
Cationic Pd(II)-catalyzed cyclization of N-tosyl-aniline tethered alkynyl ketones initiated by hydropalladation of alkynes: a facile way to 1,2-dihydro or 1,2,3,4-tetrahydroquinoline derivatives†
Kun
Shen
,
Xiuling
Han
*,
Guoqin
Xia
and
Xiyan
Lu
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: xlhan@mail.sioc.ac.cn; xylu@mail.sioc.ac.cn
First published on 26th December 2014
Abstract
A cationic Pd(II)-catalyzed intramolecular alkyne-carbonyl reductive coupling reaction of N-tosyl-aniline tethered alkynyl ketones under transfer hydrogenation conditions to give hydroquinolines is developed.
Hydrogenated quinoline derivatives, 1,2-dihydroquinolines and 1,2,3,4-tertrahydroquinolines, are ubiquitous in biochemical, biological and medicinal structures. They are an important class of compounds in the pharmaceutical and agrochemical industries, as well as building blocks for the total synthesis of natural products.1 In the past few decades, numerous studies have been conducted to evaluate their potential utilities such as antifungal and anticancer activity,2 antioxidant effects,3 and their role as ligands in some asymmetric synthesis.4 Consequently, a number of strategies have been developed for the synthesis of these scaffolds including partial hydrogenation of quinolines, Lewis acids, transition-metal- or organo-catalyzed intra- and intermolecular cyclization reactions.1d,5 Although many methods for the synthesis of these hydroquinolines have been established, there still exist some drawbacks such as harsh reaction conditions, prolonged reaction time, limited scope of substrates and modest selectivity, which promote chemists to explore other novel reactions to produce them more efficiently and conveniently.
In designing a new method for the synthesis of these hydroquinolines, palladium(II)-catalyzed cyclization of N-tosyl-aniline-tethered alkynyl ketones was selected. As we know, palladium(II)-catalyzed tandem reactions have emerged as a powerful and efficient tool for the rapid construction of C–C bonds, which have been widely used in the synthesis of many complex molecules.6 For the cyclization of alkynyl ketones, there are mainly two modes for the initiation of the palladium(II)-catalyzed reactions: nucleopalladation of alkynes and transmetallation of boron reagents with palladium(II). In these reactions, the nucleopalladation of alkynes or the insertion of alkynes into the C–Pd bond (formed from the transmetallation of boron reagents with palladium(II) complexes) can produce an alkenylpalladium species which consequently adds to the carbonyl group to offer a class of hydroxyl-bearing carbo- or heterocyclic compounds in an atom economical way. Using this methodology, our group has developed some novel tandem reactions in a redox-free way.7 Recently, we have found that the Pd–H species generated from cationic palladium complexes and ethanol can also realize similar cyclization reactions8 which have not been reported in the literature.9 Following our ongoing interest in palladium(II)-catalyzed tandem reactions, we wish to report now a cationic palladium(II)-catalyzed intramolecular alkyne-carbonyl reductive coupling reaction by using N-tosyl-aniline tethered alkynyl ketones as the substrates, allowing access to functionalized 1,2-dihydro or 1,2,3,4-tetrahydroquinolines.
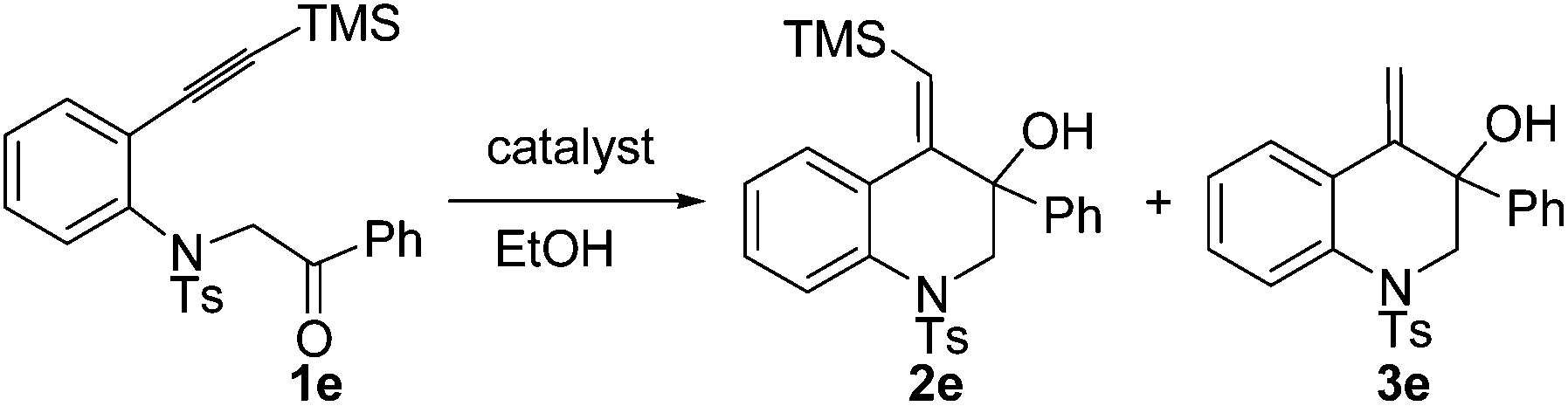
In our previous work, it was found that the steric effect of the substituents on alkynes was important for the reductive cyclization.8 Thus, the steric effect of substituent R3 in substrates 1 for this new reaction was studied first. As can be seen from Table 1, the sterically hindered group such as tBu or SiMe3 favored the transformation, which is in accordance with the previous work (Table 1, entries 1–4). Owing to the easy further transformation of the silicon moiety, compound 1e was chosen as the model substrate for reaction condition screening.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R3 | Substrate | Time (h) | Products | Yieldb (%) |
| a Conditions: reactions were carried out with 1 (0.20 mmol) and the catalyst (7.5 mol%) in EtOH (2.0 mL) at 70 °C. b Isolated yield. c Desilylation product 3e was also isolated (see Table 2, entry 1). | |||||
| 1 | H | 1a | 24 | 2a | Trace |
| 2 | n Bu | 1b | 24 | 2b | Trace |
| 3 | Ph | 1c | 24 | 2c | 17 |
| 4 | t Bu | 1d | 12 | 2d | 80 |
| 5c | SiMe3 | 1e | 6 | 2e | 62 |
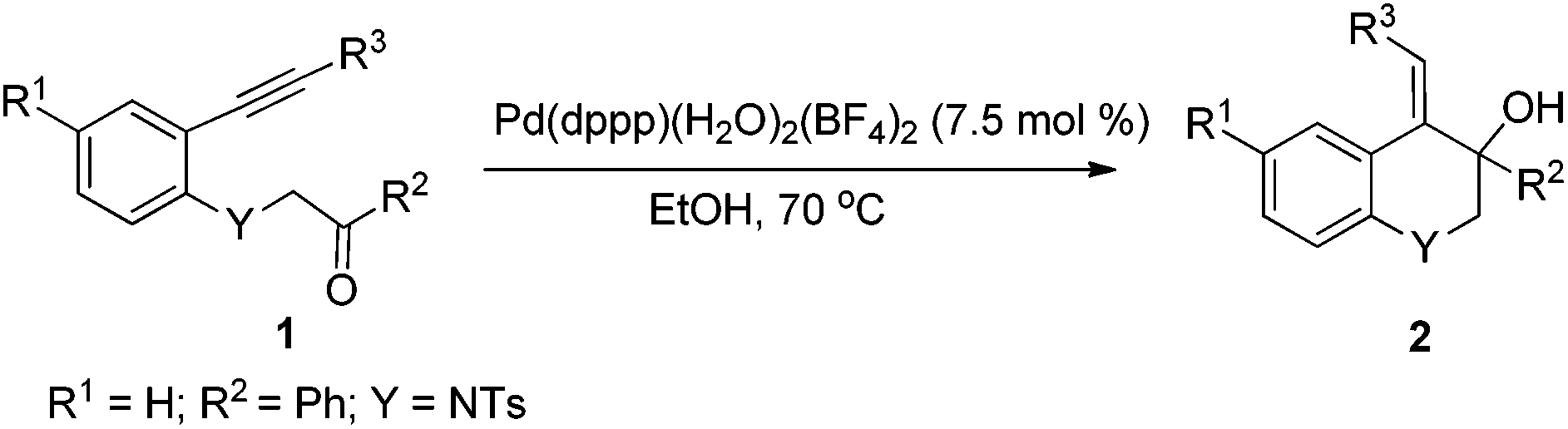
In our initial study, substrate 1e was reacted in ethanol (0.1 M) in the presence of 7.5 mol% of [Pd(dppp)(H2O)2](BF4)2 at 70 °C for 6 h (Table 2, entry 1). The reductive cyclization products 2e and 3e were isolated in 95% total yield. When the reaction temperature was decreased to 50 °C, a slightly lower yield was obtained (90%, Table 2, entry 2). iPrOH also gave a good total yield, whereas the amount of desilylation product 3e was increased (Table 2, entry 3). The use of [Pd(rac-binap)(H2O)2](BF4)2 as the catalyst lowered the total yield to 54% (Table 2, entry 4). Reducing the amount of the catalyst [Pd(dppp)(H2O)2](BF4)2 to 5 mol% did not compromise the yield (Table 2, entry 5). Interestingly, under the catalysis of [Pd(dppp)(H2O)2](OTf)2, 3e was obtained as the major product and 2e could transfer to 3e completely with a long reaction time (Table 2, entries 6 and 7). The catalyst [Pd(bpy)(μ-OH)]2(OTf)2 was totally ineffective to the reaction (Table 2, entry 8). On the basis of the above investigation, the optimal conditions for this reductive cyclization reaction are as follows: 1e (0.2 mmol) and [Pd(dppp)(H2O)2](BF4)2 (5 mol%) in EtOH (2 mL) at 70 °C (Table 2, entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalystb | Temp. (°C) | Time (h) | Yieldc (%) | ||
| 2e | 3e | Total | ||||
| a Conditions: reactions were carried out with 1e (0.20 mmol) and the catalyst (7.5 mol%) in EtOH (2 mL). b Cat. I: [Pd(dppp)(H2O)2](BF4)2; Cat. II: [Pd(rac-binap)(H2O)2](BF4)2; Cat. III: [Pd(dppp)(H2O)2](OTf)2; Cat. IV: [Pd(bpy)(μ-OH)]2(OTf)2. c Isolated yield. d i PrOH was used as the solvent. e Catalyst (5 mol%) was added. | ||||||
| 1 | Cat. I | 70 | 6 | 62 | 33 | 95 |
| 2 | Cat. I | 50 | 8 | 68 | 22 | 90 |
| 3d | Cat. I | 70 | 6 | 58 | 34 | 92 |
| 4 | Cat. II | 70 | 24 | 29 | 25 | 54 |
| 5e | Cat. I | 70 | 10 | 66 | 32 | 98 |
| 6e | Cat. III | 70 | 10 | 24 | 69 | 93 |
| 7 | Cat. III | 70 | 18 | 0 | 75 | 75 |
| 8 | Cat. IV | 70 | 24 | N.R. | ||
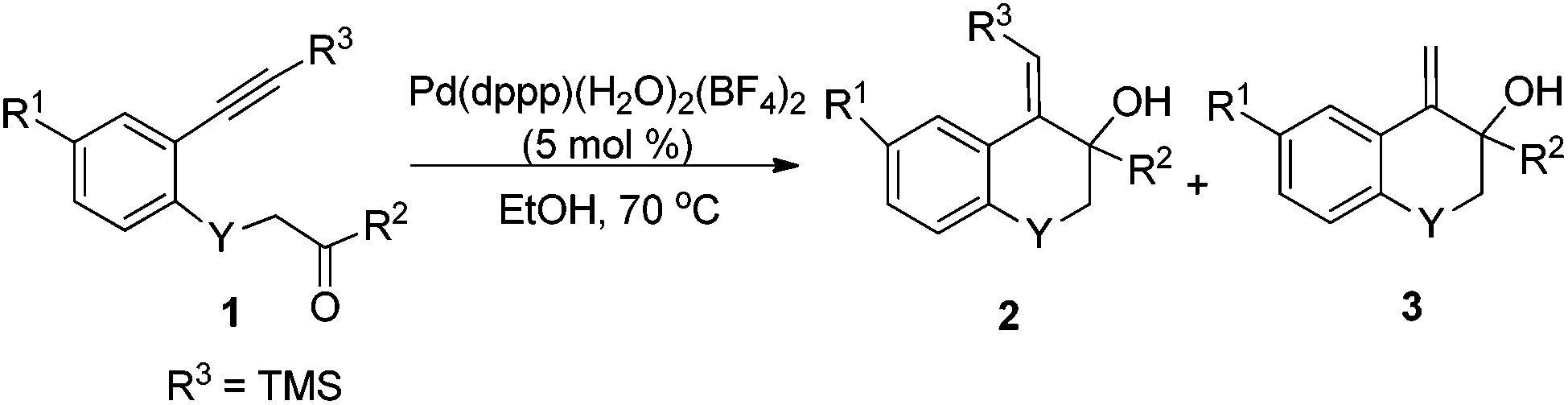
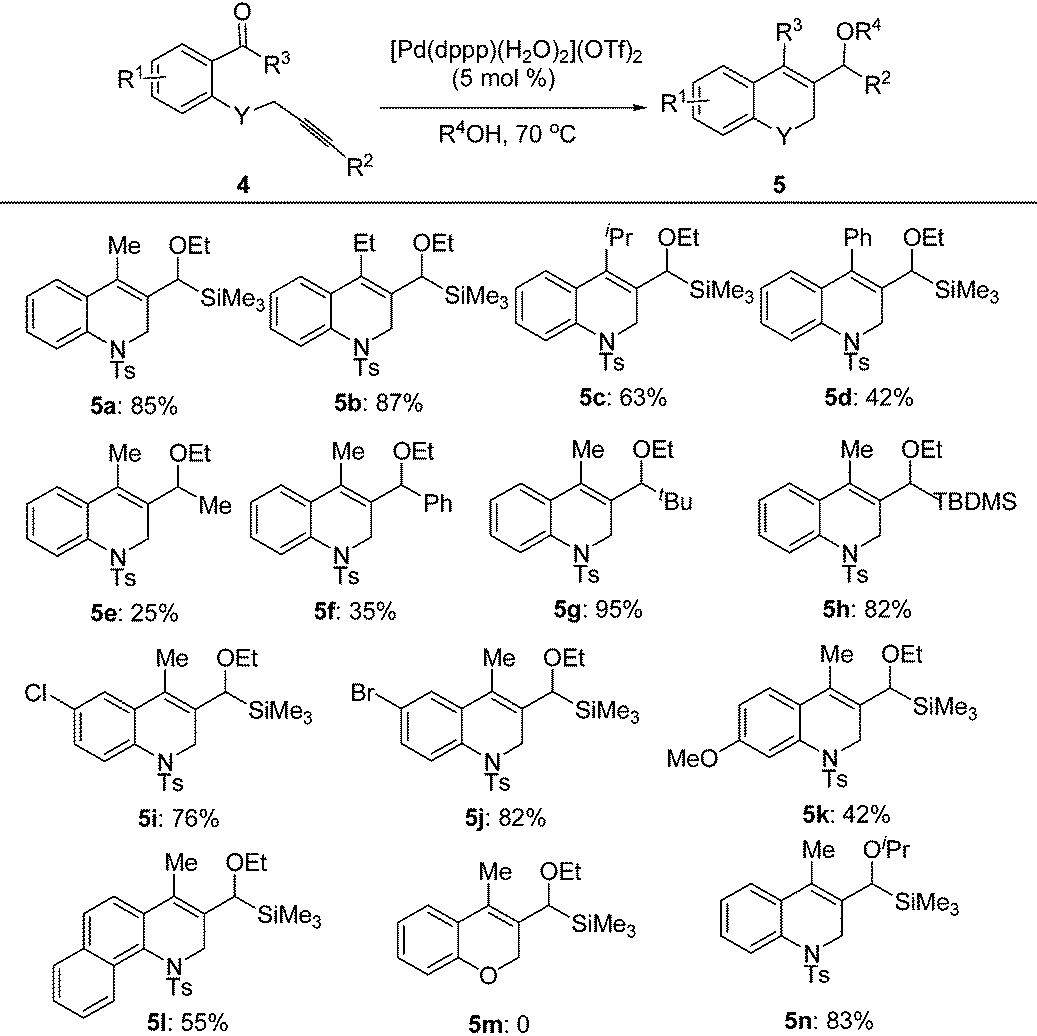
Under the optimal conditions, the substrate scope of the reaction was examined with a variety of N-tosyl-aniline tethered alkynyl ketones and the results are summarized in Table 3. Substrates bearing a methyl group, fluoro or chloro atom on the benzene ring also gave good results which were similar to that of 1e (Table 3, entries 2–4). Reactions occurred successfully when R2 was an aromatic ring bearing a nitro, methyl group or chlorine atom, whereas a methoxyl group had a deleterious effect (Table 3, entries 5–8). Ketones with a bulky group had no reactivity at all (Table 3, entries 9 and 11). Methyl ketone (1n) only provided a moderate total yield (Table 3, entry 10). NMs or O tethered substrates also reacted well for this cyclization (Table 3, entries 12 and 13). The structures of the products were confirmed by X-ray crystallography of 2g as a typical example.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | T (h) | Yieldb (%) | |||
| R1 | R2 | Y | 2 | 3 | ||
| a Conditions: reactions were carried out with 1 (0.20 mmol) and the catalyst (5 mol%) in EtOH (2 mL) at 70 °C. b Isolated yield. | ||||||
| 1 | H | Ph | NTs 1e | 10 | 66 2e | 32 3e |
| 2 | F | Ph | NTs 1f | 6 | 63 2f | 29 3f |
| 3 | Cl | Ph | NTs 1g | 8 | 76 2g | 14 3g |
| 4 | Me | Ph | NTs 1h | 10 | 75 2h | 20 3h |
| 5 | H | p-NO2-C6H4 | NTs 1i | 10 | 62 2i | 27 3i |
| 6 | H | p-Me-C6H4 | NTs 1j | 12 | 59 2j | 33 3j |
| 7 | H | p-OMe-C6H4 | NTs 1k | 8 | 46 2k | 26 3k |
| 8 | H | p-Cl-C6H4 | NTs 1l | 10 | 71 2l | 26 3l |
| 9 | H | o-Me-C6H4 | NTs 1m | 12 | Trace | 0 |
| 10 | H | Me | NTs 1n | 6 | 33 2n | 31 3n |
| 11 | H | t Bu | NTs 1o | 12 | N.R. | |
| 12 | H | Ph | NMs 1p | 4 | 47 2p | 38 3p |
| 13 | H | Ph | O 1q | 4 | 53 2q | 24 3q |
For the formation of products 3, some control experiments were then conducted. Treatment of 2e in ethanol without the catalyst did not give product 3e. However, a transformation occurred in the presence of the catalyst [Pd(dppp)(H2O)2](BF4)2 or [Pd(dppp)(H2O)2](OTf)2 and the latter could give a better result, indicating that 3e might be generated from 2e under the catalysis of the palladium(II) catalyst (Scheme 1).10
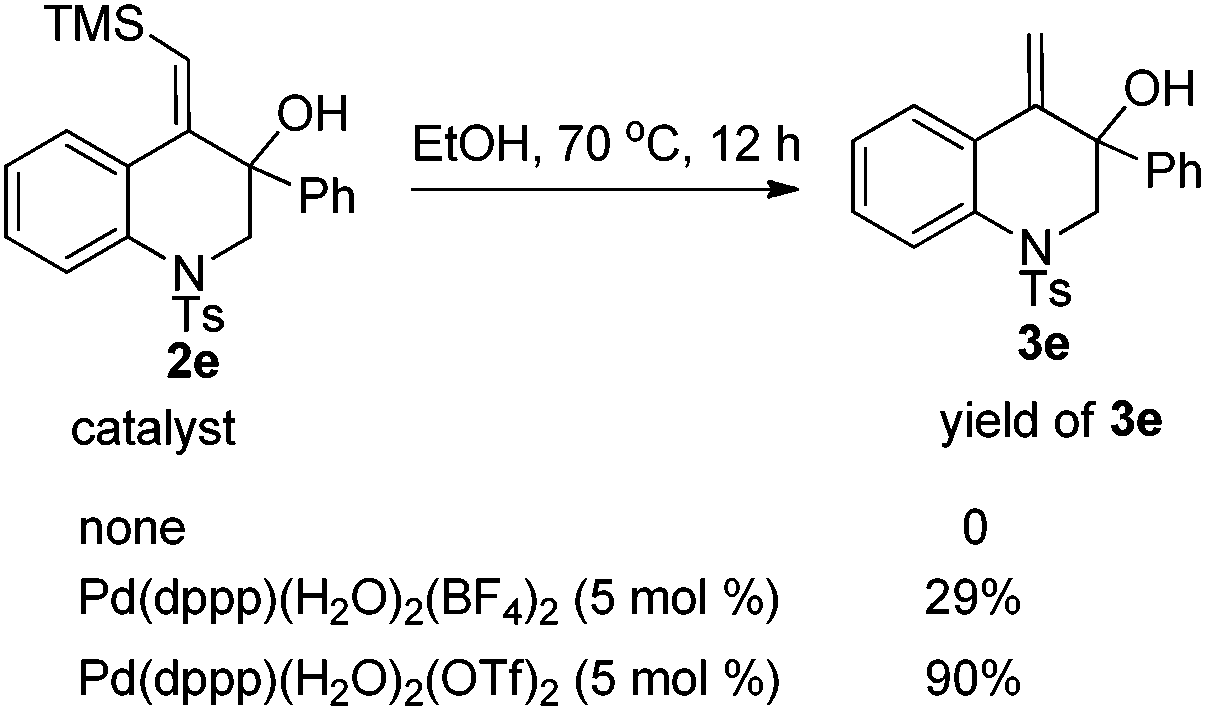 | ||
| Scheme 1 Transformation of 2e to 3e. | ||
Next, the asymmetric version of this process using chiral phosphine ligands was carried out (Scheme 2). When the reaction was catalyzed by [Pd(S,S-bdpp)(H2O)2](BF4)2, a high total yield (95%) was obtained with substrate 1e, whereas the asymmetric induction was moderate (2e, 56% ee). Under the catalysis of [Pd(R-binap)(H2O)2](BF4)2, the reaction gave 2e in a higher ee value (82%), but the yield was low (39%).
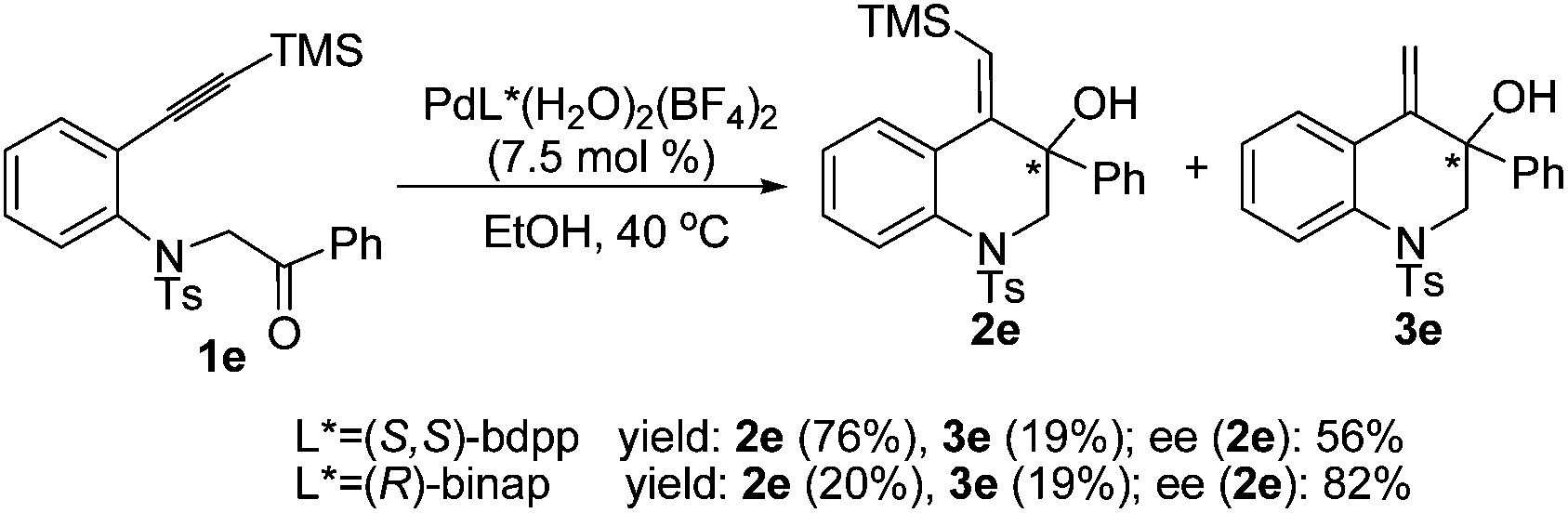 | ||
| Scheme 2 Asymmetric reductive cyclization of 1e. | ||
To gain mechanistic insight, the reaction was then conducted in deuterated ethanol. Subjecting 1e to the reductive cyclization reaction conditions in CH3CH2OD gave products 2e and 3e in 87% total yield with no deuterium incorporation into the products. However, when CH3CD2OH was used, the reaction could afford d-2e and d2-3e with deuterium at the vinyl position in 90% total yield (Scheme 3). Thus, it was concluded that the vinyl hydrogen in the products was from the α-hydrogen of ethanol.
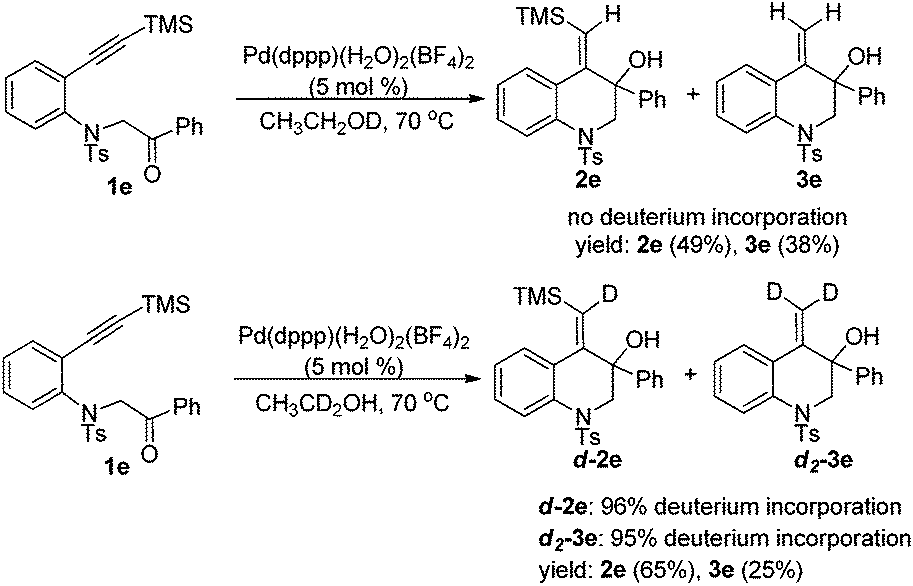 | ||
| Scheme 3 Deuterium-labeling studies. | ||
On the basis of the above results, a possible mechanism of this reductive cyclization reaction is shown in Scheme 4. The Pd hydroxy complex A is presumed to be the active catalytic species and it reacts with ethanol to generate Pd ethoxide complex B, which subsequently gives the Pd hydride complex Cvia β-hydride elimination.11 Hydropalladation of the alkyne affords a vinylpalladium intermediate D followed by intramolecular addition to the carbonyl group to give the intermediate E. The protonolysis of the newly formed intermediate E generates products 2 and the Pd(II) species A to complete the catalytic cycle.
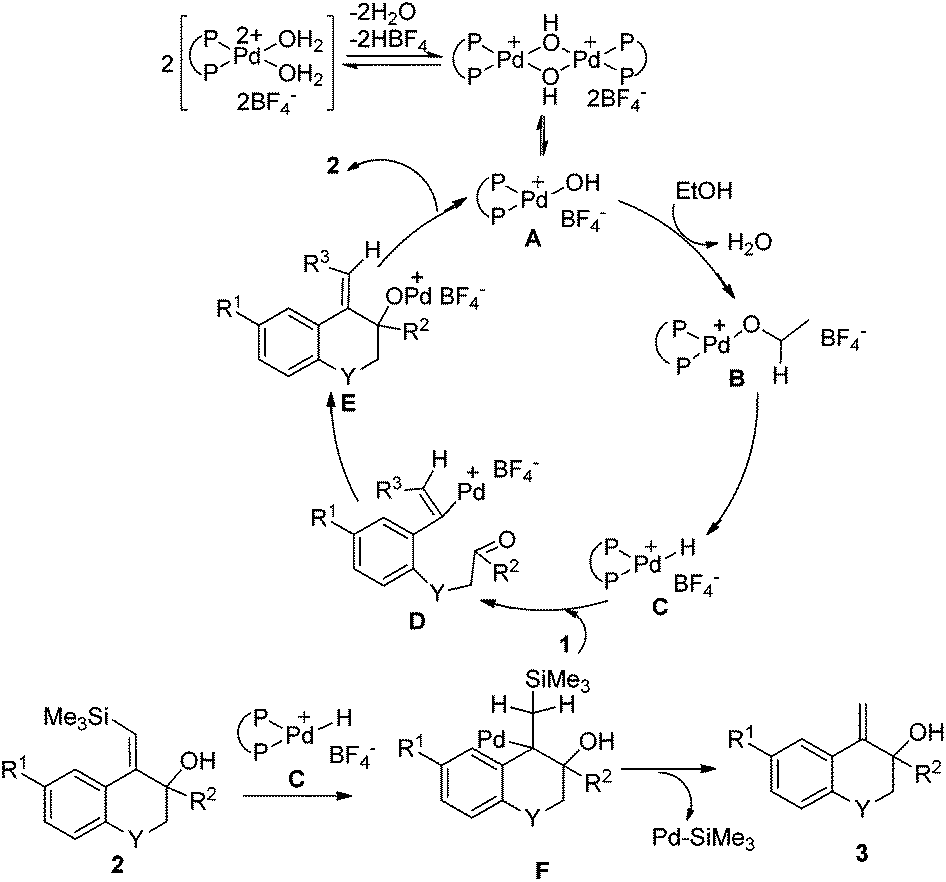 | ||
| Scheme 4 Proposed mechanism for the formation of 2 and 3. | ||
On the other hand, the insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of products 2 into Pd–H species (intermediate C) can produce intermediate F, which then undergoes β-desilylation to afford another products 3.10 In Scheme 3, when CH3CD2OH was used as the hydrogen source, both of the two vinyl hydrogens in the product 3e were replaced by deuterium, which further verified that it was a palladium-catalyzed desilylation process in our reaction.
C double bond of products 2 into Pd–H species (intermediate C) can produce intermediate F, which then undergoes β-desilylation to afford another products 3.10 In Scheme 3, when CH3CD2OH was used as the hydrogen source, both of the two vinyl hydrogens in the product 3e were replaced by deuterium, which further verified that it was a palladium-catalyzed desilylation process in our reaction.
Having synthesized some 1,2,3,4-tetrahydroquinolines successfully by using our new reactions, we next studied the cyclization of another type of aniline tethered alkynyl ketones such as substrate 4a under similar conditions. When 4a was reacted in the presence of 5 mol% of [Pd(dppp)(H2O)2](BF4)2 in ethanol (0.1 M) at 70 °C for 24 h, the cyclization product 1,2-dihydroquinoline 5a was formed, albeit in a low yield (38%, Table 4, entry 1). Excitingly, the yield of 5a was increased to 85% using [Pd(dppp)(H2O)2](OTf)2 as the catalyst (Table 4, entry 2). The catalyst [Pd(rac-binap)(H2O)2](OTf)2 can also give a moderate yield of 5a (72%, Table 4, entry 3). The yield decreased at a lower temperature (Table 4, entry 4). Only 12% yield was obtained while using 4 Å molecular sieves as the additive (Table 4, entry 5). The reaction did not occur under the catalysis of Pd(OAc)2 or Pd(CF3COO)2 (Table 4, entries 6 and 7). Thus, the optimal reductive cyclization conditions for this kind of compound were as follows: 4a (0.2 mmol) and [Pd(dppp)(H2O)2](OTf)2 (5 mol%) in EtOH (2 mL) at 70 °C.
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
| a Conditions: reactions were carried out with 4a (0.20 mmol) and the catalyst (5 mol%) in EtOH (2 mL) at 70 °C for 24 h, unless otherwise mentioned. b Isolated yield. c The reaction was performed at 50 °C. d 4 Å molecular sieves were added. | ||
| 1 | [Pd(dppp)(H2O)2](BF4)2 | 38 |
| 2 | [Pd(dppp)(H2O)2](OTf)2 | 85 |
| 3 | [Pd(rac-binap)(H2O)2](OTf)2 | 72 |
| 4c | [Pd(dppp)(H2O)2](OTf)2 | 33 |
| 5d | [Pd(dppp)(H2O)2](OTf)2 | 12 |
| 6 | Pd(OAc)2 + dppp | 0 |
| 7 | Pd(CF3COO)2 + dppp | 0 |
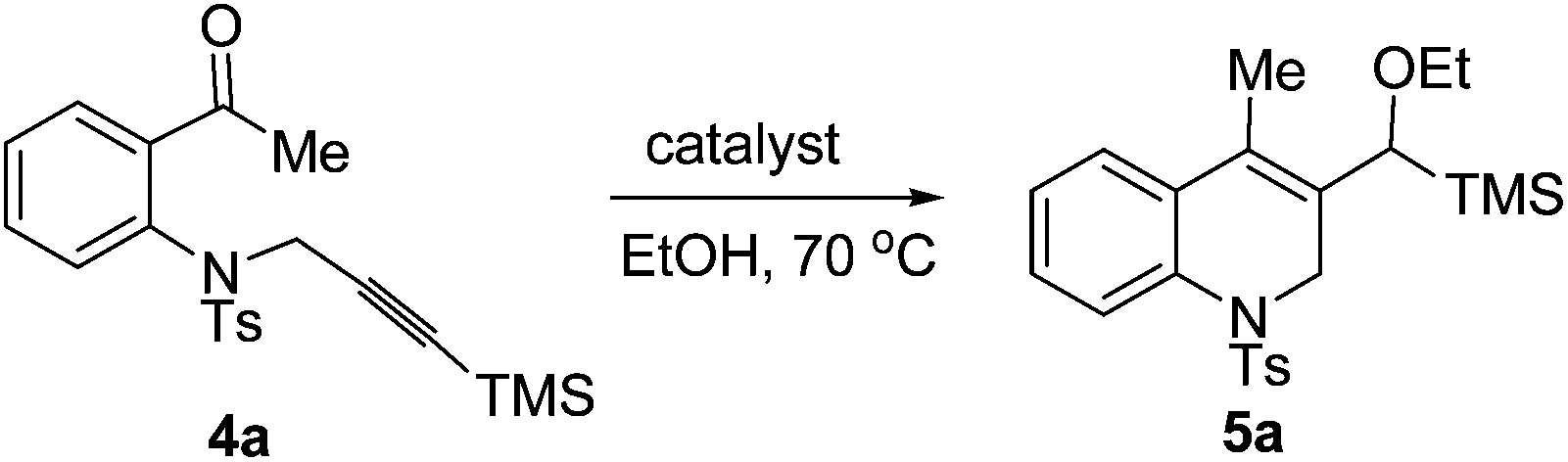
With the optimal reaction conditions in hand, we studied the scope of this cyclization reaction (Table 5). For carbonyl groups in the substrates, phenyl alkyl ketones had higher reactivity than that of diphenyl ones (Table 5, 5a–5d). Sterically hindered groups on the alkyne benefited the cyclization, which was similar to the results in Table 3 (Table 5, 5e–5h). Substrates with the benzene ring substituted by a chlorine or bromine atom gave the corresponding products in good yields, but the methoxyl group deteriorated the reaction (Table 5, 5i–5k). The naphthalene-ring tethered substrate 4l also underwent cyclization with a moderate yield (Table 5, 5l). When NTs in the substrate was changed to O, no reaction occurred at all (Table 5, 5m). Finally, a similar reactivity was found when EtOH was replaced by iPrOH, indicating that the alcohols used in the cyclization not only provided the hydrogen source, but also acted as substrates in the subsequent transformation (Table 5, 5n). The structures of the products were confirmed by X-ray crystallography of 5l as a typical example.
A plausible mechanism is outlined in Scheme 5 and the process for the formation of intermediate E′ is similar to that of intermediate E as shown in Scheme 4. Then intermediate E′ is attacked by ethanol to promote the cleavage of the C–O bond to give the product and regenerate the palladium(II) species. Comparing with the reactions in Table 3, intermediate E′ in this reaction can be attacked by the alcohol to undergo further transformation, which may be due to the stability of the conjugated double bond formed in the product.
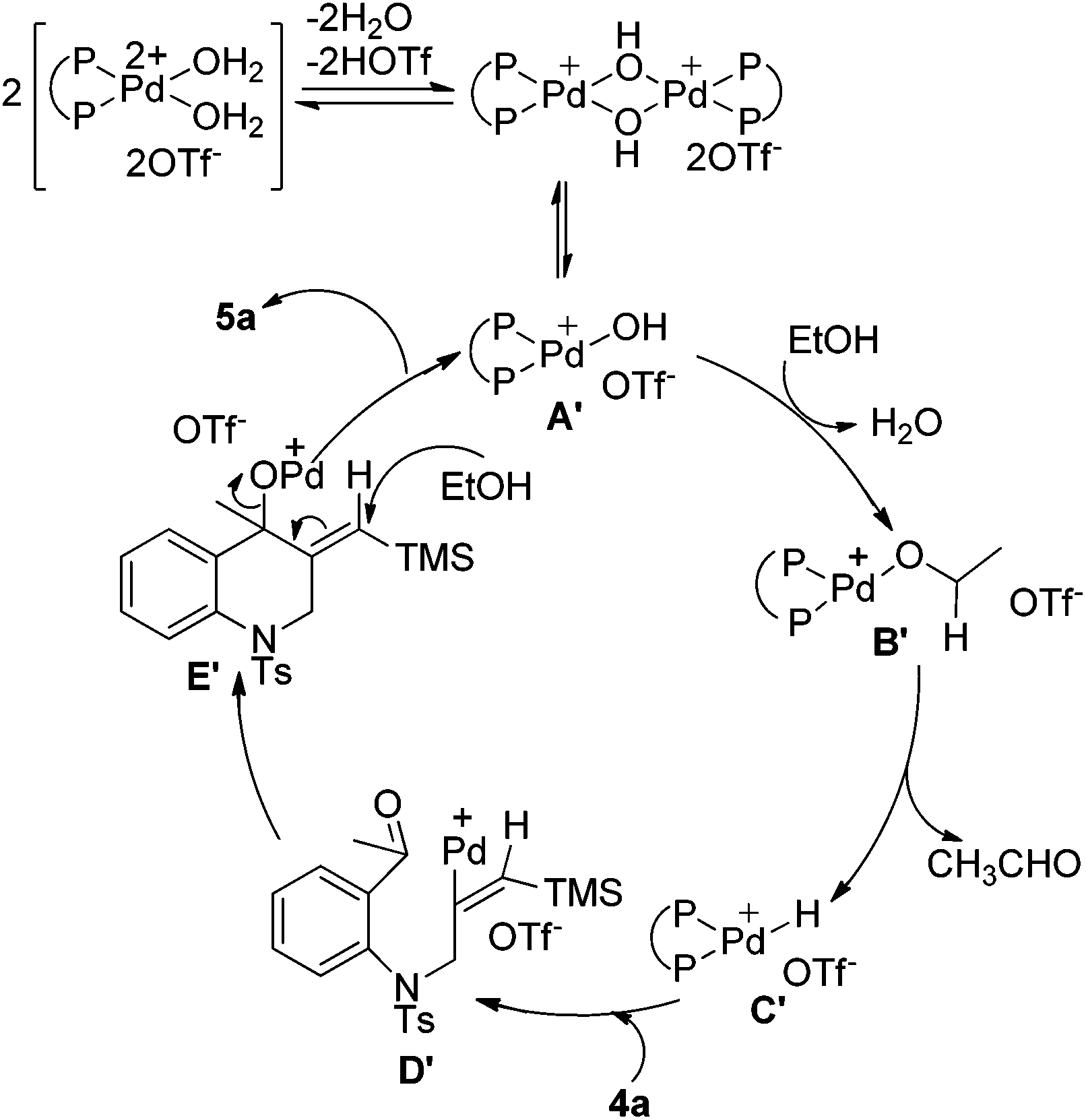 | ||
| Scheme 5 Proposed mechanism for the formation of 5a. | ||
In conclusion, two cationic Pd(II)-catalyzed intramolecular alkyne-carbonyl reductive coupling reactions under transfer hydrogenation conditions are developed. The reactions offer an environmentally benign method for the synthesis of 1,2-dihydro or 1,2,3,4-tetrahydroquinoline derivatives in high yields. Further studies on the asymmetric version of the reaction are in progress in our laboratory.
Acknowledgements
We thank the National Basic Research Program of China (2011CB808706), the National Natural Science Foundation of China (21232006, 21272249) and the Chinese Academy of Sciences for financial support.Notes and references
- For selected recent reviews, see: (a) K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245 CrossRef CAS PubMed; (b) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488 CrossRef CAS; (c) A. Garrido Montalbán, in Heterocycles in Natural Products Synthesis, eds. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, Germany, 2011, pp. 299–339 Search PubMed; (d) V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed.
- For recent examples, see: (a) V. V. Kouznetsov, C. M. Meléndez Gómez, M. G. Derita, L. Svetaz, E. del Olmo and S. A. Zacchino, Bioorg. Med. Chem., 2012, 20, 6506 CrossRef CAS PubMed; (b) V. V. Kouznetsov, F. A. Ruiz, L. Y. Vargas and M. P. Gupta, Lett. Drug Des. Discovery, 2012, 9, 680 CrossRef CAS and references cited therein.
- For the examples, see: (a) W. A. Pryor, T. Strickland and D. F. Church, J. Am. Chem. Soc., 1988, 110, 2224 CrossRef CAS; (b) A. J. de Koning, Int. J. Food Prop., 2002, 5, 451 CrossRef CAS PubMed; (c) A. Blaszczyck and J. Skolimowski, Biol. Interact., 2006, 162, 70 CrossRef PubMed.
- For the selected examples, see: (a) T. Pullmann, B. Engendahl, Z. Zhang, M. Hölscher, A. Zanotti-Gerosa, A. Dyke, G. Franciò and W. Leitner, Chem. – Eur. J., 2010, 16, 7517 CrossRef CAS PubMed; (b) W.-B. Liu, H. He, L.-X. Dai and S.-L. You, Synthesis, 2009, 2076 CAS; (c) Q.-B. Liu, C.-B. Yu and Y.-G. Zhou, Tetrahedron Lett., 2006, 47, 4733 CrossRef CAS PubMed; (d) S. Kaiser, S. P. Smidt and A. Pfaltz, Angew. Chem., Int. Ed., 2006, 45, 5194 CrossRef PubMed; (e) S. Bell, B. Wüstenberg, S. Kaiser, F. Menges, T. Netscher and A. Pfaltz, Science, 2006, 311, 642 CrossRef CAS PubMed; (f) H. Liu, M. Wang, Y. Wang, Y. Wang, H. Sun and L. Sun, Catal. Commun., 2009, 11, 294 CrossRef CAS PubMed; (g) Y. Xie, H. Huang, W. Mo, X. Fan, Z. Shen, Z. Shen, N. Sun, B. Hu and X. Hu, Tetrahedron: Asymmetry, 2009, 20, 1425 CrossRef CAS PubMed; (h) D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Org. Biomol. Chem., 2010, 8, 1081 RSC.
- For the reviews, see: (a) V. Kouznetsov, A. Palma and C. Ewert, J. Heterocycl. Chem., 1998, 35, 761 CrossRef CAS; (b) A. R. Katritzky, S. Rachwal and B. Rachwal, Tetrahedron, 1996, 52, 15031 CrossRef CAS; (c) B. Barluenga, F. Rodríguez and F. J. Fañanás, Chem. – Eur. J., 2009, 15, 1036 CrossRef PubMed.
- (a) J. Tsuji, Palladium Reagents and Catalysts: Innovations in Organic Synthesis, Wiley and Sons, New York, 1995 Search PubMed; (b) J. Tsuji, Palladium Reagents and Catalysts: New Perspectives for the 21th Century; Wiley and Sons, New York, 2003 Search PubMed; (c) M. B. Egle, B. Gianluigi, M. Michela and S. Silvia, Chem. Rev., 2007, 107, 5318 CrossRef PubMed; (d) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (e) L. F. Tietze, H. IIa and H. P. Bell, Chem. Rev., 2004, 104, 3453 CrossRef CAS PubMed; (f) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (g) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS PubMed; (h) B. M. Trost, Acc. Chem. Res., 1980, 13, 385 CrossRef CAS.
- For the cyclization reactions of alkynyl ketones or aldehydes published by our group, see: (a) L. Zhao and X. Lu, Angew. Chem., Int. Ed., 2002, 41, 4343 CrossRef CAS; (b) J. Song, Q. Shen, F. Xu and X. Lu, Org. Lett, 2007, 9, 2947 CrossRef CAS PubMed; (c) G. Liu and X. Lu, Adv. Catal., 2007, 349, 2247 CrossRef CAS; (d) F. Zhou, M. Yang and X. Lu, Org. Lett., 2009, 11, 1405 CrossRef CAS PubMed; (e) X. Han and X. Lu, Org. Lett., 2010, 12, 108 CrossRef CAS PubMed; (f) X. Han and X. Lu, Org. Lett., 2010, 12, 3336 CrossRef CAS PubMed; (g) H. Wang, X. Han and X. Lu, Tetrahedron, 2010, 66, 9129 CrossRef CAS PubMed; (h) H. Wang, X. Han and X. Lu, Synlett, 2011, 2590 CAS.
- K. Shen, X. Han and X. Lu, Org. Lett., 2013, 15, 1732 CrossRef CAS PubMed.
- For the reviews of other transition metals catalyzed hydrogen-mediated alkyne-carbonyl coupling reactions, see: (a) H.-Y. Jang and M. J. Krische, Acc. Chem. Res., 2004, 37, 635 CrossRef PubMed; (b) M.-Y. Ngai, J.-R. Kong and M. J. Krische, J. Org. Chem., 2007, 72, 1063 CrossRef CAS PubMed; (c) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394 CrossRef CAS PubMed; (d) H. Iida and M. J. Krische, Top. Curr. Chem., 2007, 279, 77 CrossRef CAS; (e) J. Montgomery and G. J. Sormunen, Top. Curr. Chem., 2007, 279, 1 CrossRef CAS; (f) J. F. Bower, I.-S. Kin, R. L. Patman and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 34 CrossRef CAS PubMed; (g) J. F. Bower and M. J. Krische, Top. Organomet. Chem., 2011, 34, 107 CrossRef CAS; (h) J. Moran and M. J. Krische, Pure Appl. Chem., 2012, 84, 1729 CrossRef CAS PubMed.
- For the example of palladium-catalyzed desilated reaction of alkenes, see: K. Karabelas and A. Hallberg, J. Org. Chem., 1986, 51, 5286 CrossRef CAS.
- (a) Y. Tsuchiya, Y. Hamashima and M. Sodeoka, Org. Lett., 2006, 8, 4851 CrossRef CAS PubMed; (b) D. Monguchi, C. Beemelmanns, D. Hashizume, Y. Hamashima and M. Sodeoka, J. Organomet. Chem., 2008, 693, 867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterisation. CCDC 1021338, 1038047. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00286e |
| This journal is © the Partner Organisations 2015 |