).51 In initial experiments aryl aldehydes without directing groups gave a mixture of various products. Intriguingly, with azetidine as the directing group the reaction not only preceded with high efficiency but delivered the azetidine ring opened product 58 with perfect enantio- and diastereoselectivity when spirochiral phosphoric acid (R)-57 was used as the catalyst. This remarkable transformation forms four bonds and four stereogenic centers in one step and the product skeleton is found in a few natural products and bioactive molecules. Selected compounds exhibited good antitumor effects against A549 and HeLa cell lines.
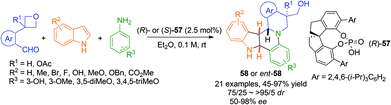 |
| | Scheme 20 Asymmetric synthesis of THIQ-containing polycycles via three component aza-hetero Diels–Alder reaction. | |
2.5 Miscellaneous reactions
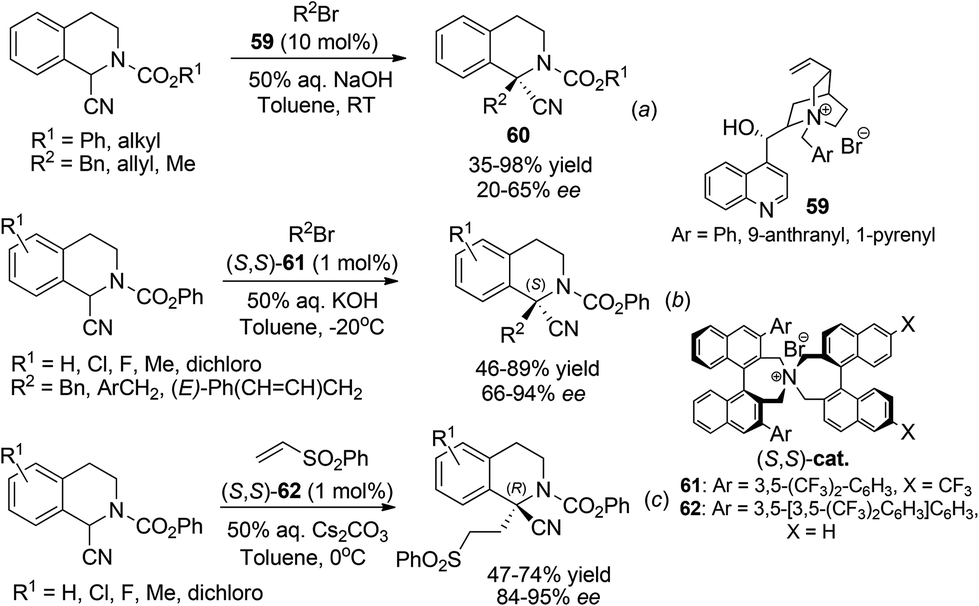
2.5.1 Direct alkylation.
Highly enantioselective catalytic processes for asymmetric synthesis of 1,1-disubstituted THIQs are much less documented compared to those for THIQs bearing tertiary C1 chiral centers. Direct asymmetric alkylation of C1-substituted THIQs provides a straightforward synthesis of these compounds. Rozwadowska et al. documented the first example of phase transfer catalytic alkylation of C1–CN substituted THIQ (the Ressert compounds) in 2006.52 Under the catalysis of chiral quaternary ammonium salts 59 derived from cinchona alkaloids, moderate enantioselectivity was achieved in the alkylation product 60 (Scheme 21a). In 2011 Maruoka and coworkers achieved high (S)-enantioselectivity in this reaction and an analogous Michael addition reaction using chiral quaternary ammonium salt catalysts (S,S)-61 (Schemes 21b and c).53 Notably, an analogous but bulkier ligand (S,S)-62 induced opposite (R)-configuration in the Michael addition products with high enantioselectivities.
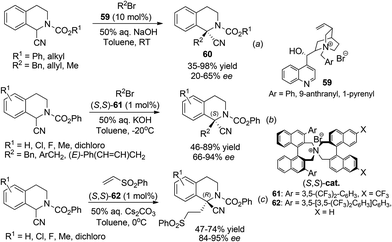 |
| | Scheme 21 Alkylation of Reissert compounds via asymmetric phase transfer catalysis. | |
2.5.2 Asymmetric destruction.
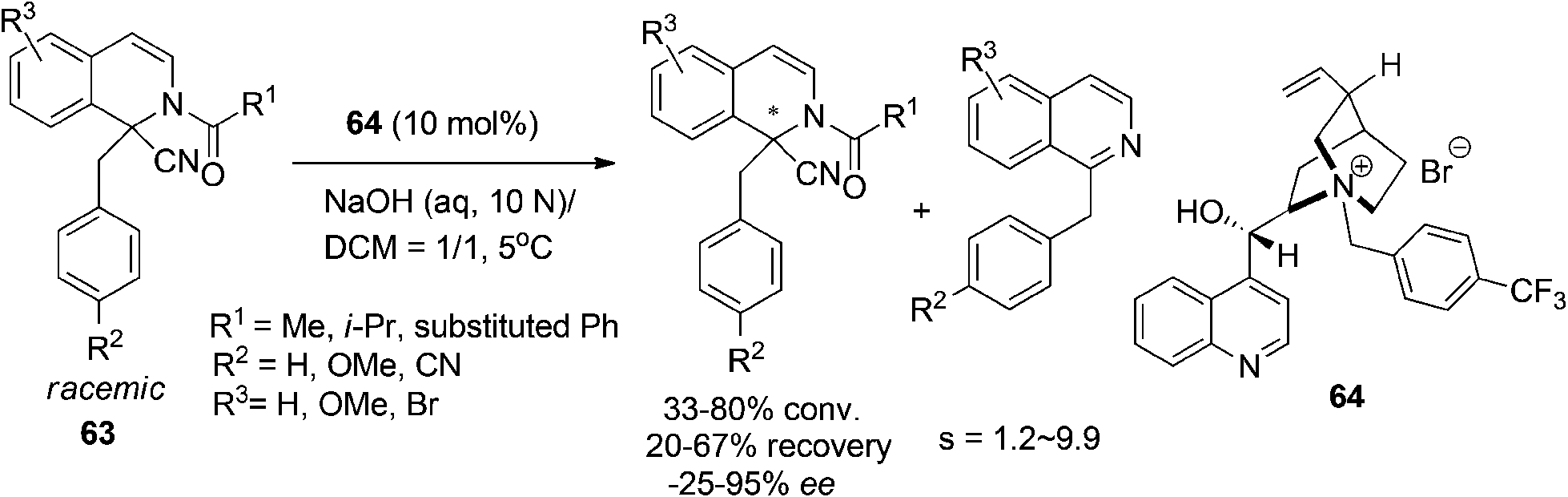
During their studies toward asymmetric alkylation of Reissert compounds, Jørgensen and coworkers observed the unexpected asymmetric decomposition of Reissert compounds into 1-substituted isoquinoline, which was named “asymmetric destruction” (Scheme 22).54 The reaction, a formal kinetic resolution of the hydrolysis of the starting 1,1-disubstituted THIQ 63, was catalyzed by chiral quaternary ammonium salt 64 as a phase-transfer catalyst and exhibited moderate enantioselectivities with selectivity factors ranging from 1.2 to 9.9.
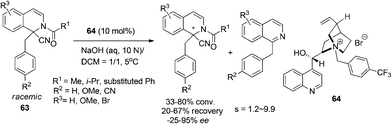 |
| | Scheme 22 Phase transfer catalyzed “asymmetric destruction” of 1,1-disubstituted DHIQ compounds. | |
2.5.3 Others.
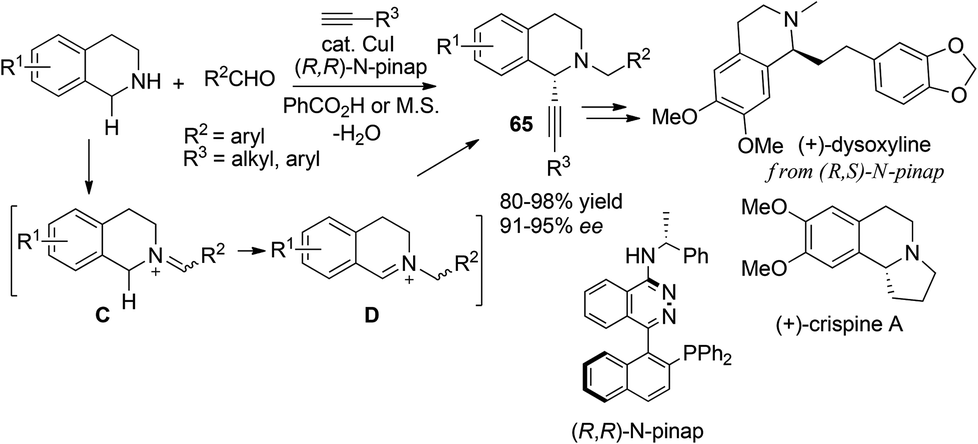
Ma55 and Yu56 independently disclosed C1-alkynylation of THIQs via three component reaction of N–H THIQ, aldehyde and alkynes (Scheme 23). The product 65 was originally isolated as a side product in the corresponding A3 reaction, according to Ma and coworkers. The addition of catalytic PPh3 along with a copper catalyst reversed the regioselectivity whereby 65 could be obtained as the major product, while Yu et al. found that this regioselectivity tuning could be realized by simply switching the catalyst from CuBr to CuI. With the CuI/(R,R)-N-pinap catalytic system Ma et al. were able to develop a catalytic asymmetric process which afforded optically enriched C1-alkynylated products with high enantioselectivities for both alkyl and aryl substituted alkynes. As a unique alternative strategy wherein the aldehyde serves as a N-alkylating reagent and iminium precursor, this methodology was utilized by Ma and coworkers in the concise synthesis of natural products (+)-dysoxyline and (+)-crispine A very recently.57
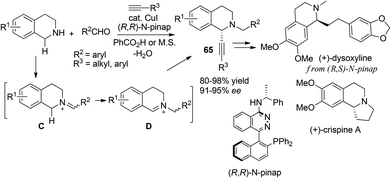 |
| | Scheme 23 Formation of N-alkylated iminium ions from N–H THIQ with aldehydes. | |
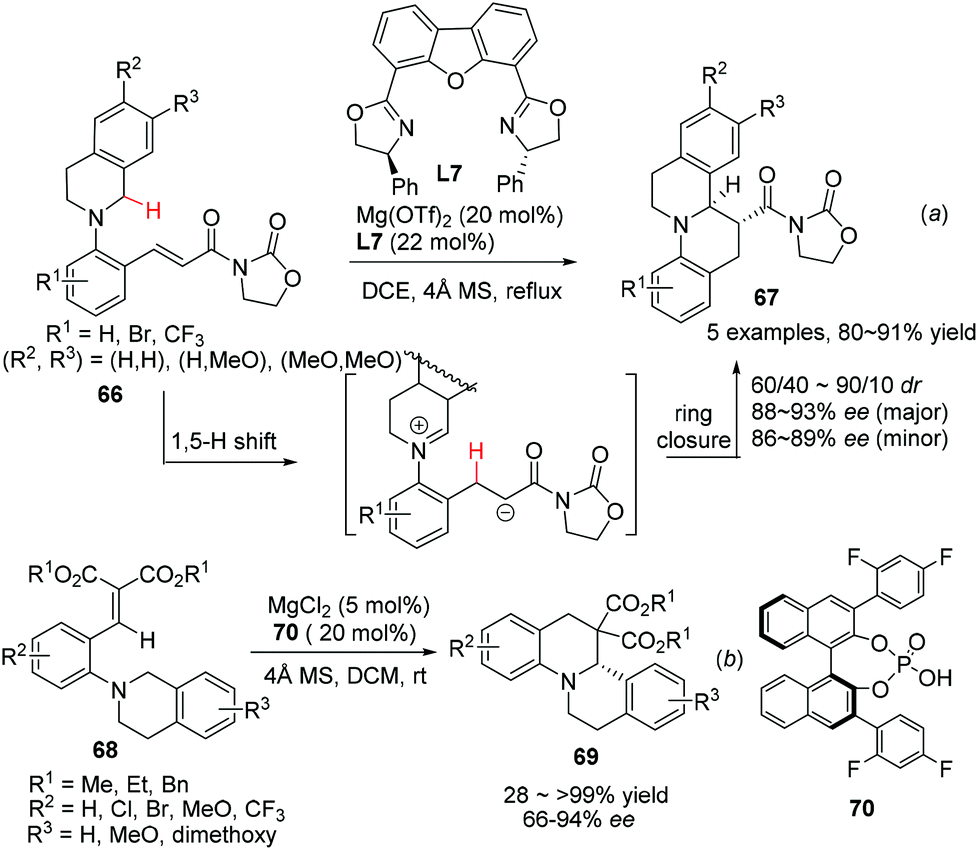
The redox neutrality feature of intramolecular redox reaction makes it an efficient greener alternative for the asymmetric construction of some nitrogen containing heterocycles.20 A catalytic enantioselective variant of this reaction was realized by Seidel and coworkers in 2009.58 The sequential 1,5-hydride transfer/Michael addition reaction of substrates 66 with a well-designed acyl oxazolidinone moiety as the chelating acceptor group afforded the annulation products 67 with high enantioselectivity and moderate diastereoselectivity (Scheme 24a). The reaction is tolerant of electron rich aromatic ring containing substrates on the THIQ moiety, but electron withdrawing substituents on the N-aryl moiety (R1) could be tolerated. A similar hydride transfer reaction was realized by Luo and coworkers with a “binary acid catalysis” strategy. One of the α-C–H bond of the tethered amine moiety in 68 undergoes a 1,5-hydride shift and the annulation follows to deliver products 69 with high selectivities (Scheme 24b).59 Not surprisingly, dramatically compromised reactivity and enantioselectivity were observed when R2 was the methoxy group. DFT calculations indicated of a transition state involving coordination of phosphoric acid 70 to magnesium Lewis acid, creating a chiral environment favoring one of the two diastereotopic hydrogen atoms to migrate.
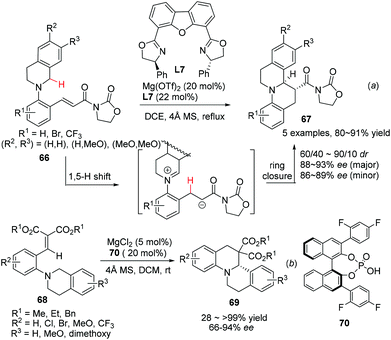 |
| | Scheme 24 Construction of C1-chiral THIQ via a Lewis acid promoted 1,5-hydride transfer/annulation process. | |
3. Conclusion and perspective
The last ten years have witnessed great advances in the catalytic asymmetric synthesis of C1-chiral THIQs in the context of continuing development of organocatalysis and transition-metal catalysis as well as the renaissance of photoredox catalysis. Novel asymmetric strategies were realized via distinct activation modes or annulation approaches. The rapidly developing non-stereoselective reactions also offer more platforms for the design of catalytic asymmetric variants. Despite these exciting advances, formidable challenges remain unsolved, among which are highly efficient construction of enantioenriched C1-aryl THIQs and 1,1-disubstituted THIQs, whose highly enantioselective synthetic methods remain scarce despite their prevalence in a good number of natural products. Moreover, examples to prepare enantioenriched THIQs with the electron-deficient aryl moiety or electron-deficient heterocycles at the core THIQ scaffold are far less documented, although such analogues were found to exhibit higher bioactivities than the parent structure in some cases.60 Nevertheless, the rapid development in this field continues to provide impetus for advancement in molecular medicine and asymmetric catalysis. We hope that this paper will attract attention from synthetic organic chemists toward overcoming the above mentioned challenges in the near future.
Acknowledgements
We gratefully acknowledge the financial support from the National Nature Science Foundation of China (21402025 and 21102016), the National Basic Research Program of China (973 Program, 2012CB720300), Northeast Normal University (for start-up funding to J.Z.), the Shanghai Rising-Star Program (14QA1400500), and the Shanghai Scientific and Technological Innovation Project (13520711500).
Notes and references
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
-
(a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed;
(b) M. Tang and G. Tang, Chin. J. Org. Chem., 2012, 32, 1568 CrossRef CAS.
-
(a) Y. Zhang, H. Fang, J. Feng, Y. Jia, X. Wang and W. Xu, J. Med. Chem., 2011, 54, 5532 CrossRef CAS PubMed;
(b) Y. Zhang, C. Liu, C. J. Chou, X. Wang, Y. Jia and W. Xu, Chem. Biol. Drug Des., 2013, 82, 125 CrossRef CAS PubMed;
(c) A. A. Orden, J. H. Schrittwieser, V. Resch, F. G. Mutti and W. Kroutil, Tetrahedron: Asymmetry, 2013, 24, 744 CrossRef CAS PubMed;
(d) V. M. Truax, H. Zhao, B. M. Katzman, A. R. Prosser, A. A. Alcaraz, M. T. Saindane, R. B. Howard, D. Culver, R. F. Arrendale, P. R. Gruddanti, T. J. Evers, M. G. Natchus, J. P. Snyder, D. C. Liotta and L. J. Wilson, ACS Med. Chem. Lett., 2013, 4, 1025 CrossRef CAS PubMed;
(e) K. Abe, K. Taguchi, T. Wasai, J. Ren, I. Utsunomiya, T. Shinohara, T. Miyatake and T. Sano, Brain Res. Bull., 2001, 56, 55 CrossRef CAS;
(f) J. N. Hanna, F. Ntie-Kang, M. Kaiser, R. Brun and S. M. N. Efange, RSC Adv., 2014, 4, 22856 RSC;
(g) J. Krauss, C. Müller, J. Kieβling, S. Richter, V. Staudacher and F. Bracher, Arch. Pharm. Chem. Life Sci., 2014, 347, 283 CrossRef CAS PubMed;
(h) F. Crestey, A. A. Jensen, M. Borch, J. T. Andreasen, J. Andersen, T. Balle and J. L. Kristensen, J. Med. Chem., 2013, 56, 9673 CrossRef CAS PubMed.
-
(a) S. K. Chakka, P. G. Andersson, G. E. M. Maguire, H. G. Kruger and T. Govender, Eur. J. Org. Chem., 2010, 972 CrossRef CAS;
(b) B. K. Peters, S. K. Chakka, T. Naicker, G. E. M. Maguire, H. G. Kruger, P. G. Andersson and T. Govender, Tetrahedron: Asymmetry, 2010, 21, 679 CrossRef CAS PubMed;
(c) T. Naicker, P. I. Arvidsson, H. G. Kruger, G. E. M. Maguire and T. Govender, Eur. J. Org. Chem., 2011, 6923 CrossRef CAS;
(d) R. B. Kawthekar, S. K. Chakka, V. Francis, P. G. Andersson, H. G. Kruger, G. E. M. Maguire and T. Govender, Tetrahedron: Asymmetry, 2010, 21, 846 CrossRef CAS PubMed;
(e) T. Naicker, K. Petzold, T. Singh, P. I. Arvidsson, H. G. Kruger, G. E. M. Maguire and T. Govender, Tetrahedron: Asymmetry, 2010, 21, 2859 CrossRef CAS PubMed.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538 CrossRef PubMed.
- D. Zhao and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 9616 CrossRef CAS PubMed.
-
(a) M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341 CrossRef CAS PubMed;
(b) E. Anakabe, D. Badia, L. Carillo and J. L. Vicario, Recent Res. Dev. Org. Chem., 2001, 5, 63 CAS;
(c)
A. L. Zein, G. Valluru and P. E. Georghiou, in Studies in Natural Products Chemistry, Elsevier B. V., Oxford, UK, 2012, vol. 38, pp. 53–80 Search PubMed.
- K. Funabashi, H. Ratni, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 10784 CrossRef CAS.
- K. Frisch, A. Landa, S. Saaby and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 6058 CrossRef CAS PubMed.
- L. Mengozzi, A. Gualandi and P. G. Cozzi, Chem. Sci., 2014, 5, 3915 RSC.
- M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700 CrossRef CAS PubMed.
- C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802 CrossRef CAS PubMed.
- T. Itoh, M. Miyazaki, H. Fukuoka, K. Nagata and A. Ohsawa, Org. Lett., 2006, 8, 1295 CrossRef CAS PubMed.
- N. Sasamoto, C. Dubs, Y. Hamashima and M. Sodeoka, J. Am. Chem. Soc., 2006, 128, 14010 CrossRef CAS PubMed.
- N. R. Amarasinghe, P. Turner and M. H. Todd, Adv. Synth. Catal., 2012, 354, 2954 CrossRef CAS.
-
(a) A. M. Taylor and S. L. Schreiber, Org. Lett., 2006, 8, 143 CrossRef CAS PubMed;
(b) Z. Li, P. D. Macleod and C. J. Li, Tetrahedron: Asymmetry, 2006, 17, 590 CrossRef CAS PubMed.
- T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 8952 CrossRef CAS PubMed.
- S. Wang and C. T. Seto, Org. Lett., 2006, 8, 3979 CrossRef CAS PubMed.
- S. Wang, M. B. Onaran and C. T. Seto, Org. Lett., 2010, 12, 2690 CrossRef CAS PubMed.
- Some of the references cited in this section are summarized in a recent review on another topic; see: Y. Qin, J. Lv and S. Luo, Tetrahedron Lett., 2014, 55, 551 CrossRef CAS PubMed.
- Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997 CrossRef CAS PubMed.
- I. Perepichka, S. Kundu, Z. Hearne and C.-J. Li, Org. Biomol. Chem., 2015, 13, 447 CAS.
- G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429 CrossRef CAS PubMed.
- O. Basléand and C.-J. Li, Org. Lett., 2008, 10, 3661 CrossRef PubMed.
- J. Zhang, B. Tiwari, C. Xing, X. Chen and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 3649 CrossRef CAS PubMed.
- G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645 RSC.
- A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044 CrossRef CAS PubMed.
- Z. Li, D. S. Bohle and C. J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS PubMed.
- G. Zhang, Y. Ma, S. Wang, Y. Zhang and R. Wang, J. Am. Chem. Soc., 2012, 134, 12334 CrossRef CAS PubMed.
- Y. Ma, G. Zhang, J. Zhang, D. Yang and R. Wang, Org. Lett., 2014, 16, 5358 CrossRef CAS PubMed.
- Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682 RSC.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed.
- Z.-J. Feng, J. Xuan, X.-D. Xia, W. Ding, W. Guo, J.-R. Chen, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Org. Biomol. Chem., 2014, 12, 2037 CAS.
- G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. Stephenson, Chem. Sci., 2014, 5, 112 RSC.
- T. Hashimoto, Y. Maeda, M. Omote, H. Nakatsu and K. Maruoka, J. Am. Chem. Soc., 2010, 132, 4076 CrossRef CAS PubMed.
- Y.-Y. Zhou, J. Li, L. Ling, S.-H. Liao, X.-L. Sun, Y.-X. Li, L.-J. Wang and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 1452 CrossRef CAS PubMed.
- S. Milosevic and A. Togni, J. Org. Chem., 2013, 78, 9638 CrossRef CAS PubMed.
- T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 3489 CrossRef CAS PubMed.
- A. Fraile, D. M. Scarpino Schietroma, A. Albrecht, R. L. Davis and K. A. Jørgensen, Chem. – Eur. J., 2012, 18, 2773 CrossRef CAS PubMed.
- W. Li, Q. Jia, Z. Du, K. Zhang and J. Wang, Chem. – Eur. J., 2014, 20, 4559 CrossRef CAS PubMed.
- W. Li, J. Wei, Q. Jia, Z. Du, K. Zhang and J. Wang, Chem. – Eur. J., 2014, 20, 6592 CrossRef CAS PubMed.
- K. Ito, S. Akashi, B. Saito and T. Katsuki, Synlett, 2003, 1809 CrossRef CAS.
- C. Shi and I. Ojima, Tetrahedron, 2007, 63, 8563 CrossRef CAS PubMed.
- J. F. Teichert, M. Fanãnás-Mastral and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 688 CrossRef CAS PubMed.
- M. Fanãnás-Mastral, J. F. Teichert, J. A. Fernández-Salas, D. Heijnen and B. L. Feringa, Org. Biomol. Chem., 2013, 11, 4521 Search PubMed.
- K. Takasu, S. Maiti and M. Ihara, Heterocycles, 2003, 59, 51 CrossRef CAS PubMed.
- S. Fustero, J. Moscardó, D. Jiménez, M. D. Pérez-Carrión, M. Sánchez-Roselló and C. del Pozo, Chem. – Eur. J., 2008, 14, 9868 CrossRef CAS PubMed.
- D. Enders, J. X. Liebich and G. Raabe, Chem. – Eur. J., 2010, 16, 9763 CrossRef CAS PubMed.
- J. Jiang, X. Ma, C. Ji, Z. Guo, T. Shi, S. Liu and W. Hu, Chem. – Eur. J., 2014, 20, 1505 CrossRef CAS PubMed.
- T. Ogata, A. Ujihara, S. Tsuchida, T. Shimizu, A. Kaneshige and K. Tomioka, Tetrahedron Lett., 2007, 48, 6648 CrossRef CAS PubMed.
- Z. Chen, B. Wang, Z. Wang, G. Zhu and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 2027 CrossRef CAS PubMed.
- D. Brózda, K. Hoffman and M. D. Rozwadowska, Heterocycles, 2006, 67, 119 CrossRef.
- S. Shirakawa, K. Liu, H. Ito, T. N. Le and K. Maruoka, Adv. Synth. Catal., 2011, 353, 2614 CrossRef CAS.
- K. Frisch and K. A. Jørgensen, Org. Biomol. Chem., 2007, 5, 2966 CAS.
- W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 277 CrossRef CAS PubMed.
- Q.-H. Zheng, W. Meng, G.-J. Jiang and Z.-X. Yu, Org. Lett., 2013, 15, 5928 CrossRef CAS PubMed.
- W. Lin and S. Ma, Org. Chem. Front., 2014, 1, 338 RSC.
- S. Murarka, I. Deb, C. Zhang and D. Seidel, J. Am. Chem. Soc., 2009, 131, 13226 CrossRef CAS PubMed.
- L. Chen, L. Zhang, J. Lv, J.-P. Cheng and S. Luo, Chem. – Eur. J., 2012, 18, 8891 CrossRef CAS PubMed.
- D. B. Horne, N. A. Tamayo, M. D. Bartberger, Y. Bo, J. Clarine, C. D. Davis, V. K. Gore, M. R. Kaller, S. G. Lehto, V. V. Ma, N. Nishimura, T. T. Nguyen, P. Tang, W. Wang, B. D. Yongblood, M. Zhang, N. R. Gavva, H. Monenschein and M. H. Norman, J. Med. Chem., 2014, 57, 2989 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.