
Two- and three-coordinate formal iron(I) compounds featuring monodentate aminocarbene ligands†
Zhenbo
Mo‡
 a,
Zhenwu
Ouyang‡
a,
Lei
Wang
a,
Kathlyn L.
Fillman
b,
Michael L.
Neidig
*b and
Liang
Deng
*a
a,
Zhenwu
Ouyang‡
a,
Lei
Wang
a,
Kathlyn L.
Fillman
b,
Michael L.
Neidig
*b and
Liang
Deng
*a
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: deng@sioc.ac.cn; Tel: (+86) 021-54925460
bDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: neidig@chem.rochester.edu
First published on 15th July 2014
Abstract
Bulky monodentate aminocarbene ligands, IMes and Me2-cAAC (IMes: 1,3-bis(2′,4′,6′-trimethylphenyl)imidazol-2-ylidene; Me2-cAAC: 3,3,5,5-tetramethyl-1-(2′,6′-diisopropylphenyl)pyrrolidine-2-ylidene), have been shown to be effective in supporting formal 13- and 11-electron iron(I) species. From the reactions of ferrous precursors and one equivalent of a reducing agent, three-coordinate complexes of the type [L2FeCl] (L = IMes or Me2-cAAC) have been synthesized in good yields. A mixed-ligand complex [(IMes)(Me2-cAAC)FeCl] was prepared from the ligand substitution reaction of [(IMes)2FeCl] with Me2-cAAC. All of the three-coordinate iron complexes can react with Na[BArF]4, from which a two-coordinate species [(Me2-cAAC)2Fe][BArF4] has been isolated. Single-crystal X-ray diffraction studies established their molecular structures to be the first examples of two- and three-coordinate formal iron(I) species supported by carbene ligands. The large solution magnetic moments, differentiated Fe–C(carbene) distances and 57Fe Mössbauer isomer shifts are indicative of their rich electronic properties.
The +1 oxidation state is an uncommon oxidation state for iron,1 but the possible involvement of iron(I) species in important chemical transformations, such as H2-activation and production by [Fe,Fe]-hydrogenases,2 N2-reduction in nitrogenases,3 and iron-catalyzed cross-coupling reactions,4 has spurred great synthetic interest. Iron(I) complexes have mostly been known for the six-, five- and four-coordinate compounds with CO, arene, alkene, and phosphine ligation.5 On the other hand, low-coordinate iron(I) species, those with coordination numbers less than four, are much less common.6 Up to now, three-coordinate iron(I) compounds have been limited to Holland's β-diketiminate complexes,3a,7 Caulton's T-shaped compound [((tBu2PCH2SiMe2)2N)Fe],8 and Jones’ dinuclear guanidionate complex [(DippN)2C(cis-2,6-Me2NC5H8)Fe]2.9 Isolable two-coordinate iron(I) compounds had remained unknown until Long's recent report on [K(crypt-222)][Fe(C(SiMe3)3)2].10
The effective stabilization of low-coordinate iron(I) species necessitates suitable supporting ligands. Recently, Cárdenas et al. reported the detection of an iron(I) intermediate in Fe(OAc)2-IMes-catalyzed alkyl–alkyl cross-coupling reactions by low temperature EPR experiments.4c Importantly, an IMes-stabilized iron(I) species such as (IMes)nFeX could be the potential active catalyst species in this chemistry. Unfortunately, iron(I) species supported by monodentate NHCs are critically absent from the literature.11 Prompted by this and recent success of NHCs in stabilizing two-coordinate first row late 3d transition metal species,12 we envisioned that NHCs might be amenable to support low-coordinate iron(I) compounds. In this context, herein we report the synthesis and structural characterization of a three-coordinate formal iron(I) NHC compound [(IMes)2FeCl] (1), as well as its analogs with cyclic (alkyl)(amino)carbene (cAAC)13 ligands, [(IMes)(Me2-cAAC)FeCl] (2) and [(Me2-cAAC)2FeCl] (3). Compounds 1–3 are the first examples of three-coordinate iron-aminocarbene species with formal iron(I) centers. Their stability at ambient temperature has enabled spectroscopic characterization by 1H NMR, solution magnetic susceptibility measurement, single crystal X-ray diffraction, and zero-field 57Fe Mössbauer spectroscopy. Moreover, we examined the reactions of 1–3 with Na[BArF4], from which the two-coordinate aminocarbene iron complex [(Me2-cAAC)2Fe][BArF4] (ArF = 3,5-bis(trifluoromethyl)phenyl) (4), as the close relative of two-coordinate NHC and cAAC complexes [L2M] (M = zinc,14 copper,15 nickel,16 manganese17), has been obtained.
Previously, we found that the reaction of [(IMes)2FeCl2], prepared from the interaction of FeCl2 with IMes (2 equiv.), with an excess amount of sodium amalgam in THF produces the cyclometallated iron(II)-NHC compound [Fe(IMes′)2] (IMes′ denoted for the cyclometallated IMes ligand) and H2.18 By carefully controlling reaction conditions, we found that a three-coordinate compound [(IMes)2FeCl] (1) can be prepared from the reaction of [(IMes)2FeCl2] with one equiv. of potassium graphite or sodium amalgam in toluene (Scheme 1).19 The solvent plays a crucial role in the preparation of 1. When the reaction was conducted in THF, iron black and [Fe(IMes′)2] were observed as the dominant products, even if only one equiv. of the reducing reagent was used. In contrast to the reaction with IMes, the one-electron reduction reactions of FeCl2 in the presence of two equiv. of IPr, SIMes, or IAd (IPr = 1,3-bis(2′,6′-diisopropylphenyl)imidazole-2-ylidene, SIMes = 1,3-bis(2′,4′,6′-trimethylphenyl)-4,5-dihydroimidazole-2-ylidene, IAd = 1,3-bis(adamantyl)imidazole-2-ylidene) generally produced iron black and free carbene ligands.
 | ||
| Scheme 1 Preparation routes for the low-coordinate formal iron(I) complexes. | ||
Single crystals of 1 were obtained by freezing its toluene solution at −30 °C. An X-ray diffraction study revealed that 1 is isostructural to [(IMes)2CoCl],12a in which the iron center adopts a distorted trigonal planar geometry with a C1–Fe1–C2 angle of 125.6(3)°, and Fe–Cl and Fe–C(IMes) distances of 2.258(3) and 2.014(9) Å, respectively (Fig. 1). Consistent with the larger atomic radius of iron versus that of cobalt, the Fe–C(IMes) distances are longer than the Co–C(IMes) bonds in [(IMes)2CoCl] (1.953(5) Å). Notably, the Fe–C(IMes) bonds in 1 are significantly shorter than previously found in three-coordinate iron(II)-NHC compounds, [(IPr2Me2)Fe(Mes)2] (2.125(3) Å),20a [(SIPr)Fe(CH2TMS)Cl] (2.128(3) Å),20b and [(ItBu)Fe(N(SiMe3)2)2] (2.152(2) Å).20c The decrease of the Fe–C(IMes) distance from the iron(II) compounds to the formal iron(I) compound is unusual and is likely induced by π-bonding between the low-valent iron center and the NHC ligands.
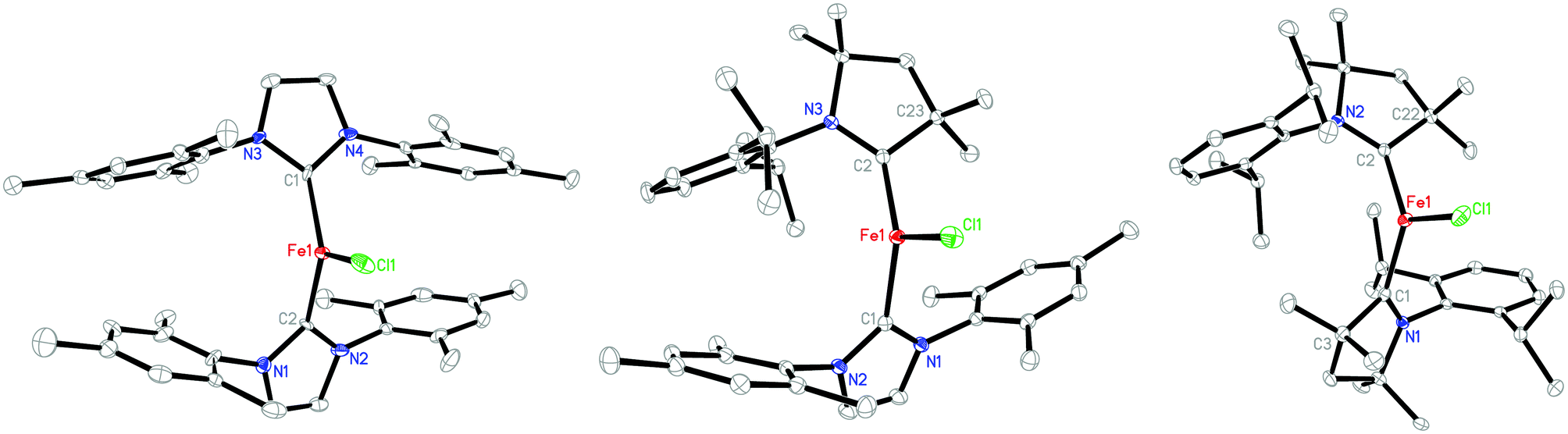 | ||
| Fig. 1 Molecular structures of 1 (left), 2 (middle), and 3 (right), showing 30% probability ellipsoids and partial atom schemes. | ||
As isolated, 1 is an air- and moisture-sensitive brown crystalline solid, but can be stored under a N2 atmosphere at room temperature for weeks without noticeable decomposition. When dissolved in C6D6, it decomposed into [(IMes)2FeCl2], IMes, iron black, and [Fe2(IMes)2]21 in 3 days at room temperature. The 1H NMR spectrum of 1 measured in C6D6 shows four paramagnetically shifted peaks at +51.73, +7.86, −12.57, and −12.77 ppm with an integrated ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:3:2, suggesting the free rotation of the NHC ligand around the Fe–C(carbene) bond. Solution magnetic susceptibility measurement in C6D6 indicated a solution magnetic moment of 5.0(1)μB for 1. This value is much larger than the spin-only value of 3.8μB for high-spin, S = 3/2 d7 ions, likely due to the presence of unquenched orbital angular momentum as found in Holland's β-diketiminato-iron(I)-alkyne compound [(DippNCtBu)2CH)Fe(η2-HCCPh)] (4.7μB).7b
:6:3:2, suggesting the free rotation of the NHC ligand around the Fe–C(carbene) bond. Solution magnetic susceptibility measurement in C6D6 indicated a solution magnetic moment of 5.0(1)μB for 1. This value is much larger than the spin-only value of 3.8μB for high-spin, S = 3/2 d7 ions, likely due to the presence of unquenched orbital angular momentum as found in Holland's β-diketiminato-iron(I)-alkyne compound [(DippNCtBu)2CH)Fe(η2-HCCPh)] (4.7μB).7b
The propensity of 1 to undergo decomposition in solution rendered attempts to access its three-coordinate derivatives by salt elimination reactions unsuccessful. However, 1 can readily undergo a ligand replacement reaction with Me2-cAAC to furnish a new three-coordinate complex [(IMes)(Me2-cAAC)FeCl] (2) and free IMes in high yield (Scheme 1).19 The conversion of 1 to 2 provides direct evidence for the stronger affinity of Me2-cAAC to transition metals versus IMes, consistent with Bertrand's determination that cAACs with small HOMO–LUMO energy-gaps are both stronger electrophiles and nucleophiles than NHCs.13 The isolation of 1 and 2 prompted attempts to access their bis(cAAC) analogs. While the ligand substitution protocol starting from either 1 or 2 was unsuccessful, the desired complex [(Me2-cAAC)2FeCl] (3) was eventually prepared by the reaction of FeCl2 with two equiv. of Me2-cAAC and one equiv. of sodium amalgam or potassium graphite in THF in 60% isolated yield (Scheme 1).19 Both 2 and 3 are stable in the solid state under an inert atmosphere. When dissolved in C6D6, 2 does not show decomposition after standing at room temperature for 3 days, whereas 3 displays partial decomposition (less than 10%). The improved thermal stability of 2 and 3versus that of 1 could be attributed to the good π-accepting ability of cAACs.13 Similar to 1, large solution magnetic moments of 5.1(1) and 4.7(1)μB for 2 and 3, respectively, have been determined by the Evans method.
The molecular structures of 2 and 3 in the solid state have been established by X-ray diffraction studies (Fig. 1). Their key structure data are compiled in Table 1 along with those of 1 for comparison. These three complexes hold similar trigonal planar FeC2Cl cores, but their Fe–C(carbene) distances differ significantly. The Fe–C(IMes) bond in 2 with the distance of 2.099(2) Å shows significant elongation over its counterparts in 1 by 0.08 Å, and is slightly shorter than those of the aforementioned three-coordinate iron(II)–NHC compounds.18 On the other hand, the Fe–C(Me2-cAAC) bond in 2 with the distance of 1.925(2) Å is substantially shorter than those observed in 3 (1.991(2) Å on average) and the iron(II) complex [Cl(cAAC)Fe(μ-Cl)2Fe(cAAC)Cl] (2.094(3) Å).22 Along with the short Fe–C(Me2-cAAC) distance, the C(carbene)–N distance of the cAAC ligand in 2 (1.360(2) Å) is found to be longer than its counterparts in 3 (1.335(2) and 1.339(2) Å), approaching the distances in [(Me2-cAAC)2Mn] (1.359(2) Å),17 but still shorter than those in [(Me2-cAAC)2Zn] (1.376(2) Å).14 The C(carbene)–N distances in 3 are close to those observed in [(Me2-cAAC)2Ni] (1.338(2) and 1.342(2) Å)16c and [(Me2-cAAC)2CoCl] (1.334 Å),23 and are longer than the ones in [Cl(cAAC)Fe(μ-Cl)2Fe(cAAC)Cl] (1.31 Å)22 and free cAAC ligands (1.32 Å).13a,b
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| a Parameters of the Fe(IMes) fragment. b Parameters of the Fe(Me2-cAAC) fragment. c Data are the average of two crystallographically independent cations in the unit cell. | ||||
| Fe–Cl | 2.258(3) | 2.267(1) | 2.264(1) | |
| Fe–C | 1.998(9) | 2.099(2)a | 1.979(2) | 1.997(3) |
| 2.030(8) | 1.925(2)b | 2.004(2) | ||
| C–N | 1.349(13)–1.404(11) | 1.360(2)a | 1.335(2) | 1.310(3) |
| 1.369(2)a | 1.339(2) | |||
| 1.360(2)b | ||||
| C–Fe–C | 125.6(3) | 126.0(1) | 121.0(1) | 180.0(1) |
| C–Fe–Cl | 116.4(3) | 116.8(1) | 119.3(1) | |
| 118.0(3) | 116.4(1) | 119.7(1) | ||
We then examined the reactions of these three-coordinate species with Na[BArF4] with the aim to obtain two-coordinate iron species. The interactions of 1 or 2 with one equiv. of Na[BArF4] in THF at −30 °C could produce red and deep blue solutions, which turned into intractable deep brown suspensions upon standing at room temperature. In contrast, the reaction of 3 with one equiv. of Na[BArF4] in THF could produce a stable deep blue suspension, from which a two-coordinate complex [(Me2-cAAC)2Fe][BArF4] (4) was isolated as a green crystalline solid in 45% yield (Scheme 1).19 The 1H NMR spectrum of 4 shows heavily broadened resonances in the range +50 to −61 ppm. Its solution magnetic moment is 5.1(2)μB, similar to the values observed for complexes 1–3.
Single-crystals of 4 were grown from its diethyl ether solution. An X-ray diffraction study revealed that the unit cell contains two sets of well-separated ion pairs ([(Me2-cAAC)2Fe]+ and [BArF4]−) and one diethyl ether molecule. The closest distance between the cations is around 12.8 Å. Since the metric data of the two cations are very similar, only one is shown as a representative example (Fig. 2). In this cation, the iron atom sits on the inversion center with a C(carbene)–Fe–C(carbene) angle of 180.0(1)°. A similar conformation has been observed in Roesky's neutral complexes [(Me2-cAAC)2M] (M = Zn, Cu, Mn).14,15,17 The Fe–C(Me2-cAAC) distance of 1.997(3) Å in 4 approximates the corresponding distances found in 3, but is shorter than its congeners in the sterically encumbered aryl complexes Fe(Ar*)2 (2.04–2.06 Å),6c [Fe(C(SiMe3)3)2] (2.05 Å),24 and [K(crypt-222)][Fe(C(SiMe3)3)2] (2.06 Å).10 The C(carbene)–N distance (1.310(3) Å) in 4 is also close to those observed in 3.
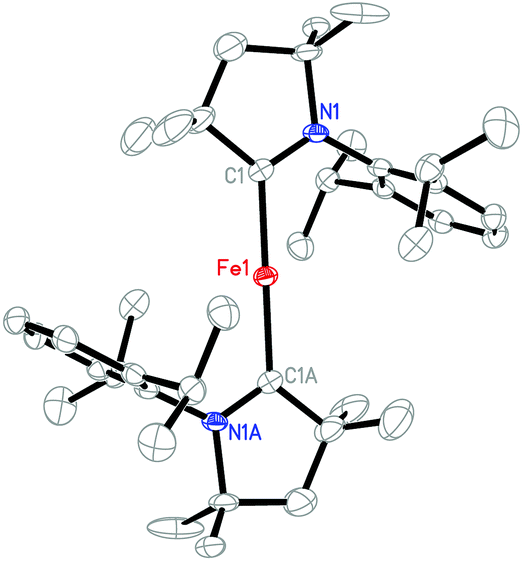 | ||
| Fig. 2 Molecular structure of the cation [(Me2-cAAC)2Fe]+ in 4 showing one of the two crystallographically independent cations in the unit cell with 30% probability ellipsoids. | ||
57Fe Mössbauer spectroscopy was utilized to further characterize complexes 1–4. The isomer shifts (δ) and quadrupole splittings (ΔEQ) determined for the 80 K spectra of 1–4 (see ESI† for spectra), as well as those of other previously reported two- and three-coordinate iron(I) compounds, are given in Table 2. The 80 K Mössbauer spectrum of 1 is well-fit to a quadrupole doublet with δ = 0.65 mm s−1 and ΔEQ = 2.63 mm s−1. These parameters compare closely to those of the S = 3/2 sulfide-bridged iron(I) complex [K(nacnac)Fe(μ-S)Fe(nacnac)K] (5),7d further indicative of its high-spin (S = 3/2) iron(I) nature. The spectra of 2–4 have δ values of 0.52, 0.49 and 0.49 mm s−1, respectively, lower than the isomer shift observed for 1, but comparable to those of 3-coordinate S = 3/2 alkyne- and hydrido-coordinated β-diketiminate iron(I) species (6 and 7).7b,c The reduced isomer shifts for 2–4 are suggestive of increased covalency in these complexes compared to 1, consistent with the observation of reduced Fe–C(carbene) bond lengths and long C(carbene)–N distances of the cAAC moieties in 2–4. Interestingly, the spectrum of the two-coordinate complex 4 shows a very large ΔEQ of 4.57 mm s−1, distinct from that observed for [K(crypt-222)][Fe(C(SiMe3)3)2] (8).25 While the isomer shifts for 4 and 8 are similar, the significantly increased ΔEQ value for 4 likely indicates a significant covalent contribution to the electric field gradient (EFG) in 4.26 Intrigued by their unique structural and spectroscopic features, we are now pursuing further spectroscopic and theoretical studies in order to develop a more complete understanding of the electronic structure and bonding in these low-coordinate iron aminocarbene compounds.
| δ/mm s−1 | ΔEQ/mm s−1 | δ/mm s−1 | ΔEQ/mm s−1 | ||
|---|---|---|---|---|---|
| a 5 = [K(nacnac)Fe(μ-S)Fe(nacnac)K], 80 K, ref. 7d. b 6 = [(nacnac)Fe(HCCPh)], 80 K, ref. 7b. c 7 = [K(crypt-222)][(nacnac)FeH], 80 K, ref. 7c. d 8 = [K(crypt-222)][Fe(C(SiMe3)3)2], 80 K, ref. 25. Mössbauer fitting errors are ±0.02 mm s−1 for δ and ±2% for ΔEQ. | |||||
| 1 | 0.65 | 2.63 | 5 | 0.67 | 2.17 |
| 2 | 0.52 | 2.04 | 6 | 0.44 | 2.05 |
| 3 | 0.49 | 2.03 | 7 | 0.40 | 1.93 |
| 4 | 0.49 | 4.58 | 8 | 0.40 | 2.55 |
Conclusions
In summary, we have achieved the synthesis of the series of three-coordinate formal iron(I) complexes [(L)(L′)FeCl] (L, L′ = IMes, Me2-cAAC) with the bulky aminocarbenes as supporting ligands. Associated with the different electronic properties of NHCs versus cAACs, these three-coordinate complexes exhibit differentiated thermal stability, Fe–C(carbene) distances, and 57Fe Mössbauer isomer shifts. The three-coordinate iron complexes can react with Na[BArF4], from which we have successfully isolated and structurally characterized a two-coordinate iron aminocarbene complex [(Me2-cAAC)2Fe][BArF4].27 The attainment of 1–4 demonstrates that both monodentate NHCs and cAACs are capable of supporting low-coordinate formal iron(I) species.Acknowledgements
We acknowledge the financial support from the National Basic Research Program of China (no. 2011CB808705), the National Natural Science Foundation of China (no. 21121062 and 21222208), and the Chinese Academy of Sciences.Notes and references
- M. V. Twigg and J. Burgess, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2005, vol. 5, p. 460 Search PubMed.
- For example, see: (a) D. Seyferth, R. S. Henderson and L. C. Song, Organometallics, 1982, 1, 125 CrossRef CAS; (b) T. Liu and M. Y. Darensbourg, J. Am. Chem. Soc., 2007, 129, 7008 CrossRef CAS PubMed; (c) J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2011, 4, 26 CrossRef PubMed.
- For example, see: (a) J. M. Smith, R. J. Lachicotte, K. A. Pittard, T. R. Cundari, K. A. Lukat-Rodgers, K. R. Rodgers and P. L. Holland, J. Am. Chem. Soc., 2001, 123, 9222 CrossRef CAS; (b) M. P. Hendrich, W. Gunderson, R. K. Behan, M. T. Green, M. P. Mehn, T. A. Betley, C. C. Lu and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17107 CrossRef CAS PubMed; (c) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780 CrossRef CAS PubMed; (d) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed.
- For example, see: (a) C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. ÁngelesCartes, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333 CrossRef CAS PubMed; (b) M. Guisán-Ceinos, F. Tato, E. Bunuel, P. Calle and D. J. Cárdenas, Chem. Sci., 2013, 4, 1098 RSC; (c) J. Kleimark, A. Hedstrçm, P.-F. Larsson, C. Johansson and P.-O. Norrby, ChemCatChem, 2009, 1, 152 CrossRef CAS PubMed.
- For recent examples, see: (a) S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322 CrossRef CAS PubMed; (b) R. P. Rose, C. Jones, C. Schulten, S. Aldridge and A. Stasch, Chem. – Eur. J., 2008, 14, 8477 CrossRef CAS PubMed; (c) C. Ni, B. D. Ellis, J. C. Fettinger, G. J. Long and P. P. Power, Chem. Commun., 2008, 1014 RSC; (d) T. Nguyen, W. A. Merrill, C. Ni, H. Lei, J. C. Fettinger, B. D. Ellis, G. J. Long, M. Brynda and P. P. Power, Angew. Chem., Int. Ed., 2008, 47, 9115 CrossRef CAS PubMed; (e) M. T. Mock, C. V. Popescu, G. P. A. Yap, W. G. Dougherty and C. G. Riordan, Inorg. Chem., 2008, 47, 1889 CrossRef CAS PubMed; (f) I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716 CrossRef CAS PubMed; (g) C. Vogel, J. S. Heinemann, C. Anthon and K. Meyer, Angew. Chem., Int. Ed., 2008, 47, 2681 CrossRef CAS PubMed; (h) Y. Nakajima, Y. Nakao, S. Sakaki, Y. Tamada, T. Ono and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 9934 CrossRef CAS PubMed; (i) J. M. Hoyt, K. T. Sylvester, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 4862 CrossRef CAS PubMed.
- For reviews on low-coordinate 3d metal species, see: (a) C. C. Cummins, Prog. Inorg. Chem., 1998, 47, 685 CrossRef CAS PubMed; (b) P. L. Holland, Acc. Chem. Res., 2008, 41, 905 CrossRef CAS PubMed; (c) P. P. Power, Chem. Rev., 2012, 112, 3482 CrossRef CAS PubMed; (d) D. L. Kays, Dalton Trans., 2011, 769 RSC.
- (a) J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756 CrossRef CAS PubMed; (b) S. A. Stoian, Y. Yu, J. M. Smith, P. L. Holland, E. L. Bominaar and E. Münck, Inorg. Chem., 2005, 44, 4915 CrossRef CAS PubMed; (c) K. P. Chiang, C. C. Scarborough, M. Horitani, N. S. Lees, K. Ding, T. R. Dugan, W. W. Brennessel, E. Bill, B. M. Hoffman and P. L. Holland, Angew. Chem., Int. Ed., 2012, 51, 3658 CrossRef CAS PubMed; (d) M. M. Rodriguez, B. D. Stubbert, C. C. Scarborough, W. W. Brennessel, E. Bill and P. L. Holland, Angew. Chem., Int. Ed., 2012, 51, 8247 CrossRef CAS PubMed.
- M. J. Ingleson, B. C. Fullmer, D. T. Buschhorn, H. Fan, M. Pink, J. C. Huffman and K. G. Caulton, Inorg. Chem., 2008, 47, 407 CrossRef CAS PubMed.
- L. Fohlmeister, S. Liu, C. Schulten, B. Moubaraki, A. Stasch, J. D. Cashion, K. S. Murray, L. Gagliardi and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8294 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577 CrossRef CAS PubMed.
- For reviews and recent reports on iron-NHC complexes: see: (a) M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 48, 3579 RSC; (b) D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19 CrossRef PubMed; (c) K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215 CrossRef CAS PubMed; (d) S. Zlatogorsky, C. A. Muryn, F. Tuna, D. J. Evans and M. J. Ingleson, Organometallics, 2011, 30, 4974 CrossRef CAS; (e) S. Meyer, C. M. Orben, S. Demeshko, S. Dechert and F. Meyer, Organometallics, 2011, 30, 6692 CrossRef CAS; (f) J. A. Przyojski, H. D. Arman and Z. J. Tonzetich, Organometallics, 2012, 31, 3264 CrossRef CAS; (g) T. Hashimoto, S. Urban, R. Hoshino, Y. Ohki, K. Tatsumi and F. Glorius, Organometallics, 2012, 31, 4474 CrossRef CAS; (h) B. Blom, G. Tan, S. Enthaler, S. Inoue, J. D. Epping and M. Driess, J. Am. Chem. Soc., 2013, 135, 18108 CrossRef CAS PubMed; (i) H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Angew. Chem., Int. Ed., 2013, 52, 8045 CrossRef CAS PubMed.
- For example, see: (a) Z. Mo, D. Chen, X. Leng and L. Deng, Organometallics, 2012, 31, 7040 CrossRef CAS; (b) C. A. Laskowski, D. J. Bungum, S. M. Baldwin, S. A. Del Ciello, V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2013, 135, 18272 CrossRef CAS PubMed; (c) M. Ito, T. Matsumoto and K. Tatsumi, Inorg. Chem., 2009, 48, 2215 CrossRef CAS PubMed.
- (a) V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; (b) V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569 CrossRef CAS PubMed; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (d) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304 CrossRef CAS PubMed.
- A. P. Singh, P. P. Samuel, H. W. Roesky, M. C. Schwarzer, G. Frenking, N. S. Sidhu and B. Dittich, J. Am. Chem. Soc., 2013, 135, 7324 CrossRef CAS PubMed.
- D. S. Weinberger, N. Amin SK, K. C. Mondal, M. Melaimi, G. Bertrand, A. Claudia Stückl, H. W. Roesky, B. Dittrich, S. Demeshko, B. Schwederski, W. Kaim, P. Jerabek and G. Frenking, J. Am. Chem. Soc., 2014, 136, 6235 CrossRef CAS PubMed.
- (a) A. J. Arduengo III, S. F. Gamper, J. C. Calabrese and F. Davidson, J. Am. Chem. Soc., 1994, 116, 4391 CrossRef; (b) R. C. Poulten, M. J. Page, A. J. Algarra, J. J. Le Roy, I. López, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640 CrossRef CAS PubMed; (c) K. C. Mondal, P. P. Samuel, Y. Li, H. W. Roesky, S. Roy, L. Ackermann, N. S. Sidhu, G. M. Sheldrick, E. Carl, S. Demoshko, S. De, P. Parameswaran, L. Ungur, L. F. Chiboyaru and D. M. Andrada, Eur. J. Inorg. Chem., 2014, 818 CrossRef CAS PubMed.
- P. P. Samuel, K. C. Mondal, H. W. Roesky, M. Hermann, G. Frenking, S. Demeshko, F. Meyer, C. Stuckl, J. H. Christian, N. S. Dalal, L. Ungur, L. F. Chibotaru, K. Pröpper, A. Meenta and B. Dittrich, Angew. Chem., Int. Ed., 2013, 52, 11817 CrossRef CAS PubMed.
- Z. Ouyang and L. Deng, Organometallics, 2013, 32, 7268 CrossRef CAS.
- For detailed information, please see ESI.†.
- (a) L. Xiang, J. Xiao and L. Deng, Organometallics, 2011, 30, 2018 CrossRef CAS; (b) A. A. Danopoulos, P. Braunstein, N. Stylianides and M. Wesolek, Organometallics, 2011, 30, 6514 CrossRef CAS; (c) B. M. Day, T. Pugh, D. Hendriks, C. F. Guerra, D. J. Evans, F. M. Bickelhaupt and R. A. Layfield, J. Am. Chem. Soc., 2013, 135, 13338 CrossRef CAS PubMed.
- T. Hashimoto, R. Hoshino, T. Hatanaka, Y. Ohki and K. Tatsumi, Organometallics, 2014, 33, 921 CrossRef CAS.
- S. Pelties and R. Wolf, Z. Anorg. Allg. Chem., 2013, 639, 2581 CrossRef CAS PubMed.
- K. C. Mondal, P. P. Samuel, H. W. Roesky, E. Carl, R. Herbst-Irmer, D. Stalk, B. Schwederki, W. Kaim, L. Ungur, L. F. Chibotaru, M. Hermann and G. Frenking, J. Am. Chem. Soc., 2014, 136, 1770 CrossRef CAS PubMed.
- A. M. Lapointe, Inorg. Chim. Acta, 2003, 345, 359 CrossRef CAS.
- J. M. Zadrozny, D. J. Xiao and J. R. Long, Inorg. Chem., 2013, 52, 13123 CrossRef CAS PubMed.
- (a) E. J. Hawrelak, W. H. Bernskoetter, E. Lobkovsky, G. T. Yee, E. Bill and P. J. Chirik, Inorg. Chem., 2005, 44, 3103 CrossRef CAS PubMed; (b) P. A. Rudd, S. Liu, N. Plana, E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2013, 52, 4449 CrossRef CAS PubMed.
- During the submission of this manuscript, a study on three- and two-coordinate iron complexes with Et2-cAAC (3,3-diethyl-5,5-dimethyl-1-(2′,6′-diisopropylphenyl)pyrrolidine-2-ylidene) appeared: G. Ung, J. Rittle, M. Soleihavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201404078.
Footnotes |
| † Electronic supplementary information (ESI) available: Text gives the experimental procedures, Mössbauer spectra, and characterization for 1–4. CCDC 1010108–1010111 for 1–4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00175c |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2014 |