
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold trifluoromethyl complexes
Juan
Gil-Rubio
* and
José
Vicente
Grupo de Química Organometálica, Departamento de Química Inorgánica, Facultad de Química, Universidad de Murcia, E-30100 Murcia, Spain. E-mail: jgr@um.es; Web: http://www.um.es/gqo/
First published on 14th July 2015
Abstract
This article reviews the synthesis, reactivity and applications of gold trifluoromethyl complexes, which are the only isolated perfluoroalkyl complexes of gold. The most reported examples are neutral Au(I) complexes of the type [Au(CF3)L], whereas only two Au(II) trifluoromethyl complexes have been reported, both being diamagnetic and containing a strong Au–Au bond. A number of Au(III) trifluoromethyl complexes have been prepared by oxidative addition of halogens or iodotrifluoromethane to Au(I) complexes or, in a few cases, by transmetallation reactions. Owing to the limitations of the available synthetic methods, a lower number of examples is known, particularly for the oxidation states (II) and (III). Gold trifluoromethyl complexes present singular characteristics, such as thermal stability, strong Au–C bonds and, in some cases, reactive α-C–F bonds. Some of the Au(III) complexes reported, show unusually easy reductive elimination reactions of trifluoromethylated products which could be applied in the development of gold-catalyzed processes for the trifluoromethylation of organic compounds.
 Juan Gil-Rubio | Juan Gil-Rubio obtained his PhD in Chemistry at the University of Murcia under the supervision of Prof. José Vicente and Dr José A. Abad. After this, he was awarded with a Marie Curie fellowship to work in Würzburg (Germany) with Prof. Helmut Werner. In 2001 he was awarded with a Ramón y Cajal grant, and in 2010 he became an associate professor at the University of Murcia. His main research interests include the study of new reactions of fluorinated organometallic complexes of the platinum-group metals and gold, and the study of self-assembly processes involving organometallic and coordination complexes. |
 José Vicente | José Vicente received his Ph.D. in Chemistry from the University of Zaragoza in 1973 under the supervision of Profs Rafael Usón and Victor Riera. He was a postdoctoral fellow at Bristol University (1976–7) with Prof. F. Gordon A. Stone. He was appointed as a Lecturer in Zaragoza (1978), a Reader (1980) in the University of Murcia, later (1981) a Full Professor, and currently he is an Emeritus Professor. He was awarded in 2001 with the Research Medal of the Real Sociedad Española de Química. He has supervised 37 PhD theses. His past and current research is focused on the syntheses, structural characterization and applications of Au, Ag, Pd, Pt, Rh, Hg, Tl, and Sn complexes. |
Introduction
Perfluoroalkyl complexes form a special class among transition metal organometallic compounds. After the first examples were synthesized in the middle of the past century, it has been realized that their properties are remarkably different from those of their non-fluorinated analogues.1,2 Thus, it is commonly accepted that they are generally more stable than their alkyl counterparts and that, owing to this stability, the metal–perfluoroalkyl bond presents low reactivity, particularly in reductive elimination or migratory insertion reactions,3–5 which are the key steps in metal-promoted coupling reactions.Efficient synthetic methods for the introduction of fluorinated substituents into organic molecules are currently demanded because fluoroorganic compounds are scarce in nature,6–8 and they offer industrial applications as advanced materials,9–12 in fluorous chemistry13–17 and, most importantly, in medicine.18,19 Owing to their pharmaceutical applications, trifluoromethylated organic compounds have received special attention. This demand has stimulated the search for reactive perfluoroalkyl metal complexes, giving rise to the recent development of a plethora of new metal mediated or catalyzed trifluoromethylation reactions.20–27
Most metal-promoted synthetic routes for trifluoromethylated organic compounds make use of copper trifluoromethyl complexes,22,24,26,28–31 some of which were used as trifluoromethylating agents in organic synthesis for a long time.32,33 Methods based on other metals, such as silver34–39 or palladium,40–47 have also been developed, although they are less used. Overall, these advances have revealed an unforeseen synthetic potential for perfluoroorganotransition metal compounds, which should be further developed by the investigation of new types of transition metal perfluoroalkyl complexes.
There is still a limited knowledge of the chemistry of gold perfluoroalkyl complexes, which contrasts with the remarkable progress made in the organometallic and catalytic chemistry of gold over the last 25 years.48–55 The first gold perfluoroalkyls were prepared rather late (1973) by Puddephatt and coworkers.56,57 Since then, a relatively low number of trifluoromethyl complexes has been reported. For instance, no higher perfluoroalkyls of gold have been described, with the exception of [Au(C2F5)(PEt3)], which was spectroscopically observed but not isolated.58 It is also noteworthy that the synthetic routes to simple complexes such as [Au(CF3)x]− (x = 2, 4) have been only recently reported.59–61 Interestingly, Au(III) complexes present a low activation barrier for the reductive elimination of R–R′ products,62–66 including those with R = CF3.67 This is of considerable interest, because reductive elimination is one of the key steps in metal mediated- or catalyzed coupling reactions and its activation barrier is usually very high for perfluoroalkyl complexes, limiting their applicability in this field.40–42,68
The pioneering work on the synthesis of fluorocarbon metal complexes was compiled in three reviews by Stone et al.1,2,69 The field of transition metal trifluoromethyl complexes has been reviewed by Morrison (1993),5 and Menjón et al. (2012).70 In this Perspective, we have reviewed the synthesis, properties and reactivity of gold trifluoromethyl compounds, putting emphasis on recently reported reactions with potential synthetic interest.
Structure, bonding and spectroscopic properties of gold trifluoromethyl complexes
The available experimental evidence suggests that, as observed for other metal perfluoroalkyls, gold trifluoromethyl complexes are more stable than their methyl analogues. For instance, the compounds [Au(CF3)(CNR)] (R = Me,71tBu61) melt without decomposition at 110 and 119 °C, respectively, whereas their methyl analogues decompose at 95 and 62 °C, respectively.72 The salts [PPh4][Au(CF3)x] (x = 2, 4) start to decompose at 275 °C (x = 2) and 370 °C (x = 4),61 whereas di- and tetramethylaurates Q[AuMen] (n = 2, Q = Li(MeN(CH2CH2NMe2)2); n = 4, Q = NnBu4) readily decompose at 140 °C.73,74 This stability can be attributed to a stronger Au–CF3 bond, as evidenced by the generally shorter Au–CF3 distances compared to those for the Au–CH3 bonds in analogous compounds (Table 1).| Compounds | d(Au–C)/Å | |
|---|---|---|
| X = H | X = F | |
| a IPr = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene. | ||
| Ph3P–Au–CX3 | 2.124(28)77 | 2.045(10)79 |
| 2.065(10)78 | ||
IPr–Au–CX3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
2.042(10)80 | 2.042(10), 2.030(14)81 |
| Q[Au(CX3)2] | 2.075 (average, Q = NnBu4)82 | 2.033(2) (Q = PPh4)61 |
| Q[Au(CX3)4] | 2.100 (average, Q = NMe2Bn2)82 | 2.059(6), 2.072(6) (Q = PPN)60 |
| 2.075(6), 2.085(7) (Q = NnBu4)61 | ||
In addition, gold trifluoromethyl complexes show two characteristic structural features usually found in other trifluoromethyl complexes: (i) the elongation of the MC–F bonds with respect to the C–F distances observed in non-metallic trifluoromethyl derivatives,75 and (ii) the decrease of the F–C–F bond angle and the increase of the M–C–F bond angle76 with respect to the ideal tetrahedral angle. Interestingly, whereas the feature (ii) is always found in all reported structures, the feature (i) is not observed in some Au(II) and Au(III) trifluoromethyl complexes.
The stability and special structural features of Mn, Rh, Ni and Pt perfluoroalkyl complexes have been addressed by theoretical studies. Thus, in 1972 Hall and Fenske reported the molecular orbital calculations of complexes [Mn(CX3)(CO)5] (X = H, F),83 and in 2012 Macgregor and Grushin reported Natural Bonding Orbital (NBO) analyses of Mn, Rh, Pt and Ni trifluoromethyl and methyl complexes.3 These studies show that: (i) the C 2s character of the M–C bonding orbitals and the strength of the σ(M–C) interaction are higher for the CF3 complexes than for the analogous methyl complexes, which accounts for the observed M–C bond shortening; (ii) the contribution of the metal-to-CF3 π-backbonding to the overall M–CF3 bonding is small, as also noted by Graham84 and Harvey,85 and it alone cannot account for the observed bond strengthening; (iii) the lone pair of the CF3− anion86,87 has antibonding character with respect to the C-F bond, and therefore the degree of donation of this lone pair to a metal or non-metal acceptor significantly influences the strength of the C–F bonds; (iv) the high positive charge on the CF3 carbon stabilizes the metal-based orbitals, conferring stability to the whole molecule.
Although NBO analyses have not been reported for Au trifluoromethyl complexes, similar effects could account for their increased stability as well as for the Au–C bond shortening and C–F bond weakening observed in these complexes. Accordingly, a comparison of the mean Au–CF3 and C–F distances for the reported structures of Au(I) and Au(III) trifluoromethyl complexes reveals that in Au(III) derivatives, the Au–C distances are longer, and the C–F ones are shorter, than those in Au(I) complexes by about 0.05 Å. This difference could be attributed to rehybridization of the orbitals involved in the Au–C and C–F bonds upon increasing both the coordination number and the oxidation state of the metal. Thus, the Au–C bond distance should be longer for tetracoordinate Au(III) complexes than that for dicoordinate Au(I) complexes, whereas the Au–C orbital should decrease its C 2s character in the Au(III) complexes. Consequently, the C–F orbitals should increase their C 2s character.
19F NMR spectroscopy is the most helpful technique to structurally characterize trifluoromethyl complexes in solution. The 19F chemical shift of the Au(I) trifluoromethyl complexes is rather insensitive to the nature of the other ligand (Fig. 1) and, therefore, all the reported values for Au(I) complexes fall in the narrow range of −24.0 to −31.5 ppm. In contrast, the chemical shift range of the Au(III) complexes is wider (from −7.9 to −44.7 ppm). As noted by Sanner and coworkers,88 in square-planar Au(III) halo trifluoromethyl complexes the 19F chemical shift is strongly influenced by the nature of the cis halide ligand. Thus, in complexes of the type trans-[Au(CF3)X2L] (X = halogen, L = phosphine, nitrogen heterocyclic carbene (NHC), CF3−), δ(19F) increases in the sequence Cl < Br < I (Fig. 1). This effect is also observed in complexes of the type cis-[Au(CF3)2X(PR3)] (Fig. 1), wherein a variation of the halogen X has a stronger impact on the cis-CF3 than on the trans-CF3 ligand.
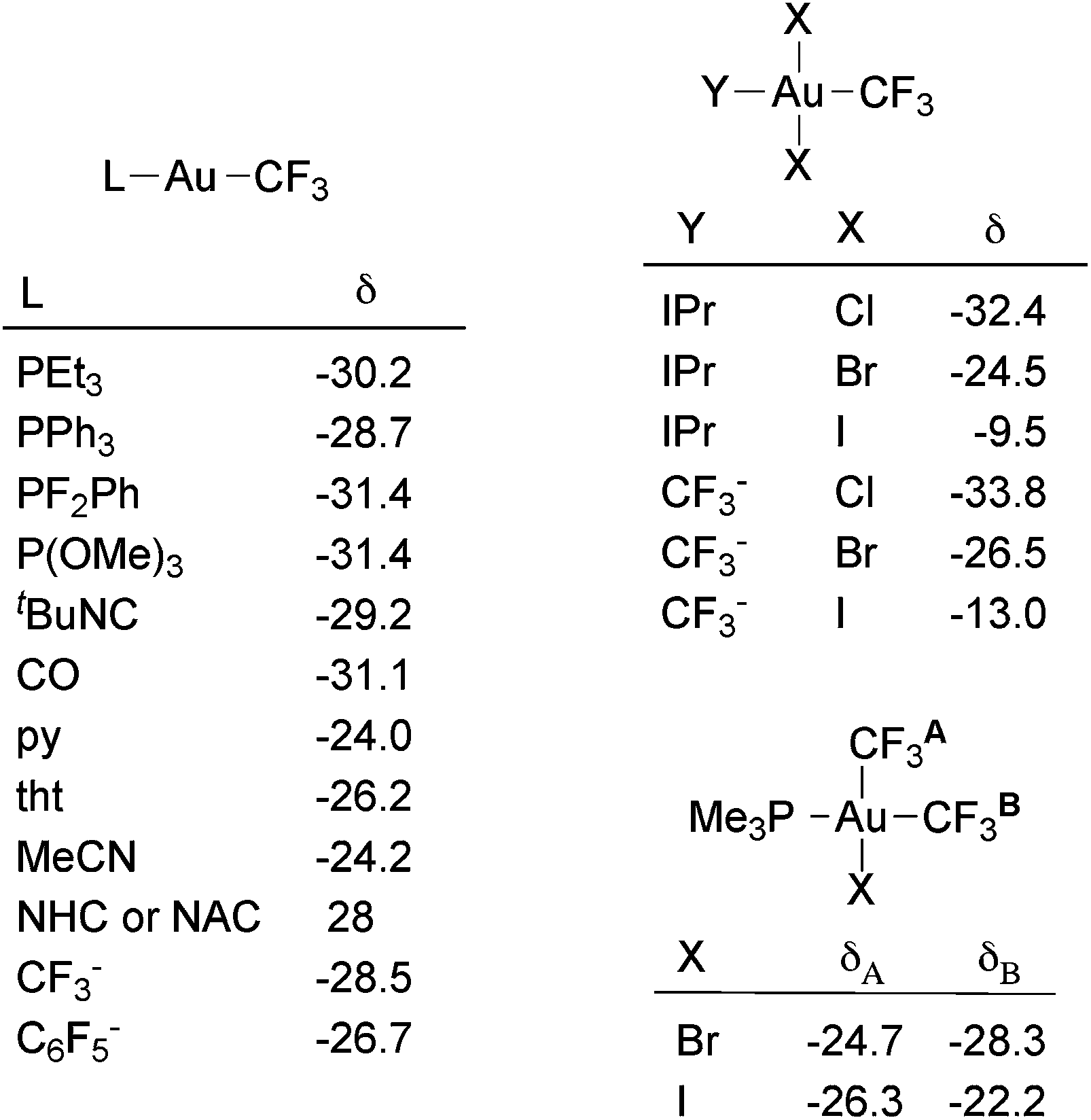 | ||
| Fig. 1 19F chemical shifts (ppm) in gold trifluoromethyl complexes.60,61,81,88 IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene. | ||
Overview of the synthetic methods used for the formation of Au–CF3 bonds
Transmetallation
Owing to the instability of trifluoromethyl lithium or magnesium derivatives,86,87 compounds containing less electropositive metals such as Cu, Zn, Cd and Hg have been used as CF3-transfer agents in earlier times.5 Among these derivatives, the reagent Cd(CF3)2·DME (DME = 1,2-dimethoxyethane) was most popular because of its well-defined composition and high reactivity, which allowed the synthesis of a series of trifluoromethyl complexes of transition metals in low oxidation states,5 including Au(I) complexes (see below). However, owing to the toxicity of Cd compounds and the need to synthesize the reagent, it was replaced by the commercially available Ruppert–Prakash reagent, Me3SiCF3.89,90 This reagent is typically used in the presence of hard Lewis bases, usually F− or RO−, to generate pentacoordinate Si(IV) species, which act as the nucleophilic CF3-transfer agents.91,92 The volatile Me3SiX (X = F, OR) by-products can be easily separated. A convenient variation of this method is the reaction of Me3SiCF3 with a metal fluoro,93–95 hydroxo,96 or alkoxo97,98 complex.The reactions of the Au(I) chloro complexes with trifluoromethyl silver have been recently used for the synthesis of Au(I) trifluoromethyl complexes (see below). The trifluoromethylating reagent can be in situ generated by using Cd(CF3)2 and a silver salt99 or, more conveniently, by the reaction of AgF and Me3SiCF3 in a N-donor solvent, as reported by Naumann, Tyrra and coworkers.100,101 The NMR studies carried out by the same authors showed that these solutions contain equilibrium mixtures of [Ag(CF3)(solvent)] and [Ag(solvent)2][Ag(CF3)2].99,100 Trifluoromethyl silver has also been used in the synthesis of trifluoromethyl derivatives of various elements of the groups 12–16,100,102 and in the trifluoromethylation of organic substrates.34–36,38,39,103
A few Au(III) trifluoromethyl complexes have been prepared by transmetallation reactions between Cd(CF3)2·DME or Me3SiCF3 and Au(III) precursors,61,88,98 although mixtures of Au(I) and Au(III) complexes frequently result from these reactions (see below).61,88
Oxidative addition of ICF3 to Au(I) complexes
This reaction has been reported for complexes of the type [Au(R)L] (R = Me,56,57 aryl,67 CF3;58,88 L = phosphine). When R = Me or CF3, it gives mixtures of the products resulting from (i) R by CF3 ligand exchange between the reaction product [Au(CF3)(R)(I)L] and unreacted [Au(R)L], and (ii) reductive elimination to give RI and [Au(CF3)L]. In all these reactions, evidence for the involvement of free radical intermediates has been obtained (see below).Methods based on the condensation of Au atoms with ˙CF3 radicals or XCF3 (X = Cl, Br)
These methods enabled the synthesis of highly reactive AuCF3 species (see below), although they were obtained in small amounts and the syntheses require special equipment which is not available in common synthetic laboratories.104Au(I) trifluoromethyl complexes
Puddephatt and coworkers isolated the first gold trifluoromethyl complexes by reacting [Au(Me)L] (L = phosphine) with ICF3 (Scheme 1).56,57 Thus, when L = PPh3 the complex [Au(CF3)(PPh3)] was formed, likely by the reductive elimination of MeI from the expected oxidative addition product, [Au(Me)(CF3)(I)L], which was not detected. When L is PMe3 or PMe2Ph, the reactions gave Au(III) trifluoromethyl complexes, and for L = PMePh2 both Au(I) and Au(III) derivatives were obtained (see below). Evidence to show that these reactions occur at least in part through radical intermediates was obtained. Glockling and coworkers reported the formation of [Au(CF3)(PPh3)] in the reaction of [Au{CH(SiMe3)2}(PPh3)] with ICF3 (Scheme 1).105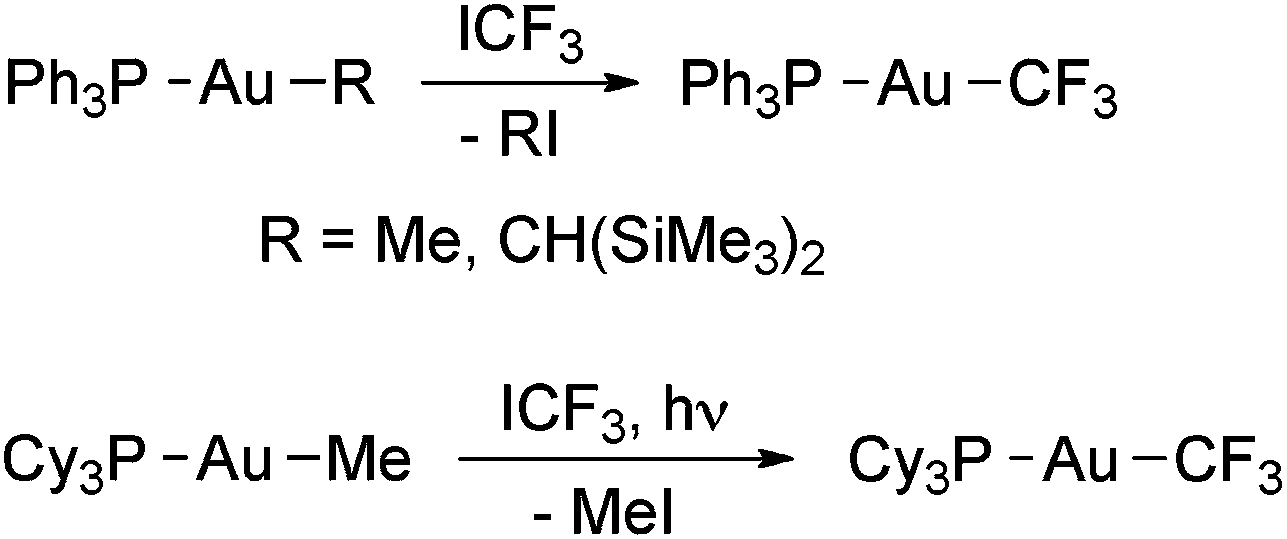 | ||
| Scheme 1 Synthesis of [Au(CF3)L] complexes (L = PPh3,56,57,105 PCy367) by the reaction of Au(I) alkyls with ICF3. | ||
Toste and coworkers have reported that [Au(Me)(PCy3)] does not react with ICF3 in the dark. However, the complex [Au(CF3)(PCy3)] was quantitatively formed upon irradiating the mixture with ambient light (Scheme 1).67 The reaction was fast and no Au(III) intermediates were detected by NMR spectroscopy. Interestingly, HCF3 was formed when the same reaction was run in THF, which may be attributed to H˙ abstraction from the solvent, suggesting that the ˙CF3 radicals are formed by photoactivation of ICF3 in the presence of the Au(I) complex.
A series of [Au(CF3)L] complexes (L = phosphine or fluorophosphine) was obtained by the reactions of [Au(Cl)L] with Cd(CF3)2·DME as reported by the groups of Morrison,58 Sanner88 and Kruck106 (Scheme 2). The volatile complex [Au(CF3)(CNMe)] was similarly prepared by Puddephatt and coworkers and then used for the preparation of gold films by chemical vapour deposition (Scheme 2).71 Decomposition of this complex takes place by CNMe dissociation, followed by Au and C2F6 formation. The only higher gold perfluoroalkyl reported, [Au(C2F5)(PEt3)], was detected by NMR spectroscopy in the mixture of compounds resulting from the reaction between [Au(CF3)(PEt3)] and IC2F5, although it was not isolated.58
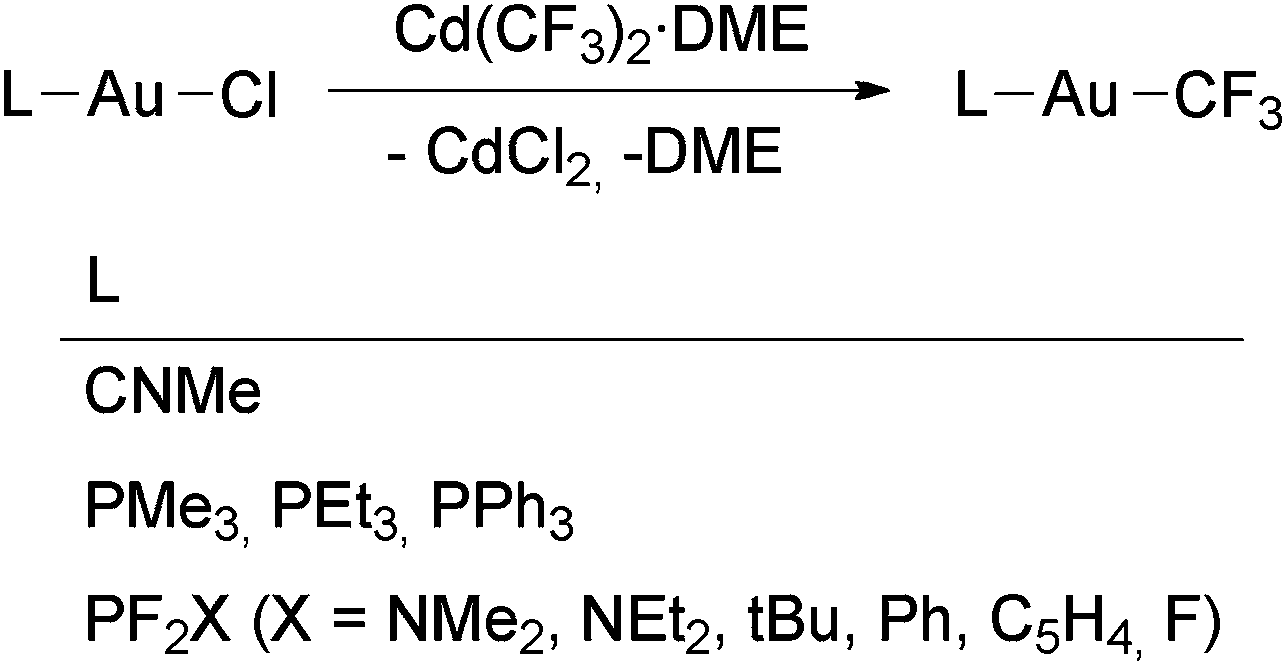 | ||
| Scheme 2 Synthesis of [Au(CF3)L] complexes (L = PR3,58,88 PF2X,106 CNMe71) by transmetallation using Cd(CF3)2·DME. | ||
The reactions of Me3SiCF3 with [Au(OR)L] (R = CH(CF3)2, L = PMe3, PCy3, PPh3, PMe2Ph; R = Ph, L = PCy3, PPh3) were used by Komiya and coworkers to prepare a series of trifluoromethyl complexes of the type [Au(CF3)(phosphine)] (Scheme 3).98 In contrast, the complex [Au(OH)(IPr)] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene)107 did not react with Me3SiCF3 at room temperature or at 80 °C.108
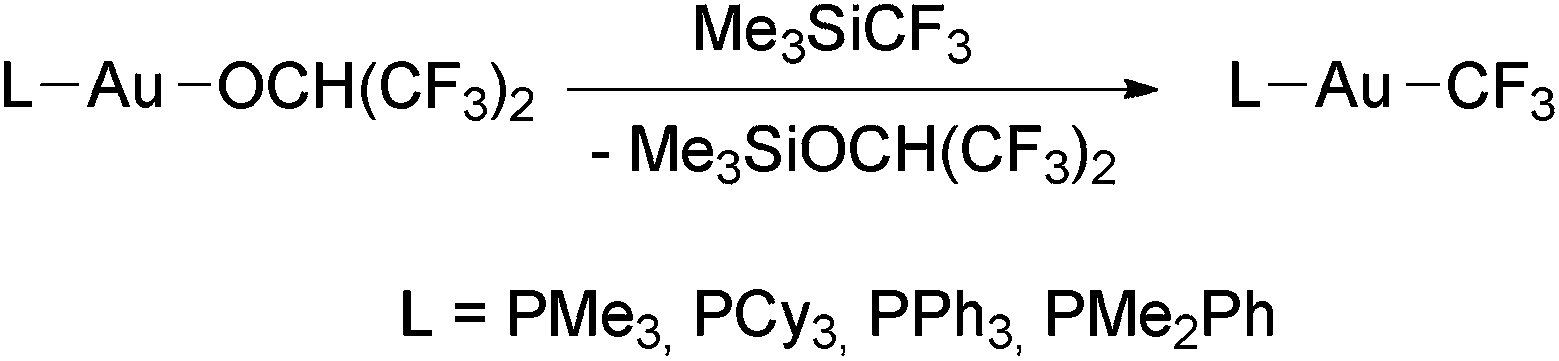 | ||
| Scheme 3 Synthesis of [Au(CF3)L] complexes by the reaction of Au(I) alkoxo complexes with Me3SiCF3.98 | ||
Recently, we reported the synthesis of a family of Au(I) trifluoromethyl complexes by the reaction of Au(I) chloro complexes with AgF and Me3SiCF3 (Scheme 4).81 This method allows the synthesis of new examples of [Au(CF3)L] complexes, where L is phosphine, phosphite, or isonitrile, and the first Au(I) trifluoromethyl complexes containing NHC ligands. The analogous reaction with PPN[AuCl(C6F5)] (PPN+ = Ph3PNPPh3+) gave the anionic complexes [Au(CF3)(C6F5)]−, [Au(CF3)2]− and [Au(C6F5)2]−. Single crystals of PPN[Au(CF3)(C6F5)]− were isolated and used for a crystal structure determination (Fig. 2). Notably, this is the only transition metal complex reported that contains a CF3 and a C6F5 ligand attached to the same metal. The isonitrile complexes reacted with NHEt2 to give Au(I) trifluoromethyl complexes containing nitrogen acyclic carbene (NAC) ligands (Scheme 5).
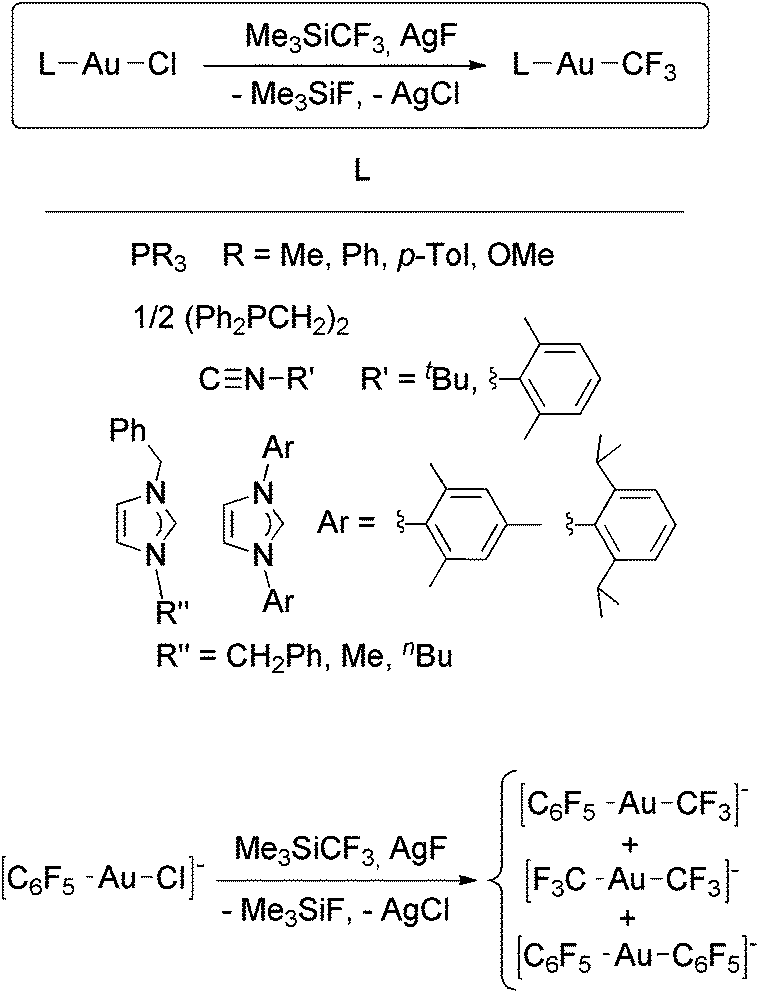 | ||
| Scheme 4 Synthesis of Au(I) trifluoromethyl complexes by the reaction of Au(I) chloro complexes with in situ generated AgCF3.81 | ||
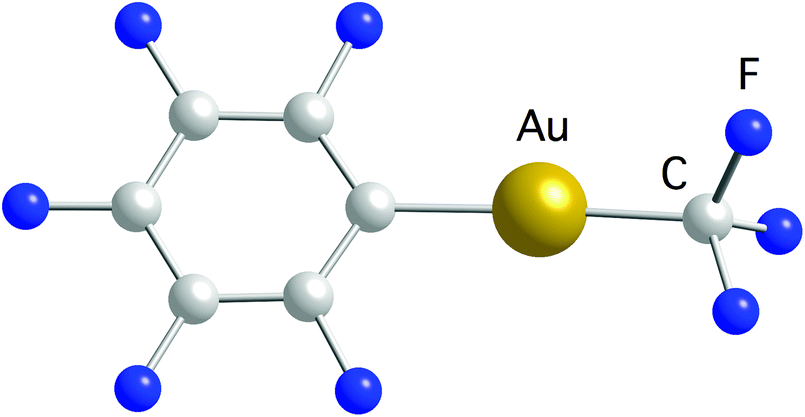 | ||
| Fig. 2 Molecular structure of the anion in the crystal structure of the PPN[Au(CF3)(C6F5)] salt.81 | ||
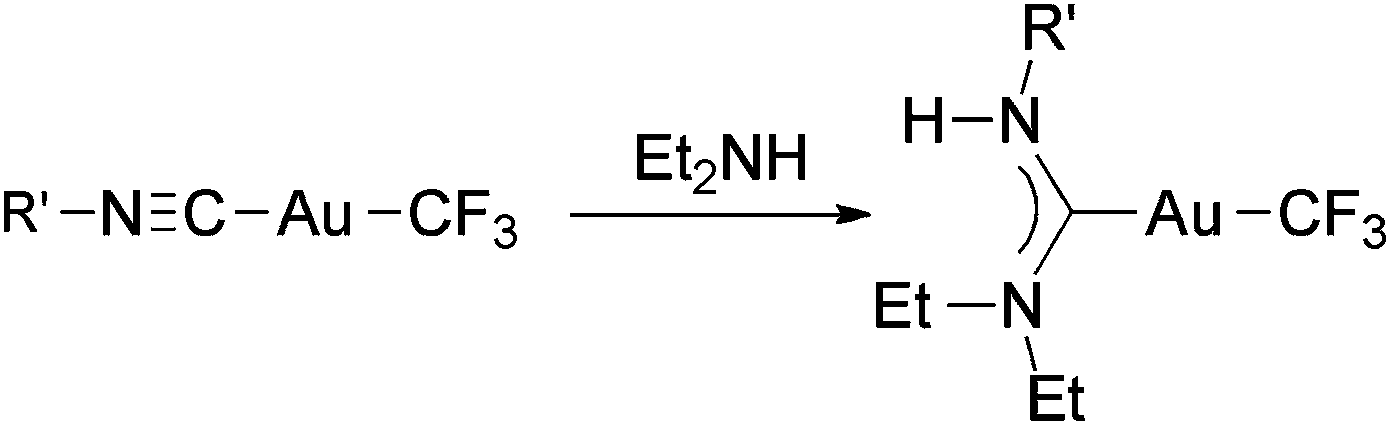 | ||
| Scheme 5 Synthesis of Au(I) trifluoromethyl complexes containing NAC ligands by the reaction of (trifluoromethyl)isocyanide complexes with Et2NH.81 | ||
The groups of Tyrra,60 Forniés59 and Menjón59,61 independently reported the synthesis of the complex [Au(CF3)2]− by using the reactions of AuCl or [AuCl(tht)] with Me3SiCF3 and F− (Scheme 6). This is the only anionic Au(I) trifluoromethyl complex prepared in practical amounts, and it was obtained as PPh4+, NMe4+, PNP+, K(18-crown-6)+ or [Ag(py)2]+ salts. Remarkably, this complex is sensitive to hydrolysis, decomposing to form Au nanoparticles. Tyrra and coworkers reported that the treatment of [Au(CF3)2]− with (CF3CO)2O led to a mixture of CF3COF, [Au(CF3)(CF2OC(O)CF3)]− and [Au(CF2OC(O)CF3)2]−, which were detected by NMR spectroscopy (Scheme 6).60 On the other hand, Forniés, Menjón and coworkers reported that BF3-assisted fluoride abstraction of [Au(CF3)2]− led to nearly quantitative formation of [Au(CF3)(CO)] (Scheme 6).59,61 These reactions probably proceed through the extremely electrophilic Au(I) difluorocarbene109 CF3–Au![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2, which was not detected.
CF2, which was not detected.
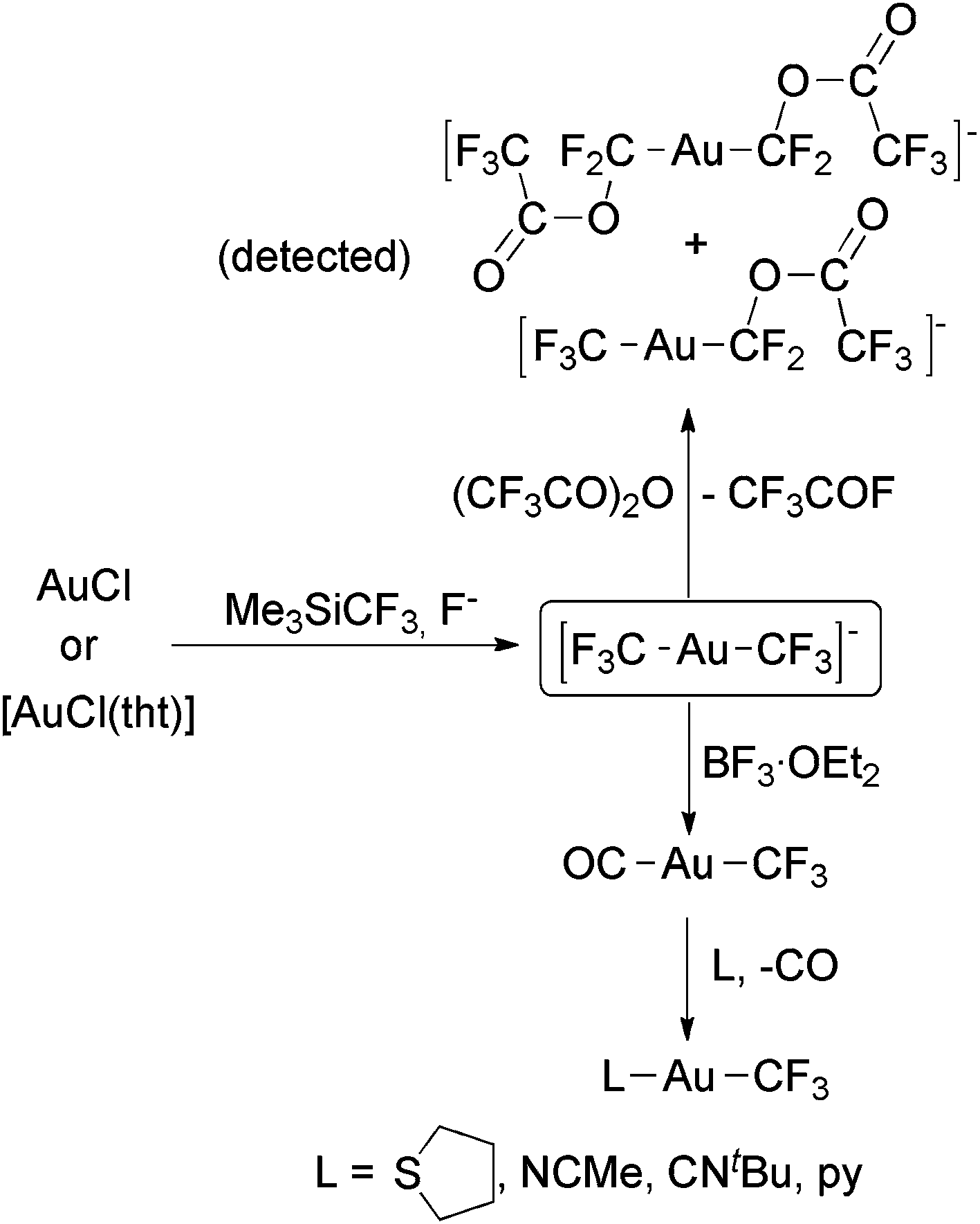 | ||
| Scheme 6 Synthesis and fluoride abstraction reactions of [Au(CF3)2]−.59–61 Substitution reactions of [Au(CF3)(CO)].61 | ||
Despite its thermal instability and extreme moisture-sensitivity, the carbonyl [Au(CF3)(CO)] was isolated and structurally characterized.59 The crystal structure shows a rare trigonal arrangement of molecules connected by aurophilic interactions (Fig. 3). The weak nature of these interactions, the bond distances and angles, and the position of the ν(CO) band in the IR spectrum suggest that the CO ligand acts as a weak σ-donor coordinated to a highly electrophilic AuCF3 fragment. Thus, the CO ligand is very labile, being immediately substituted at 0 °C by tetrahydrothiophene (tht), MeCN, pyridine or tBuNC (Scheme 6). The more thermally robust and air-stable complex [Au(CF3)(tht)] has been proposed as a convenient source for the “Au(CF3)” synthon,61 in analogy to [AuR(tht)] being a source for complexes containing the “AuR” fragment.110–114
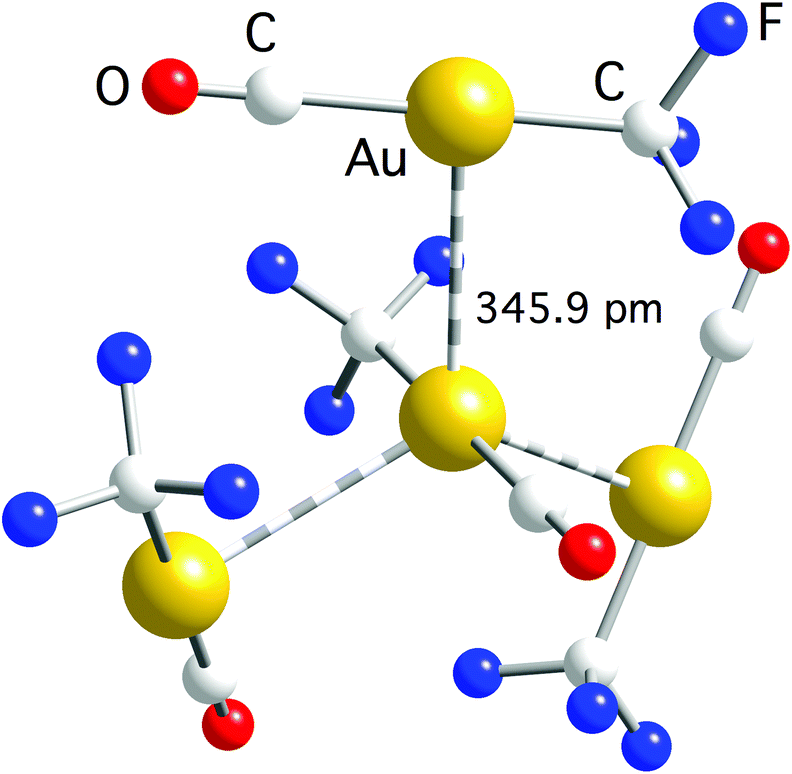 | ||
| Fig. 3 Trigonal arrangement of molecules connected by aurophilic interactions in the crystal structure of [Au(CF3)(CO)]. Owing to the C3 symmetry axis, the three Au–Au bond distances are equal to the indicated one.59 | ||
The anions [Au(CF3)X]− (X = F, OC(O)CF3) have been detected by mass spectrometry in the collision-induced dissociation of the [Au(OC(O)CF3)2]− ion.115 [Au(CF3)Cl]− was detected by IR spectroscopy in the reaction of laser-ablated Au atoms with ClCF3.116
Au(II) trifluoromethyl complexes
Only two Au(II) trifluoromethyl complexes have been reported to date (Scheme 7), both being diamagnetic and displaying strong Au–Au bonds.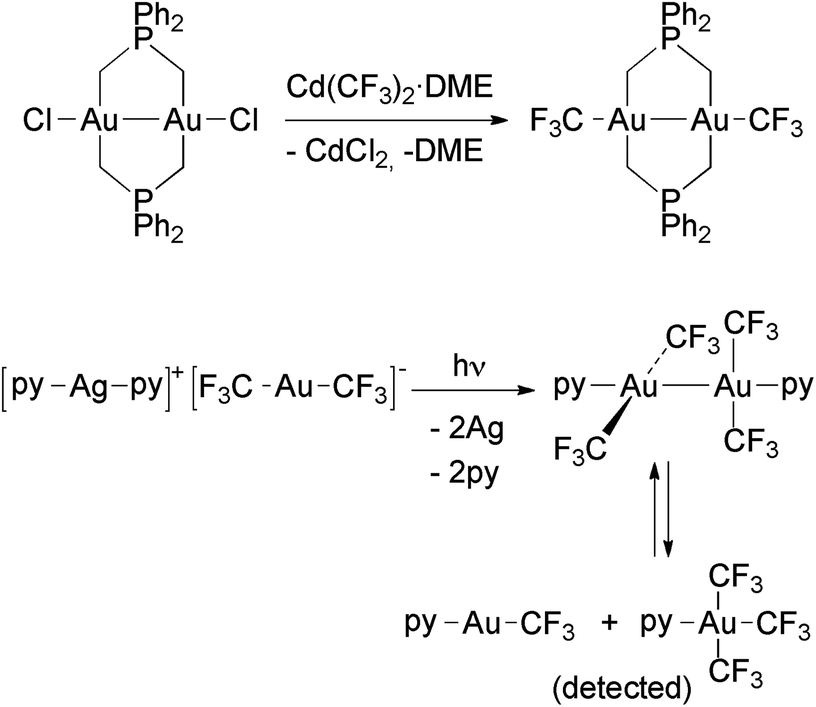 | ||
| Scheme 7 Formation of Au(II) trifluoromethyl complexes by (i) transmetallation in an Au(II) chloro complex,117 and (ii) photo-disproportionation of [Ag(py)2][Au(CF3)2].118 The unstable Au(II) complex [Au2(CF3)4(py)2] disproportionates in solution to an Au(I) and an Au(III) complex. | ||
The first example is [Au2(CF3)2{μ-Ph2P(CH2)2}2], prepared by using the reaction of [Au2Cl2{μ-Ph2P(CH2)2}2] with Cd(CF3)2·DME, which contains two bridging bis(ylide) ligands.117 The other compound is [Au2(CF3)4(py)2], obtained after irradiating a solution of [Ag(py)2][Au(CF3)2] with UV light. In this remarkable reaction, the [Ag(py)2]+ cation is reduced to metallic Ag and oxidizes [Au(CF3)2]− to Au(II).118 This compound is a rare example of a dinuclear Au(II) complex where the Au–Au bond is not supported by the bridging ligands, and it has been the object of a theoretical study.119 The computational results showed that the bond between both the Au(II) centers is of covalent nature. Moreover, the analysis of the bonding orbitals suggested that they can be described by an uncommon 6s6pz5dxy hybridization type. [Au2(CF3)4(py)2] was characterized in the solid state by single crystal X-ray diffraction (Fig. 4), but no data could be obtained in solution because it spontaneously disproportionates into a 1:1 mixture of [Au(CF3)(py)] and [Au(CF3)3(py)], both being detected by NMR spectroscopy.
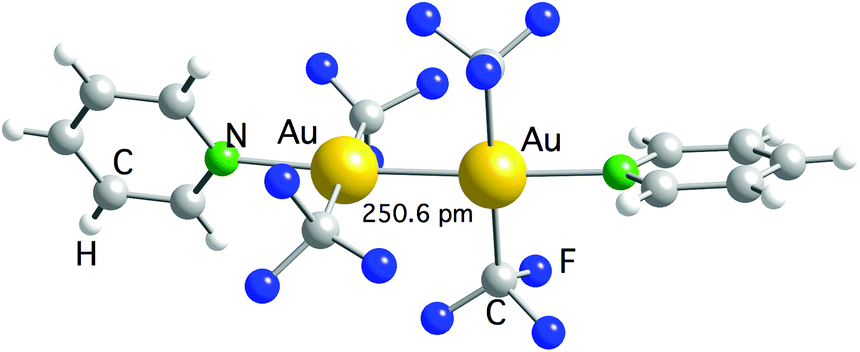 | ||
| Fig. 4 Molecular structure of [Au(CF3)4(py)2]. The Au–Au bond distance is indicated.118 | ||
Au(III) trifluoromethyl complexes
Au(III) trifluoromethyl complexes have been prepared either (i) by oxidative addition of ICF3 to Au(I) complexes, (ii) by halogenation of Au(I) trifluoromethyl complexes, or (iii) by trifluoromethylation of Au(III) complexes.The first reported Au(III) perfluoroalkyls were obtained by Puddephatt and coworkers in the reaction of [Au(Me)L] with ICF3 (Scheme 8), which gave mixtures of cis- and trans-[Au(CF3)Me2L] (L = PMe3, PMe2Ph) in variable ratios depending on the solvent and the nature of the ligand L.56,57 From these mixtures, only the trans isomers were isolated. These reactions seem to proceed by the oxidative addition of ICF3 to the Au(I) complexes to give a [Au(CF3)(Me)IL] intermediate, which undergoes Me for I ligand exchange with the unreacted [Au(Me)L]. The intermediate complex was detected in certain experiments and independently prepared by the reaction of trans-[Au(CF3)Me2(PMe3)] with HCl and NaI. Evidence of a radical chain mechanism for these oxidative addition reactions was obtained (Scheme 9).
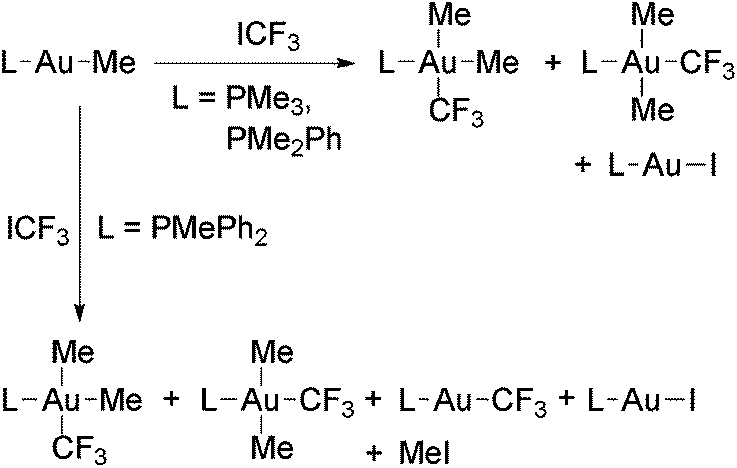 | ||
| Scheme 8 Reactions of Au(I) methyl complexes with ICF3 which lead to mixtures of Au(I) and Au(III) complexes.56,57 | ||
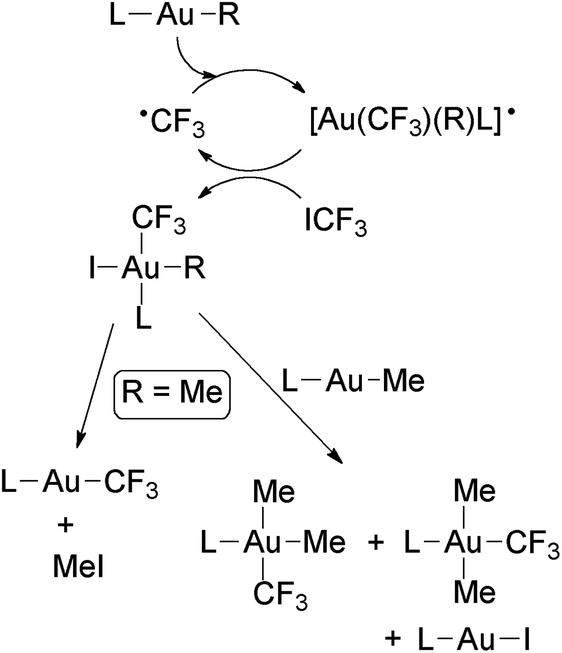 | ||
| Scheme 9 Proposed radical chain mechanism for the oxidative addition of ICF3 to Au(I) complexes. Subsequent evolution of the formed Au(III) trifluoromethyl complexes with R = Me to give mixtures of Au(I) and Au(III) complexes.67 | ||
The photooxidative addition of ICF3 to [Au(Ar)L] (L = PPh3, Ar = 4-X-C6H4 (X = Me, F); L = PCy3, Ar = 4-X-C6H4 (X = H, Me, F, MeO), 3,5-F2C6H4) has been reported by Toste's group (Scheme 10).67 The complexes [Au(CF3)(Ar)I(PR3)] were isolated in high yields as single isomers. Detailed mechanistic studies support a radical chain mechanism as depicted in Scheme 9. The radical chain is initiated by electron transfer from the Au(I) complex or PCy3 to a photoexcited molecule of ICF3, giving rise to I− and a ˙CF3 radical. However, in contrast to the analogous Au(III) complexes containing a methyl ligand (see above), the complexes [Au(CF3)(Ar)I(PR3)] are stable against reductive elimination and do not undergo R for I ligand exchange with the starting [Au(R)L] complexes.
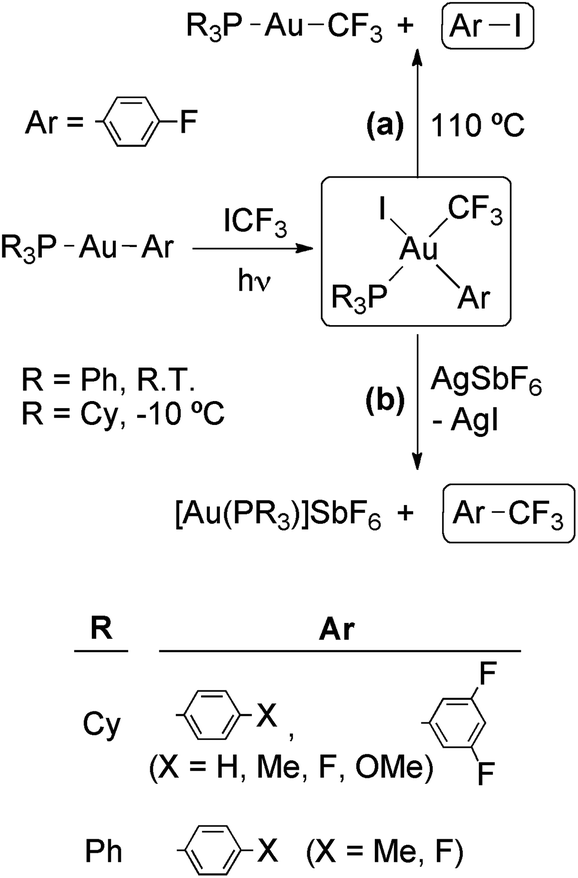 | ||
| Scheme 10 Photooxidative addition of ICF3 to Au(I) aryl complexes. The resulting Au(III) complexes may undergo (a) thermal reductive elimination of ArI or (b) fast reductive elimination of ArCF3 after removal of an I− ligand.67 | ||
Interestingly, the complexes [Au(CF3)(Ar)I(PR3)] underwent thermal reductive elimination to afford ArI and [Au(CF3)(PPh3)] (Scheme 10, reaction (a)). The complete inhibition of this reaction in the presence of added PPh3 suggests that it proceeds through PPh3 dissociation to give a neutral tricoordinate intermediate which eliminates ArI. In contrast, a very fast reductive elimination of ArCF3 was observed when the complexes [Au(CF3)(Ar)I(PR3)] were treated with AgSbF6 to abstract an I− anion and generate a cationic tricoordinate intermediate (Scheme 10, reaction (b)). The last process is remarkable taking into account (i) the known reluctance of transition-metal trifluoromethyl complexes to undergo reductive elimination of trifluoromethylated products, and (ii) the potential synthetic utility for a gold-catalyzed aromatic trifluoromethylation reaction. In this respect, the authors of this study have demonstrated the feasibility of each individual step in a hypothetical catalytic cycle for the gold-catalyzed photoactivated trifluoromethylation of an arylstannane.
The reactions of the Au(I) trifluoromethyl complexes [Au(CF3)L] (L = phosphine) or [Au(CF3)2]− with an excess of Cl2, Br2 or I2 afforded Au(III) complexes resulting from the oxidative addition of the halogens (Scheme 11, reactions (a) and (c)).61,88 In these complexes, the halo ligands are disposed mutually in trans (only small amounts of the cis isomers were observed for L = PMe3 and PEt3). When equimolar amounts of [Au(CF3)L] and halogen were used, the additional products resulting from CF3/X exchange with the remaining starting complex were also observed (Scheme 11, reaction (b)). The Au(III) complexes obtained in these reactions are stable against thermal reductive elimination.
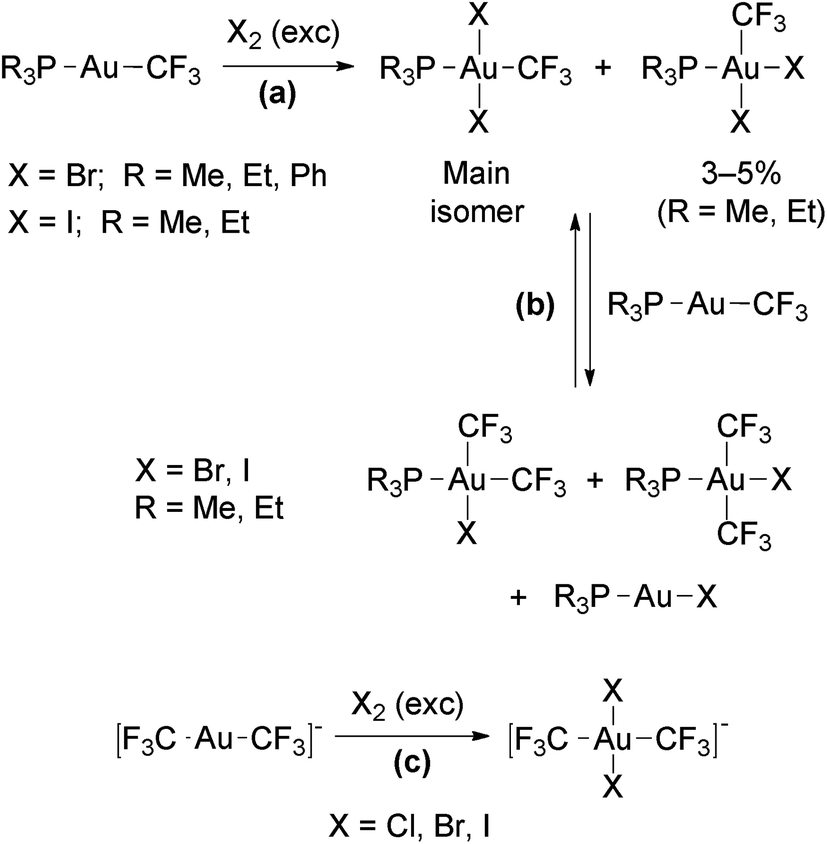 | ||
| Scheme 11 Oxidative addition of halogens to neutral122 and anionic98 Au(I) trifluoromethyl complexes. | ||
The complexes [Au(CF3)(IPr)] react similarly with PhICl2, Br2, I2 or ICl, affording exclusively the isomers with the halogens disposed mutually in trans (Scheme 12). These complexes are thermally stable except in the case of trans-[Au(CF3)I2(IPr)], which reductively eliminates ICF3 even at low temperature. However, on irradiating with UV light the compounds [Au(CF3)XY(IPr)] (X = Y = Cl or Br; X = Cl, Y = I) underwent reductive elimination of halotrifluoromethane (Scheme 12). Remarkably, the chloro(iodo) complex selectively eliminated ICF3 instead of ClCF3.81
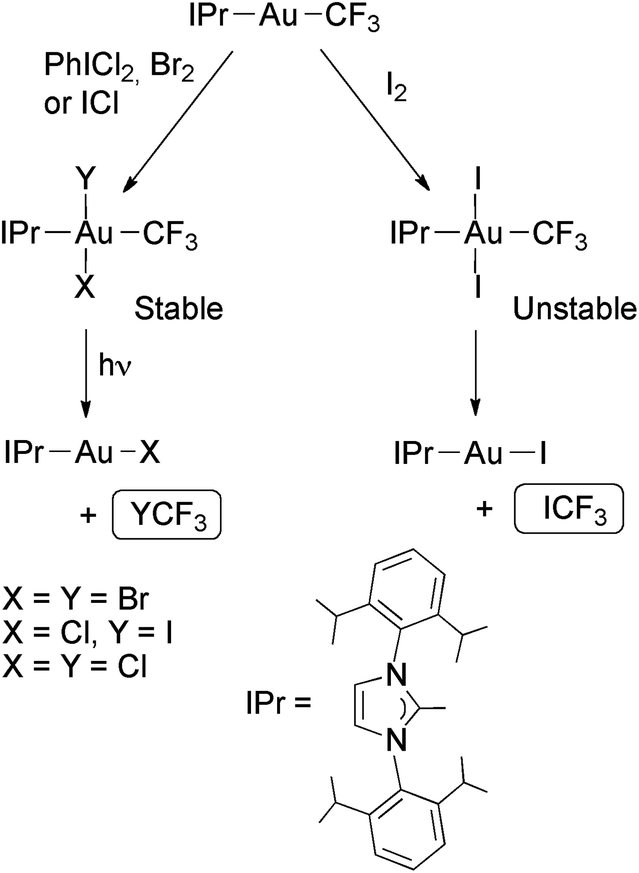 | ||
| Scheme 12 Oxidative addition of halogens to Au(I) trifluoromethyl complexes containing a bulky NHC ligand. Photochemical or thermal reductive eliminations of halotrifluoromethanes.81 | ||
The oxidative addition of ICF3 to [Au(CF3)(PR3)] (R = Me,58,88 Et88) gave mixtures of cis- and trans-[Au(CF3)2I(PR3)], where the cis isomer is the predominant product (Scheme 13).88 Inhibition of the reactions by a radical scavenger (galvinoxyl) suggests that they also proceed through a radical mechanism. The partial reactions between [Au(CF3)2I(PEt3)] and [Au(CF3)(PR3)] to give mixtures of [Au(CF3)3(PR3)] and [AuI(PR3)] have been also reported (Scheme 13).58 No reductive elimination of ICF3 or CF3–CF3 from these Au(III) complexes was described.
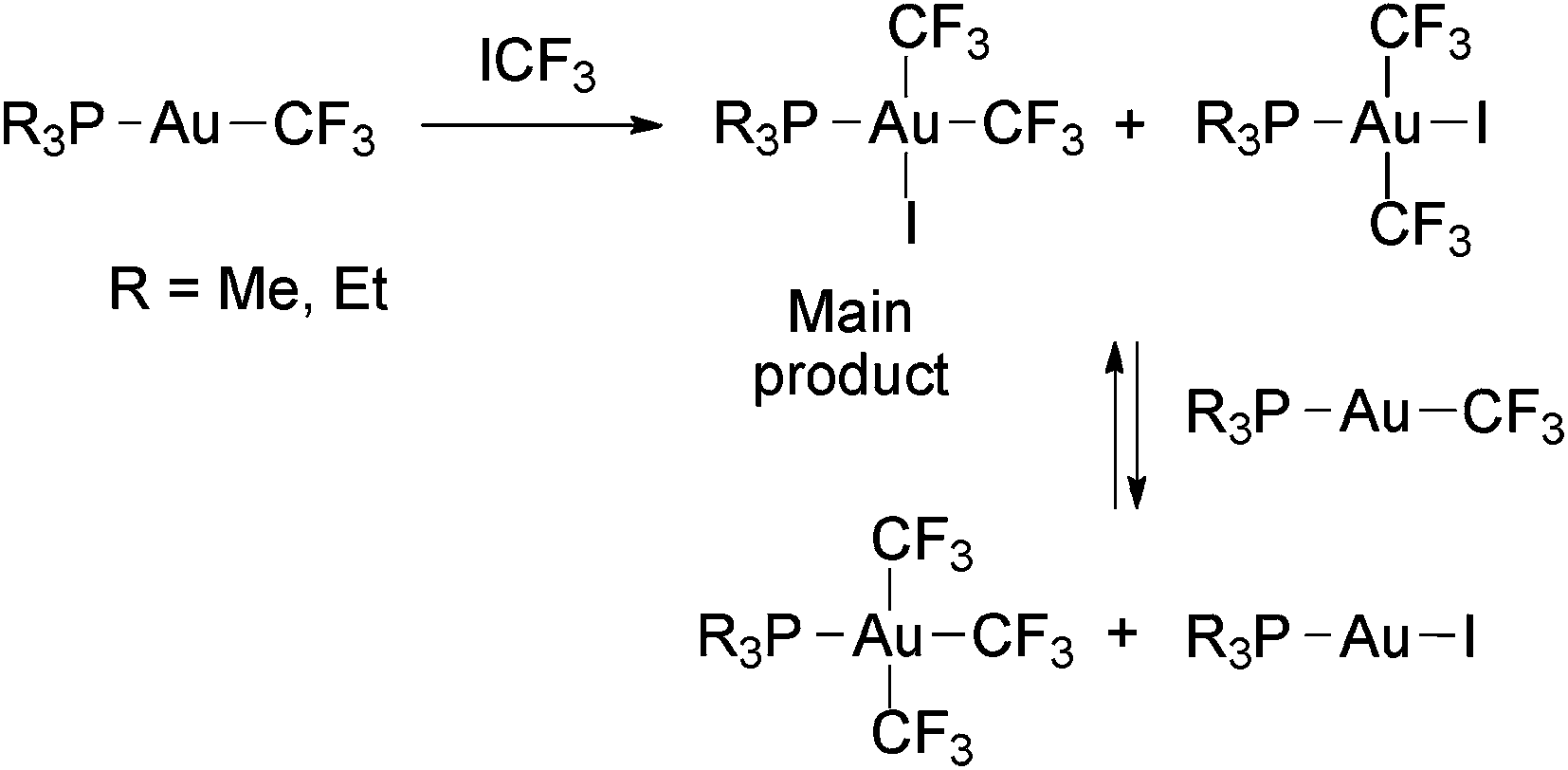 | ||
| Scheme 13 Oxidative addition of ICF3 to Au(I) trifluoromethyl complexes. A further ligand exchange with the remaining starting material leads to tris(trifluoromethyl) complexes.58,88 | ||
The dinuclear complexes [Au2(CF3)4(μ-X)2] (X = Br, I) were prepared by condensation of Au atoms with XCF3 (Scheme 14). The isolated compounds are moderately stable and have been characterized by single crystal X-ray diffraction and NMR spectroscopy.120
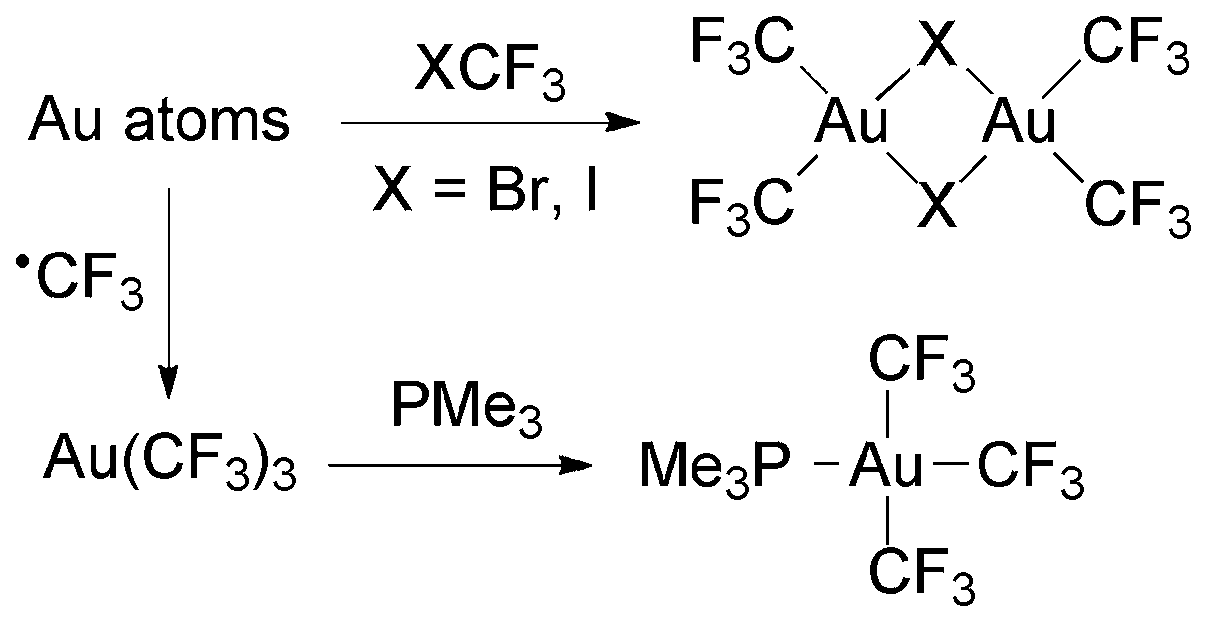 | ||
| Scheme 14 Synthesis of Au(III) trifluoromethyl complexes by the reaction of Au atoms with halotrifluoromethanes or trifluoromethyl radicals.120,121 | ||
An unstable compound formulated as Au(CF3)3 was isolated from the reaction of trifluoromethyl radicals and Au atoms (Scheme 14).121 The NMR data of its PMe3 adduct are in agreement with those reported for [Au(CF3)3(PMe3)]88 (see below).
Most attempts to obtain Au(III) trifluoromethyls by transmetallation reactions of Au(III) complexes and Cd(CF3)2·DME resulted in the formation of Au(I) complexes as the side or main products, although the desired Au(III) trifluoromethyls are stable once isolated.88 For instance, the reaction of [Au(CF3)I2(PMe3)] with Cd(CF3)2·DME gave [Au(CF3)(PMe3)], ICF3 and CdI2 instead of the expected [Au(CF3)2I(PMe3)]. The reaction of [Au(CF3)2I(PMe3)] (prepared from [Au(CF3)(PMe3)] and ICF3) with Cd(CF3)2·DME gave [Au(CF3)3(PMe3)] in high yield (Scheme 15), although the reaction had to be carried out under an ICF3 atmosphere, because otherwise [Au(CF3)(PMe3)] and ICF3 could also be formed.88 The tris(trifluoromethyl) complex is a thermally, air and moisture stable crystalline solid, which can be purified by sublimation.58,88 The complexes cis-[Au(CF3)(Me)2L] (L = PMe3, PEt3, PMe2Ph or PMePh2) have been also obtained by Komiya and coworkers in the reaction of cis-[Au(OPh)(Me)2L] with Me3SiCF3 (Scheme 15).98
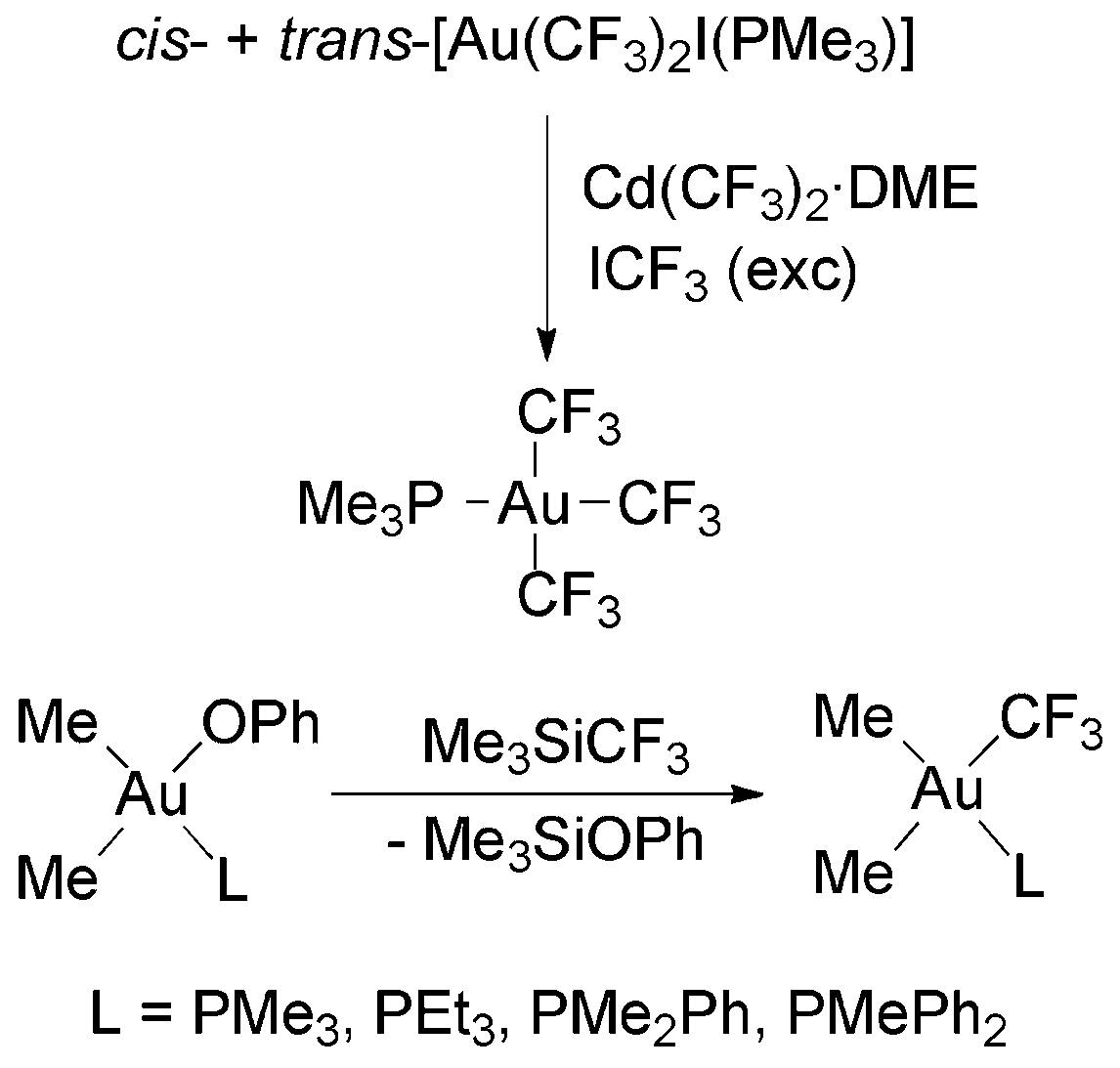 | ||
| Scheme 15 Synthesis of Au(III) trifluoromethyl complexes by transmetallation.88,98 | ||
[Au(CF3)4]− was firstly prepared by Neumann and coworkers as its PPN+ salt, although the synthetic method was not reported.122 Recently, Menjón and coworkers described the synthesis of this complex by the reaction of AuCl3, Me3SiCF3 and CsF (Scheme 16).61 During the reaction, partial reduction of the metal took place to give a mixture of [Au(CF3)2]− and [Au(CF3)4]−. Fortunately, the mixture could be separated by using PPh4+ as the countercation due to the different solubilities of the Au(I) and Au(III) salts. (PPh4)[Au(CF3)4] is remarkably stable, because it melts at 125 °C and does not show significant weight loss until it is heated at 370 °C. In addition, delocalization of the negative charge over 12 fluorine atoms, makes [Au(CF3)4]− an uncommon weakly coordinating anion with a flat shape,123 which has been used as a component of organic superconductors.122,124–127
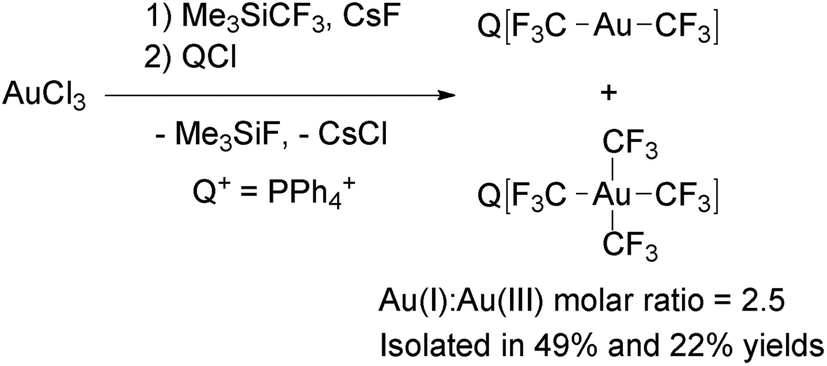 | ||
| Scheme 16 Synthesis of complexes [Au(CF3)x]− (x = 2, 4) from AuCl3. The PPh4+ salts were separated by crystallization.61 | ||
Finally, the reaction of [NnBu4][Au(CN)4] with ClF afforded a mixture of complexes of the type [AuFxCly(CF3)4−x−y]− (x = 0–4, y = 0–2). Metathesis of this mixture with Me3SiX (X = Cl or CN) gave Me3SiF and new mixtures of the composition [AuXx(CF3)4−x]− (x = 0–4). The components of these mixtures were identified by NMR spectroscopy but could not be separated.128
Summary and outlook
Au(I), Au(II) and Au(III) trifluoromethyl complexes have been reported being, in general, more thermally stable than their methyl analogues. All known Au(I) complexes are of the [Au(CF3)L] type, except [Au(CF3)2]− and [Au(CF3)(C6F5)]−. Most of them have been obtained by using Me3SiCF3 as the nucleophilic trifluoromethylating agent in the presence of fluoride salts as activators of the silane. Among all the methodologies tested, the combination of Me3SiCF3 and AgF is the most versatile one. Only two Au(II) trifluoromethyl complexes have been described, both displaying an Au–Au bond. Au(III) complexes containing 1, 2, 3 or 4 trifluoromethyl ligands have been reported. Most of them have been prepared by the oxidative addition of ICF3 or halogens to Au(I) complexes.Overall, the available synthetic methods still suffer from important drawbacks, such as the use of expensive reagents, the low yields in selected cases and the narrow scope. Thus, the development of efficient methodologies for the introduction of trifluoromethyl and, specially, higher perfluoroalkyl groups in the coordination sphere of gold is highly desirable, and it should boost the application of these compounds in different fields.
Potentially interesting applications may emerge from the singular structural properties and reactivity of these compounds. Thus, the observation of unusually fast reductive eliminations of trifluoromethylated compounds in the Au(III) trifluoromethyl complexes could be applied in the catalytic trifluoromethylation of organic compounds. Other opportunities for the application of these compounds in homogeneous catalysis may arise from the strong electron-withdrawing ability of the trifluoromethyl groups, which enhances the Lewis acidity of the metal center, or from the α-fluoride abstraction reactions, which generate highly reactive difluorocarbene intermediates. In addition, the development of efficient methods for the synthesis of the anions [Au(CnF2n+1)4]− would improve their use as flat, weakly coordinating, fluorophilic anions in synthesis and materials science.
Note added in proof
After submission of this review, Toste and coworkers have reported a study of the reductive elimination of ArX and ArCF3 in complexes [Au(CF3)(Ar)X(PPh3)] (X = F, Cl, Br, I). See M. S. Winston, W. J. Wolf, F. D. Toste J. Am. Chem. Soc. 2015, 137, 7921.Acknowledgements
We thank the Spanish Ministerio de Economía y Competitividad (grant CTQ2011-24016, with FEDER support) and Fundación Séneca (grant 04539/GERM/06) for financial support.Notes and references
- F. G. A. Stone, J. Fluorine Chem., 1999, 100, 227 CrossRef CAS.
- M. I. Bruce and F. G. A. Stone, in Prep. Inorg. React., ed. W. L. Jolly, Interscience Publishers, Bristol, 1968, vol. 4, p. 177 Search PubMed.
- A. G. Algarra, V. V. Grushin and S. A. MacGregor, Organometallics, 2012, 31, 1467 CrossRef CAS.
- R. P. Hughes, Adv. Organomet. Chem., 1990, 31, 183 CrossRef CAS.
- J. A. Morrison, Adv. Organomet. Chem., 1993, 35, 211 CrossRef CAS.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS.
- G. W. Gribble, Acc. Chem. Res., 1998, 31, 141 CrossRef CAS.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed.
- T. Hiyama, in Organofluorine Compounds: Chemistry and Applications, Springer-Verlag, Berlin, 2000 Search PubMed.
- R. C. Chambers, in Fluorine in Organic Chemistry, Blackwell, Oxford, UK, 2004 Search PubMed.
- P. Kirsch, in Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed.
- P. M. Murphy, C. S. Baldwin and R. C. Buck, J. Fluorine Chem., 2012, 138, 3 CrossRef CAS PubMed.
- I. T. Horváth, Acc. Chem. Res., 1998, 31, 641 CrossRef.
- L. P. Barthel-Rosa and J. A. Gladysz, Coord. Chem. Rev., 1999, 190–192, 587 CrossRef CAS.
- M. A. Úbeda and R. Dembinski, J. Chem. Educ., 2006, 83, 84 CrossRef.
- W. Zhang and C. Cai, Chem. Commun., 2008, 5686 RSC.
- W. Zhang, Tetrahedron, 2003, 59, 4475 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- R. J. Lundgren and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 9322 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed.
- O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed.
- Z. Jin, G. Hammond and B. Xu, Aldrichimica Acta, 2012, 45, 6783 Search PubMed.
- C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed.
- T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed.
- B. Lantaño, M. R. Torviso, S. M. Bonesi, S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2015, 285, 76 CrossRef PubMed.
- T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed.
- J.-J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z.-J. Liu, X. Lu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 8436 CrossRef CAS PubMed.
- H. Serizawa, K. Aikawa and K. Mikami, Chem. – Eur. J., 2013, 19, 17692 CrossRef CAS PubMed.
- N. O. Ilchenko, P. G. Janson and K. J. Szabó, Chem. Commun., 2013, 49, 6614 RSC.
- M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628 CrossRef CAS PubMed.
- D. M. Wiemers and D. J. Burton, J. Am. Chem. Soc., 1986, 108, 832 CrossRef CAS.
- D. J. Burton and L. Lu, Top. Curr. Chem., 1997, 193, 45 CrossRef CAS.
- Y. Ye and M. Sandford, Synlett, 2012, 2005 CAS.
- Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955 CrossRef CAS PubMed.
- X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330 CrossRef CAS PubMed.
- S. Seo, J. B. Taylor and M. F. Greaney, Chem. Commun., 2013, 49, 6385 RSC.
- A. Hafner and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 3713 CrossRef CAS PubMed.
- M. M. Kremlev, A. I. Mushta, W. Tyrra, D. Naumann, H. T. M. Fischer and Y. L. Yagupolskii, J. Fluorine Chem., 2007, 128, 1385 CrossRef CAS PubMed.
- N. D. Ball, J. B. Gary, Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 7577 CrossRef CAS PubMed.
- E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CrossRef CAS PubMed.
- V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2006, 128, 12644 CrossRef CAS PubMed.
- A. Maleckis and M. S. Sanford, Organometallics, 2014, 33, 2653 CrossRef CAS.
- R. N. Loy and M. S. Sanford, Org. Lett., 2011, 13, 2548 CrossRef CAS PubMed.
- N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 2878 CrossRef CAS PubMed.
- Y. Ye, N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 14682 CrossRef CAS PubMed.
- X. Mu, S. Chen, X. Zhen and G. Liu, Chem. – Eur. J., 2011, 17, 6039 CrossRef CAS PubMed.
- T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC.
- L.-P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC.
- Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed.
- A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed.
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2012, 51, 12935 CrossRef CAS PubMed.
- H. G. Raubenheimer and H. Schmidbaur, J. Chem. Educ., 2014, 91, 2024 CrossRef CAS.
- A. Johnson and R. J. Puddephatt, Inorg. Nucl. Chem. Lett., 1973, 9, 1175 CrossRef CAS.
- A. Johnson and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 1976, 1360 RSC.
- H. K. Nair and J. A. Morrison, J. Organomet. Chem., 1989, 376, 149 CrossRef CAS.
- S. Martínez-Salvador, J. Forniés, A. Martín and B. Menjón, Angew. Chem., Int. Ed., 2011, 50, 6571 CrossRef PubMed.
- D. Zopes, S. Kremer, H. Scherer, L. Belkoura, I. Pantenburg, W. Tyrra and S. Mathur, Eur. J. Inorg. Chem., 2011, 273 CrossRef CAS PubMed.
- S. Martínez-Salvador, L. R. Falvello, A. Martín and B. Menjón, Chem. – Eur. J., 2013, 19, 14540 CrossRef PubMed.
- W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159 CrossRef CAS PubMed.
- J. Vicente, M. D. Bermúdez, J. Escribano, M. P. Carrillo and P. G. Jones, J. Chem. Soc., Dalton Trans., 1990, 3083 RSC.
- J. Vicente, M. D. Bermúdez and F. J. Carrión, Inorg. Chim. Acta, 1994, 220, 1 CrossRef CAS.
- J. Vicente, M. D. Bermúdez, F. J. Carrión and P. G. Jones, Chem. Ber., 1996, 129, 1395 CrossRef CAS PubMed.
- J. Vicente, M. D. Bermúdez and J. Escribano, Organometallics, 1991, 10, 3380 CrossRef CAS.
- M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777 CrossRef CAS PubMed.
- D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398 CrossRef CAS.
- P. M. Treichel and F. G. A. Stone, Adv. Organomet. Chem., 1964, 1, 143 CrossRef CAS.
- M. A. García-Monforte, S. Martínez-Salvador and B. Menjón, Eur. J. Inorg. Chem., 2012, 4945 CrossRef PubMed.
- N. H. Dryden, J. G. Shapter, L. L. Coatsworth, P. R. Norton and R. J. Puddephatt, Chem. Mater., 1992, 4, 979 CrossRef CAS.
- R. J. Puddephatt and I. Treurnicht, J. Organomet. Chem., 1987, 319, 129 CrossRef CAS.
- S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1977, 99, 8440 CrossRef CAS.
- S. Komiya, S. Ochiai and Y. Ishizaki, Inorg. Chem., 1992, 31, 3168 CrossRef CAS.
- The mean C–F distances in Au(I), Au(II) and Au(III) compounds are 1.368, 1.306 and 1.323 Å, respectively, whereas the mean XF2C–F (X ≠ metal) distance is 1.33 Å (see A. Yokozeki and S. H. Bauer, Top. Curr. Chem., 1973, 53, 71–119 CrossRef CAS and F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC ).
- The mean Au–C–F and F–C–F angles are 115.4° and 102.9° (Au(I)), 116.4° and 101.6° (Au(II)), 113.6° and 105.1° (Au(III)).
- P. D. Gavens, J. J. Guy, M. J. Mays and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 137 CrossRef.
- P. Tasker, A. Parkin, D. Coventry, S. Parsons and D. Messenger, personal communication.
- U. Florke, H.-J. Haupt and P. G. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 609 CrossRef.
- M. T. Johnson, J. M. J. van Rensburg, M. Axelsson, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2011, 2, 2373 RSC.
- M. Blaya, D. Bautista, J. Gil-Rubio and J. Vicente, Organometallics, 2014, 33, 6358 CrossRef CAS.
- D. Zhu, S. V. Lindeman and J. K. Kochi, Organometallics, 1999, 18, 2241 CrossRef CAS.
- M. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768 CrossRef CAS.
- W. A. G. Graham, Inorg. Chem., 1968, 7, 315 CrossRef CAS.
- T. Leyssens, D. Peeters, A. G. Orpen and J. N. Harvey, Organometallics, 2007, 26, 2637 CrossRef CAS.
- G. K. S. Prakash, F. Wang, Z. Zhang, R. Haiges, M. Rahm, K. O. Christe, T. Mathew and G. A. Olah, Angew. Chem., Int. Ed., 2014, 53, 11575 CrossRef CAS PubMed.
- W. B. Farnham, Chem. Rev., 1996, 96, 1633 CrossRef CAS PubMed.
- R. D. Sanner, J. H. Satcher Jr. and M. W. Droege, Organometallics, 1989, 8, 1498 CrossRef CAS.
- G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757 CrossRef CAS PubMed.
- X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed.
- N. Maggiarosa, W. Tyrra, D. Naumann, N. V. Kirij and Y. L. Yagupolskii, Angew. Chem., Int. Ed., 1999, 38, 2252 CrossRef CAS.
- W. Tyrra, M. M. Kremlev, D. Naumann, H. Scherer, H. Schmidt, B. Hoge, I. Pantenburg and Y. L. Yagupolskii, Chem. – Eur. J., 2005, 11, 6514 CrossRef CAS PubMed.
- J. Vicente, J. Gil-Rubio and D. Bautista, Inorg. Chem., 2001, 40, 2636 CrossRef CAS.
- J. Vicente, J. Gil-Rubio, J. Guerrero-Leal and D. Bautista, Organometallics, 2004, 23, 4871 CrossRef CAS.
- F. L. Taw, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2003, 125, 14712 CrossRef CAS PubMed.
- J. Vicente, J. Gil-Rubio, J. Guerrero-Leal and D. Bautista, Organometallics, 2005, 24, 5634 CrossRef CAS.
- G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600 CrossRef CAS PubMed.
- Y. Usui, J. Noma, M. Hirano and S. Komiya, Inorg. Chim. Acta, 2000, 309, 151 CrossRef CAS.
- D. Naumann, W. Wessel, J. Hahn and W. Tyrra, J. Organomet. Chem., 1997, 547, 79 CrossRef CAS.
- W. Tyrra, J. Fluorine Chem., 2001, 112, 149 CrossRef CAS.
- W. Tyrra and D. Naumann, J. Fluorine Chem., 2004, 125, 823 CrossRef CAS PubMed.
- C. Hegemann, W. Tyrra, J.-M. Neudörfl and S. Mathur, Organometallics, 2013, 32, 1654 CrossRef CAS.
- W. Wessel, W. Tyrra and D. Naumann, Z. Anorg. Allg. Chem., 2001, 627, 1264 CrossRef CAS.
- K. J. Klabunde, Angew. Chem., Int. Ed. Engl., 1975, 14, 287 CrossRef PubMed.
- F. Glockling and V. B. Mahale, J. Chem. Res., 1978, 170 CAS.
- A. Graefe and T. Kruck, J. Organomet. Chem., 1996, 506, 31 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- M. Blaya, J. Gil-Rubio and J. Vicente, unpublished work.
- P. J. Brothers and W. R. Roper, Chem. Rev., 1988, 88, 1293 CrossRef CAS.
- R. Usón, A. Laguna and J. Vicente, J. Chem. Soc., Chem. Commun., 1976, 353 RSC.
- R. Uson, A. Laguna and J. Vicente, J. Organomet. Chem., 1977, 131, 471 CrossRef CAS.
- R. Usón, A. Laguna, J. Vicente, J. García, B. Bergareche and P. Brun, Inorg. Chim. Acta, 1978, 28, 237 CrossRef.
- R. Usón, A. Laguna, J. Vicente, J. García, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1981, 655 RSC.
- J. Vicente, M. T. Chicote, J.-A. Cayuelas, J. Fernández-Baeza, P. G. Jones, G. M. Sheldrick and P. Espinet, J. Chem. Soc., Dalton Trans., 1985, 1163 RSC.
- N. J. Rijs and R. A. J. O'Hair, Dalton Trans., 2012, 41, 3395 RSC.
- H.-G. Cho and L. Andrews, Organometallics, 2014, 33, 4315 CrossRef CAS.
- H. H. Murray, J. P. Fackler Jr., L. C. Porter, D. A. Briggs, M. A. Guerra and R. J. Lagow, Inorg. Chem., 1987, 26, 357 CrossRef CAS.
- D. Zopes, C. Hegemann, W. Tyrra and S. Mathur, Chem. Commun., 2012, 48, 8805 RSC.
- X.-G. Xiong and P. Pyykkö, Chem. Commun., 2013, 49, 2103 RSC.
- J. L. Margrave, K. H. Whitmire, R. H. Hauge and N. T. Norem, Inorg. Chem., 1990, 29, 3252 CrossRef CAS.
- M. A. Guerra, T. R. Bierschenk and R. J. Lagow, J. Organomet. Chem., 1986, 307, C58 CrossRef CAS.
- J. A. Schlueter, J. M. Williams, U. Geiser, J. D. Dudek, S. A. Sirchio, M. E. Kelly, J. S. Gregar, W. H. Kwok and J. A. Fendrich, et al. , J. Chem. Soc., Chem. Commun., 1995, 1311 RSC.
- U. Preiss and I. Krossing, Z. Anorg. Allg. Chem., 2007, 633, 1639 CrossRef CAS PubMed.
- J. A. Schlueter, U. Geiser, A. M. Kini, H. H. Wang, J. M. Williams, D. Naumann, T. Roy, B. Hoge and R. Eujen, Coord. Chem. Rev., 1999, 190–192, 781 CrossRef CAS.
- J. A. Schlueter, U. Geiser, H. H. Wang, M. E. Kelly, J. D. Dudek, J. M. Williams, D. Naumann and T. Roy, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 284, 195 CrossRef CAS PubMed.
- J. A. Schlueter, J. M. Williams, U. Geiser, H. H. Wang, A. M. Kini, M. E. Kelly, J. D. Dudek, D. Naumann and T. Roy, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 285, 43 CrossRef CAS PubMed.
- J. A. Schlueter, U. Geiser, J. M. Williams, J. D. Dudek, M. E. Kelly, J. P. Flynn, R. R. Wilson, H. I. Zakowicz, P. P. Sche, D. Naumann, T. Roy, P. G. Nixon, R. W. Winter and G. L. Gard, Synth. Met., 1997, 85, 1453 CrossRef CAS.
- E. Bernhardt, M. Finze and H. Willner, J. Fluorine Chem., 2004, 125, 967 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |