
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First synthesis of naphthalene annulated oxepins†
L.
Dobelmann
ab,
A. H.
Parham
b,
A.
Büsing
b,
H.
Buchholz
b and
B.
König
*a
aInstitute of Organic Chemistry, University of Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: burkhard.koenig@chemie.uni-regensburg.de
bPerformance Materials Division, Merck KGaA, Frankfurter Straße 250, 64293 Darmstadt, Germany
First published on 6th November 2014
Abstract
Highly condensed oxepins have been prepared in good yields from their corresponding diols by etherification using p-toluenesulfonic acid. Their intriguing twisted structures were unambiguously determined by X-ray crystallography. Substitution effects of a novel highly aromatic naphthalene annulated oxepin indicate that structural and conjugative effects have an influence on the optoelectronic properties.
Introduction
Organic π-conjugated materials have been a focus of research in both academia and industry for the past few decades. Owing to their unique optical and charge transporting properties they play an important role in the development of organic semiconductors for applications such as organic light-emitting diodes (OLEDs),1,2 organic field-effect transistors (OFETs)3–6 and organic photovoltaics (OPVs).7 Constant progress has been achieved in these research areas by modulating the materials properties towards the specific demands of each application. Typically heteroatoms are incorporated into the carbon frameworks to tailor and improve the electronic properties.8–11 Other than sulphur- and nitrogen-containing organic semiconductors, which have been studied extensively in the last decades, oxygen-containing compounds have received less attention. Still polycyclic aromatic hydrocarbons comprising furan12,13 and pyran14 cores have been reported and showed good carrier mobilities and strong fluorescence, both desirable properties for materials in organic optoelectronic devices.As part of our investigation of novel compounds for applications in organic electronics we were interested in π-conjugated systems containing twisted oxepin cores as an approach to develop soluble π-electronic materials.15 We focused on naphthalene- and benzene-fused derivatives as their energy levels were predicted by calculations to be interesting for use as host or emissive materials in OLED applications. The calculated energy levels of naphthalene-fused oxepins were comparable to those of anthracene derivatives, which have extensively been used as materials in fluorescent blue OLEDs because of their excellent optical and electronic properties.16 However, only few polyaromatic compounds carrying oxepin core structures have been described in the literature and general protocols for their synthesis are not available.17–20 For the synthesis of the target oxepin derivative an acid mediated cyclization, which was previously used for the formation of furan-containing systems, was considered.21 Applying this strategy, the seven-membered cyclic ethers were obtained in high yields and their structures confirmed by X-ray diffraction studies. Electrochemical and optoelectronic studies as well as quantum chemical calculations of dinaphthoxepin and some derivatives confirmed their interesting physical properties.
Results and discussion
Synthesis
Scheme 1 summarizes the synthesis of oxepin 3. Compound 2 was synthesized from 1-methoxynaphthalene (1) according to a literature known procedure.22 Lithiation using tert-BuLi was followed by an oxidative carbon–carbon bond formation with Fe(acac)3. Deprotection of the methoxy groups with BBr3 gave diol 2 in 54% yield. The acid mediated cyclization using p-toluenesulfonic acid (p-TsOH) in refluxing toluene21 converted diol 2 into oxepin 3 in 87% yield.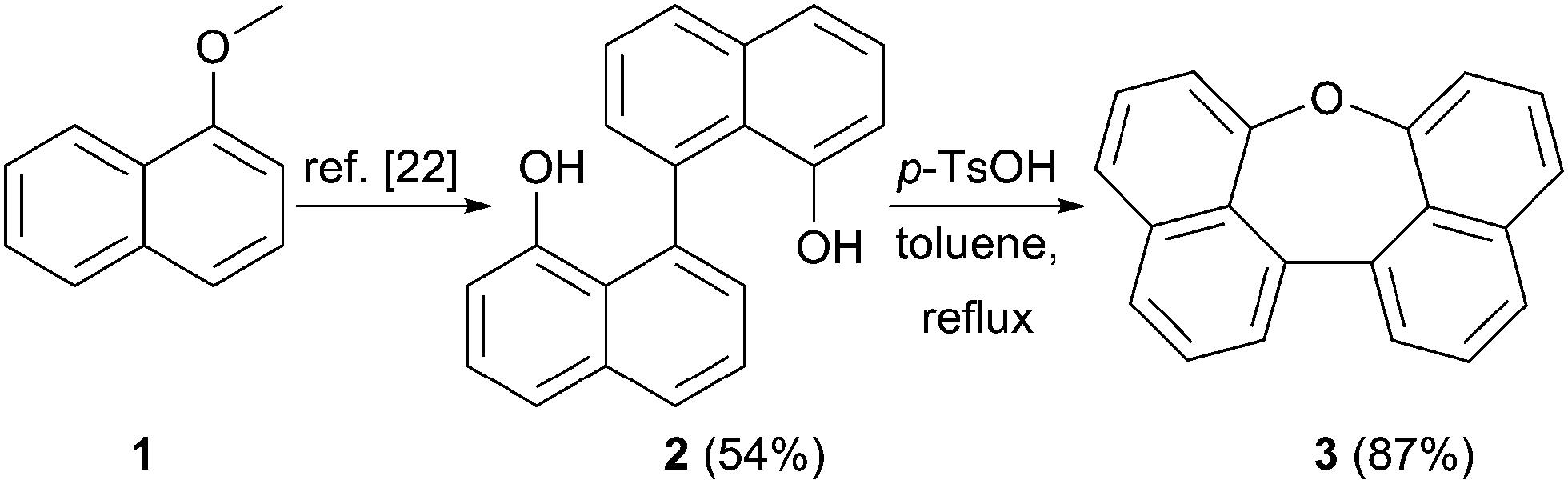 | ||
| Scheme 1 Synthesis of compound 3. | ||
The bromination of 3 using NBS in DMF at 40 °C was used to further functionalize the compound. The dibromo-compound 4 was formed exclusively in 87% yield (Scheme 2). Palladium-catalyzed coupling of dibromide 4 with phenyl- and m-terphenyl-boronic acid, respectively, gave 5a and 5b in moderate yields.‡ Further bromination of 5a was achieved using NBS at higher temperatures, but was less selective and product 6 could only be obtained in 44% yield. Tetra-phenyl substituted oxepin 7 was obtained in moderate yield via palladium-catalyzed coupling reaction. All synthesized derivatives are highly soluble in common organic solvents and have a good thermally stability as shown by differential scanning calorimetry (DSC) for 5a and 5b (DSC curves are shown in Fig. S1 and S2 in the ESI†).
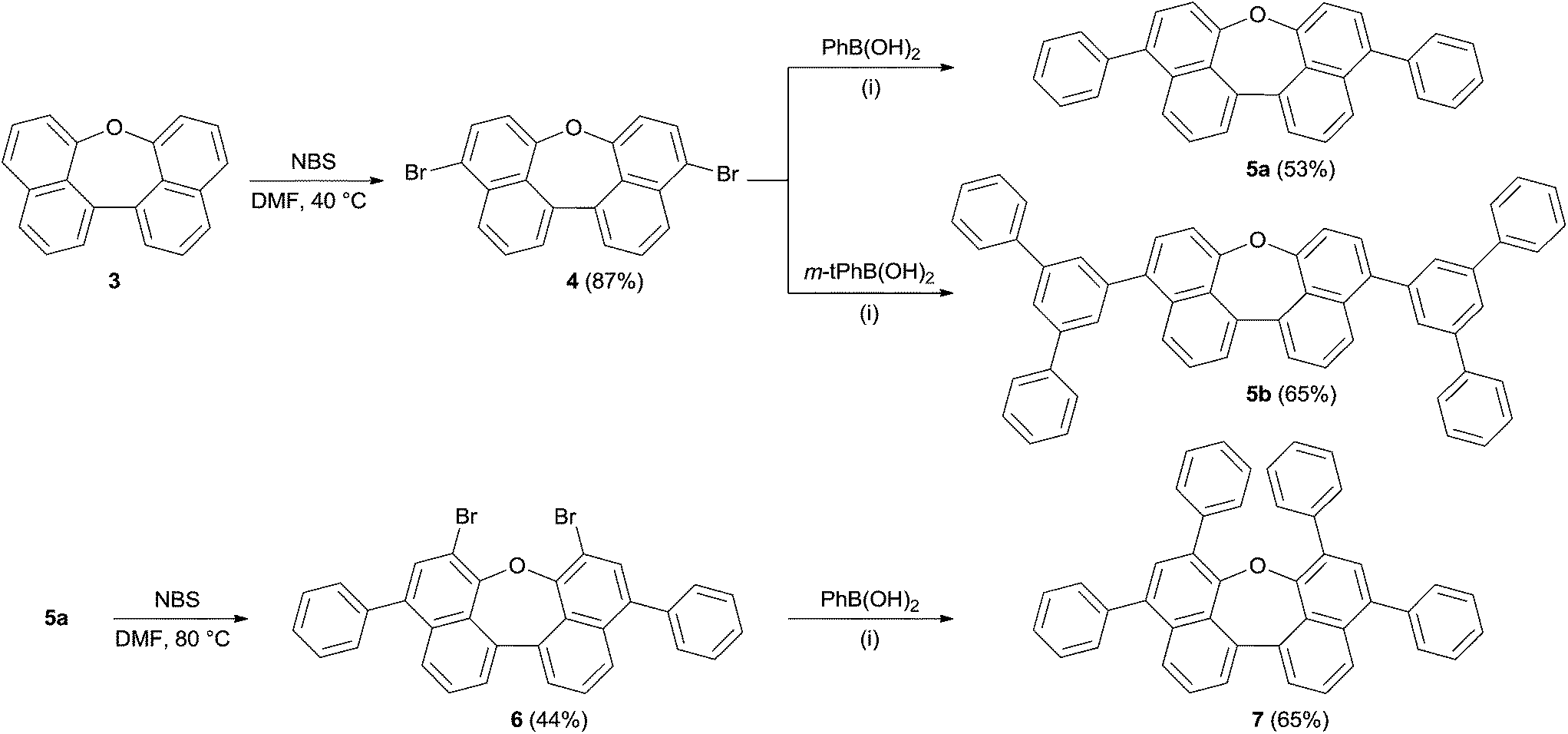 | ||
| Scheme 2 Synthesis of compounds 5–7. (i) K3PO4, Pd(OAc)2, P(o-Tol)3, toluene/dioxane/water, 70 °C. | ||
X-ray structure analyses
Crystals of dinaphthoxepins 3 and 7 suitable for X-ray crystallographic analysis were obtained by slow evaporation from CH2Cl2 and heptane at room temperature.The central distorted seven-membered ring shows an envelope-like conformation with the oxygen-atom lying out-of-plane (Fig. 1). The rigid C11–O1–C12 bond angle (118.26°) forces the naphthalene rings to adapt a bent geometry with an angle of 33.8°. Owing to the repulsion of the H-atoms attached to C18 and C19 the molecule exhibits a dihedral angle of 12.6° between the two naphthalene planes (C3–C4–C5–C6). Sandwich-herringbone packing was observed with pairs of stacked molecules, probably driven by π–π intermolecular interactions (3.67 Å) between two adjacent naphthalene planes.
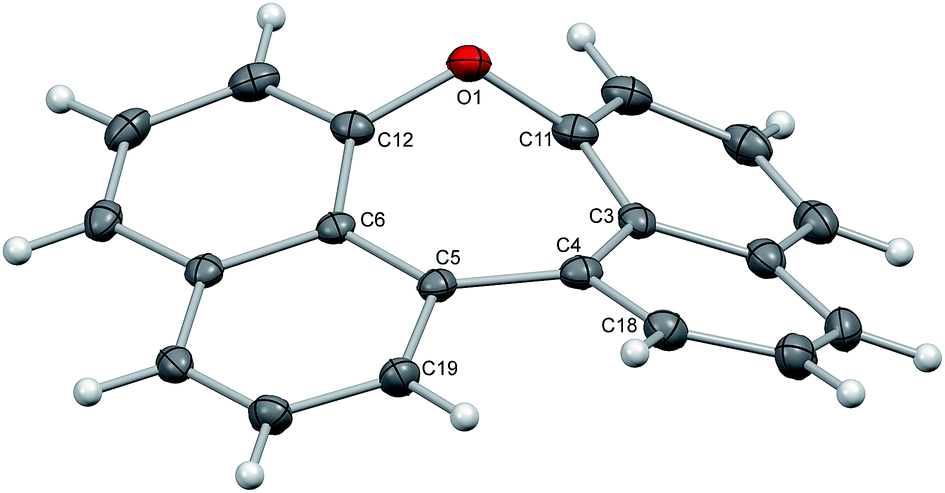 | ||
| Fig. 1 Structure of compound 3 in the solid state. The thermal ellipsoids correspond to 50% probability. | ||
The structure of 7 (Fig. 2) is strongly affected by the two phenyl substituents in ortho-positions to the ether bridge. Their alignment with an average distance of 3.84 Å pushes the naphthalene moieties apart and confines the oxepin ring in a twisted conformation. The dihedral angle between the naphthalene units increases to 44.6° (C11–C13–C17C18), just as the C10–O1–C23 bond angle (131.9°). The naphthalene units of adjacent molecules in the crystal are displaced and the distance between them is 4.48 Å on average.
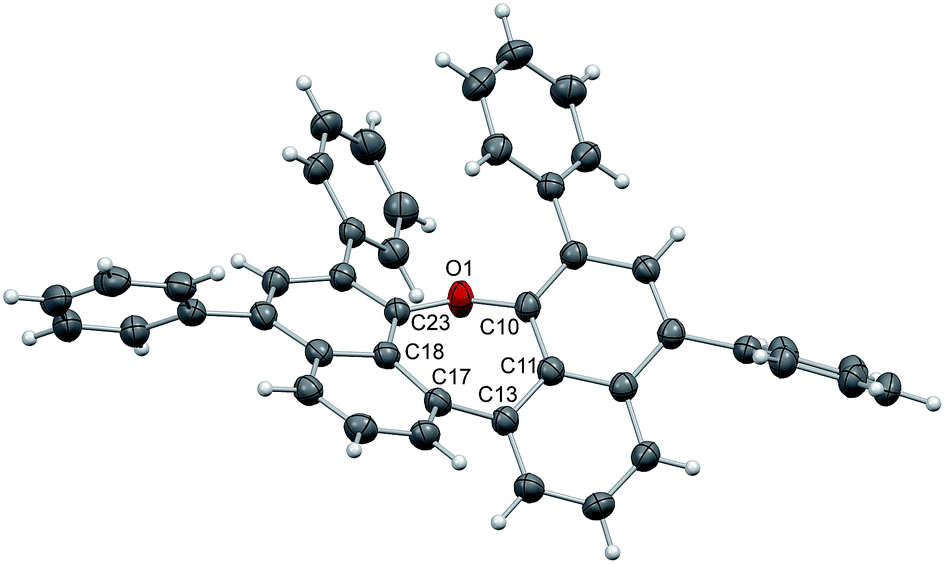 | ||
| Fig. 2 Structure of compound 7 in the solid state. The thermal ellipsoids correspond to 50% probability. | ||
Electrochemical measurements and quantum mechanical calculations
The redox potentials of the naphthalene-annulated oxepins were measured by cyclic voltammetry (CV). As depicted in Fig. 3, compounds 3, 5a, 5b and 7 exhibit reversible first reduction potentials. No reversible oxidation is detected for compound 3, while 5a and 5b show a reversible oxidation. The oxidation of 7 occurs at a lower potentials and a second reversible oxidation is observed, indicating the formation of a dication.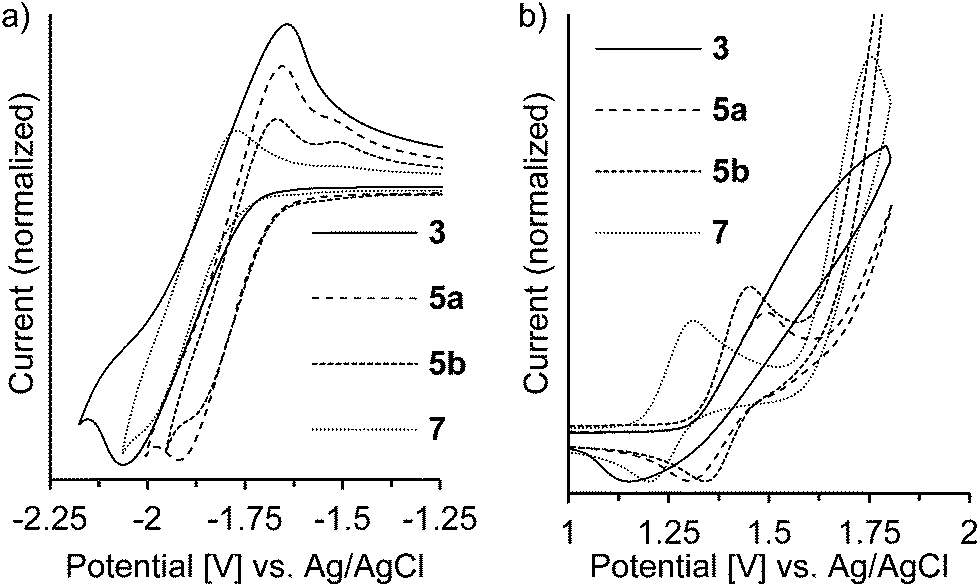 | ||
| Fig. 3 Cyclic voltammogram of dinaphthoxepins 3, 5a, 5b and 7 in (a) THF- (reduction) and (b) CH2Cl2-solution (oxidation) at room temperature. Measured at a scan rate of 500 mV s−1 with a gold working electrode. | ||
The HOMO and LUMO energy levels were calculated from the measured CV-potentials, by comparison with ferrocene (4.8 eV), used as the internal standard. Due to the irreversible oxidation of 3, the HOMO energy value was obtained by using the LUMO energy and the optical bandgap taken from the absorption spectrum (absorption edge). HOMO and LUMO distributions in the molecules as well as geometrical structures of 5a and 5b were determined by quantum mechanical calculations, performed at the TD-DFT/B3-LYP/6-31G(d) level of theory. The geometric parameters for the calculations for compounds 3 and 7 were obtained from X-ray diffraction data. The results are presented in Fig. 4.
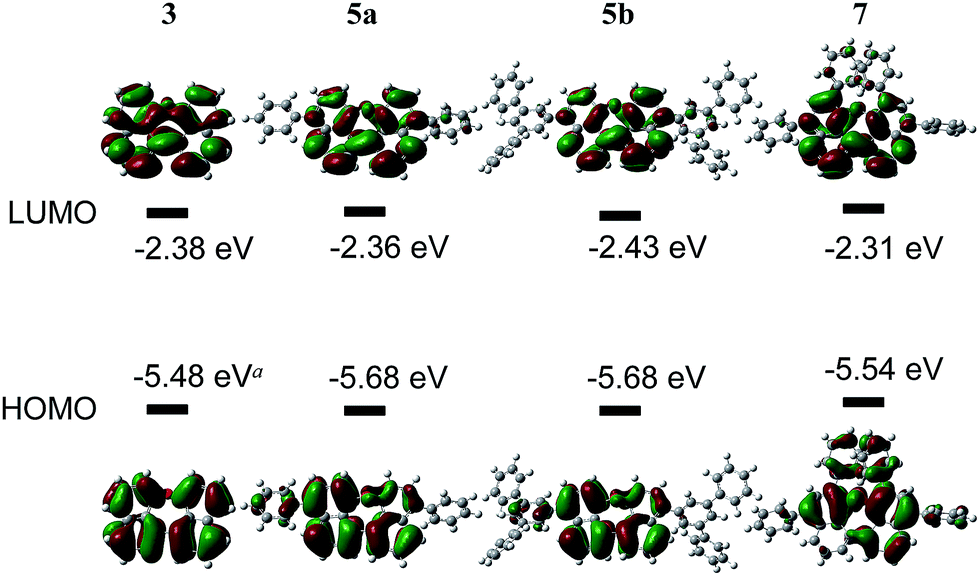 | ||
| Fig. 4 Optimized geometry, HOMO–LUMO-energies and -distribution in dinaphthoxepin derivatives. (a) Calculated using optical band gap and LUMO level, decomposes in CV. (b) Optical band gap was determined from the edge of absorption tail in toluene solution. | ||
The molecular orbitals in 3 (dihedral angle: 12.6°) delocalize symmetrically over the whole molecule with periodical changes in the sign of the wave function. Its HOMO and LUMO energy-levels were determined to −5.48 and −2.39 eV respectively. Based on the computationally optimized geometry for 5a and 5b, the introduction of substituents on the naphthalene in para-position has a strong influence on the dihedral angle in the molecule, which increases to 32.8°. A result of this is a slightly asymmetric distribution of the HOMO and a reduced density of the LUMO along the ether bond. The energies of the HOMOs are identical, but comparably lower than the calculated value for 3.
In compound 7 the π-conjugated system is extended by two additional phenyl groups, which leads to an increased dihedral angle of 44.6°. The twisted conformation perturbs the delocalization leading to a multiple periodic change in the sign of the wave function. HOMO and LUMO energies are both higher as in the other derivatives, which could be attributed to the substituents in ortho-positions to the ether bond. Two factors influence the electronic properties of dinaphthoxepins: The dihedral angle affects the HOMO and LUMO distribution and energy-values among the two naphthalene moieties, while the extension of the π-conjugated system depends on the phenyl substituents. The HOMO energy-levels for 3, 5a, 5b and 7 are in the range of −5.5 to −5.7 eV and lower than typical p-type organic semiconductors, but slightly higher than reported for other oxygen-containing bisnaphthalene structures.15
Optical properties
Compounds 3, 5a, 5b and 7 show strong fluorescence under UV-light. Their optical properties were characterized by both UV-Vis and fluorescence spectroscopy in solution and solid state; their fluorescence quantum yield was determined in solution. Electronic absorption spectra of oxepin derivatives were measured in toluene (Fig. 5). The absorption spectra show broad unstructured lowest-energy bands ranging from 300 to 400 nm with maximum absorption at wavelengths of 360 (3), 366 (5a) and 373 nm (5b) thus demonstrating the extension of the π-conjugation. For 7 a much lower absorption of 349 nm is observed. This is consistent with the observation from the crystal structure analysis, which revealed a strong deformation of the naphthalene system in 7 resulting in a lower electron delocalization around the aromatic core and consequently leading to absorption at lower wavelengths. The UV-Vis data of thin films were comparable with the solution absorption spectra (Table 1), except for the featureless broadened absorption spectrum of 3, as seen in Fig. S4 in the ESI.† The remarkable red shift of the longest wavelength absorption bands compared with that in solution may, in accord with the crystal structure, be due to transannular π-electronic interaction between chromophores in the solid state.23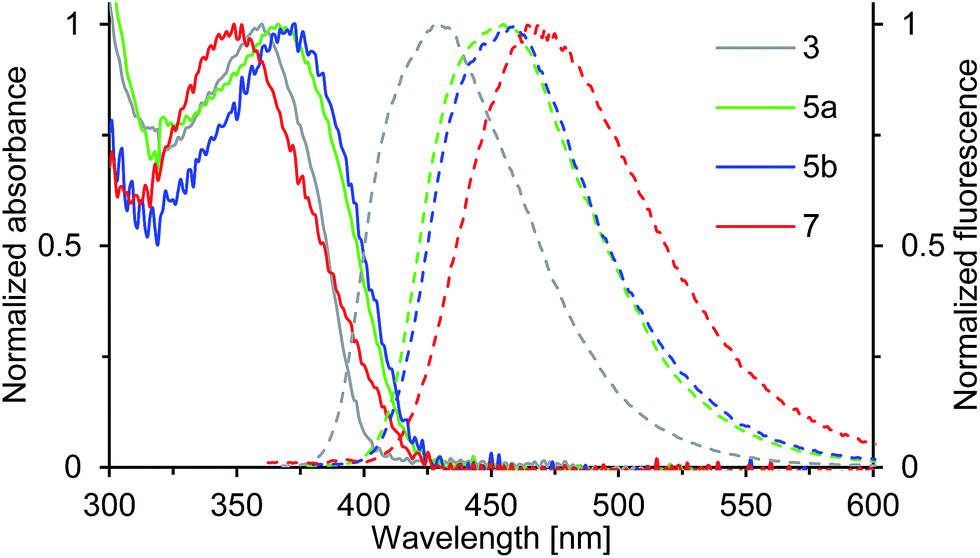 | ||
| Fig. 5 Normalized absorption (solid line) and emission spectra (dashed line) of solutions (c = 0.02 mM) of compounds 3, 5a, 5b and 7 in toluene (recorded at T = 298 K). | ||
| Cpd. | Solution | Film | |||
|---|---|---|---|---|---|
| λ a,max a [nm] | λ fl,max a [nm] | Φ F b | λ a,max c [nm] | λ fl,max c [nm] | |
| a Measured in toluene (c = 0.01 mg ml−1). b Absolute fluorescence quantum yield determined by calibrated integrating sphere, measured in toluene (non-degassed, c ≈ 0.1 mM), excited at 350 nm. c Spincoated film (50 nm) prepared from a toluene solution. | |||||
| 3 | 360 | 427 | 0.38 | 380 | 465 |
| 5a | 366 | 455 | 0.47 | 373 | 489 |
| 5b | 373 | 457 | 0.48 | 370 | 481 |
| 7 | 349 | 465 | 0.24 | 350 | 471 |
The emission spectra of compounds 3, 5a, 5b and 7 (Fig. 5) in toluene solution consist of broad unstructured emission bands. Substitution of the π-conjugated system in para-position to the ether bridge shifts the emission wavelength bathochromically. The relatively flat dinaphthoxepin 3 emits at 427 nm, whereas the emission of more twisted compounds 5a and 5b shift to around 456 nm resulting in a large Stokes shift ranging from 67 nm in 3 to 89 nm in 5a (Table 1). Compound 7 emits at 465 nm and has a Stokes shift of 116 nm. The large Stokes shifts indicate the ground states (S0) to be inherently different in geometry from the excited states (S1), which is probably caused by the high flexibility of the dinaphthoxepin core, leading to a relaxation of the molecular framework upon photoexcitation and reduction of the emission energy.24,25 The emission properties of thin films of oxepins were determined by photoluminescence spectroscopy (Table 1). Emission peaks shift bathochromically in the solid state for 3, 5a and 5b, which may be due to π–π stacking and formation of excimers or solid-state solvation effects.26 In contrast to that, the crystal structure of compound 7 suggests the presence of rather isolated and only weakly coupled chromophores with large π-stacking distances. In the emission spectra, this is reflected by the minimum red shifts of the emission peak for compound 7 between solid state and solution.
The fluorescence quantum yields of 3, 5a, 5b and 7, recorded in toluene solution are given in Table 1. The ΦF values rise from 0.38 in 3 to around 0.47 in 5a and 5b, which can be attributed to the enlarged π-conjugated system. The quantum yield of compound 7 is smaller with 0.24. Thus the S1-state of 7 is more susceptible to deactivation by radiationless decay. This is probably because the spin–orbit coupling is enhanced by the degree of nonplanarity of 7, which allows intersystem crossing to the non-emissive triplet state.27–29
Synthesis of tribenz[b,d,f]oxepin (9)
To provide further application of the acid mediated cyclization route for the synthesis of sterically congest oxygen heterocycles, the synthesis of the phenylene annulated oxepin 9 was targeted. Using the acid mediated cyclization conditions of the cyclization of 1,1'-bi-8-naphtol (2), diol 8 was converted into tribenz[b,d,f]oxepin 9 (Scheme 3). Diol 8 was synthesized in 80% over two steps according to a literature procedure.30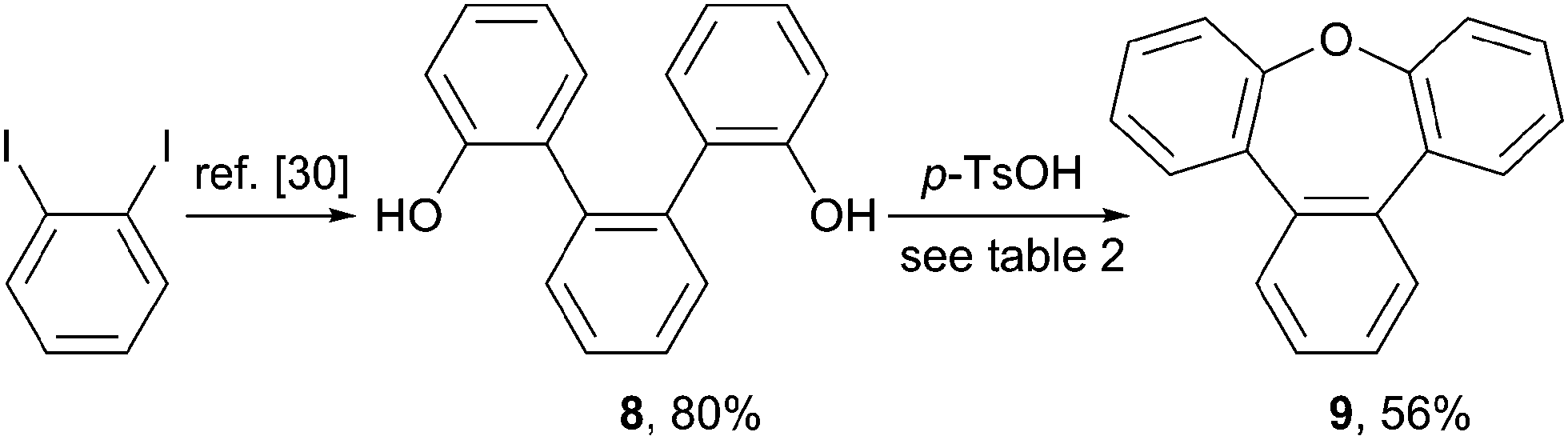 | ||
| Scheme 3 Synthesis of tribenz[b,d,f]oxepin 9. | ||
The reaction conditions used for the formation of dinaphthoxepin 3 had to be optimized to yield the desired product (Table 2). Removal of water from the reaction mixture or using a higher boiling solvent were not suitable. Using neat reaction conditions and higher temperatures gave the desired product 9 in 56% yield (entry 4).
The synthesis of compound 9 has been reported previously.17,18 Colourless crystals of 9 suitable for X-ray analysis were obtained from heptane. Two crystallographically independent molecules are found in the unit cell (Fig. 6). In the present oxepin, the central seven-membered ring adapts a boat-like conformation with the phenylene groups arranged in an alternating up/down fashion. The whole molecule is in good approximation symmetric with a mirror plane passing through the oxygen atom.
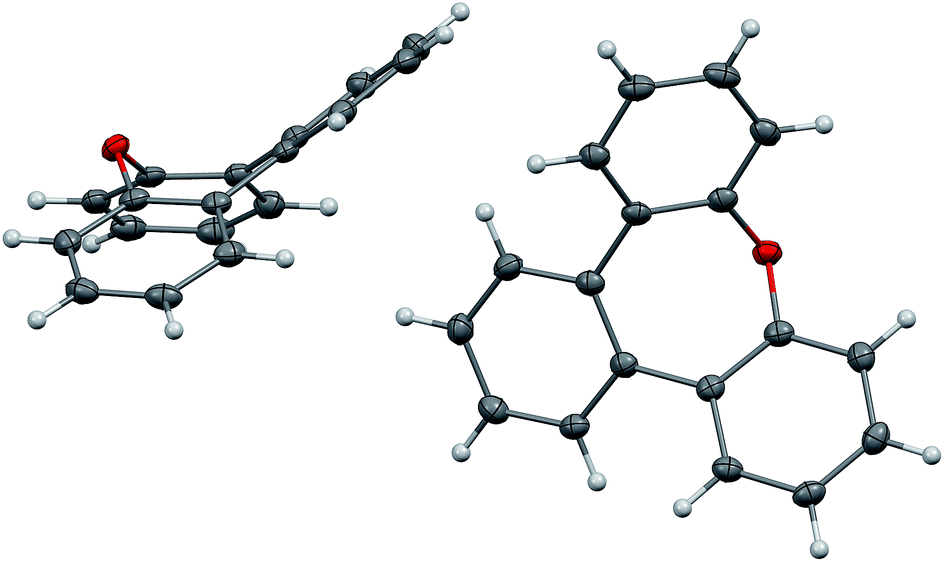 | ||
| Fig. 6 Structure of compound 9 in the solid state. The thermal ellipsoids correspond to 50% probability. | ||
Conclusions
Several aromatic oxepins were synthesized in moderate to good yields from diols by a dehydration reaction. Subsequent bromination and Suzuki coupling lead to functionalized compounds. X-ray crystallographic analysis confirmed their highly distorted conformations, induced by the seven-membered ring. The HOMO and LUMO energy levels were determined by electrochemical analysis and quantum chemical calculations. The physical properties of dinaphthoxepins make them an interesting class of compounds for application in optical and electronic materials.Acknowledgements
We thank Dr C. Kühn (Merck KGaA) and Dr H. Schubert (University of Tübingen) for performing crystal structure analysis.Notes and references
- R. Muangpaisal, W.-I. Hung, J. T. Lin, S.-Y. Ting and L.-Y. Chen, Tetrahedron, 2014, 70, 2992–2998 CrossRef CAS PubMed.
- B. Wei, J.-Z. Liu, Y. Zhang, J.-H. Zhang, H.-N. Peng, H.-L. Fan, Y.-B. He and X.-C. Gao, Adv. Funct. Mater., 2010, 20, 2448–2458 CrossRef CAS.
- W. Zhang, Y. Liu and G. Yu, Adv. Mater., 2014, 26, 6898–6904 CrossRef CAS PubMed.
- C. Mitsui, J. Soeda, K. Miwa, H. Tsuji, J. Takeya and E. Nakamura, J. Am. Chem. Soc., 2012, 134, 5448–5451 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- N. Kaur, M. Singh, D. Pathak, T. Wagner and J. M. Nunzida, Synth. Met., 2014, 190, 20–26 CrossRef CAS PubMed.
- X.-D. Xiong, C.-L. Deng, X.-S. Peng, Q. Miao and H. N. C. Wong, Org. Lett., 2014, 16, 3252–3255 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- C. Mitsui, T. Okamoto, H. Matsui, M. Yamagishi, T. Matsushita, J. Soeda, K. Miwa, H. Sato, A. Yamano, T. Uemura and J. Takeya, Chem. Mater., 2013, 25, 3952–3956 CrossRef CAS.
- M. Nakano, K. Niimi, E. Miyazaki, I. Osaka and K. Takimiya, J. Org. Chem., 2012, 77, 8099–8111 CrossRef CAS PubMed.
- K. Nakahara, C. Mitsui, T. Okamoto, M. Yamagishi, H. Matsui, T. Ueno, Y. Tanaka, M. Yano, T. Matsushita, J. Soeda, Y. Hirose, H. Sato, A. Yamano and J. Takeya, Chem. Commun., 2014, 50, 5342–5344 RSC.
- K. Nakanishi, T. Sasamori, K. Kuramochi, N. Tokitoh, T. Kawabata and K. Tsubaki, J. Org. Chem., 2014, 79, 2625–2631 CrossRef CAS PubMed.
- N. Kobayashi, M. Sasaki and K. Nomoto, Chem. Mater., 2009, 21, 552–556 CrossRef CAS.
- K. Nakahara, C. Mitsui, T. Okamoto, M. Yamagishi, K. Miwa, H. Sato, A. Yamano, T. Uemura and J. Takeya, Chem. Lett., 2013, 42, 654–656 CrossRef CAS.
- J. Huang, J.-H. Su and H. Tian, J. Mater. Chem., 2012, 22, 10977–10989 RSC.
- E. Dimitrijevic, M. Cusimano and M. S. Taylor, Org. Biomol. Chem., 2014, 12, 1391–1394 CAS.
- S. Kumar, H. Ila and H. Junjappa, Tetrahedron, 2007, 63, 10067–10076 CrossRef CAS PubMed.
- S. v. Angerer and W. Tochtermann, in Houben-Weyl, Georg Thieme Verlag, Stuttgart, 1998, vol. E 9 d Search PubMed.
- A. Rosowsky, in Chemistry of Heterocyclic Compounds, John Wiley & Sons, Inc., New York, 1972, vol. 26 Search PubMed.
- J. Areephong, N. Ruangsupapichart and T. Thongpanchang, Tetrahedron Lett., 2004, 45, 3067–3070 CrossRef CAS PubMed.
- S. P. Artz, M. P. DeGrandpre and D. J. Cram, J. Org. Chem., 1985, 50, 1486–1496 CrossRef CAS.
- T. Otsubo, F. Ogura and S. Misumi, Tetrahedron Lett., 1983, 24, 4851–4854 CrossRef CAS.
- X. Liu, Z. Xu and J. M. Cole, J. Phys. Chem. C, 2013, 117, 16584–16595 CAS.
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag, Weinheim, 2001 Search PubMed.
- V. Bulović, R. Deshpande, M. E. Thompson and S. R. Forrest, Chem. Phys. Lett., 1999, 308, 317–322 CrossRef.
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef PubMed.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS PubMed.
- M. Sapir and E. V. Donckt, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef CAS.
- R. Shukla, S. V. Lindeman and R. Rathore, Chem. Commun., 2009, 5600–5602 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, electronic spectra and copies of the 1H NMR and 13C NMR spectra of the prepared intermediates and final products. CCDC 1024839–1024841. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra10652k |
| ‡ The yields of compounds 5a and 5b and 7 reflect unoptimized reaction conditions. |
| This journal is © The Royal Society of Chemistry 2014 |