
Copper-catalysed reductive amination of nitriles and organic-group reductions using dimethylamine borane†
Dominic van der Waals*a,
Alan Pettmanb and
Jonathan M. J. Williamsa
aDepartment of Chemistry, University of Bath, Claverton Down, BA2 7AY, Bath, UK. E-mail: dvdw20@bath.ac.uk; J.M.J.Williams@bath.ac.uk
bChemical R & D, Pfizer Global Research & Development, Sandwich, Kent CT13 9NJ, UK. E-mail: alan.pettman@pfizer.com
First published on 9th October 2014
Abstract
A heterogeneous copper catalyst, formed in situ, has been shown to dehydrocouple commercially available amine boranes whilst transferring hydrogen for the reduction of selected organic functional groups in an aqueous medium. The catalytic system has also been shown to promote the reductive amination of aryl nitriles.
The use of stoichiometric metallic reagents for the reduction and interconversion of organic functional groups is a common approach in organic synthesis. Well known literature surrounding the use of hydride reagents, such as LiAlH4 and NaBH4, has long been established. Their use however involves intrinsic detriments including stoichiometric metallic waste and hazardous reactivity which suggests a need for an alternative and ideally catalytic method for organic functional group reductions.
Amine boranes have attracted a lot of wide spread interest due to their high hydrogen to molecular mass ratios, affording a light weight but dense energy source, the intrinsic stability of the amine boranes however means that catalytic methods to facilitate the release of hydrogen have been required. Pioneering investigations focused initially on group IV metallocenes as catalysts.1,2 These however were superseded by the use of platinum group metals which showed greater rates of hydrogen release. Particular emphasis of investigation was given to heterogeneous Rh catalysts3 as well as homogeneous Ir based catalysts; this included an interesting report by Stevens et al. investigating the use of an Ir(III) compound alongside computational studies.4 A rare example of a non-precious metal in the catalysis of amine borane dehydrocoupling was reported where the combined use of Ni(cod)2 along with a range of NHCs with kinetic experiments showing that both the B–H and the N–H bonds are broken in the rate determining step.5 Alternatively, heterogeneous methods have also been reported focussing on the use of copper nano-particles and copper oxides6 and this was extended to investigate the bis-chloride salts of copper, nickel and cobalt for dehydrogenation.7 The amino-borane polymeric product was seen to stabilise the nano-particles formed, a feature that has also been used in other reductively formed nano-particles such as gold and silver.8
Amine boranes are less reported for their use in organic synthesis than as a source of hydrogen. Notable exceptions to this is the application of Au/TiO2 nano-particles for the catalytic reduction of nitro-aromatics, this proceeds via the very rapid production of the aryl hydroxylamine which is then slowly reduced to the aniline product.9 There are also several reports of the use of precious metals such as that by Burke et al. regarding the reduction of olefins with a variety of Re catalysts. These catalysed the hydrogen transfer from dimethylamine borane to reduce a range of alkenes via a homogeneous mechanism.10 The use of Rh colloids for the reduction of alkenes and nitro-aryl compounds has also been reported.11
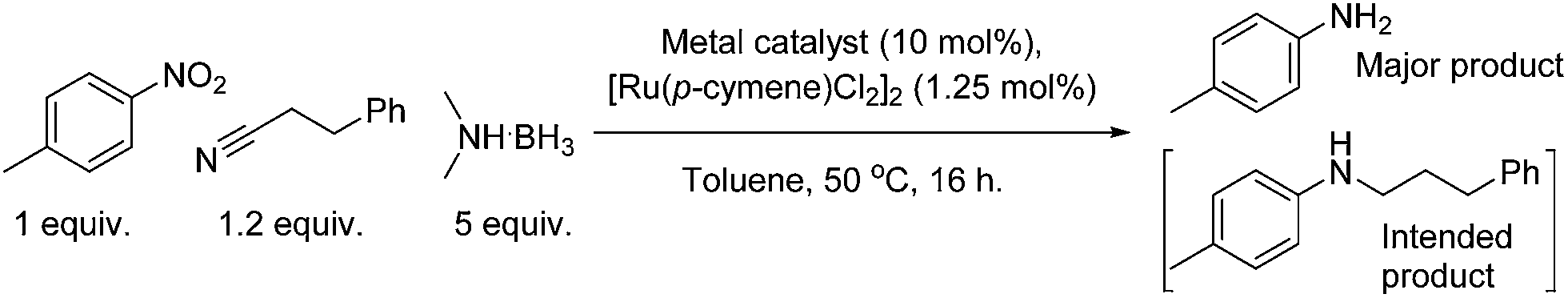
Within our own group, the catalytic reduction of a wide scope of organic functional groups using a ruthenium catalyst has been reported. The introduction of enantiomerically pure ligands to the ruthenium centre was shown to enable the stereoselective reduction of aromatic ketones with moderate enantiomeric excesses.12 Further work to expand this methodology to enable the reductive amination of nitriles was considered interesting, as nitriles are widely available yet there is very little literature concerning their reductive amination. Rare examples include our own report of the heterogeneous reductive amination of nitriles, focussing on the use of a Pt/C13 catalyst. A Pd/C system was also reported by Rylander et al., alongside the use of a Rh/C catalyst.14 Recently the use of Pt nanowires has been reported which showed rapid amine–nitrile addition followed by slow reduction of the intermediate.15 Following from these publications, a methodology for the reductive amination of nitriles with amines or amine precursors, building upon previous work involving the use of amine boranes, was considered desirable.
Research into the ruthenium catalysed reductive amination of nitriles with nitro aromatics showed only moderate success initially. Investigations revealed that higher equivalencies of dimethylamine borane produced the expected higher conversions of nitro aromatics to anilines; the selectivity for the amination of 3-phenylpropionitrile however decreased with greater levels of the reductant. The use of excesses of amine borane was shown not to increase the production of the aminated product. Alternative methods to enhance selectivity were therefore investigated. One of these methods, the results of which are shown in Table 1, included the addition of a co-catalyst in order to increase the electrophilicity of the nitrile species.
|
|
|---|---|
| Co-catalyst | Conversion of Nitro by 1H NMR |
| Sc(OTf)3 | 72% |
| Zn(OTf)2 | 79% |
| ZnCl2 | 73% |
| Ti(OiPr)4 | 79% |
| Cu(OAc)2 | 100% |
| FeCl3 | 68% |
| No co-catalyst | 75% |
Considering the significant improvement in the reaction rate with the addition of a copper co-catalyst to the reaction, it was thought that a separate copper mediated reaction might be progressing. The next stage of optimisation, once this was identified, was to determine if the identity of the copper salt in the absence of ruthenium had an effect on the reaction. The results for the copper salt screen are shown in Table 2, and what is clear is that almost all of the salts and oxides investigated reduced the nitro aromatic with a good degree of success in toluene, the only exception being copper(I) iodide.
|
|
|---|---|
| Catalyst | Conversion by 1H NMR (%) |
| Cu(OAc)2 | 66 |
| CuCl2 | 66 |
| Cu(NO3) | 44 |
| Cu2O | 73 |
| CuO | 84 |
| Cu(OTf)2 | 52 |
| CuBr | 73 |
| CuI | 0 |
| CuCl | 50 |
| No catalyst | 0 |
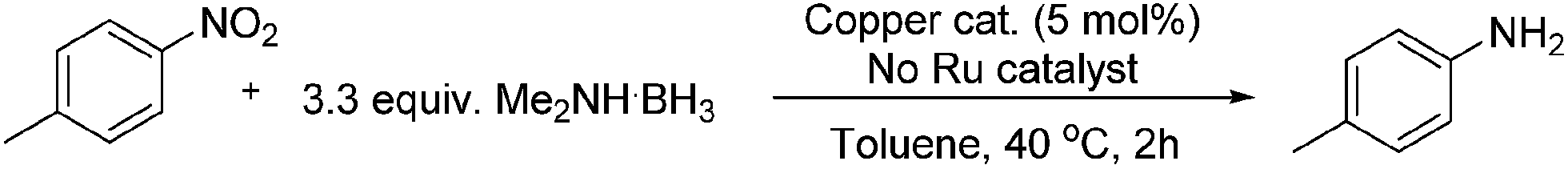
A solvent screen was conducted for the reduction of benzonitrile by dimethylamine borane, catalysed by copper triflate. It was seen that although the reaction worked to some degree in all of the solvents tested it was found that water was the most suitable solvent. Further optimisation showed that copper triflate was the most successful catalyst for aqueous reactions showing the greatest conversions for the reduction of benzonitrile. Dimethylamine borane showed a slightly improved reactivity when compared to ammonia borane whilst the tert-butylamine borane gave no conversion.
Investigating the scope of aromatic nitro reductions, the results of which are given in Table 3, it is apparent that some steric bulk around the nitro group could be tolerated as could aromatic nitros without the inductive effect of the para-methyl group. Tolerance of a free aniline group was also compatible as might be expected; the isolated yield of the product however was considerably diminished and this is thought to be due to product degradation. Aromatic halogens were also tolerated without showing signs of reduction under the reaction conditions.
|
|
|---|---|
| Aniline formed | Isolated yield (%) |
 |
86 |
 |
100 |
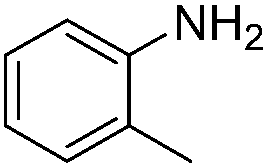 |
86 |
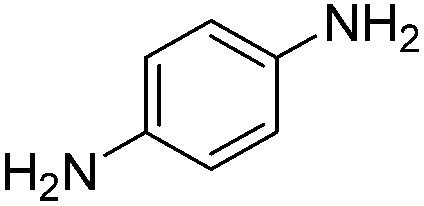 |
34 |
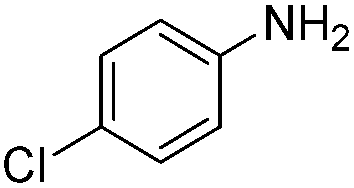 |
94 |
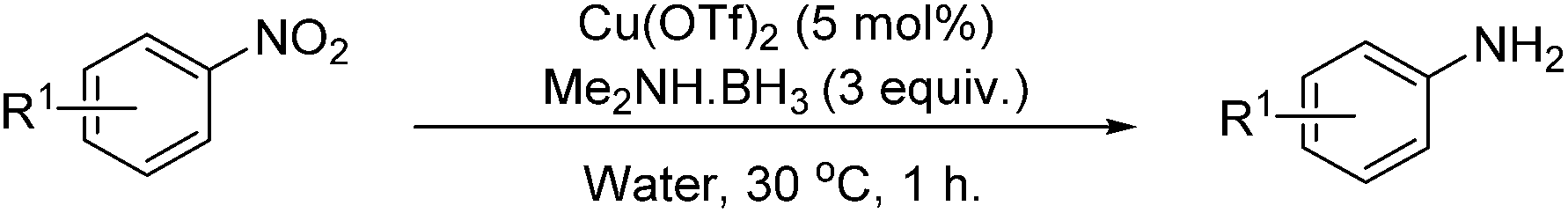
Carbonyls were also investigated, primarily aromatic ones, although the aliphatic 2-heptanone also showed good potential for reduction. As the results in Table 4 show both electron poor and electron rich aromatic ketones can be reduced as well as the aromatic aldehyde, benzaldehyde.
|
|
|---|---|
| Alcohol formed | Isolated yield (%) |
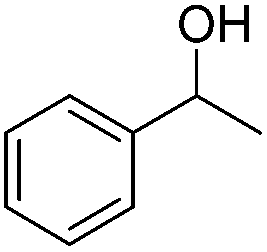 |
99 |
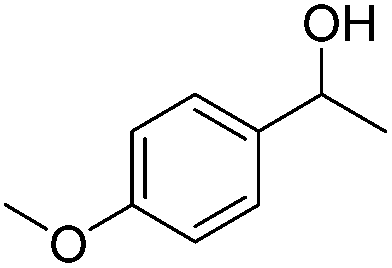 |
79 |
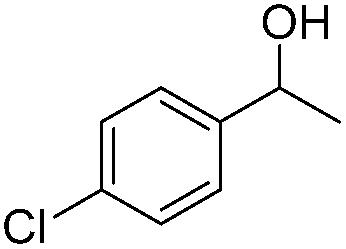 |
100 |
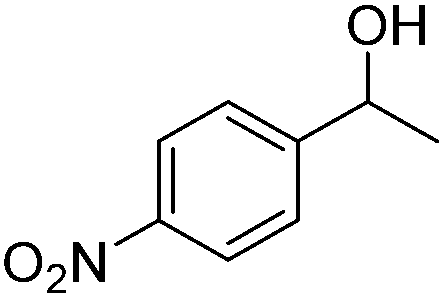 |
39 |
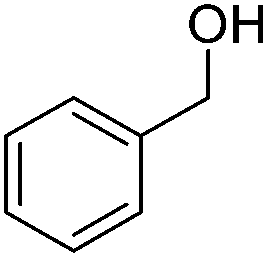 |
75 |
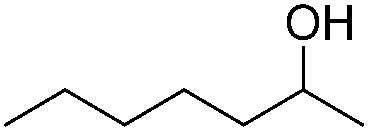 |
84 |
|
||
|---|---|---|
| Substrate | Product | Isolated yield (%) |
| a Conversion based on 1H NMR. Reactions run on 3 mmol scale. | ||
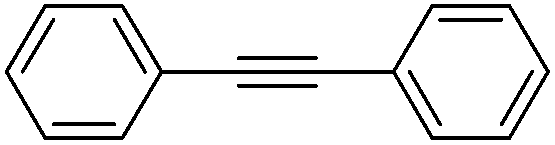 |
 |
97 |
 |
 |
98 |
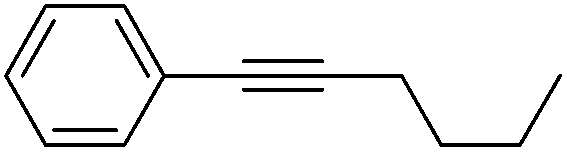 |
 |
88 |
 |
 |
71 |
 |
 |
72 |
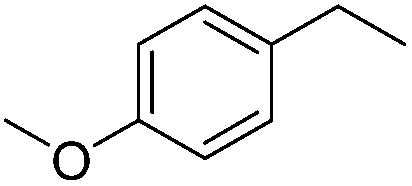
Whilst the presence of an aromatic halogen was tolerated, compounds containing an aromatic nitro group produced a range of side products. Both the solely nitro-reduced product was seen as well as the product formed when both functional groups are reduced, resulting in the low isolated yield of the desired alcohol.
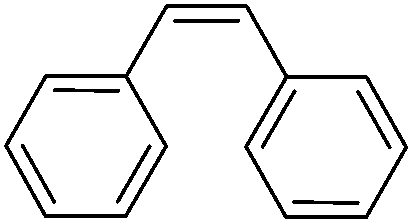
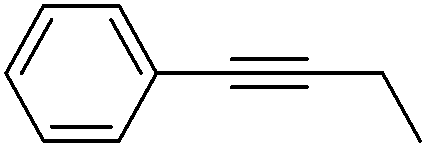
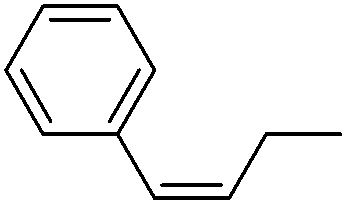
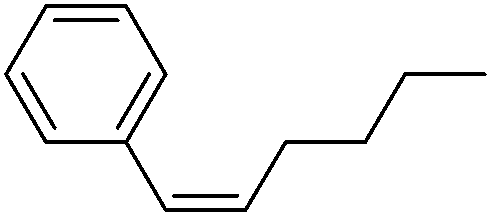
More challenging substrates were then looked at to determine the scope of the reaction with unsaturated hydrocarbons the next to be investigated, (Table 5). Interestingly it was seen that alkynes were considerably easier to reduce than alkenes which enables selective reduction of the alkynes to the corresponding cis-alkene only as determined by 1H NMR. The reaction was tolerant for aromatic alkynes such as the sterically crowded bis-phenylacetylene, from which cis-stilbene could be isolated in quantitative yields, as well as internal alkynes with various aliphatic groups attached. In contrast when purely aliphatic alkynes were used, a mixture of products was seen with both the terminal 1-octyne and internal 2-octyne producing intractable mixtures.
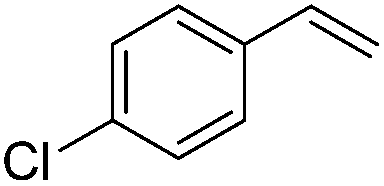
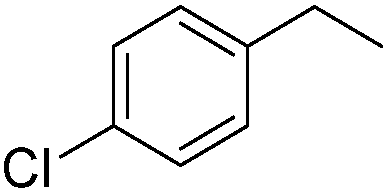
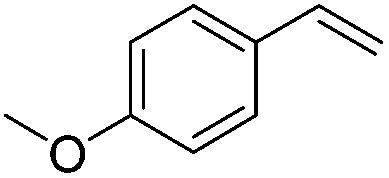
The reduction of alkenes was also briefly investigated and again the aromatic styrene systems produced the best results with 4-chlorostyrene and 4-methoxystyrene showing high conversions to the alkylated analogues. Sterically hindered alkenes such as both cis- and trans-stilbene showed themselves to be unreactive under the reaction conditions.
A variety of imines were shown to undergo rapid and clean reductions. In a similar fashion to the results seen for the reduction of other functionalities the reaction conditions tolerated the presence of aromatic halogens but could not tolerate the additional presence of an aromatic nitro group, producing a range of side products. Both N-benzylic and N-anilinic imines showed good reduction, Table 6, as did the N-isopropyl aliphatic analogue. The isolated yields are significantly lower than the conversions seen; this is due to the method of isolation whereby acidification with ethereal HCl produced the salt of the amine with some loss during filtration.
|
|
|---|---|
| Amine salt formed | Isolated yield (%) |
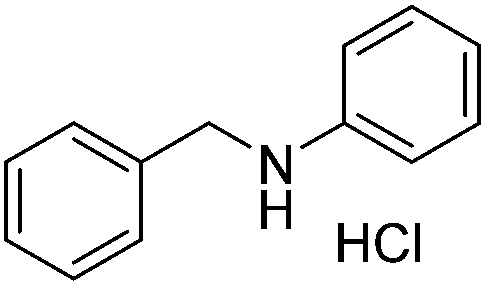 |
83 |
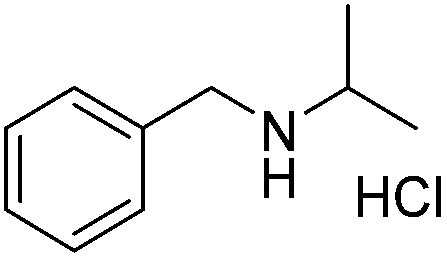 |
70 |
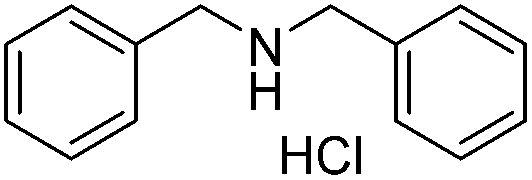 |
65 |
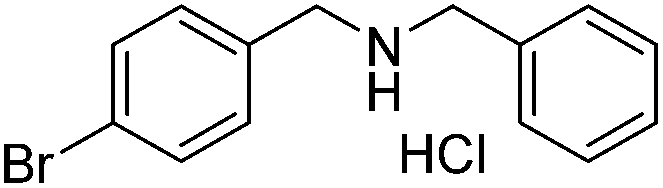 |
87 |

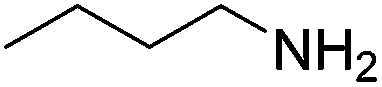
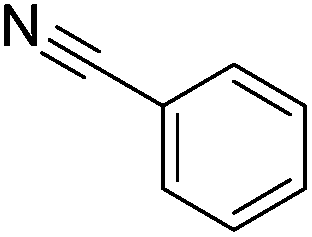
With the methodology having proved successful for the reduction of a range of functional groups it was decided to investigate whether the reaction could be expanded to selectively reduce the intermediate formed from the attack of a nitrile by an amine. In order to determine which nitrile substrates were the most suitable, butylamine was added to a solution of copper triflate and a range of nitriles followed by the reductive amine borane. The results for this showed that the aromatic nitriles are far more susceptible to amination with heptyl cyanide showing only minimal conversion under the reaction conditions. This could be either due to a greater intrinsic reactivity of the nitrile as an electrophile due to the electron withdrawing effects of the aromatic ring or due to the ability to resonance stabilise the intermediate formed from the attack of the amine, prolonging the lifetime of this reactive intermediate.
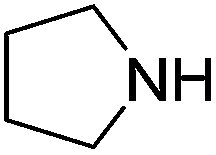
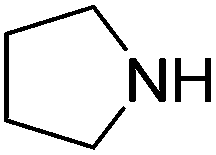
An initial selection of nitriles was aminated with simple, highly nucleophilic amines as shown in Table 7. Other amines, such as piperidine, were also investigated but conversions were seen to decrease markedly with a decrease in nucleophilicity. Further work to determine the extents of the methodology is being undertaken.
|
||
|---|---|---|
| Amine | Nitrile | Conversion (yield) % |
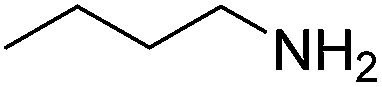 |
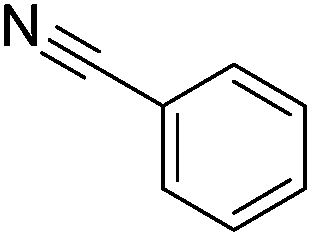 |
78 (47) |
 |
 |
76 (60) |
 |
 |
72 (72) |
 |
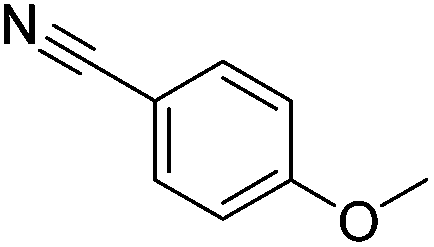 |
100 (80) |
During the reactions the appearance of a black precipitate was noted which suggested that the reaction may be proceeding through a heterogeneous pathway, several different techniques were therefore employed to determine the reaction mechanism. One of the most common methods for testing the phase of a catalyst is the mercury drop test whereby a large excess of elemental mercury is added to the reaction mixture. In the case of homogeneous reactions this should have little effect on the reaction rate whereas the addition of the mercury to heterogeneous reactions can lead to either coating of the catalyst surface or the formation of metal amalgams, both of which result in significant loss of catalytic activity. The results, shown in Table 8, show that the addition of mercury reduces the conversion of the reaction down to the levels seen when no catalyst had been added. This indicates that the reaction is heterogeneous in nature as the mercury would be expected to interact with any metal particulates formed yet not significantly interfere with a homogeneous reaction mixture.
|
|
|---|---|
| Catalyst system | Conversion to product (%) |
| (CuOAc)2 | 88 |
| (CuOAc)2 with Hg | 22 |
| Neither (CuOAc)2 or Hg | 22 |
In order to further elucidate the phase of the catalyst, NMR studies were conducted to follow the conversion of one of the reactions over time. For homogeneous reactions there is not usually expected to be an induction period; heterogeneous reactions, by contrast, invariably see an induction period followed by a period of rapid conversion which subsequently falls away. NMR spectra were taken every minute to investigate the copper(II) triflate catalysed reduction of 4-nitrotoluene with dimethylamine borane in deuterated water (further details in the ESI†). There is an initial delay of around 10 minutes before any reaction occurs, indicative of a reaction proceeding through a heterogeneous pathway. This is followed by conversion of the starting aryl nitro group into an intermediate which is postulated to be either the corresponding N-4-methylphenylhydroxylamine or the nitroso compound. This intermediate is then rapidly reduced to the anilinic species followed by further rapid reduction of the remaining starting nitro group.
Conclusions
It has been shown that the use of a variety of copper salts as well as elemental copper and its oxides can be used to catalyse the dehydrogenation of a range of amine boranes with dimethylamine borane showing high levels of reactivity to the catalyst. These catalytic systems can be used reduce a range of organic functional groups in good conversions and under milder reaction conditions compared with some of those previously reported. Copper triflate proved the most versatile catalyst and was seen to reduce several groups chemoselectively and of particular note was the reduction of a range of alkynes selectively to the corresponding cis-alkenes.The reaction is believed to proceeding through a heterogeneous mechanism. This hypothesis is supported by two separate analytical methods; the first focussing on the addition of elemental mercury led to a poisoning of the reaction. The second supporting piece of evidence focussed on the reaction kinetics which showed the existence of an induction period which is indicative of formation of the metal particulates.
Acknowledgements
The authors would like to thank the EPSRC and Pfizer for funding.Notes and references
- T. Clark, K. Lee and I. Manners, Chem.–Eur. J., 2006, 12, 8634 CrossRef CAS PubMed.
- D. Pun, E. Lobkovsky and P. Chirik, Chem. Commun., 2007, 3297 RSC.
- C. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 2698 CrossRef CAS PubMed.
- C. Stevens, R. Dallanegra, A. Chaplin, A. Weller, S. Macgregor, B. Ward, D. McKay, G. Alcaraz and S. Sabo-Etienne, Chem.–Eur. J., 2011, 17, 3011 CrossRef CAS PubMed.
- R. Keaton, J. Blacquiere and T. Baker, J. Am. Chem. Soc., 2007, 1844 CrossRef CAS PubMed.
- S. Kalidindi, U. Sanyal and B. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870 RSC.
- S. Kalidindi, J. Joseph and B. Jagirdar, Annu. Rev. Energy, 2009, 2, 1274 CAS.
- S. Kalidindi, U. Sanyal and B. Jagirdar, Inorg. Chem., 2010, 49, 3965 CrossRef CAS PubMed.
- E. Vasilikogiannaki, C. Gryparis, V. Kotzabasaki, I. Lykakis and M. Stratakis, Adv. Synth. Catal., 2013, 355, 907 CrossRef CAS.
- Y. Jiang, O. Blacque, T. Fox, C. Frech and H. Berke, Organometallics, 2009, 28, 5493 CrossRef CAS.
- M. Sloan, A. Staubitz, K. Lee and I. Manners, Eur. J. Org. Chem., 2011, 672 CrossRef CAS.
- T. Nixon, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 6652 CrossRef CAS PubMed.
- S. K. Sharma, J. Lynch, A. M. Sobolewska, P. Plucinski, R. J. Watson and J. M. J. Williams, Catal. Sci. Technol., 2013, 3, 85 CAS.
- P. Rylander, L. Hasbrouck and I. Karpenko, Ann. N. Y. Acad. Sci., 1973, 5, 100 CrossRef PubMed.
- S. Lu, J. Wang, X. Cao, X. Li and H. Gu, Chem. Commun., 2014, 50, 3512 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11193a |
| This journal is © The Royal Society of Chemistry 2014 |