
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative rearrangements during fungal biosynthesis
Russell
Cox
*ab
aInstitute for Organic Chemistry, Leibniz University of Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
bSchool of Chemistry, University of Bristol, Cantock's Close, Clifton, Bristol, BS8 1TS, UK. E-mail: r.j.cox@bristol.ac.uk
First published on 25th July 2014
Abstract
Covering: 1942–2014
Oxidative rearrangements are key reactions during the biosyntheses of many secondary metabolites in fungi. This review highlights the most important examples of these reactions and aims to draw together key mechanistic themes to allow a better understanding and future exploitation of this key class of fungal catalysts.
 Russell Cox | Russell Cox was born in 1967 in the New Forest in the UK where he grew up. He studied chemistry at the University of Durham, and then worked with Prof. David O'Hagan at the same institution for his Ph.D., studying the biosynthesis of fungal metabolites. Post-doctoral periods with Professor John Vederas FRS in Edmonton Alberta, and Professors David Hopwood FRS and Tom Simpson FRS at Norwich and Bristol in the UK were followed by his appointment as a lecturer in the School of Chemistry at the University of Bristol where he rose to become Professor of Organic and Biological Chemistry. Russell Cox moved to become Professor of Microbiological Chemistry at the Leibniz Universität Hannover in Germany in 2013. He served as an editorial board member of Natural Product Reports until 2012, and is currently an editorial board member of RSC Advances. |
1 Introduction and scope
Fungi produce natural products with arguably the greatest range of structural complexity of all micro-organisms. They achieve this using much the same ‘standard’ machinery as other organisms, using: polyketide synthases (PKS); ribosomal synthesis and non-ribosomal peptide synthetases (NRPS); and terpene cyclases etc. These basic skeletons are then diversified using highly selective tailoring reactions. In fungi these tailoring steps are very often oxidative, and an important sub-set of these oxidative reactions are those which also include a skeletal rearrangement. Such reactions are known in synthetic chemistry, but their scope and selectivity are limited compared to those observed in fungi. This review will cover the most important of these biosynthetic transformations – it does not set out to be comprehensive, but to illustrate the relationships between the best known oxidative rearrangements. It is no coincidence that the review appears in this particular issue Natural Product Reports since the contributions of Professors Simpson, Vederas and Townsend have been seminal to development of the subject.2 Cephalosporins
The production of β-lactams provides one of the most compelling examples of the power of selective oxidations during biosynthesis. The tripeptide ACV 1 is first oxidised twice by a single equivalent of dioxygen, catalysed by the enzyme isopenicillin-N-synthase (IPNS) to form the bicyclic penicillin skeleton 2. While the reaction catalysed by IPNS is oxidative, it is not strictly a rearrangement and will not be discussed here (IPNS has been recently and comprehensively reviewed elsewhere).1 A distal epimerisation of 2 giving penicillin N 3 is then followed by an oxidative rearrangement to provide the cephalosporin C skeleton (Scheme 1).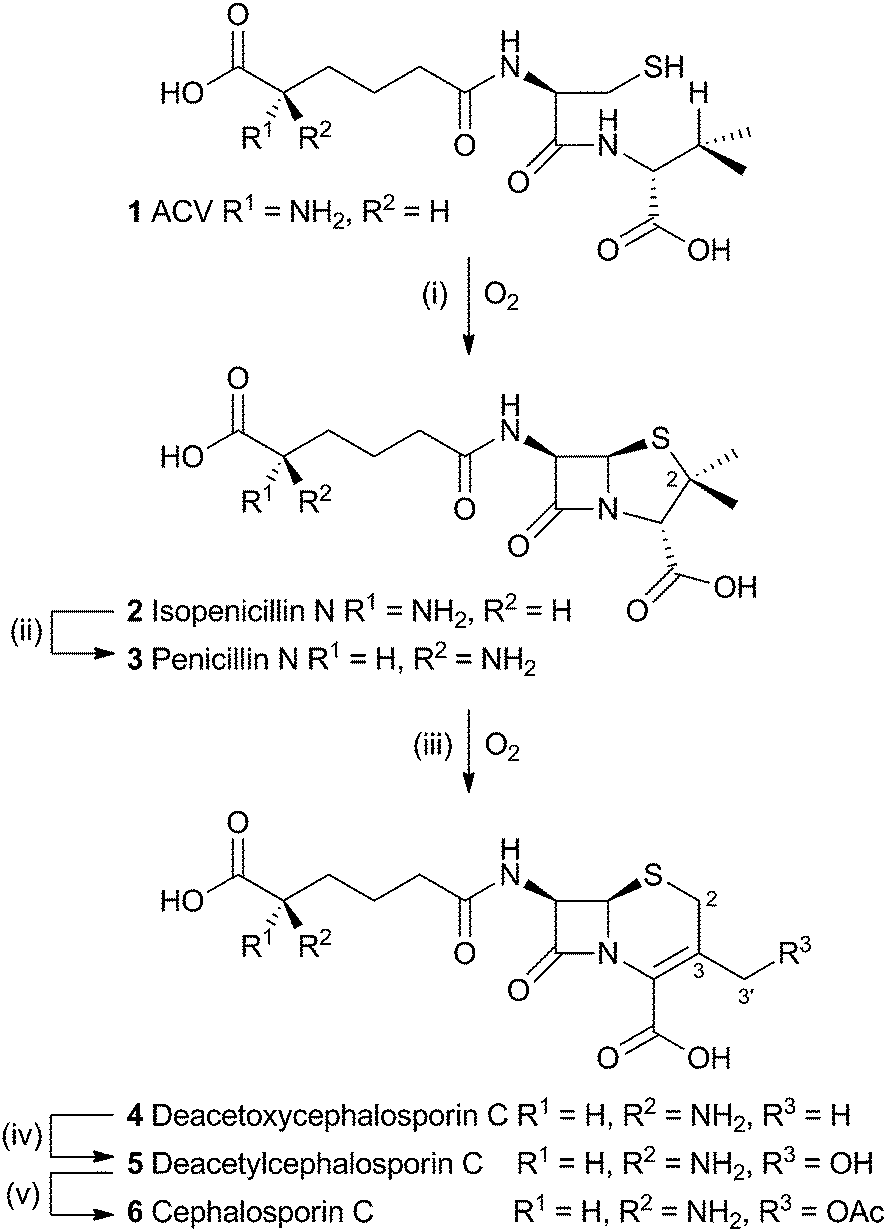 | ||
| Scheme 1 Overall pathway from ACV 1 to cephalosporin C 6. Enzymes: (i), isopenicillin N synthase; (ii), isopenicillin N epimerase; (iii) deacetoxycephalosporin C synthase; (iv) deacetylcephalosporin C synthase; (v) acetyl-coenzyme A:DAC O-acetyltransferase. | ||
The rearrangement is known to occur both in fungi (e.g. Acremonium chrysogenum, also known as Cephalosporium acremonium) and bacteria (e.g. Streptomyces clavuligerus). In bacteria the rearrangement is catalysed by an enzyme known as deacetoxycephalosporin C synthase (DAOCS) – a second oxidase then catalyses hydroxylation at the 3′ methyl position (deacetylcephalosporin C synthase, DACS). In fungi both transformations are performed by a single bifunctional enzyme (known as DAOCS/DACS).
2.1 Isotopic labelling experiments
Early work using cell-free extracts of A. chrysogenum2 showed that an impure enzyme fraction could be prepared which possessed the ring-expanding activity and which was stimulated by iron, α-ketoglutarate3 and ascorbate – typical of non-heme iron dioxygenases.4 Isopenicillin N 2 was shown to be the direct substrate, ruling out parallel pathways from ACV to 3 and 6.5 Pioneering synthesis by Baldwin and coworkers of (2RS,3R)-[4-13C]-valine 7a allowed the first investigations of the stereoselectivity of the ring expansion to be performed.613C NMR was used to show that the labelled valine was incorporated into both the penicillin skeleton and into cephalosporin C 6. Specifically, the 13C label was incorporated into the β-methyl position of penicillin V isolated from Penicillium chrysogenum and into the 2-position of Cephalosporin C 6 from A. chrysogenum suggesting that the ring expansion must selectively incorporate the β-methyl group into the thiopyran ring of cephalosporin.7 Later the same year Sih and Abraham reported the synthesis of (2S,3S)-[4-13C]-valine 7b and showed that the label is incorporated into the α-methyl of penicillin N 3 and the 3′-methylene of cephalosporin C 6 (Scheme 2A).8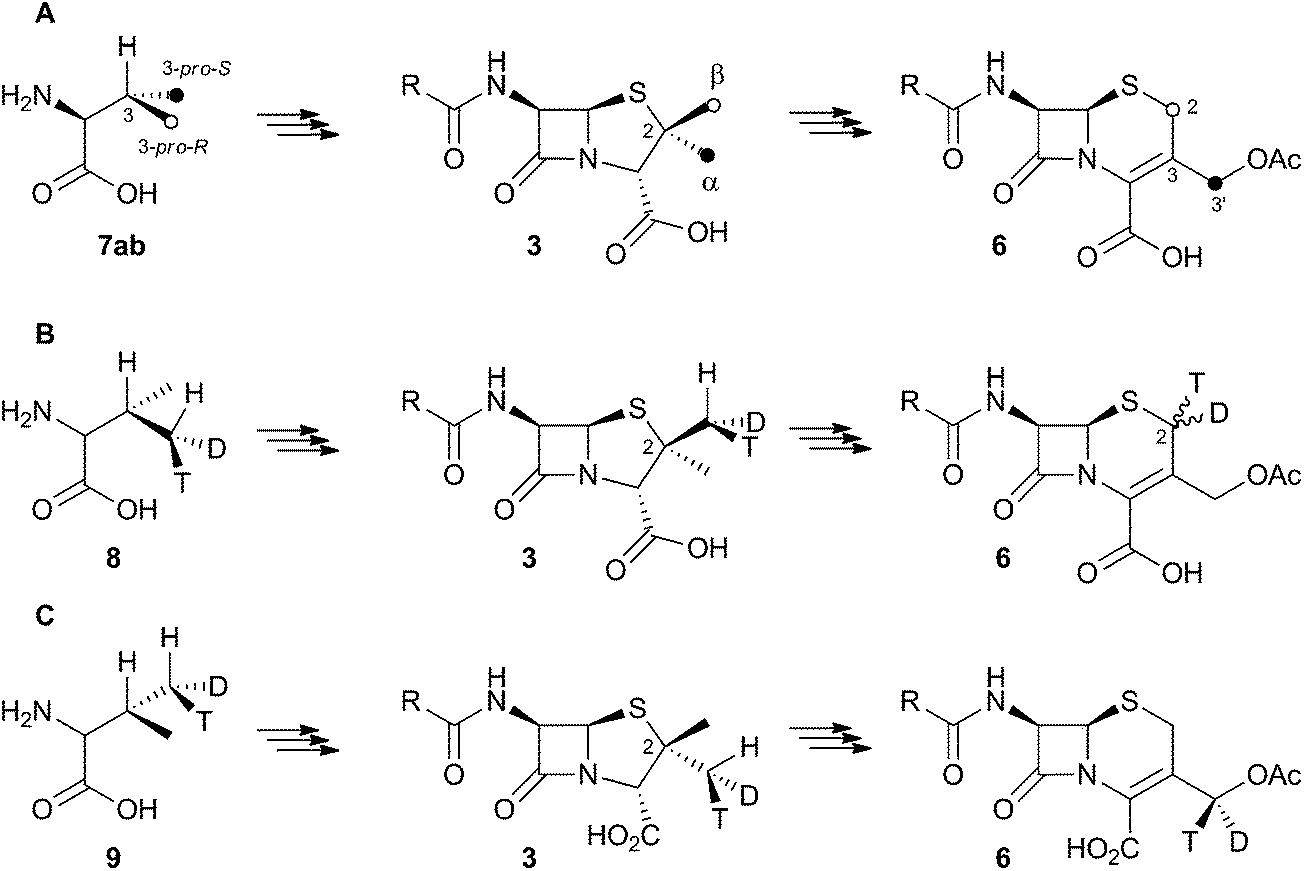 | ||
| Scheme 2 Results of isotopic feeding studies using chiral valines. | ||
Townsend and coworkers investigated the stereochemical fate of the 3-pro-R methyl group of valine during the ring expansion reaction (Scheme 2B and C). They synthesised both (2RS,3R,4R)8 and (2RS,3R,4S)-[4-2H,3H]valine99 and fed them to growing cultures of Acremonium strictum. The prior work of Baldwin and others already showed that the 3-pro-R methyl of valine is incorporated into the ring during expansion with clean retention of configuration at C-3. Townsend's results showed that, in contrast, stereochemical integrity at the β-2-methyl of penicillin N 3 is lost, showing that the ring expansion proceeds with complete epimerization (Scheme 2B).10 This loss of stereochemical integrity, however, is not a feature intrinsic to the fungal DAOCS/DACS enzyme because in a parallel series of experiments it was shown that hydroxylation at the 3′ carbon of deacetoxycephalosporin C 5, performed by the same enzyme, proceeds with retention of stereochemistry (Scheme 2C).11
These results were interpreted as an indication that the first step of the rearrangement is the abstraction of a hydrogen atom from the β-methyl group of 3 by a typical iron(IV) oxo species, and formation of a methylene radical 10 which rapidly rotates (thus scrambling the label to 11) before the next step. In the case of abstraction of a hydrogen atom from the 3′ methyl of 4, the formed methylene radical 12 cannot rotate (or at least rotates more slowly than competing hydroxylation), probably because of conjugation of the methylene radical to the adjacent olefin, and thus hydroxylation occurs with retention of stereochemistry giving 5 (Scheme 3).
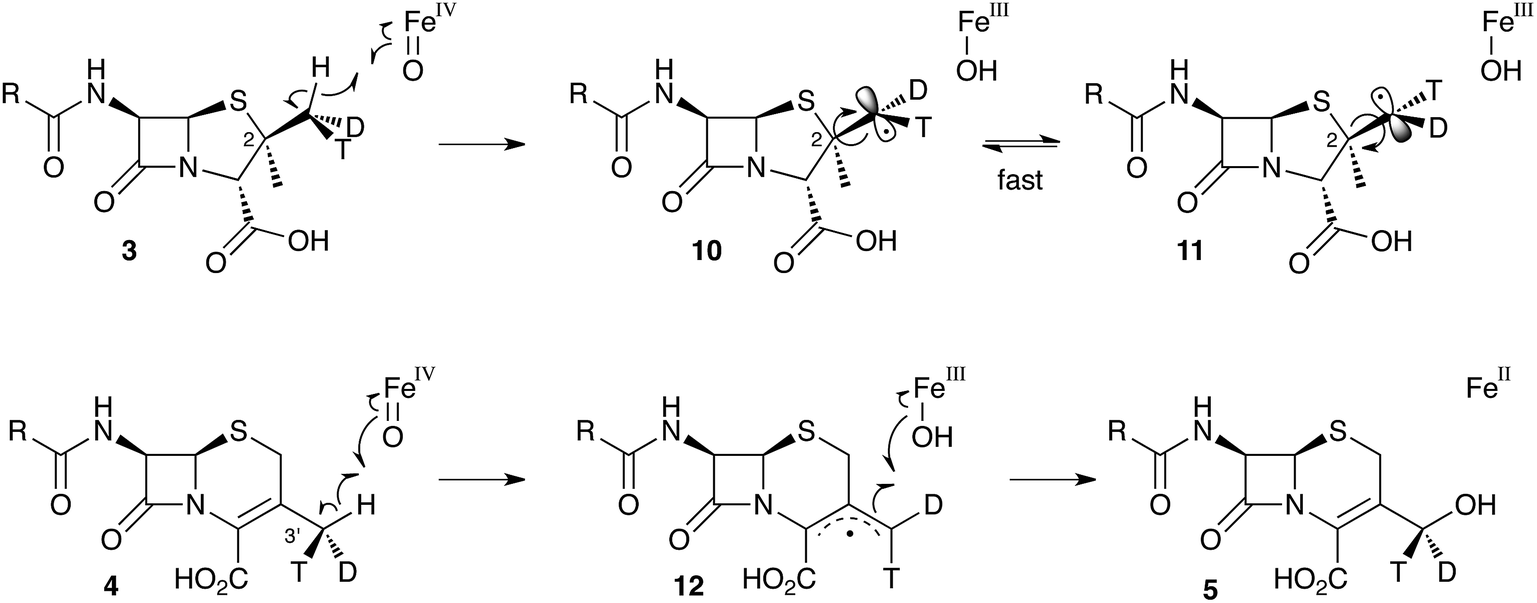 | ||
| Scheme 3 Hydrogen abstraction reactions catalysed by DAOCS/DACS. Iron ligands not shown for clarity. See Scheme 6 for a more detailed proposed mechanism of the ring expansion. | ||
It was already known that growing cultures of C. acremonium produced the hydrated deacetoxycephalosporin C analogue 13 in low titre (Scheme 4). This compound is also produced in vitro using purified DAOCS/DACS (around 1.6% of the total ring-expanded products) showing that it is produced by the enzyme rather than by another activity of the whole organism. Baldwin and coworkers reported an intriguing observation when they used [3-2H] penicillin N 3a as the in vitro substrate of DAOCS/DACS.12 In this case 13 was produced as 35% of the total ring expanded product. This result was interpreted as indicating the presence of a thiranyl cation or radical intermediate such as 14 (Scheme 4). Elimination of the 3-hydrogen would lead to the major product 4, but operation of a significant isotope effect could slow this route and allow hydroxylation to give 13. Hydroxylation on the top face of the cepham skeleton is consistent with the proposed position of the enzyme's iron hydroxyl species. Labelling with 18O2 showed that the introduced oxygen of 13 does indeed come from this source. 13 is not a substrate for further 3′ oxidation by DAOCS/DACS, and although not explicitly reported, seems unlikely to be an intermediate on the pathway to 6 itself.
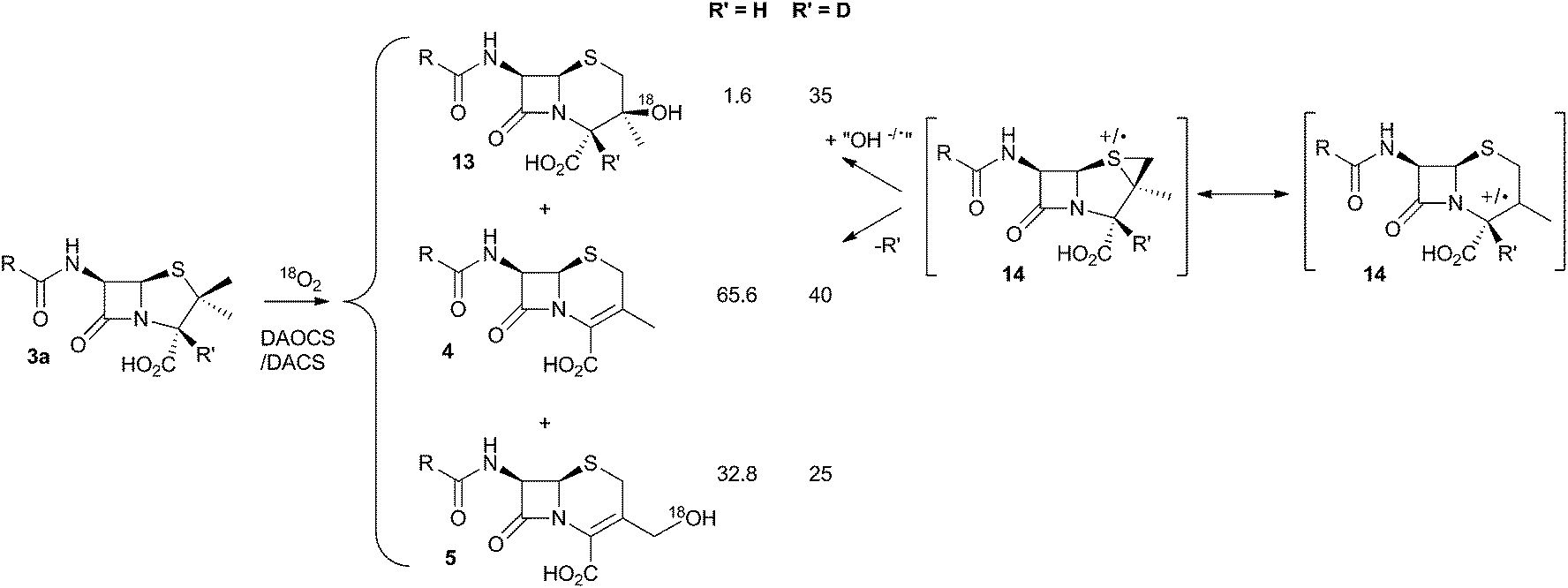 | ||
| Scheme 4 Effect of 2H at the 3-position of penicillin N 3a during the ring expansion. | ||
Further mechanistic insight into the ring expansion is scant – but information from two sources has been used to formulate a plausible hypothesis. First, biomimetic reactions have been performed in vitro which provide information about possible mechanisms. Second, and more latterly, structural data has been obtained from the bacterial DAOCS.
2.2 Biomimetic reactions
The chemical conversion of penicillin N 3 to cephalosporin C 6 was first achieved by Morin and coworkers at Lilly.13 The Morin rearrangement begins with oxidation of the sulfur atom of 3-methyl ester to sulfoxide 15. Treatment of this with a trace of acid in refluxing toluene then gives the cepham skeleton. The most likely mechanism involves initial thermal elimination of a sulfenic acid to provide an olefinic intermediate 16. This then recombines and displaces water to form the cepham cation 17, and final elimination of a proton provides the observed product 4 (Scheme 5A). Baldwin and coworkers later investigated related one-electron processes by synthesising a variety of radical precursors (Scheme 5B). In all cases initiation of homolysis led to the same mixture of products (roughly 35% of each diastereomer of 21) when Ph3SnH was used. The allyl thiol 22 was formed in the presence of allyltriphenyl tin.14 It thus appears that regardless of the starting radical, the intermediates probably rearrange to the most stable radical 23c which then abstracts a hydrogen from Ph3SnH to form diastereomers of 21. The more nucleophilic radical 23b reacts instead with allyltriphenyl tin to give 22. In principle, thermal heterolytic processes could potentially form the same cationic intermediates suggested for the Morin reaction in these reactions (Scheme 5A), but in the absence of radical initiators no products were formed ruling out this mechanistic possibility.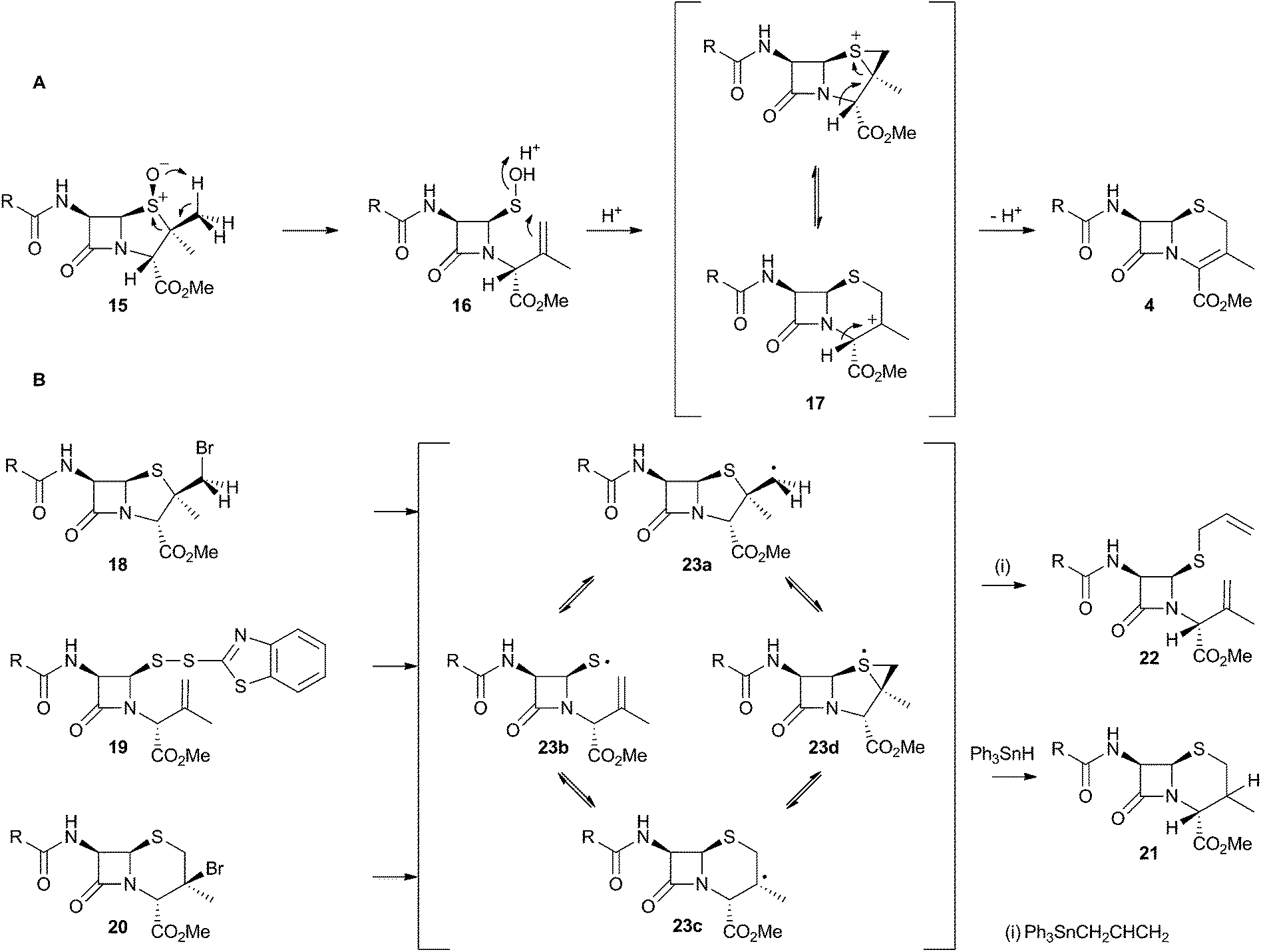 | ||
| Scheme 5 Likely mechanistic courses of the Morin rearrangement and radical reactions reported by Baldwin and coworkers. | ||
2.3 Structural information
The crystal structure of the fungal DACS enzyme has not yet been reported, but the structure of the analogous bacterial DAOCS enzyme was determined by Baldwin and coworkers and reported in 1998.15 Although the enzyme is trimeric in the crystal structures it is thought to function in solution as a monomer. The active site contains the expected iron atom, bound by two histidines (H183 and H243) and an aspartate (D185) (Fig. 1). The structure also clearly shows α-ketoglutarate (αKG) bound to the iron, ready for reaction with molecular oxygen. Crystals containing substrate analogues such as ampicillin show that the αKG and O2 must bind and react first, before expulsion of succinate followed by binding of the substrate ready for expansion. As expected, the β-methyl group comes within 3 Å of the likely position of the ferryl oxygen atom, rationalising the high selectivity of the oxidation.16,17 Thus the currently accepted mechanism (Scheme 6) involves the generation of the iron(IV) oxo species by reaction of the Fe(II) enzyme with α-ketoglutarate and O2 (steps 1–4). Loss of succinate (step 5) allows substrate binding and the positioning of the β-methyl close to the iron(IV) oxo center. Hydrogen atom abstraction (step 6) is then followed by rearrangement (steps 7 and 8). It is suggested that the final steps involve electron transfer directly to the Fe(III) center, and elimination of a proton to form the product (step 9). Rebound oxygenation prior to this stage, would lead to the shunt product 13 (Scheme 4). Final dissociation of the product (step 10) allows another cycle to begin.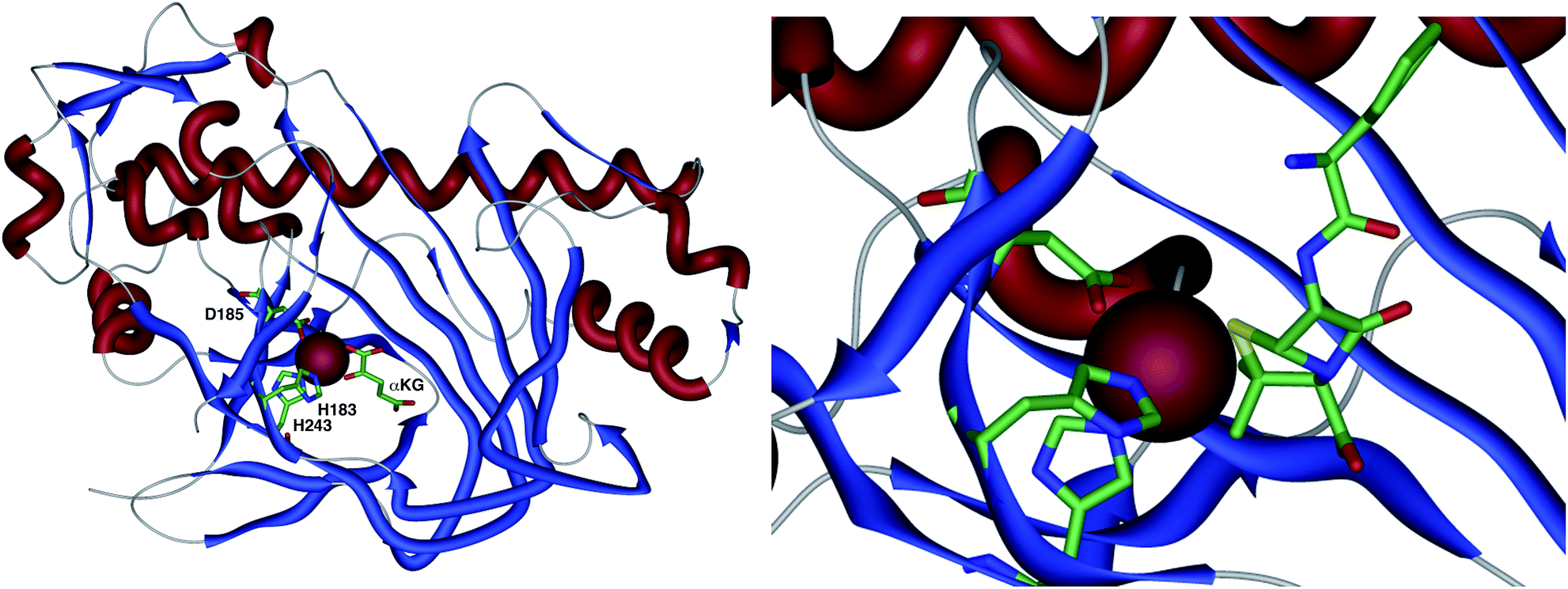 | ||
| Fig. 1 The overall structure of DAOCS showing the active site iron atom, key residues and substrates. Expansion shows ampicillin bound in active site. Coordinates from 1RXG and 1UNB respectively. | ||
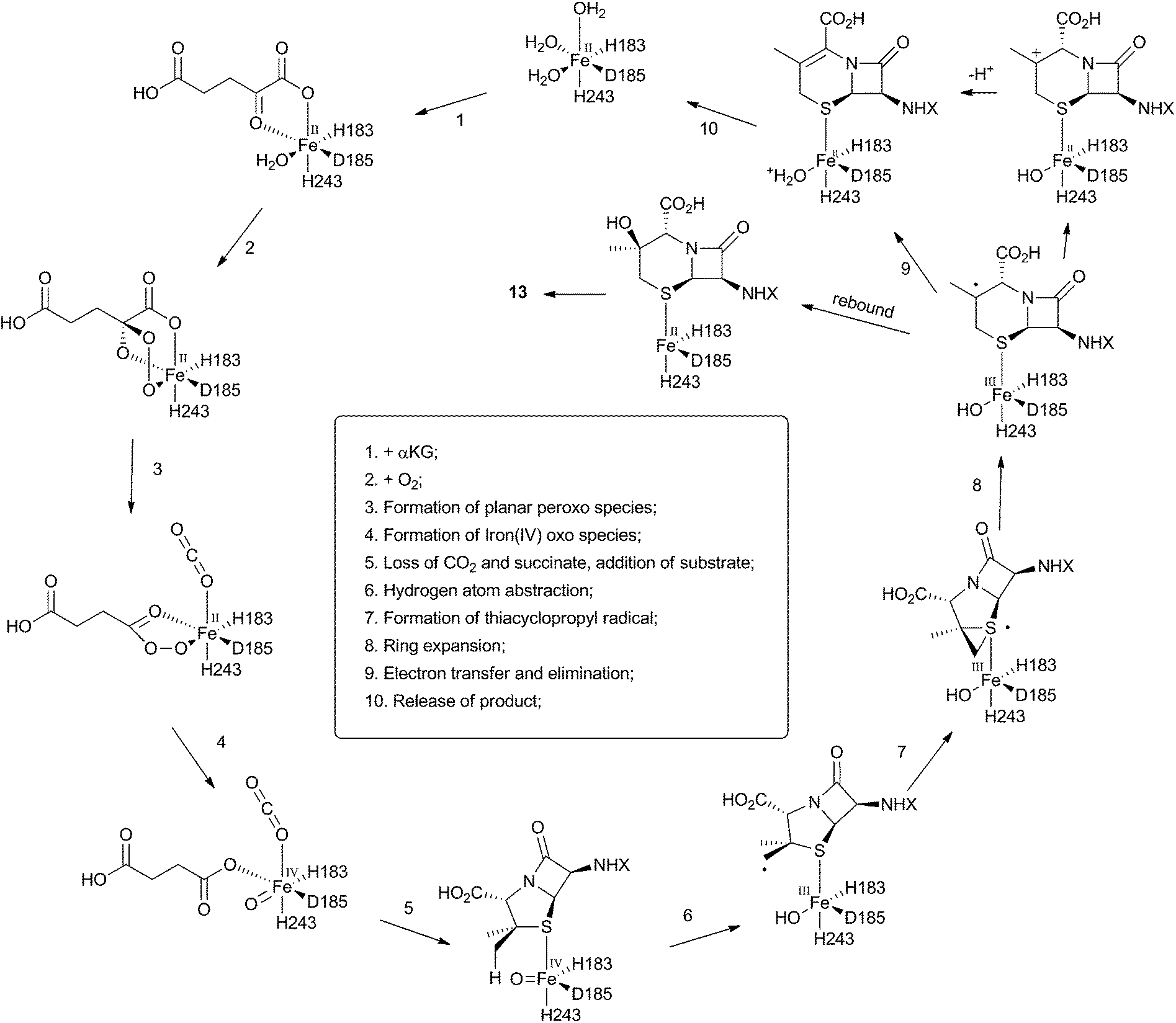 | ||
| Scheme 6 Mechanism of DAOCS deduced from structural analysis of its active site. | ||
3 Aflatoxins and related systems
Aflatoxin B1 24 is a potent environmental carcinogen produced by various species of Aspergilli from the polyketide intermediate norsolorinic acid 25. Its name derives from ‘Aspergillus flavus toxin’ and A. flavus and other producing-species such as A. parasiticus have been studied in depth. Aflatoxin B1 24 and related compounds such as sterigmatocystin, produced by A. versicolor have been the focus of detailed biosynthetic study. Over the years classical isotope feeding,18 genetic19 and enzymological20 studies have provided a detailed view of the overall biosynthetic pathway and several of the key steps. Like many other secondary metabolite pathways in fungi, the tailoring steps are mostly oxidative. The biosynthesis of the aflatoxins, however, is remarkable in requiring three oxidative rearrangements. The first of these sets up the key benzobisfuran motif which mediates DNA interaction; the second rearranges the polyketide-derived anthraquinone; and the final rearrangement is a remarkable oxidative ring contraction to form the cyclopentanone of 24. Indeed the number and complexity of the rearrangements undergone renders 24 almost unrecognisable as a polyketide and it is a testament to the skill of early workers that much of the pathway was deduced from isotopic feeding experiments and the study of blocked mutants.3.1 Formation of the benzobisfuran
Vederas performed labelling studies with a blocked mutant of A. parasiticus and showed that averufin is derived from acetate and atmospheric oxygen (Scheme 7A).21 Complementary feeding experiments from the groups of Simpson22 and Townsend23–26 using synthetic selectively isotope-labelled averufins then proved that averufin 26 is a precursor of aflatoxin B1 24 (Scheme 7B). These experiments also showed that the two tail (C-5′ and C-6′) carbons of averufin 26 are lost during biosynthesis, and that C-2 of averufin migrates from an acetate C-1 derived carbon in 26 to an acetate C-2 derived carbon in 27 during the rearrangement.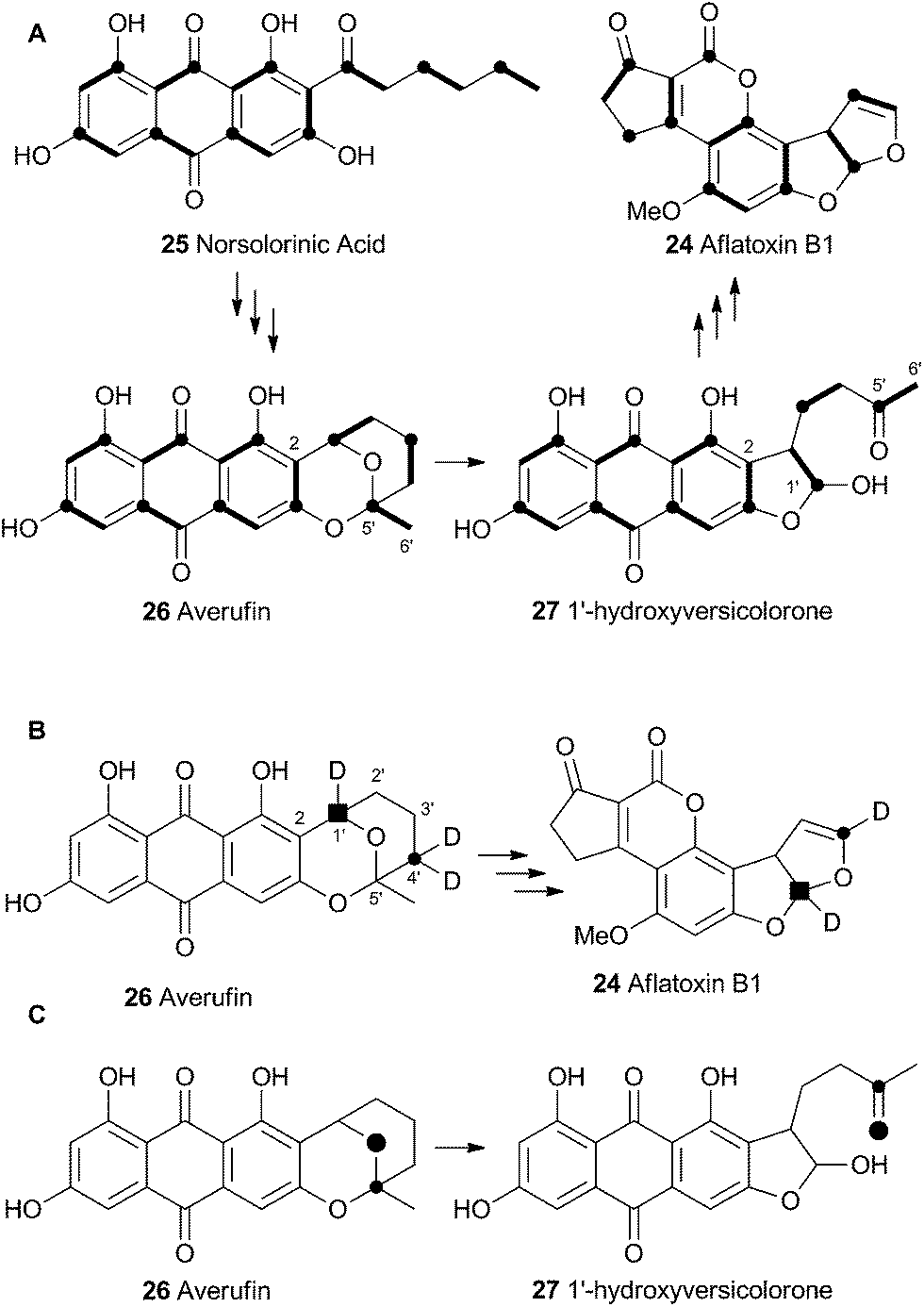 | ||
| Scheme 7 (A) Labelling patterns observed during the biosynthesis of aflatoxin B1. Bold bonds indicate intact acetate. Black circles indicate C-1 of acetate. (B) Selective labelling pattern during construction of the benzobisfuran system from averufin – results from 3 independent experiments shown together. C, retention of intact C–O bond observed via dual 13C–18O labelling. | ||
Townsend investigated the rearrangement using a model system in vitro which involved solvolysis of 6,8-di-O-methylnidurufin 28b and its 2′ epimer 6,8-di-O-methylpseudonidurufin 29b (Scheme 8).27,28 Activation of the 2′ hydroxyl to mesylates 28c and 29c and treatment at elevated temperature led to a smooth rearrangement in the case of 28c itself, but not in the case of its epimer 29c. Neither nidurufin 28a itself or pseudonidurufin 29a can be converted to aflatoxin by various strains of A. parasiticus (despite being shown to penetrate growing cells) and so neither are intermediates.25 It is thus concluded that oxidation of averufin 26 at the 2′ carbon is required for the rearrangement, but that oxygenation does not occur before the rearrangement.
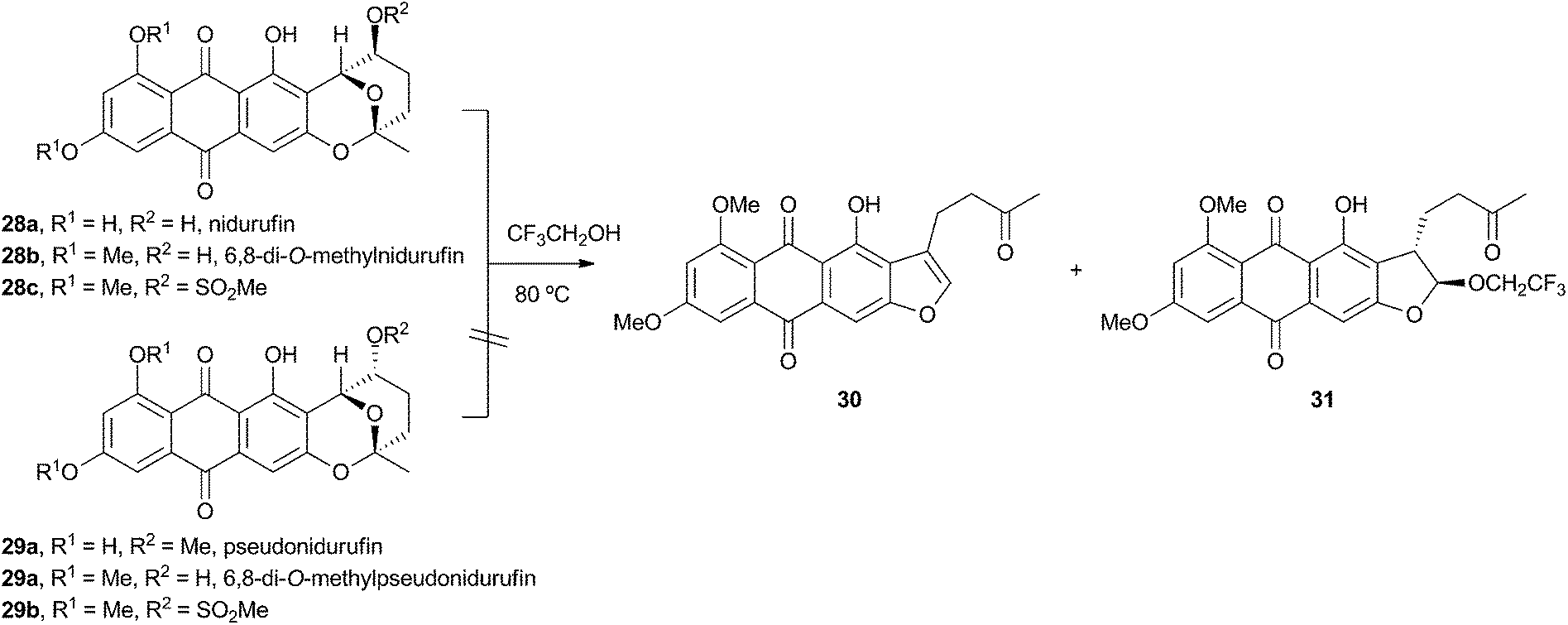 | ||
| Scheme 8 Rearrangement of activated and protected 6,8-di-O-methylnidurufin epimers. | ||
Attempts to investigate the possibility of a radical rearrangement mechanism met with failure starting with 2′-exo-iodo averufin 32, but treatment of 32 with silver acetate in organic solvent led to a high yield of 33, which, in turn, could be hydrolysed to 34 under mild conditions (Scheme 9). These observations are consistent with a rearrangement mechanism involving formation of the oxocarbenium ion 35 (Scheme 9B), and it was hypothesised that the biosynthetic reaction is likely to be very similar.
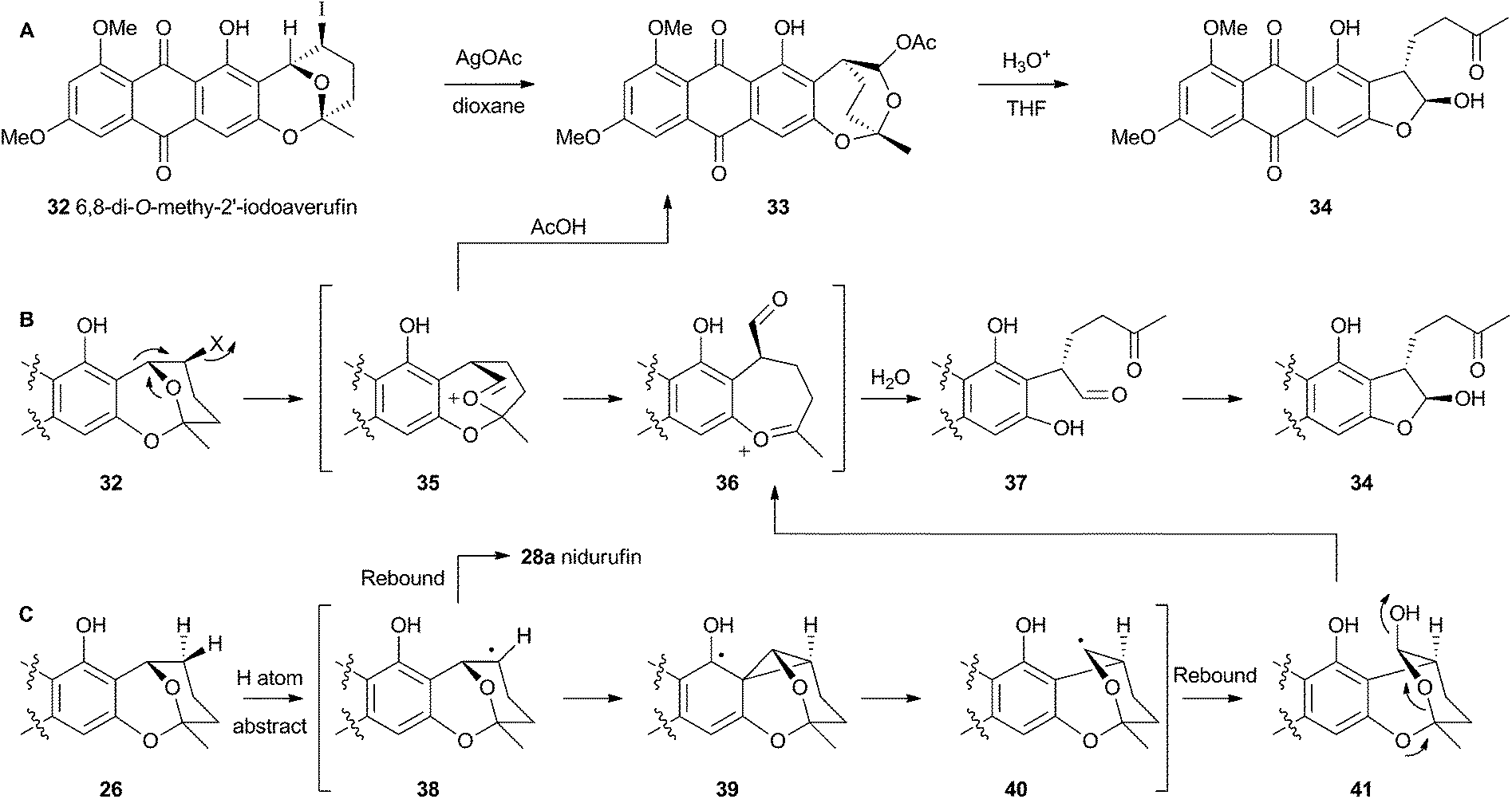 | ||
| Scheme 9 (A) In vitro reactions of 2′-iodoaverufin leading to the hydroxyversicolorone skeleton. (B) Proposed mechanism accounting for the stereoselectivity of the rearrangement. (C) Proposed reaction manifold stemming from cytochrome P450 catalysed hydrogen atom abstraction, followed by rearrangement and hydroxylation. | ||
Despite the evidence for a two-electron cationic process obtained from the model reactions in vitro, it has more recently been discovered that the enzyme responsible for the conversion of averufin 26 to hydroxyversicolorin 27in vivo is a cytochrome P450 enzyme (StcB)29 – a class of catalyst most frequently associated with hydrogen atom abstraction (and thus one electron) reactions and oxygenation via the accepted oxygen rebound mechanism.30 These observations can be rationalised if the rearrangement is initialised by hydrogen atom abstraction (Scheme 9C) followed by formation of a transient spirocyclopropane 39 stabilised by delocalisation of the radical into the phenolic system. This could rearomatise to give the oxymethylene radical 40 which could then be hydroxylated to 41. Elimination of water giving 36 would lead back to the previously proposed 2-electron reaction manifold (Scheme 9B). Early hydroxylation of the initially formed radical 38 would form nidurufin – a minor shunt metabolite of the pathway31 which is itself not a precursor of hydroxyversicolorone 27.
3.2 Oxidative rearrangement of versicolorin: formation of the sterigmatocystin skeleton
Hydroxyversicolorone 27 undergoes a series of conventional reactions including Baeyer–Villiger oxidation to form versiconal acetate,32 hydrolysis to versiconal24 and finally dehydration to versicolorin B 42.20 Versicolorin B is oxidised to Versicolorin A 43. Both anthraquinones 42 and 43 can undergo oxidative rearrangement to form the xanthone skeleton. The pathways are identical and the discussion here will focus on the rearrangement of 43 (Scheme 10). Two genes have been implicated in this process, aflN and aflM which encode a cytochrome P450 oxygenase and an NADPH dependent oxidoreductase respectively.33 Since the overall conversion of 43 to 44 requires two oxidations and one reduction it appeared likely that AflN should act twice, but the order of the reactions was not obvious from the knockout experiments. Townsend and coworkers synthesised 45, the putative product if the reduction occurred first, but this was not transformed to aflatoxin B1 24 by whole cells of A. parasiticus (Scheme 10A). Likewise, the products of putative early Baeyer–Villiger reaction (carboxy benzophenones such as 46) were not transformed either.34 Facile incorporation of demethyl sterigmatocystin 44a, but not of its 9-hydroxylated analogue showed that 44a was on the direct pathway to aflatoxin B1 24, and that reduction could not occur as the final step of the rearrangement. | ||
| Scheme 10 Deduced oxidative rearrangement of the versicolorin A anthraquinone 43 to the demethyl sterigmatocystin xanthone 44a. A, synthetic compounds not incorporated into the pathway; B, deduced pathway consistent with all evidence; C, Results of conversion of versicolorin to methylsterigmatocystin 44c in deuterated media showing high exchange into the A-ring but not the C-ring. | ||
These observations led to the development of a new hypothesis in which initial oxidative dearomatisation of the anthraquinone provides the first intermediate (47) during the rearrangement (Scheme 10B).35 Second step reduction to 49 and tautomerisation to 50 provides the substrate for a more conventional Baeyer–Villiger type oxidation as the final redox step giving 51. This must then be followed by hydrolysis, ring closing, decarboxylation and consequent rearomatisation to form the xanthone skeleton of 44a. While significant synthetic chemistry was achieved to access various putative intermediates, none was ever positively incorporated into aflatoxin B1 24in vivo. Furthermore it proved impossible to synthesise or observe any intermediate between 43 and 44a in Scheme 10. However, evidence for the pathway was accumulated from observations of incorporations of deuterium from labelled media specifically at C-8, C-9 and C-10 of 44c, supporting dearomatisation of the A-ring ring (Scheme 10C). Furthermore, Baeyer–Villiger reaction of the deraomatised intermediate 50 is likely to be energetically more feasible than that of the anthraquinone 43 – a species which is known as a very poor substrate for this reaction in vitro.36
3.3 Formation of the aflatoxin B1 and G1 skeletons
Unremarkable methyltransferase steps form sterigmatocystin 44b and O-methylsterigmatocystin 44c before the final oxidative rearrangements. Sankawa and coworkers showed that deuterium from [2-2H3] acetate labels sterigmatocystin 44b as shown in Scheme 11.37 Simpson and coworkers then followed the incorporation of [2-2H3] acetate into aflatoxin B1 24 and showed that C-5 of 24 becomes deuterated. Since this carbon derives from C-1 of acetate there must have been a 1,2-deuterium migration during biosynthesis and this was rationalised as being due to an NIH-shift (Scheme 11).38 This work also implicated 11-hydroxy-O-methylsterigmatocystin (HOMST) 54b as a pathway intermediate, but at this stage this could not be proven.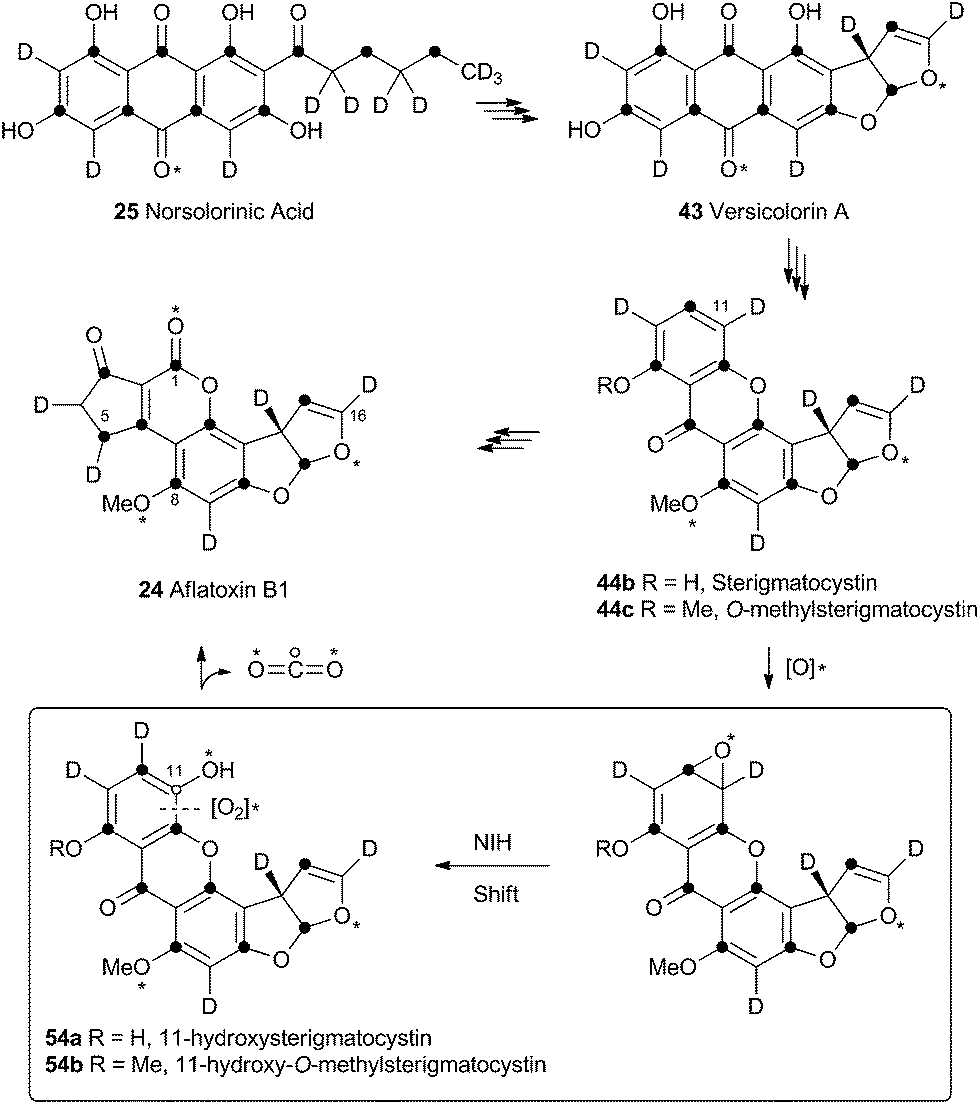 | ||
| Scheme 11 Rationalisation of results obtained when feeding [2-2H3] acetate to A. parasiticus. Dark circles indicate positions of acetate C-1-derived carbons. Hollow circle shows fate of C-11. * = atmospheric oxygen. | ||
Further evidence for the intermediacy of HOMST 54b came from experiments by Townsend and coworkers who produced sterigmatocystin 44b from [2-14C]-acetate and chemically converted it to O-methylsterigmatocystin 44c which was then used in cell-free assays with a mutant of A. parasiticus blocked in the early part of the pathway.39 The exogenous compound was converted to aflatoxin B1 24, and 14CO2 was released (Scheme 11). Rigorous controls showed that the label was not released as formate or formaldehyde. Radiochemical quantification of the CO2 and 24 produced showed the expected ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7. The results showed that the released carbon must be oxidised twice as shown in Scheme 11. Further isotopic labelling experiments directly probed the source and fate of the oxygen atoms used in the rearrangement with 18O2. Aflatoxin B1 24 produced under these conditions contained 18O at carbons 1, 8 and 16 in accord with the pathway set out in Scheme 11.40
:7. The results showed that the released carbon must be oxidised twice as shown in Scheme 11. Further isotopic labelling experiments directly probed the source and fate of the oxygen atoms used in the rearrangement with 18O2. Aflatoxin B1 24 produced under these conditions contained 18O at carbons 1, 8 and 16 in accord with the pathway set out in Scheme 11.40
No further progress was made in understanding this step until the gene ord1 was identified by knockout and expression as being involved in the conversion of O-methylsterigmatocystin 44c to aflatoxin B1 24 in 1997.41 The ord1 gene encodes a cytochrome P450 enzyme, and Townsend and coworkers showed that the Ord1 protein is the sole catalyst required for the remarkable transformation of 44c to 24.42 The ord1 gene was expressed in yeast and its product shown to be highly active in its ability to convert O-methylsterigmatocystin 44c to 24 in both whole cells and cell-free preparations. HOMST 54a was synthesised and also shown to be an efficient precursor to 24, placing it firmly on the direct pathway. Although no other intermediates have been isolated, the mechanism shown in Scheme 12 has been deduced from all the available evidence.41
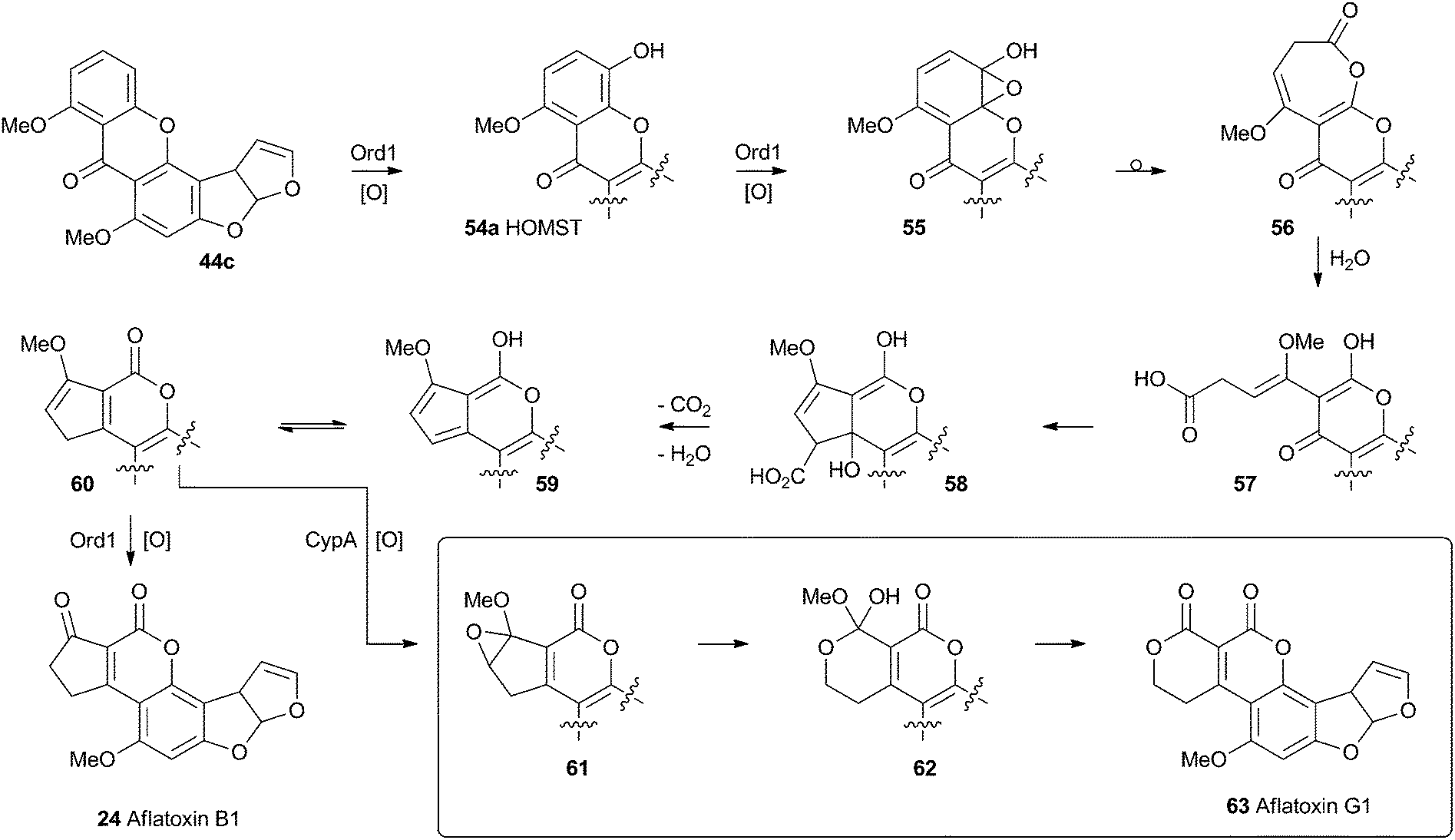 | ||
| Scheme 12 Proposed final oxidative rearrangement pathways during the formation of aflatoxins B1 24 and G1 63. HOMST 54a is the only intermediate which has been isolated and characterised. | ||
A final oxidative rearrangement during the formation of aflatoxin G1 63 has also recently been investigated. Evidence for the involvement of another cytochrome P450 enzyme, CypA, has been gathered by extensive KO and expression studies, as well as in vitro experiments. The evidence suggests that 60, or one of its precursors 58 or 59, is epoxidised leading to the formation of 61. Rearrangement and loss of methanol then leads to Aflatoxin G1 63.43
4 Spirocycles and related systems
Filamentous fungi produce a wide range of spirocyclic polyketides, meroterpenoids and alkaloids, exemplified by geodin 64, griseofulvin 65, austinol 66 and notoamide B 67.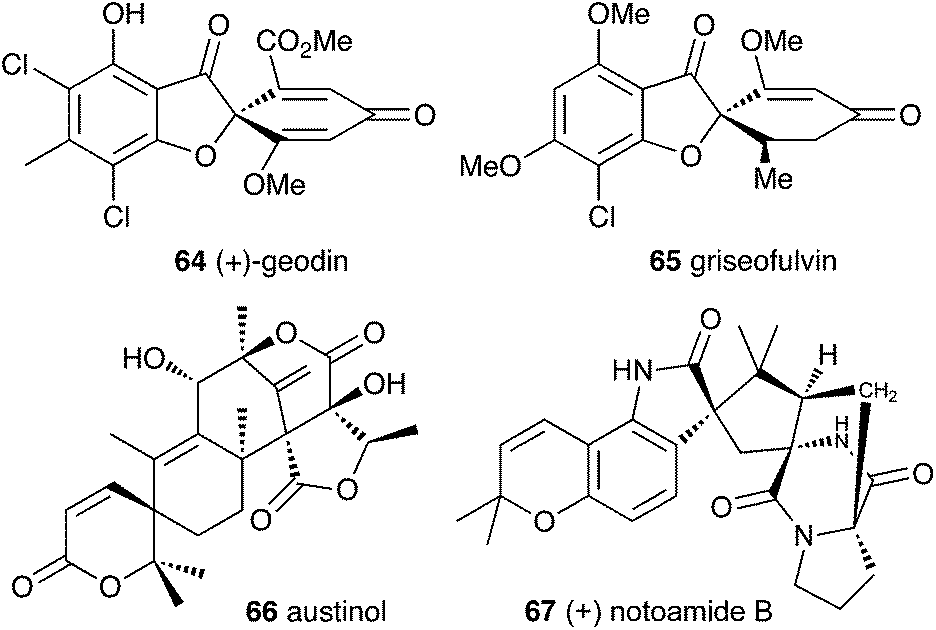
4.1 Geodin
Townsend and Henry speculated about the biosynthesis of geodin based upon the conclusions gained from the study of the xanthone formation during aflatoxin biosynthesis.34 It was already known that the polyketide emodin 68 is methylated to form questin 69.44 It was proposed that questin 69 then undergoes and AflN-like oxidative dearomatisation and Baeyer–Villiger/hydrolysis sequence to form cyclohexadienone 72, which is then reduced in a reaction analogous to that performed by AflM. The produced alcohol 73 cannot simply dehydrate like 49 (Scheme 10) because the hydroxyl is methylated, but an alternative dehydration gives desmethylsulochrin 74. O-Methylation and aromatic halogenation is then followed by a final oxidative step to form (+)-geodin 64 (Scheme 13). The oxidative spirocycle-formation is catalysed by a rare blue-copper enzyme.45 All key features of this pathway have been supported by the recent work of Larsen, Mortensen and coworkers who identified a putative geodin biosynthetic gene cluster in Aspergillus nidulans which contains the expected aflN homologue (gedK = ATEG_08459).46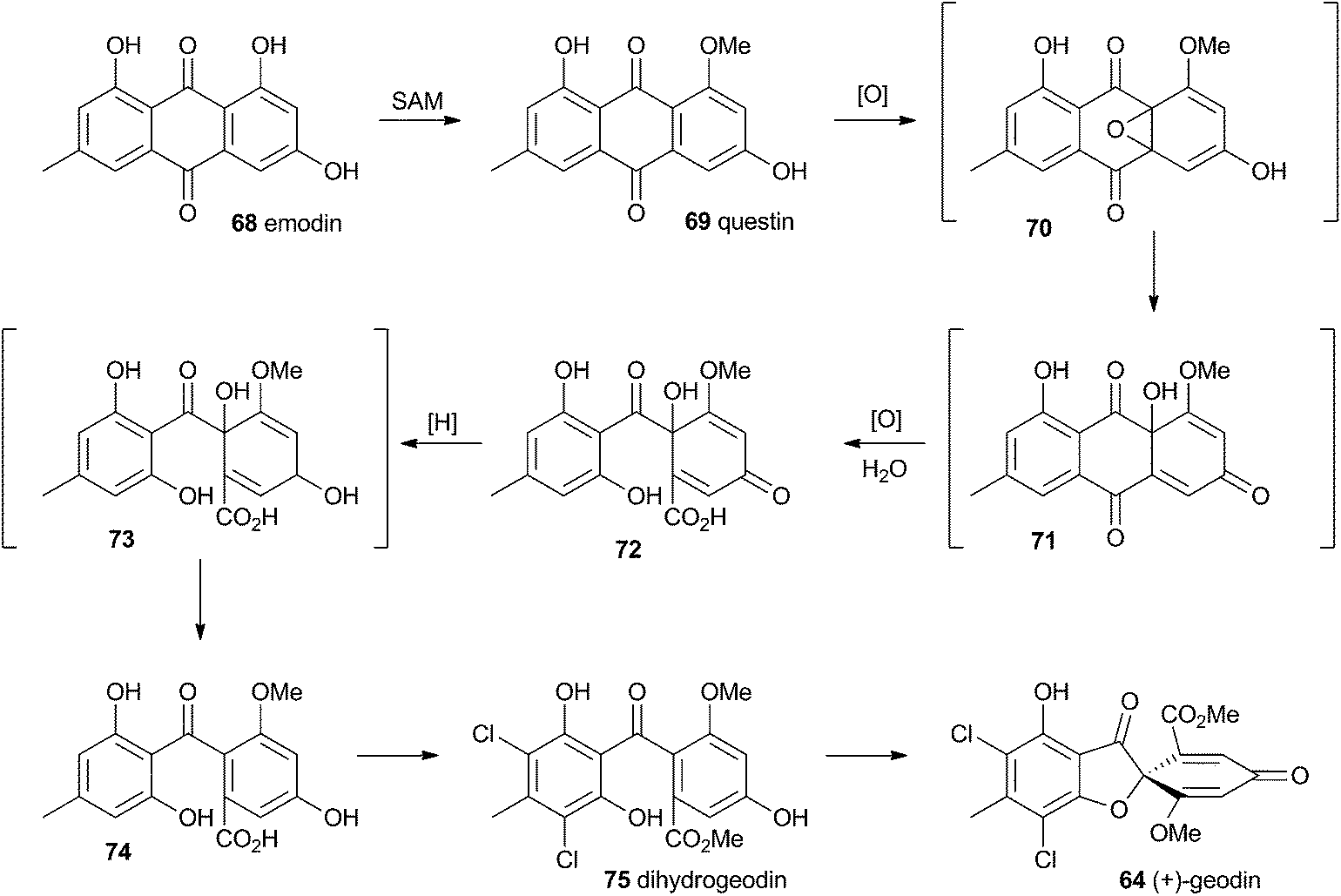 | ||
| Scheme 13 Biosynthesis of geodin 64 in Aspergillus nidulans. | ||
4.2 Griseofulvin
At first-glance the structure of griseofulvin 65 would suggest a biosynthetic pathway similar to that of geodin 64. However Tang and coworkers recently completely elucidated the biosynthetic pathway to 65 and showed that yet another oxidative process is used to form the core spirocycle – although oxidative dearomatisation once again plays a key role.47 In this case the carbon atoms are again provided by a polyketide synthase (GsfA), but rather than an anthraquinone, the PKS forms a benzophenone (griseophenone) skeleton 76 directly. This is doubly O-methylated and chlorinated, before a cytochrome P450 enzyme, GsfF, catalyses probable epoxidation, spirocycle formation and final dehydration to 81 (Scheme 14). Biosynthesis is then completed by a third O-methylation and reduction of the dienone.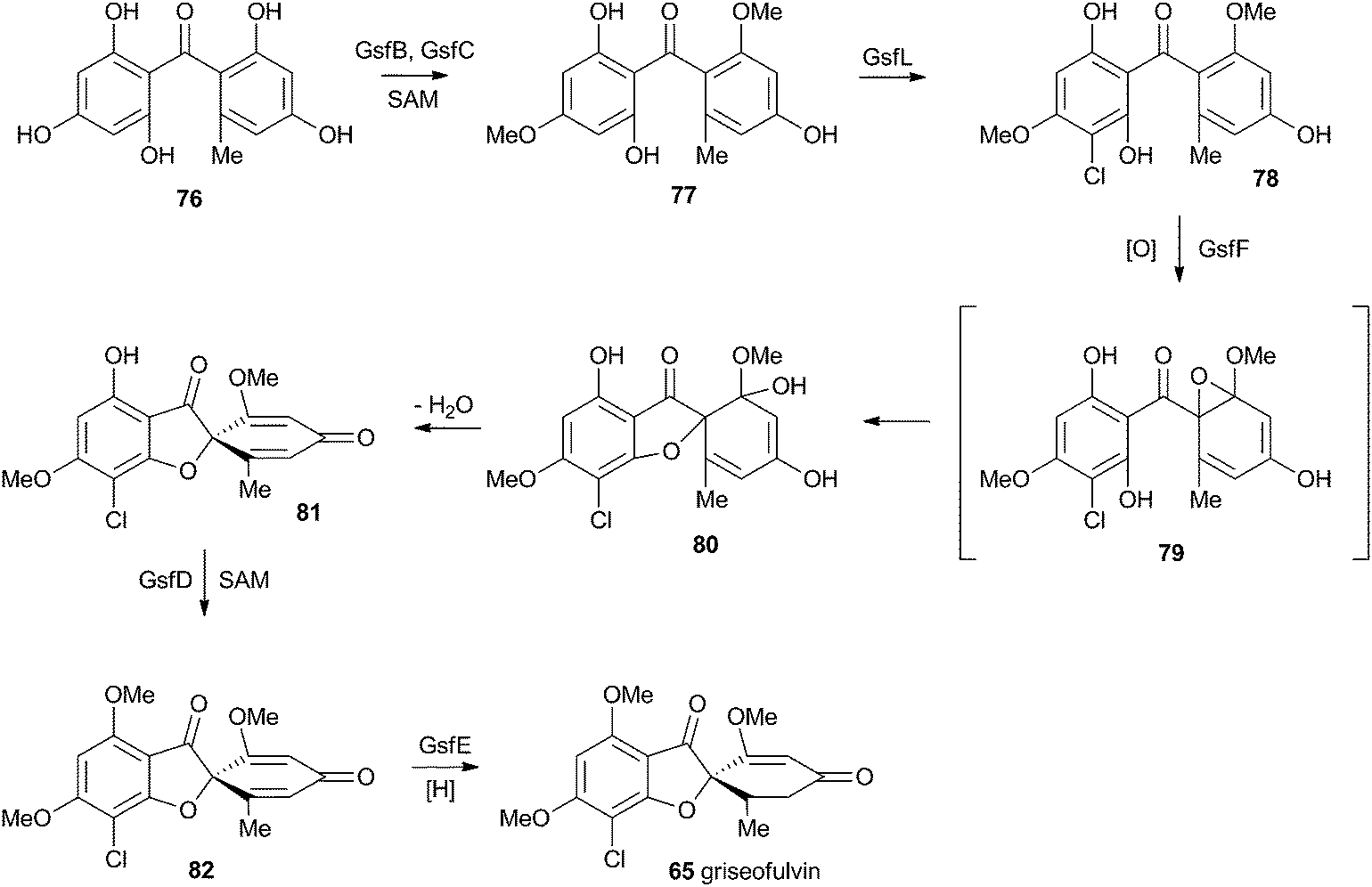 | ||
| Scheme 14 Biosynthesis of griseofulvin 65 in Penicillium aethiopicum. | ||
4.3 Austin and related compounds
In the case of austinol 66a, and related compounds such as the andibenins,48 extensive isotopic labelling studies, principally by the groups of Simpson and Vederas,49 revealed the meroterpenoid nature of the carbon skeleton. The labelling evidence shows that dimethylorsellinic acid, produced by a non-reducing fungal PKS from acetate and methionine, condenses with a sesquiterpenoid, and a typical oxidative cyclisation probably leads to the early intermediate 84a (Scheme 15). Oxidative rearrangement of the orsellinate-derived ring then sets up the D-ring of 85, ready for a Baeyer–Villiger type oxidation. The A-ring of 85 must also be rearranged, and subjected to another Baeyer–Villiger type oxidation.Genetic investigations in various aspergillus species have revealed the gene cluster(s) involved in the biosynthesis of austin 66b and related metabolites.50,51 Very recently Abe and coworkers have discovered the molecular details of the oxidative rearrangements during the construction of the key spirocyclic A-ring of austinol 66a.52 Protoaustinoid A 86 was supplemented to cultures of Aspergillus oryzae in which various oxidative genes from the biosynthetic cluster had been heterologously expressed. In a control reaction it was shown that 86 was not transformed by any endogenous A. oryzae enzymes, but incubation with A. oryzae/ausB led to the formation of 87 (Scheme 15). Addition of ausE resulted in the production of the shunt metabolite austinoid C 92, but further addition of ausC led to formation of preaustinoid A3 91. The reconstructed pathway thus appears to involve initial 5′ hydroxylation (AusB), followed by oxidation of the 3-hydroxyl to a ketone (AusE). AusC then catalyses a Baeyer–Villiger oxidation to give preaustinoid A1 89, but this is then followed by two more oxidations catalysed by AusE which form the αβ-unsaturated system of preaustinoid A2 90 and then the spirocycle itself (Scheme 15). AusE encodes a non-heme iron oxygenase, and this is clearly multifunctional, catalysing 3 separate reactions around the A-ring.
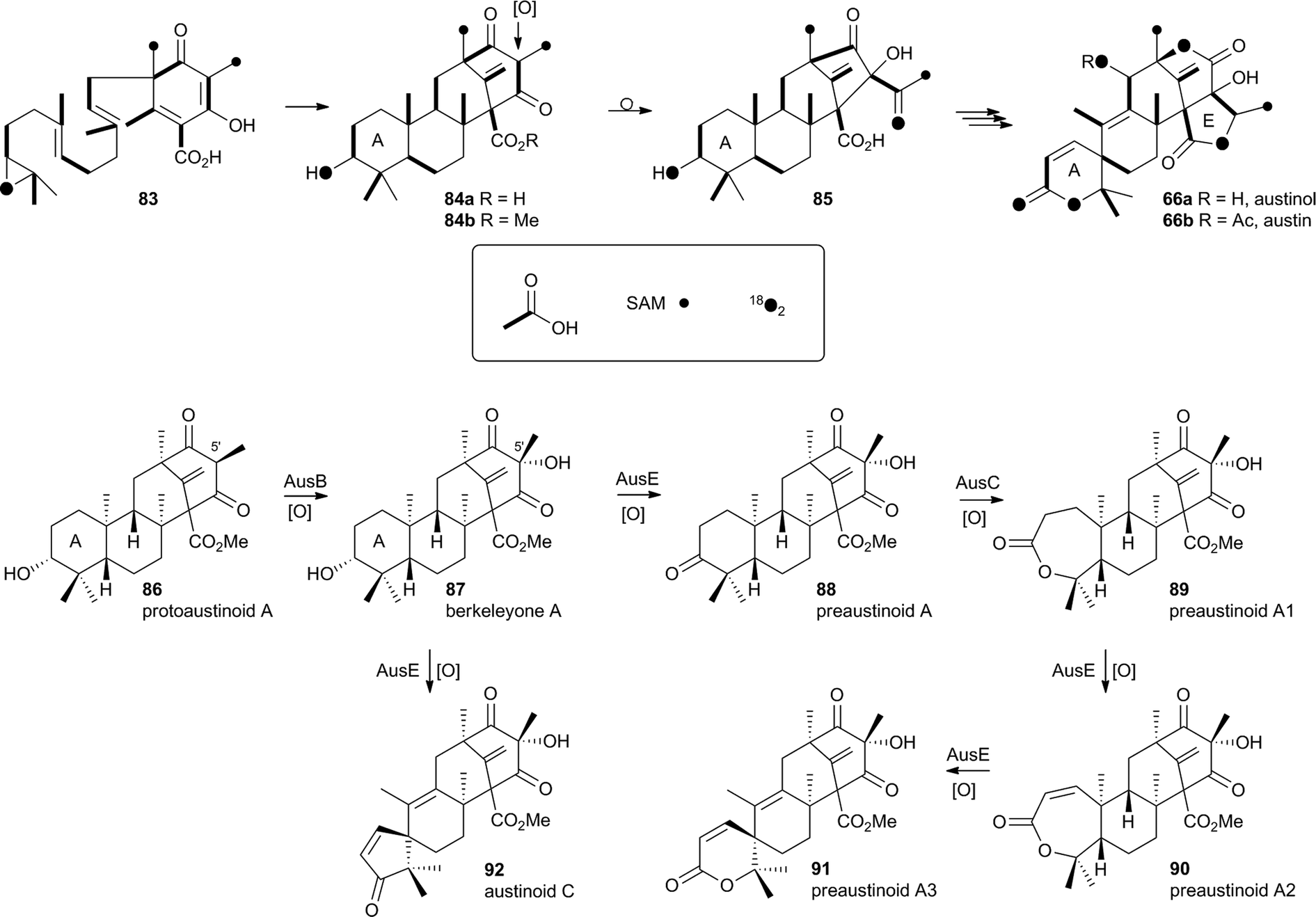 | ||
| Scheme 15 Origin of carbon and oxygen atoms during the synthesis of austinol 66a, and deduced chemical transformations during the oxidative rearrangement of the A-ring. | ||
The mechanism proposed for the spirocycle formation (Scheme 16) involves presumed hydrogen atom abstraction at the 5-position of preaustinoid A2 90, followed by top-face hydroxylation. Top-face hydroxylation is assumed because the oxidative iron-oxo center of AusE must be above the ring for it to successfully oxidise berkeleyone A 87 in the first reaction. Suprafacial elimination/bond migration are then proposed to cause the ring contraction (Scheme 16). This mechanism appears problematic because of stereoelectronic considerations regarding the syn-arrangement of the migrating bond and the oxy-iron leaving group. An alternative mechanism could involve initial rearrangement of the 5-radical via a methylene cyclopropane intermediate 95, followed by later rebound hydroxylation to form 97. This could then undergo allowed syn elimination to form 91 (Scheme 16). However, current evidence does not allow these mechanistic alternatives to be distinguished.
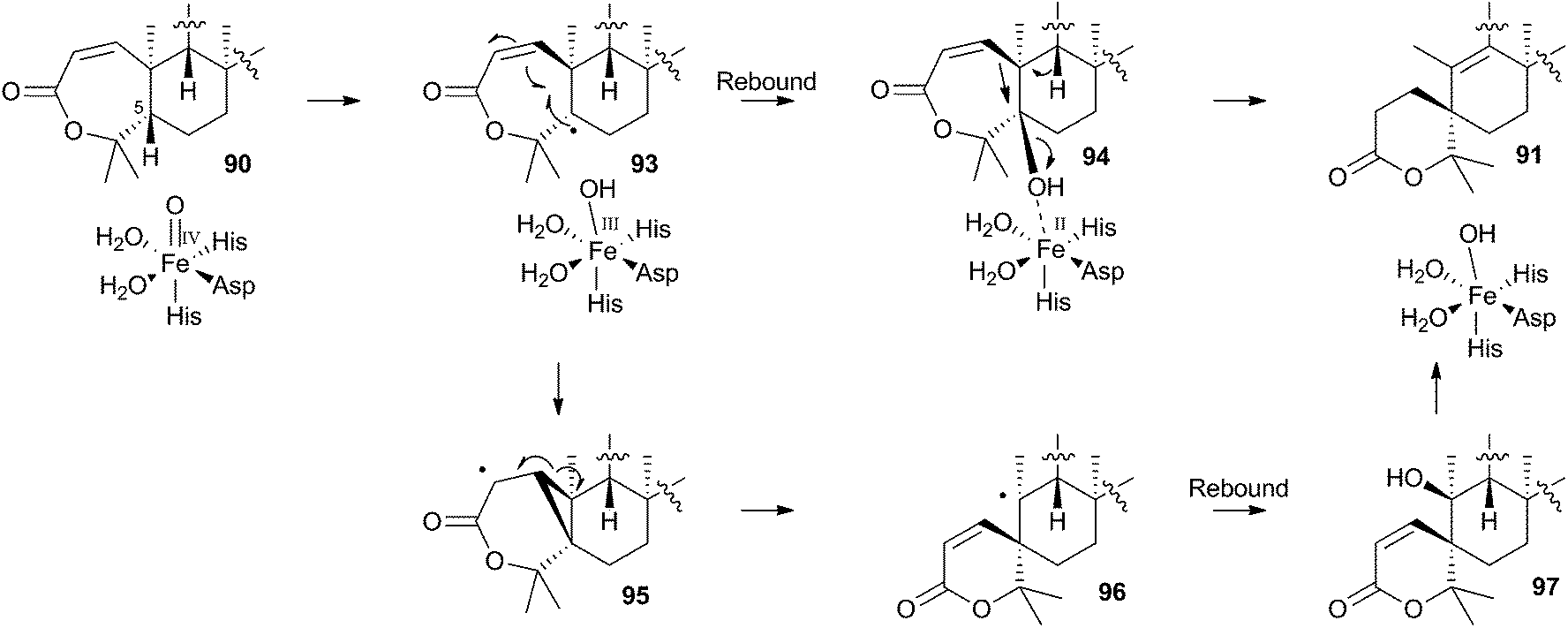 | ||
| Scheme 16 Possible mechanisms leading to the formation of the austinol A-ring. | ||
Finally, AusJ has been identified as the enzyme responsible for the ring contraction of the D-ring in the pathway. Directed knockout of ausJ gave preaustinoid A3 91 as the product,50 although further mechanistic studies have not yet been reported.
4.4 Prenylated indole alkaloids
Recent advances in genome sequencing technology have provided rapid access to the biosynthetic gene clusters responsible for the production of fungal prenylated indole alkaloids such as the notoamides, spiroprostatins, fumitremorgins and tryprostatins (Scheme 17). Genetic studies have been supported by in vitro assays which have delineated the activities of the enzymes responsible for the key oxidative rearrangements. The common intermediate in both pathways is the diketopiperazine brevianamide F 98 which is produced by a non-ribosomal peptide synthetase from L-tryptophan and L-proline.53 The key rearrangement reactions in the notoamide pathway were reported by Sherman, Williams and coworkers in 2012, while Watanabe and coworkers reported the related spirotryprostatin rearrangements in 2013.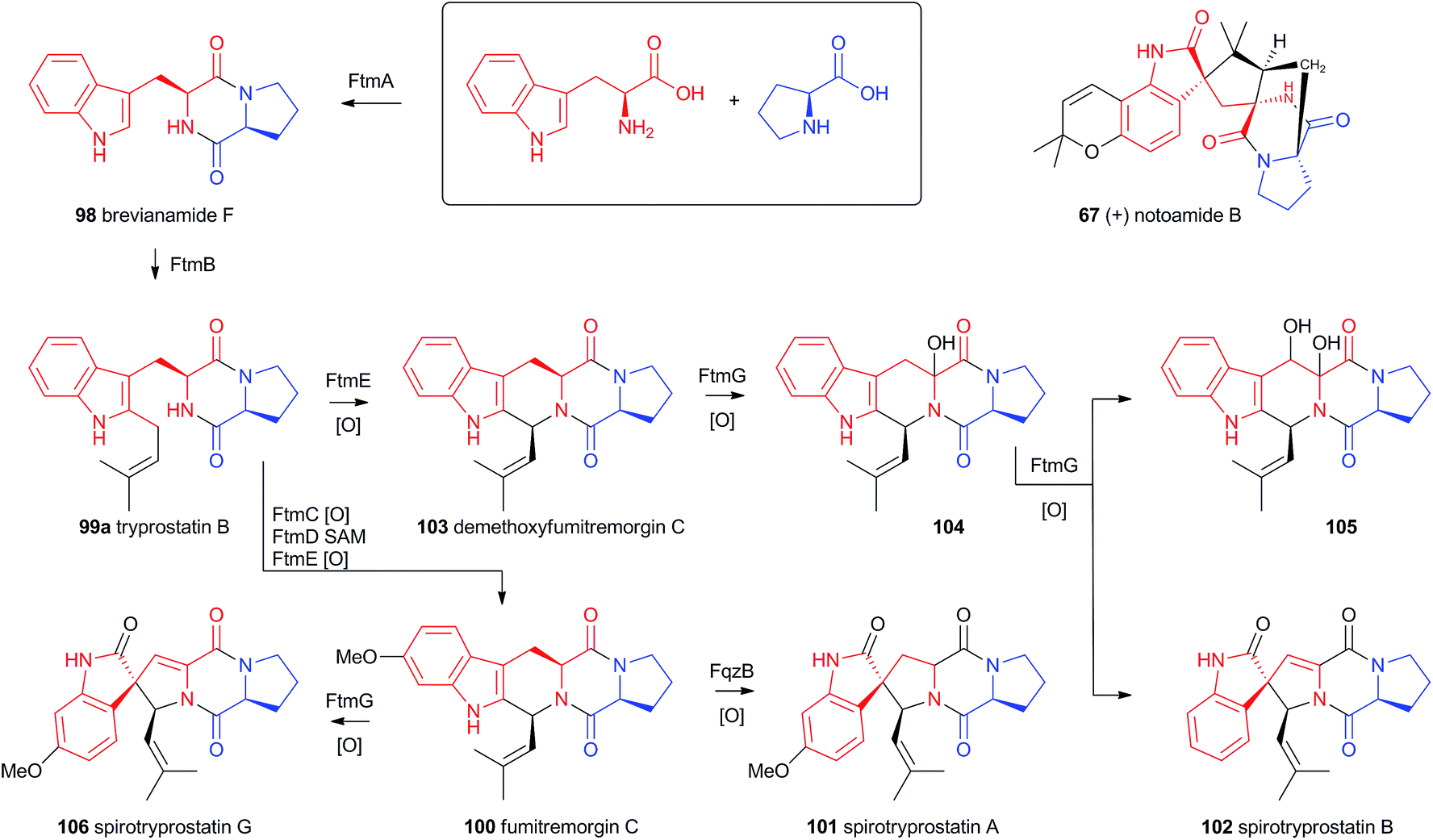 | ||
| Scheme 17 Compounds derived from brevianamide F and the spirotryprostatin pathways. | ||
Heterologous expression of five genes from the fumitremorgin gene cluster in Aspergillus niger (ftmA–E, encoding the NRPS, prenyltransferase, tryptophan hydroxylase, O-methyltransferase and piperidine synthase respectively) yielded fumitremorgin C 100 (Scheme 17). Intermediates for in vitro studies were produced by appropriate expression of combinations of the genes in yeast. Expression of ftmA–E plus the gene fqzB, which encodes an FAD-dependent monooxygenase, led to the production of spirotryprostatin A 101 (Scheme 17) indicating that this is the spirocycle forming enzyme.54 The mechanism is proposed to involve epoxidation of the indole 2,3-olefin, followed by pinacol-type rearrangement (Scheme 18A/B). In vitro, tryprostatin B 99a gave only dihydroxy products when it was incubated with FqzB, resulting from epoxide hydrolysis (Scheme 18A). However the p-methoxy analogue tryprostatin A 99b underwent a mixture of FqzB-catalyzed oxidative rearrangement and dihydroxylation showing that participation of the p-methoxy group is required for the rearrangement.
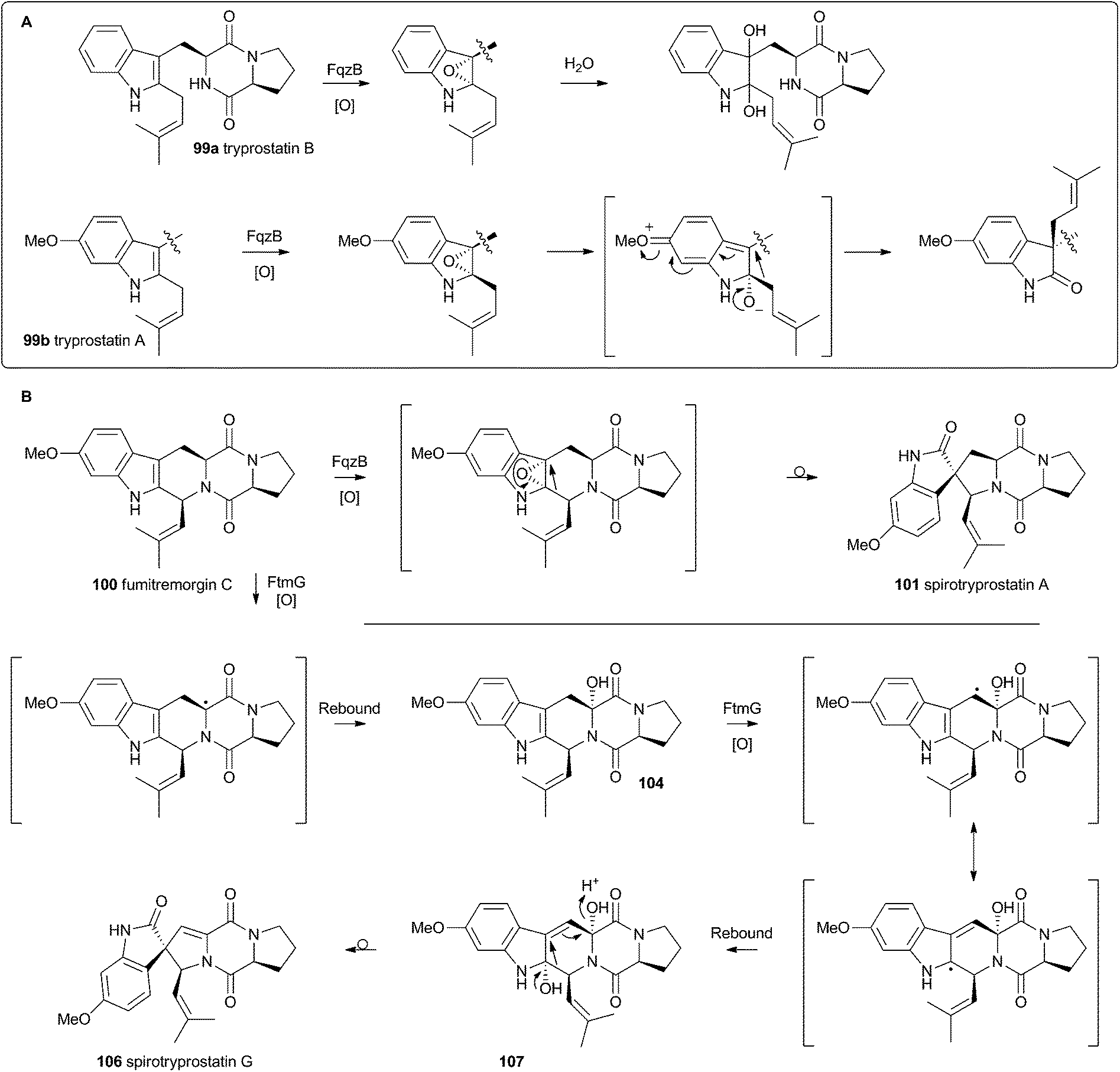 | ||
| Scheme 18 Proposed oxidative rearrangement mechanisms during spirotryprostatin biosynthesis. | ||
These results suggested that FqzB cannot be involved in the biosynthesis of spirotryprostatin B 102 which lacks a p-methoxy group. The gene ftmG (encoding a cytochrome P450 monooxygenase) was identified as providing the catalysis for this conversion: heterologous expression of ftmABE provided demethoxyfumitremorgin C 103 (Scheme 17), and addition of ftmG provided mono- and dihydroxylated shunt products 104 and 105, as well as the spirocyclic spirotryprostatin B 102 (Scheme 17). In this case the mechanism of rearrangement must be more complex, probably involving a double oxidation series, by the usual hydrogen atom abstraction/oxygen rebound route, before a concerted rearrangement and elimination (Scheme 18B). Interestingly, while FqzB is selective in that it requires the p-methoxy for correct reaction, FtmG can process both substrate series.
The notoamides differ from the spirotryprostatins in being doubly prenylated. Importantly C-2 alkylation, while occurring with the same regiochemistry with respect to the indole, occurs with inverted regioselectivity55 at the dimethylallyl unit and notoamide E 108 is the key intermediate in the pathway.56,57 Feeding studies have been performed which showed the intermediacy of a pinacol-type rearrangement.58 The notB gene encodes an FAD-dependent monooxygenase which has been identified at the catalyst for this step.59In vitro, NotB converts notoamide E 108 to notoamides C 113 and D 111 (Scheme 19A). The mechanisms of these transformations are again likely to involve epoxide intermediates, with the migration assisted by the para-oxygen. Interestingly these compounds are not precursors of the more complex family members such as notoamide B 67 – here the oxidative rearrangement occurs later. In this pathway the achiral azadiene intermediate 114 is believed to undergo intramolecular Diels–Alder reaction. Different fungi appear to possess different selectivities for this cyclo-addition and from this point enantiomeric pathways are known in different organisms.60 The pathway is thought to proceed via stephacidin A 115 which undergoes oxidative rearrangement catalysed by NotI (FAD monooxygenase) to give notoamide B 67 (Scheme 19B).
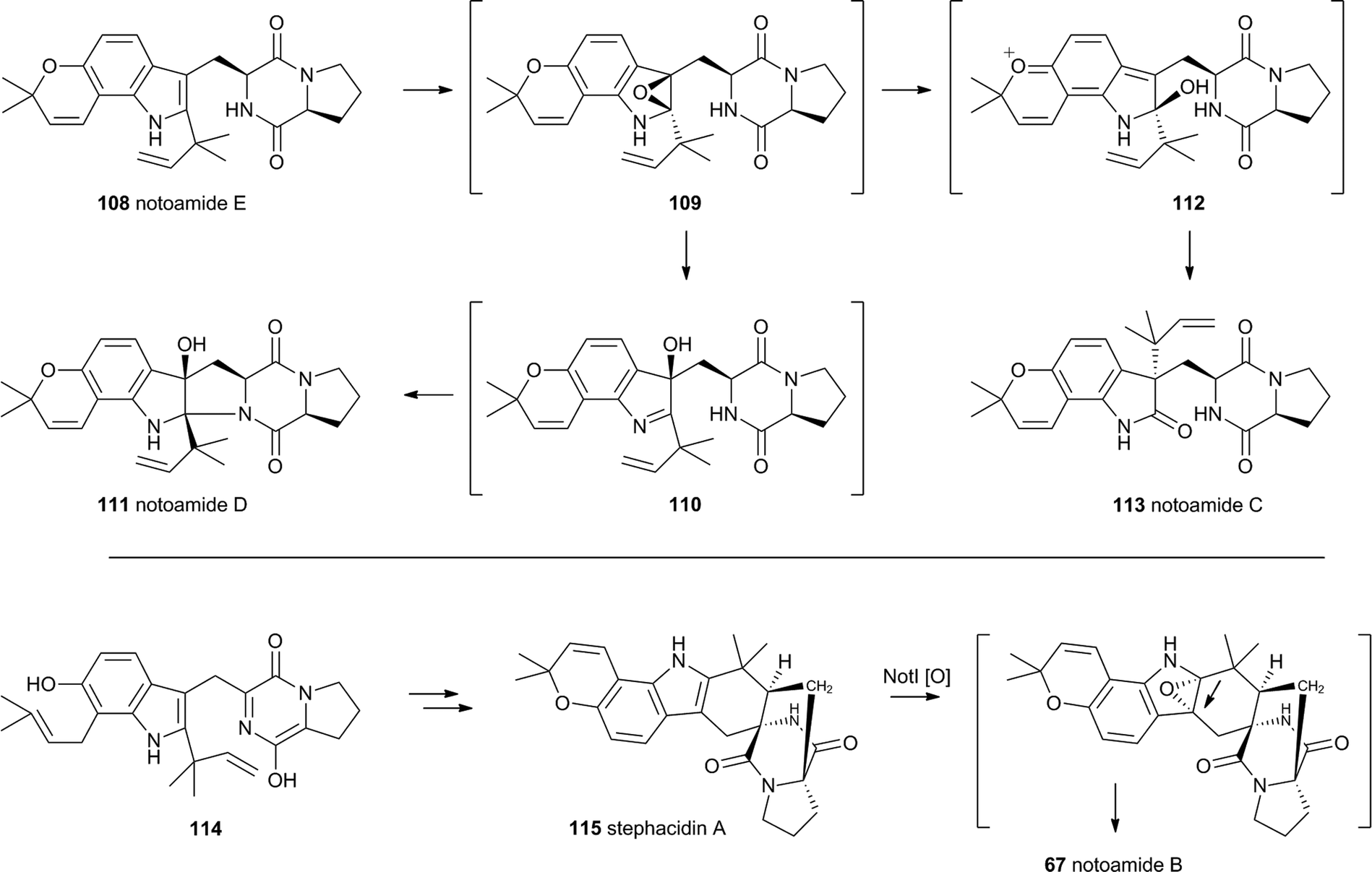 | ||
| Scheme 19 Oxidative rearrangements involved in notoamide biosynthesis. | ||
5 Tropolones and related systems
Tropolones are relatively common fungal secondary metabolites – indeed the first natural product discovered to be a tropolone was stipitatic acid 116 from Talaromyces stipitatus which was reported by Raistrick and coworkers in 1942,61 although the structure was not elucidated until Dewar's seminal publication of 1945.62 The biosynthesis has been extensively investigated using isotopic labels which showed that oxidative rearrangement of an aromatic polyketide derived from acetate and methionine formed the tropolone ring.63 However the molecular details of the pathway have been only recently elucidated.64Xenovulene A 117 (Scheme 20) from the fungus Acremonium strictum is a potent antagonist of the GABAA receptor.65 Isotopic labelling studies showed it is derived from a methylated polyketide fused to humulene. Simpson and coworkers suggested a route involving oxidative ring expansion to a tropolone, followed by sequential ring contractions to 117 (Scheme 20).66 Although a full genome sequence of the producing strain is not yet available, a traditional genome library-probing approach afforded a ca 22 Kb genomic dna fragment containing genes encoding a non-reducing PKS (aspks1), an FAD-dependent monooxygenase, a non-heme iron monooxygenase and a cytochrome P450 monooxygenase. Expression of aspks1 showed it produced methylorcinaldehyde 129, but further elucidation of the xenovulene gene cluster was not possible because of the difficulty in obtaining targeted knockouts.67
 | ||
| Scheme 20 Proposed biosynthesis of xenovulene A 117via a tropolone intermediate and consecutive ring contractions. | ||
However, knowledge of the sequence of the xenovulene PKS allowed the sequenced genome of T. stipitatus to be mined. Although four T. stipitatus PKS genes are closely related to aspks1, only one (tropA) is clustered with a similar array of monooxygenases. Targeted knockout of tropA proved it was involved in the biosynthesis of stipitatic acid 116, and heterologous expression of tropA showed it encoded the synthesis of methylorcinaldehyde 129 (Scheme 21).68 The next catalyst in the sequence is TropB which is an FAD-dependent monooxygenase which oxidatively dearomatises the ring to afford the cyclohexadione 130. Once the ring is dearomatised it is then subject to a second oxidation catalysed by TropC, a non-heme iron monooxygenase. This enzyme must oxidise the methyl group and possible mechanisms could include a 1-electron rearrangement pathway, followed by oxygen rebound and elimination, or a hydroxylation, followed by a two-electron rearrangement and iron assisted elimination (Scheme 21). TropC thus provides stipitaldehyde 134 as the first tropolone. A cascade of further oxidations to form the maleic anhydride of stipitatonic acid 136 is then set in train by TropD, a cytochrome P450 monooxygenase. Discovery of the activity of TropB revealed that oxidative dearomatisation also lies behind the biosynthesis of other well-known fungal metabolites including the sorbicillinoids69 and the azaphilones.70
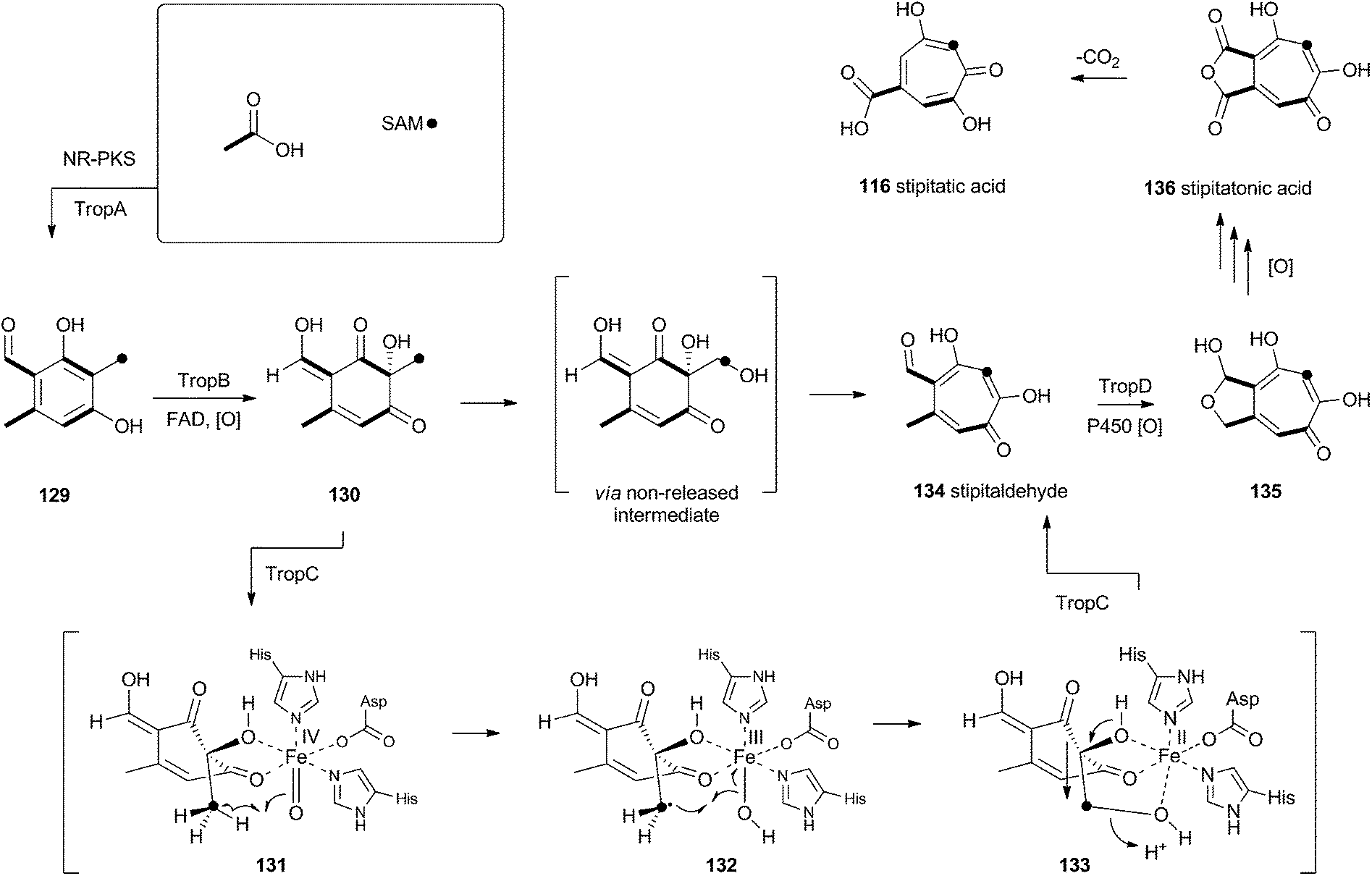 | ||
| Scheme 21 Biosynthesis of tropolones. | ||
6 2-Pyridones
Acyl tetramic acids are common fungal metabolites, derived from amino acids and polyketides. Fungal 2-pyridones, such as tenellin 137 (Scheme 22), are also derived from amino acids and polyketides. Leete, Vining, Wright and coworkers showed through elegant double 13C labelling experiments that in the case of tenellin the amino acid undergoes intramolecular rearrangement, but that this could occur either before, or after linkage to the polyketide.71 O'Hagan, Cox and coworkers showed that the rearrangement likely occurs after linkage of the amino acid and polyketide, implicating the tetramic acid pretenellin A 138a as an intermediate.72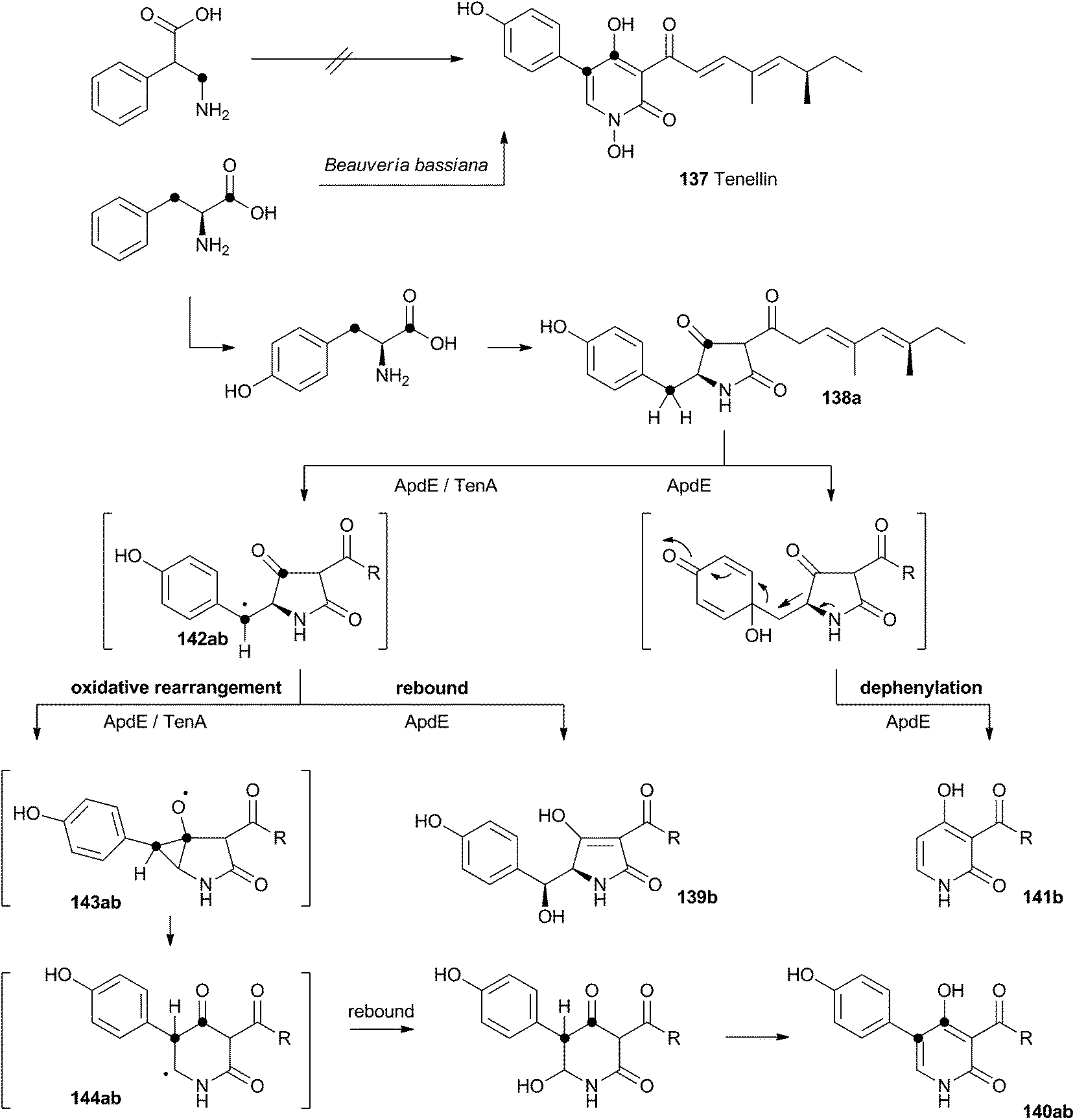 | ||
| Scheme 22 Oxidative rearrangements during the biosynthesis of 2-pyridones. (a) (Tenellin) series: R = CHCHC(Me)CHCH(Me)CH2Me; (b) (aspyridone) series R = CH(Me)CH2CH(Me)CH2Me. | ||
More recently, cloning of the tenellin biosynthetic gene cluster73 has revealed that the iterative PKS–NRPS hybrid TenS synthesises pretenellin A 138a.74 This then undergoes clean oxidative rearrangement catalysed by the cytochrome P450 monooxygenase TenA to form pretenellin B 140a.75 The related compound aspyridone is produced in a similar fashion – the hybrid PKS–NRPS ApdS produces the tetramic acid preaspyridone A 138b which undergoes oxidative ring expansion catalysed by ApdE.76 However, in this case ApdE catalysis leads to the formation of three compounds. It appears that while ApdE can catalyse the same ring expansion as TenA giving 140b, it can also produce two other compounds. The first of these is the benzylic alcohol 139b which presumably arises through simple hydroxylation, while the second compound 141b is dephenylated.
These observations are consistent with a mechanism in which the cytochrome P450 initially abstracts a hydrogen atom to give a benzylic radical 142ab. This may be long-lived enough to rearrange via an oxy-cyclopropane radical intermediate 143ab. The final step may be oxygen rebound followed by elimination to form the aromatic pyridone ring. In the case of 139b, simple early oxygen rebound can give the alcohol. A related oxygenation at the ring quaternary carbon would lead to a compound which could undergo a concomitant ring expansion and dephenylation. Further details about the mechanisms of TenA and ApdE have not been elucidated since the proteins cannot yet be obtained in soluble form.
7 Strobilurins
Strobilurins (and the closely-related mucidins and oudemansins) such as strobilurin A 145 are potent antifungal compounds produced by basidiomycetes. Numerous compounds are known, but all feature the unusual β-methoxyacrylate moiety – and this has been shown to be the pharmacophore.77 Many commercially valuable fungicides have been produced based on these structures and the class has become prevalent worldwide in agriculture.78 Labelling studies have shown that strobilurin A 145 is composed of a polyketide, using an unusual (for fungi) phenylalanine-derived starter unit.79 The β-methoxyacrylate appears to be derived by an oxidative rearrangement of a putative polyunsaturated polyketide precursor (Scheme 23), followed by O-methylation. The mechanism may involve epoxidation, followed by pinacol-type rearrangement, however no further details are currently available.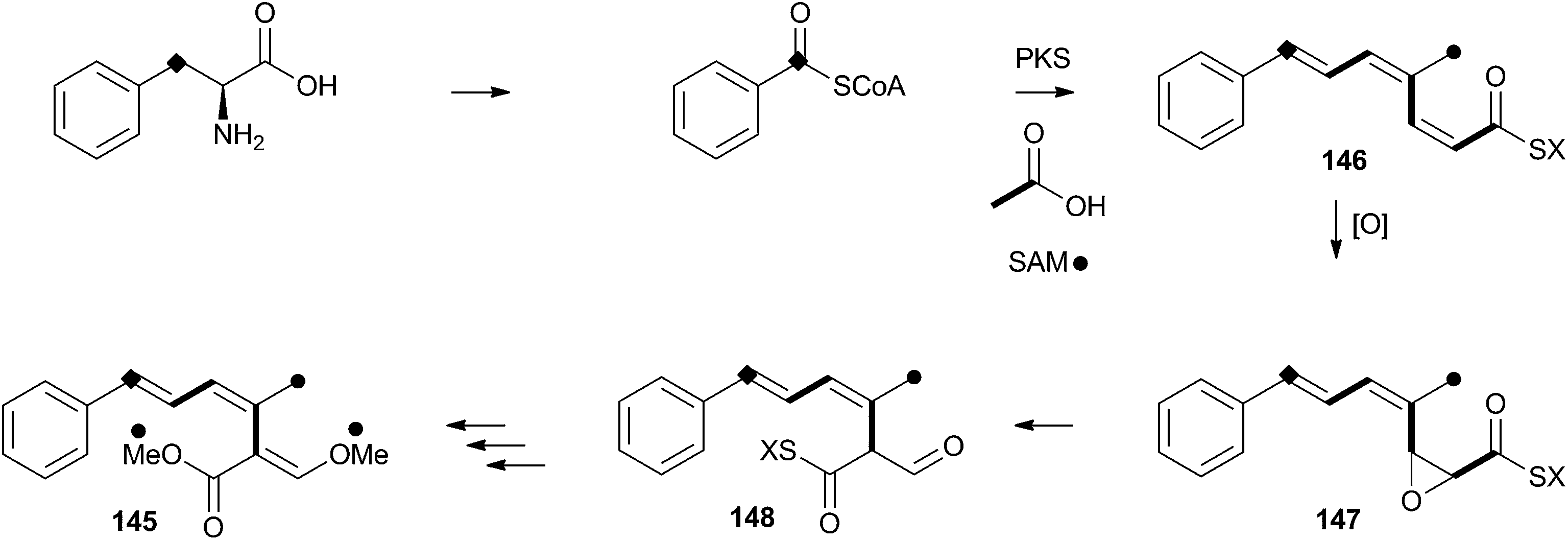 | ||
| Scheme 23 Proposed oxidative rearrangement during the biosynthesis of strobilurin A 145. | ||
8 Perspective
Oxidative rearrangements are found in all classes of fungal secondary metabolites: polyketides (e.g. tropolones, Section 5.0), peptides (e.g. β-lactams, Section 2.0), terpenes (e.g. austin, Section 4.3) and alkaloids (e.g. notoamides, Section 4.4) are all known substrates. The reactions are catalysed in the main by cytochrome P450 and non-heme iron oxygenases, but participation by FAD-dependent oxygenases is also known. Many of the reactions occur on aromatic substrates, and oxidative dearomatisation is a key theme in many reactions where the stability of the aromatic system is broken to enable the rearrangement. These reactions probably occur by epoxidation of the ring (evidenced in some cases by observation of NIH shifts) and often occur in rings which are already electron-rich (e.g. sterigmatocystin biosynthesis, Scheme 10). In many other cases the reactions appear to be initiated by hydrogen atom abstraction, as in the classical cases of cephalosporin biosynthesis (e.g.Scheme 6). In these reactions formation of a radical allows rearrangement through adjacent double bonds or heteroatoms, probably followed by late oxygen rebound and elimination (e.g. Scheme 9C) – direct electron transfer as observed in the cephalosporin case appears to be rare. It is mechanistically difficult to distinguish various possible reaction manifolds since it is often the case that intermediates cannot be isolated or easily observed, and a key linking theme is that one- and two-electron rearrangement pathways are difficult to discriminate. For example in the cases of hydroxyversicolorone 27, model reactions in vitro suggest a cationic reaction manifold instead of a radical pathway, while conversely genetic data suggest a P450 (and thus possible radical) route. In the case of cephalosporin 6 biosynthesis, in vitro model reactions show that both radical and cationic pathways could be possible. In the absence of definitive data in many of these systems it may be better to envisage a continuum of possible mechanisms from cationic routes at one extreme, to radical pathways at the other. What is certain is that the electron deficiency caused by the oxidation is a necessary requirement for the rearrangement itself.Early investigation of the fungal cephalosporin synthase showed that it catalyses sequential oxidations and again this is often the case – AusE catalyses 3 sequential oxidations including alcohol to ketone, desaturation and ring contraction reactions, during the biosynthesis of austinol 66a. Finally, the range of chemistry precipitated by these enzymes is highly diverse, and it remains extremely challenging to predict the chemical rearrangements from genome sequence data – but rapidly increasing knowledge about many diverse fungal pathways is making such predictions easier and it seems highly likely that oxidative rearrangements will find a place in the toolbox of synthetic biology as they offer rapid routes to highly complex natural products which are still significantly challenging for synthetic chemistry.
9 Notes and references
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, C. Ducho, M. A. McDonough and C. J. Schofield, Nat. Prod. Rep., 2013, 30, 21–107 RSC.
- M. Kohsaka and A. L. Demain, Biochem. Biophys. Res. Commun., 1976, 70, 465–473 CrossRef CAS.
- D. J. Hook, L. T. Chang, R. P. Elander and R. B. Morin, Biochem. Biophys. Res. Commun., 1979, 87, 258–265 CrossRef CAS.
- Y. Sawada, N. A. Hunt and A. L. Demain, J. Antibiot., 1979, 32, 1303–1310 CrossRef CAS.
- J. E. Baldwin, J. W. Keeping, P. D. Singh and C. L. Vallejo, Biochem. J., 1981, 194, 649–651 CAS.
- J. E. Baldwin, J. Löliger, W. Rastetter, N. Neuss, L. L. Huckstep and N. De La Higuera, J. Am. Chem. Soc., 1973, 95, 3796–3797 CrossRef CAS.
- N. Neuss, C. H. Nash, J. E. Baldwin, P. A. Lemke and J. B. Grutzner, J. Am. Chem. Soc., 1973, 95, 3797–3798 CrossRef CAS.
- H. Kluender, C. H. Bradley, C. J. Sih, P. Fawcett and E. P. Abraham, J. Am. Chem. Soc., 1973, 95, 6149–6150 CrossRef CAS.
- C. A. Townsend, A. S. Neese and A. B. Theis, J. Chem. Soc., Chem. Commun., 1982, 116 RSC.
- C. A. Townsend, A. B. Theis, A. S. Neese, E. B. Barrabee and D. Poland, J. Am. Chem. Soc., 1985, 107, 4760–4767 CrossRef CAS.
- C. A. Townsend, J. Nat. Prod., 1985, 48, 708–724 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, C. J. Schofield, N. J. Turner and R. T. Aplin, Tetrahedron, 1991, 47, 9881–9900 CrossRef CAS; J. E. Baldwin, R. M. Adlington, R. T. Aplin, N. P. Crouch, G. Knight and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1987, 1651–1654 RSC.
- R. B. Morin, B. G. Jackson, R. A. Mueller, E. R. Lavagnino, W. B. Scanlon and S. L. Andrews, J. Am. Chem. Soc., 1963, 85, 1896–1897 CrossRef CAS; R. B. Morin, B. G. Jackson, R. A. Mueller, E. R. Lavagnino, W. B. Scanlon and S. L. Andrews, J. Am. Chem. Soc., 1969, 91, 1401–1407 CrossRef.
- J. E. Baldwin, R. M. Adlington, T. W. Kang, E. Lee and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1987, 104 RSC.
- K. Valegard, A. van Scheltinga, M. D. Lloyd, T. Hara, S. Ramaswamy, A. Perrakis, A. Thompson, H. J. Lee, J. E. Baldwin, C. J. Schofield, J. Hajdu and I. Andersson, Nature, 1998, 394, 805–809 CrossRef CAS PubMed.
- L. M. Öster, A. C. T. van Scheltinga, K. Valegård, A. M. Hose, A. Dubus, J. Hajdu and I. Andersson, J. Mol. Biol., 2003, 343, 157–171 CrossRef PubMed.
- K. Valegård, A. C. T. van Scheltinga, A. Dubus, G. Ranghino, L. M. Öster, J. Hajdu and I. Andersson, Nat. Struct. Mol. Biol., 2003, 11, 95–101 Search PubMed.
- K. Pachler, P. Steyn, R. Vleggaar, P. Wessels and D. Scott, J. Chem. Soc., Perkin Trans. 1, 1976, 1182–1195 RSC.
- G. Feng, F. Chu and T. Leonard, Appl. Environ. Microbiol., 1992, 58, 455–460 CAS.
- R. Minto and C. Townsend, Chem. Rev., 1997, 97, 2537–2555 CrossRef CAS PubMed.
- J. C. Vederas and T. T. Nakashima, J. Chem. Soc., Chem. Commun., 1980, 183 RSC.
- T. J. Simpson, A. E. de Jesus, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1982, 631 RSC.
- C. A. Townsend, S. B. Christensen and S. G. Davis, J. Am. Chem. Soc., 1982, 104, 6152–6153 CrossRef CAS.
- C. A. Townsend, S. B. Christensen and S. G. Davis, J. Am. Chem. Soc., 1982, 104, 6154–6155 CrossRef CAS.
- C. A. Townsend and S. B. Christensen, J. Am. Chem. Soc., 1985, 107, 270–271 CrossRef CAS.
- C. A. Townsend, S. B. Christensen and S. G. Davis, J. Chem. Soc., Perkin Trans. 1, 1988, 839–861 RSC.
- C. A. Townsend, S. G. Davis, M. Koreeda and B. Hulin, J. Org. Chem., 1985, 50, 5428–5430 CrossRef CAS.
- C. A. Townsend, Y. Isomura, S. G. Davis and J. A. Hodge, Tetrahedron, 1989, 45, 2263–2276 CrossRef CAS.
- Y. Wen, H. Hatabayashi, H. Arai, H. K. Kitamoto and K. Yabe, Appl. Environ. Microbiol., 2005, 71, 3192–3198 CrossRef CAS PubMed.
- J. T. Groves and G. A. Mcclusky, J. Am. Chem. Soc., 1976, 98, 859–861 CrossRef CAS.
- D. Kingston, P. N. Chen and J. R. Vercellotti, Phytochemistry, 1976, 15, 1037–1039 CrossRef CAS.
- S. M. McGuire and C. A. Townsend, Bioorg. Med. Chem. Lett., 1993, 3, 653–656 CrossRef CAS.
- N. P. Keller, C. M. Watanabe, H. S. Kelkar, T. H. Adams and C. A. Townsend, Appl. Environ. Microbiol., 1999, 66, 359–362 CrossRef.
- K. M. Henry and C. A. Townsend, J. Am. Chem. Soc., 2005, 127, 3300–3309 CrossRef CAS PubMed.
- K. M. Henry and C. A. Townsend, J. Am. Chem. Soc., 2005, 127, 3724–3733 CrossRef CAS PubMed.
- B. Franck, V. Radtke and U. Zeidler, Angew. Chem., Int. Ed. Engl., 1967, 6, 952–953 CrossRef CAS.
- U. Sankawa, H. Shimada, T. Kobayashi, Y. Ebizuka, Y. Yamamoto, H. Noguchi and H. Seto, Heterocycles, 1982, 19, 1053–1058 CrossRef CAS.
- T. J. Simpson, A. E. de Jesus, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1983, 338 RSC.
- M. Chatterjee and C. A. Townsend, J. Org. Chem., 1994, 59, 4424–4429 CrossRef CAS.
- C. Watanabe and C. A. Townsend, J. Org. Chem., 1996, 61, 1990–1993 CrossRef CAS.
- R. Prieto and C. P. Woloshuk, Appl. Environ. Microbiol., 1997, 63, 1661–1666 CAS.
- D. W. Udwary, L. K. Casillas and C. A. Townsend, J. Am. Chem. Soc., 2002, 124, 5294–5303 CrossRef CAS PubMed.
- H. Zeng, H. Hatabayashi, H. Nakagawa, J. Cai, R. Suzuki, E. Sakuno, T. Tanaka, Y. Ito, K. C. Ehrlich, H. Nakajima and K. Yabe, Appl. Microbiol. Biotechnol., 2011, 90, 635–650 CrossRef CAS PubMed.
- Z. G. Chen, I. Fujii, Y. Ebizuka and U. Sankawa, Arch. Microbiol., 1992, 158, 29–34 CrossRef CAS.
- K. X. Huang, I. Fujii, Y. Ebizuka, K. Gomi and U. Sankawa, J. Biochem., 1995, 270, 21495–21502 CAS; I. Fujii, H. Iijima, S. Tsukita, Y. Ebizuka and U. Sankawa, J. Biochem., 1987, 101, 11–18 Search PubMed.
- M. T. Nielsen, J. B. Nielsen, D. C. Anyaogu, D. K. Holm, K. F. Nielsen, T. O. Larsen and U. H. Mortensen, PLoS One, 2013, 8, e72871 CAS.
- R. A. Cacho, Y.-H. Chooi, H. Zhou and Y. Tang, ACS Chem. Biol., 2013, 8, 2322–2330 CrossRef CAS PubMed.
- T. J. Simpson, S. A. Ahmed, C. R. McIntyre, F. E. Scott and I. H. Sadler, Tetrahedron, 1997, 53, 4013–4034 CrossRef CAS.
- S. A. Ahmed, F. E. Scott, D. J. Stenzel, T. J. Simpson, R. N. Moore, L. A. Trimble, K. Arai and J. C. Vederas, J. Chem. Soc., Perkin Trans. 1, 1989, 807 RSC.
- C.-J. Guo, B. P. Knox, Y.-M. Chiang, H.-C. Lo, J. F. Sanchez, K.-H. Lee, B. R. Oakley, K. S. Bruno and C. C. C. Wang, Org. Lett., 2012, 14, 5684–5687 CrossRef CAS PubMed; A. B. Rodríguez-Urra, C. Jimenez, M. I. Nieto, J. Rodríguez, H. Hayashi and U. Ugalde, ACS Chem. Biol., 2012, 7, 599–606 CrossRef PubMed; M. L. Nielsen, J. B. Nielsen, C. Rank, M. L. Klejnstrup, D. K. Holm, K. H. Brogaard, B. G. Hansen, J. C. Frisvad, T. O. Larsen and U. H. Mortensen, FEMS Microbiol. Lett., 2011, 321, 157–166 CrossRef PubMed.
- H.-C. Lo, R. Entwistle, C.-J. Guo, M. Ahuja, E. Szewczyk, J.-H. Hung, Y.-M. Chiang, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 4709–4720 CrossRef CAS PubMed.
- Y. Matsuda, T. Awakawa, T. Wakimoto and I. Abe, J. Am. Chem. Soc., 2013, 135, 10962–10965 CrossRef CAS PubMed.
- S. Maiya, A. Grundmann, S.-M. Li and G. Turner, ChemBioChem, 2006, 7, 1062–1069 CrossRef CAS PubMed.
- Y. Tsunematsu, N. Ishikawa, D. Wakana, Y. Goda, H. Noguchi, H. Moriya, K. Hotta and K. Watanabe, Nat. Chem. Biol., 2013, 9, 818–825 CrossRef CAS PubMed.
- Y. Ding, J. R. de Wet, J. Cavalcoli, S. Li, T. J. Greshock, K. A. Miller, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, S. Tsukamoto, R. M. Williams and D. H. Sherman, J. Am. Chem. Soc., 2010, 132, 12733–12740 CrossRef CAS PubMed.
- S. Tsukamoto, H. Kato, T. J. Greshock, H. Hirota, T. Ohta and R. M. Williams, J. Am. Chem. Soc., 2009, 131, 3834–3835 CrossRef CAS PubMed.
- J. M. Finefield, T. J. Greshock, D. H. Sherman, S. Tsukamoto and R. M. Williams, Tetrahedron Lett., 2011, 52, 1987–1989 CrossRef CAS PubMed.
- H. Kato, Y. Nakamura, J. M. Finefield, H. Umaoka, T. Nakahara, R. M. Williams and S. Tsukamoto, Tetrahedron Lett., 2011, 52, 6923–6926 CrossRef CAS PubMed.
- S. Li, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, R. M. Williams and D. H. Sherman, J. Am. Chem. Soc., 2012, 134, 788–791 CrossRef CAS PubMed.
- S. Li, K. Srinivasan, H. Tran, F. Yu, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, S. Tsukamoto, R. M. Williams and D. H. Sherman, Med. Chem. Commun., 2012, 3, 987 RSC.
- J. Birkinshaw, A. Chambers and H. Raistrick, Biochem. J., 1942, 36, 242–251 CAS.
- M. Dewar, Nature, 1945, 155, 50–51 CrossRef CAS.
- M. O'Sullivan and J. Schwab, Bioorg. Chem., 1995, 23, 131–143 CrossRef.
- R. J. Cox and A. Al Fahad, Curr. Opin. Chem. Biol., 2013, 17, 532–536 CrossRef CAS PubMed.
- A. Ainsworth, M. Chicarelli-Robinson, B. Copp, U. Fauth, P. Hylands, J. Holloway, M. Latif, G. Obeirne, N. Porter, D. Renno, M. Richards and N. Robinson, J. Antibiot., 1995, 48, 568–573 CrossRef CAS.
- M. Raggatt and M. Chicarelli-Robinson, Chem. Commun., 1997, 2245–2246 RSC.
- A. M. Bailey, T. J. Simpson, R. J. Cox, K. Harley and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC; A. M. Bailey, K. M. Fisch, E. Skellam, D. Ivison, R. J. Cox, C. M. Lazarus and T. J. Simpson, Chem. Commun., 2010, 46, 5331–5333 RSC.
- J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS PubMed.
- A. Al Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2013, 5, 523–527 RSC.
- A. O. Zabala, W. Xu, Y.-H. Chooi and Y. Tang, Chem. Biol., 2012, 19, 1049–1059 CrossRef CAS PubMed.
- E. Leete, N. Kowanko, R. Newmark, L. Vining, A. Mcinnes and J. Wright, Tetrahedron Lett., 1975, 4103–4106 CrossRef CAS.
- R. J. Cox and D. O’Hagan, J. Chem. Soc., Perkin Trans. 1, 1991, 2537–2540 RSC; M. Moore, R. J. Cox, G. Duffin and D. O'Hagan, Tetrahedron, 1998, 54, 9195–9206 CrossRef CAS.
- K. L. Eley, T. J. Simpson, A. M. Bailey, L. M. Halo, Z. Song, H. Powles, R. J. Cox and A. M. Bailey, ChemBioChem, 2007, 8, 289–297 CrossRef CAS PubMed.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS PubMed.
- L. M. Halo, M. N. Heneghan, A. A. Yakasai, Z. Song, K. Williams, A. M. Bailey, R. J. Cox, C. M. Lazarus and T. J. Simpson, J. Am. Chem. Soc., 2008, 130, 17988–17996 CrossRef CAS PubMed.
- Z. Wasil, K. A. K. Pahirulzaman, C. Butts, T. J. Simpson, C. M. Lazarus and R. J. Cox, Chem. Sci., 2013, 4, 3845 RSC.
- J. Clough, Nat. Prod. Rep., 1993, 10, 565–574 RSC.
- H. Sauter, W. Steglich and T. Anke, Angew. Chem., Int. Ed., 1999, 38, 1329–1349 CrossRef CAS.
- F. Nerud, P. Sedmera, Z. Zouchová, V. Musílek and M. Vondráček, Collect. Czech. Chem. Commun., 1982, 47, 1020–1025 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |